 ,1,**, 焦悦,2,**, 杨江涛1, 王旭静,1,*, 王志兴,1,*
,1,**, 焦悦,2,**, 杨江涛1, 王旭静,1,*, 王志兴,1,*Molecular characterization identification by genome sequencing of transgenic glyphosate-tolerant rice G2-7
MA Shuo,1,**, JIAO Yue,2,**, YANG Jiang-Tao1, WANG Xu-Jing,1,*, WANG Zhi-Xing,1,*通讯作者:
收稿日期:2020-01-14接受日期:2020-06-2网络出版日期:2020-11-12
| 基金资助: |
Received:2020-01-14Accepted:2020-06-2Online:2020-11-12
| Fund supported: |
作者简介 About authors
马硕, E-mail:
焦悦, E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (7988KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
马硕, 焦悦, 杨江涛, 王旭静, 王志兴. 基因组测序技术解析耐除草剂转基因水稻G2-7的分子特征[J]. 作物学报, 2020, 46(11): 1703-1710. doi:10.3724/SP.J.1006.2020.02002
MA Shuo, JIAO Yue, YANG Jiang-Tao, WANG Xu-Jing, WANG Zhi-Xing.
分子特征是转基因植物的身份象征, 是转基因植物安全评价的基础, 也是转基因植物检测监测的先决条件和主要依据。在我国转基因植物安全评价中, 自环境释放阶段开始, 研发者需要提供外源DNA片段在受体基因组中的拷贝数、整合位点、插入位点处的侧翼序列等方面的分子特征信息。
分析转基因植物分子特征的传统方法有多种, 其中分析基因拷贝数的常用方法有Southern杂交、荧光定量PCR和数字PCR等[1,2,3], 分析外源基因的整合位点和侧翼序列的常用方法为基于PCR的染色体步移技术, 包括TAIL-PCR[4,5]、反向PCR[6,7]、RSE PCR (restriction site extension PCR)[8]和接头介导的PCR (adapter ligated PCR)[9]等。这些方法虽然已在实践中得以验证和应用[10,11,12,13,14], 但每种方法都有其自身的优缺点和局限性, 有时很难获得理想结果。
近年来, 随着高通量测序技术的发展, 全基因组测序已成为解析转基因植物分子特征的一种新方法, 并建立起SBS (southern by sequence)、T-DNA捕获测序和全基因组测序等不同的分析转基因植物分子特征的技术体系[15,16,17], 用于解析转基因拟南芥[16,18]、玉米[19]、水稻[20,21,22]和大豆[23]中外源DNA片段在受体基因组中的整合情况及拷贝数分析, 且用此方法发现在转基因水稻[24]和玉米[25]中存在非预期插入。
G2-7为转G2-aroA基因的耐草甘膦水稻, 是通过农杆菌介导法将G2-aroA基因导入粳稻品种中花11 (ZH11)获得的独立转化事件, 具有很好的耐受草甘膦的能力[26]。目前G2-7已申请了中间试验, 通过Southern杂交证明外源基因在受体基因组中为单拷贝插入, 但通过TAIL-PCR、反向PCR等方法未能获得外源DNA片段插入位点处的侧翼序列[27]。因此, 本研究利用全基因组测序技术结合生物信息学分析, 解析外源DNA片段在受体基因组中的插入位点及其侧翼序列, 明确转基因水稻G2-7的分子特征, 为其后期的安全评价提供数据支撑。
1 材料与方法
1.1 植物材料
所用植物材料为耐草甘膦转基因水稻G2-7及其受体中花11。G2-7是通过农杆菌介导法将耐草甘膦基因G2-aorA导入中花11而获得, 转化时所用植物表达载体为p13UG2 [26]。p13UG2质粒DNA序列作为后期测序数据的参考序列。1.2 试验方法
1.2.1 DNA提取及全基因组测序 利用基因组提取试剂盒(北京博迈德基因技术有限公司)提取水稻叶片基因组DNA, 琼脂糖凝聚电泳对提取的基因组DNA进行质量检测。用Covaris破碎仪将基因组DNA处理成350 bp左右的DNA片段, 利用NEB Next Ultra DNA Library Prep 试剂盒进行建库, 并用NGS3K/Caliper对文库的DNA片段大小进行检测, 用qPCR方法对文库的有效浓度(3×10-9 mol L-1)进行准确定量。本研究共构建了G2-7、中花11 (ZH11)和中花11+1拷贝质粒DNA (ZH11-p) 3个基因组文库。构建好的文库采用PE150双末端测序法在Illuminia NovaSeq 6000平台进行测序, 测序深度为70×。1.2.2 测序数据质量控制及比对 分析测序原始数据碱基错误率, 并对测序原始数据(raw reads)进行过滤(去除带接头、单端测序read中N的比例大于10%、以及单端测序read中含有的低质量碱基数超过其长度比例50%的reads)处理获得clean reads。
利用BWA-0.7.17软件, 经MEM算法将Clean reads与质粒DNA序列进行比对。比对分析后, 将clean reads分成3组, 即完全与参考基因组序列匹配的reads, 完全与质粒DNA序列匹配的reads, 结合区(部分与质粒DNA序列匹配, 部分与基因组序列匹配)序列的reads。匹配到质粒DNA和结合区的reads将用于后期外源基因拷贝数、整合位点及侧翼序列分析。
1.2.3 外源基因拷贝数、整合位点及侧翼序列分析
将匹配到质粒DNA和结合区的reads通过IGV-2.5.0 (integrative genomic viewer)进行可视化分析, 通过分析结合位点的数量来确定外源DNA插入的拷贝数, 通过与质粒DNA比对分析是否有载体骨架插入, 其中ZH11+P和ZH11测序数据分别作为阳性对照和阴性对照。
接合区reads物理位置指示了T-DNA在受体基因组上的整合位点。编写Python脚本对接合区序列进行提取, 使用SOAPdenovo进行侧翼序列拼接, 拼接完后与转化载体p13UG2序列和日本晴 (O. sativa L. spp. japonica, var. nipponbare) 参考基因组进行分别比对, 获得外源基因在受体基因组中整合位点及侧翼序列。
1.2.4 转基因水稻G2-7分子特征验证 根据基因组测序获得的T-DNA 5′端和3′端侧翼序列设计引物5F (5′-GGTGGCTGGGCGATGTGC-3′)和3F (5′- ACTTCAAACAAGTGTGACAA-3′), 根据T-DNA LB端和RB端序列设计引物5R (5′-GTACTCGCCG ATAGTGGAAACCG-3′)和3R (5′-CATTGTCAAATC ATAGAGCAATT-3′)。以G2-7叶片DNA为模板, 分别以5F/5R和3F/3R为引物对进行PCR扩增。PCR反应程序: 为95℃预变性8 min; 95℃ 30 s, 54℃ 30 s, 72℃ 1 min, 35个循环; 72℃延伸5 min。PCR扩增得到的DNA片段进行Sanger测序, 并与全基因组测序获得的侧翼序列进行比对分析。
2 结果与分析
2.1 全基因组测序数据分析
Illumina NovaSeq 6000高通量测序得到的原始图像数据文件经CASAVA碱基识别(base calling)分析转化为原始测序序列(raw bases)。对获得的原始测序数据进行质量控制, 过滤掉带接头(adapter)的读序、单端测序读序中N数量超过此读序长度比例10%的读序、及单端测序读序中含有的低质量(Q≤5)碱基数超过该条读序长度比例50%的等低质量读序, 获得Clean Bases和Clean Reads。本研究获得样品的原始测序量为41.16~47.13 G, 有效读序为274,371,776~314,171,200, Q30大于90%, 说明获得的测序数据丰富可靠(表1)。Table 1
表1
表1高通量测序数据质量控制统计
Table 1
| 样品名称 Sample | 原始数据量 Raw bases | 原始读序量 Raw reads | 有效数据量 Clean bases | 有效读序量Clean reads | Q20 (%) | Q30 (%) | GC含量 GC content (%) |
|---|---|---|---|---|---|---|---|
| ZH11 | 41,196,003,900 | 274,646,422 | 41,155,766,400 | 274,371,776 | 96.22 | 90.36 | 43.04 |
| ZH11-p | 41,266,820,400 | 275,112,136 | 41,228,215,500 | 274,854,770 | 97.73 | 93.69 | 42.93 |
| G2-7 | 47,246,859,300 | 314,990,174 | 47,125,680,000 | 314,171,200 | 96.58 | 91.15 | 44.27 |
新窗口打开|下载CSV
2.2 外源基因拷贝数分析
利用BWA-0.7.17软件将获得clean reads与质粒DNA序列进行比对, 获得了比对到载体骨架、T-DNA和结合区的DNA读序(表2)。分析发现, G2-7中匹配到结合区的读序有348条, 其中匹配到3′端结合区的读序有156条, 匹配到5′端结合区的读序有192条, 外源DNA片段与受体基因组的结合位点为2个(图1); G2-7中完全匹配到T-DNA区的读序有6130条, ZH11-p中完全匹配到T-DNA区的读序有10,487条(图2)。以上结果说明, 外源DNA在受体基因组中为单位点单拷贝插入。图1
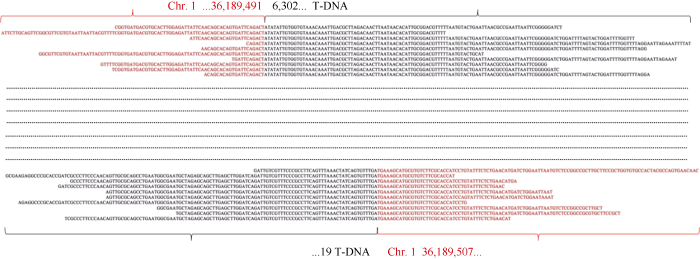 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1G2-7中外源插入片段与受体基因组结合位点分析(部分结合区序列的比对结果)
Fig. 1Analysis of the binding site between the inserted fragments in G2-7 and the receptor genome (comparison results of some binding region sequences)
图2
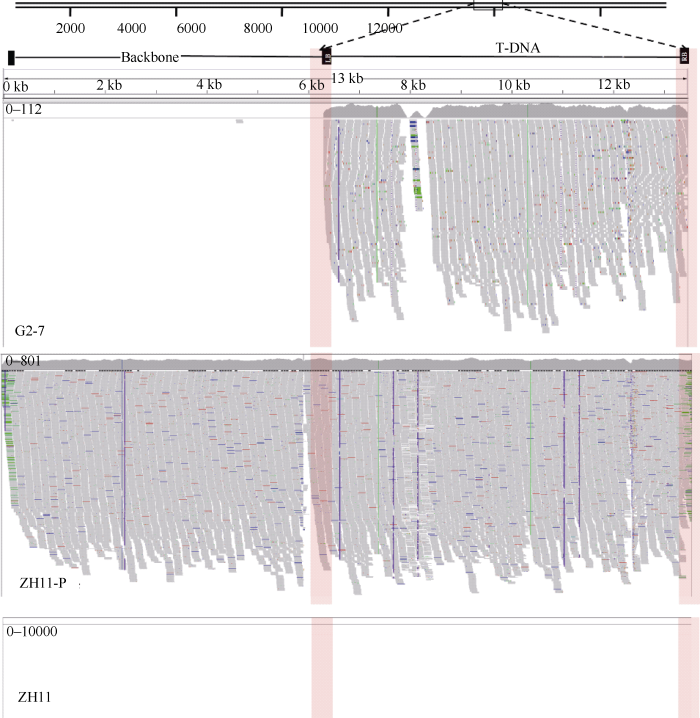 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2测序数据与质粒DNA比对结果的可视化
Fig. 2Visualization of sequencing data and plasmid DNA comparison results
为了明确是否有载体骨架插入, 本研究分析测序数据与载体骨架序列的匹配情况, 发现G2-7中匹配到载体骨架的读序有3条, 分别定位在载体参考序列的171~233、4576~4725和4586~4735位置, ZH11-p中完全匹配到载体骨架区的读序有35,163条, ZH11中匹配到载体骨架上的读序为0。对载体骨架4483~5036位置设计引物对进行PCR扩增, 在G2-7水稻中未见该段序列存在(图3)。且G2-7与单拷贝对照ZH11-p在该区域的测序深度相差非常大, 由此认为G2-7中无载体骨架的插入, G2-7中匹配到载体骨架上的读序为污染所致。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3载体骨架匹配读序的PCR验证
1: G2-7; 2: ZH11; 3: p13UG2.
Fig. 3PCR verification of reads aligned to backbone
2.3 外源基因在受体基因组中的整合位点及侧翼序列分析
将G2-7中匹配到结合区序列用SOAPdenovo进行拼接, 获得了插入位点处3′端接合区序列780 bp和5′端接合区序列823 bp。其中, 3′端接合区有353 bp为水稻基因组序列, 有427 bp为T-DNA序列, T-DNA序列在3′端缺失42 bp; 5′端接合区序列有375 bp为水稻基因组, 有448 bp为T-DNA序列, T-DNA序列在5′端缺失7 bp。通过与已知的水稻基因组序列进行Blast比对分析, 确定T-DNA插入到受体基因组的1号染色体36,189,491~36,189,507 bp处, 在T-DNA与受体基因组整合过程中造成16 bp基因组DNA缺失(图4)。图4
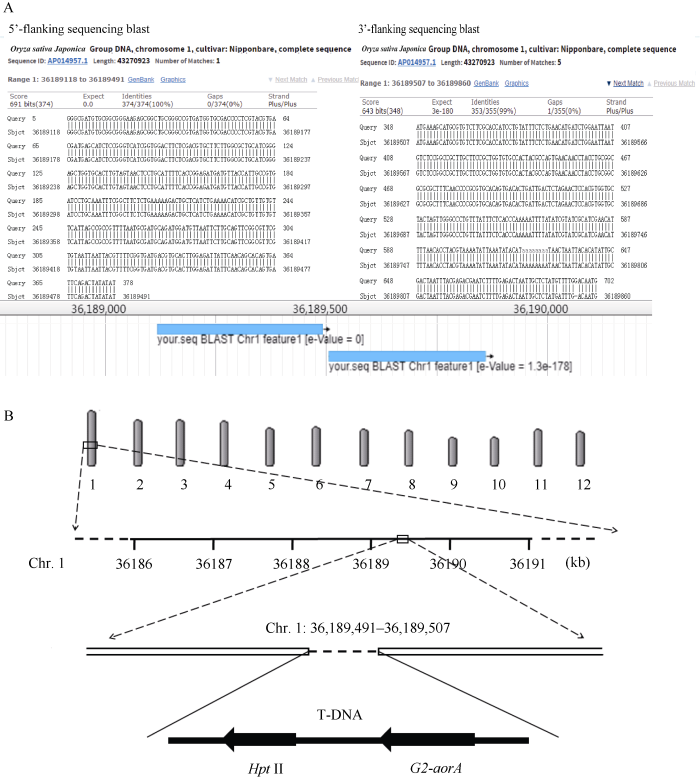 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4外源DNA片段在受体基因组中的整合位点及侧翼序列分析
A: G2-7转化体侧翼序列和整合位点分析; B: G2-7转化体插入序列整合情况示意图。
Fig. 4Analysis of integration site and flanking sequence of insert DNA fragment in receptor genome
A: analysis of flanking sequence and integration site of G2-7; B: sketch map of insert DNA integration in G2-7.
2.4 侧翼序列的Sanger测序验证
根据全基因组测序获得的5′端侧翼序列和T-DNA的LB端序列, 3′端侧翼序列和T-DNA的RB端序列设计2对引物, 以G2-7叶片基因组DNA为模板进行PCR扩增, 结果得到预期目标DNA条带, Sanger测序证明获得的3′和5′段侧翼序列与全基因组测序获得的序列一致(图5)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5PCR电泳图及序列比对
A: 水稻G2-7转化体侧翼序列扩增; 1: G2-7-5F/5R; 2: ZH11-5F/5R; 3: G2-7-3F/3R; 4: ZH11-3F/3R。B: G2-7 5′端序列比对验证。C: G2-7 3′端序列比对验证。
Fig. 5Agarose gel electrophoresis and sequence alignment results
A: amplification of rice transformant G2-7 flanking sequence; 1: G2-7-5F/5R; 2: ZH11-5F/5R; 3: G2-7-3F/3R; 4: ZH11-3F/3R. B: G2-7 5′ end sequence comparison and verification. C: G2-7 5′ end sequence comparison and verification.
3 讨论
伴随大数据时代的到来, 全基因组测序已成为解析转基因植物分子特征的一种新技术。与传统转基因植物分子特征解析方法(如Southern杂交和染色体步移技术等)相比, 全基因组测序不但具有高通量、标准化程度高、灵敏度高、重复性好和准确度高等特点, 而且能够提供插入的DNA序列信息、小片段DNA的非预期插入和DNA重排等信息, 如SBS技术能检测到50 bp小片段DNA在玉米基因组的插入[15], 全基因组测序技术能检测到100 bp 单拷贝DNA片段在玉米基因组的插入[28]。本研究在利用反向PCR、TAIL-PCR等方法无法获取到G2-7转基因水稻中外源插入序列两端的侧翼序列的情况下, 通过高通量全基因组测序技术结合生物信息学分析, 发现G2-7中外源DNA片段以单拷贝形式整合到水稻1号染色体上, 无载体骨架插入、无DNA重排, 并获得了外源DNA片段在受体基因组中插入位置的侧翼序列, 从而成功解析G2-7的分子特征。数据分析过程中, 比对到载体骨架序列的读序可能来自于遗传转化过程中造成的载体骨架插入, 或者是受体基因组中同源序列和建库过程中细菌质粒及其他污染所造成的假阳性[15,29]。建库过程中细菌质粒或其他因素污染所造成的假阳性现象非常普遍, 而且难以完全避免[17]。判断比对到载体骨架的序列是否为假阳性, 可以通过分析这些读序的测序深度和覆盖度, 及将这些读序与受体基因组序列和已知的污染源进行比对来确定[2]。在本研究中, 将G2-7测序数据与载体序列进行比对, 发现有少量读序比对到载体骨架上, 这些读序的测序深度很低, 将非转基因水稻中花11的基因组序列与载体序列进行比对时未发现有读序比对到载体骨架上(图4), 结合PCR验证, 认为比对到载体骨架上的少量读序不是来自于载体骨架真正插入, 而是来源于建库过程中细菌质粒或其他因素污染所造成的假阳性。
在将G2-7测序数据与载体序列进行比对过程中, 发现比对到T-DNA 7800~8400位置的读序很少, 测序深度比较低, 出现gap现象, 推测可能是由于建库过程中此段DNA片段缺失或测序因素造成。为了对此进行验证, 本研究根据T-DNA的相关序列设计引物, 以G2-7叶片DNA为模板进行PCR, 经sanger测序后证明实际插入到受体基因组中的片段此区段并未缺失, 因此我们认为利用基因组测序分析外源插入片段的完整性时, 建议高通量测序与Sanger测序结合使用, 以保证结果的可靠性。
4 结论
利用高通量基因组测序技术结合生物信息学分析, 明确耐除草剂转基因水稻G2-7中外源DNA片段以单位点单拷贝的形式插入到水稻基因组1号染色体36,189,491~36,189,507 bp处, 造成16 bp水稻基因组DNA缺失, 获得外源插入片段3′端侧翼序列353 bp, 5′端侧翼序列375 bp。本研究结果为G2-7商业化过程中的安全评价和转化事件特异性PCR检测方法的建立提供了数据支撑。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/s0022-2836(75)80083-0URLPMID:1195397 [本文引用: 1]
URLPMID:15459795 [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
URLPMID:7759102 [本文引用: 1]
[本文引用: 1]
URLPMID:12519937 [本文引用: 1]
URLPMID:2349129 [本文引用: 1]
URLPMID:20485508 [本文引用: 1]
DOI:10.1111/j.1365-313X.2010.04119.xURLPMID:20409268 [本文引用: 1]

The large collections of Arabidopsis thaliana sequence-indexed T-DNA insertion mutants are among the most important resources to emerge from the sequencing of the genome. Several laboratories around the world have used the Arabidopsis reference genome sequence to map T-DNA flanking sequence tags (FST) for over 325,000 T-DNA insertion lines. Over the past decade, phenotypes identified with T-DNA-induced mutants have played a critical role in advancing both basic and applied plant research. These widely used mutants are an invaluable tool for direct interrogation of gene function. However, most lines are hemizygous for the insertion, necessitating a genotyping step to identify homozygous plants for the quantification of phenotypes. This situation has limited the application of these collections for genome-wide screens. Isolating multiple homozygous insert lines for every gene in the genome would make it possible to systematically test the phenotypic consequence of gene loss under a wide variety of conditions. One major obstacle to achieving this goal is that 12% of genes have no insertion and 8% are only represented by a single allele. Generation of additional mutations to achieve full genome coverage has been slow and expensive since each insertion is sequenced one at a time. Recent advances in high-throughput sequencing technology open up a potentially faster and cost-effective means to create new, very large insertion mutant populations for plants or animals. With the combination of new tools for genome-wide studies and emerging phenotyping platforms, these sequence-indexed mutant collections are poised to have a larger impact on our understanding of gene function.
URLPMID:16302741 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
URLPMID:26445462 [本文引用: 2]
[本文引用: 2]
URLPMID:23951038 [本文引用: 1]
URLPMID:20409008 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:27462336 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2017.00885URLPMID:28611804 [本文引用: 2]
Glyphosate is a widely used herbicide, due to its broad spectrum, low cost, low toxicity, high efficiency, and non-selective characteristics. Rice farmers rarely use glyphosate as a herbicide, because the crop is sensitive to this chemical. The development of transgenic glyphosate-tolerant rice could greatly improve the economics of rice production. Here, we transformed the Pseudomonas fluorescens G2 5-enolpyruvyl shikimate-3-phosphate synthase (EPSPS) gene G2-EPSPS, which conferred tolerance to glyphosate herbicide into a widely used japonica rice cultivar, Zhonghua 11 (ZH11), to develop two highly glyphosate-tolerant transgenic rice lines, G2-6 and G2-7, with one exogenous gene integration. Seed germination tests and glyphosate-tolerance assays of plants grown in a greenhouse showed that the two transgenic lines could greatly improve glyphosate-tolerance compared with the wild-type; The glyphosate-tolerance field test indicated that both transgenic lines could grow at concentrations of 20,000 ppm glyphosate, which is more than 20-times the recommended concentration in the field. Isolation of the flanking sequence of transgenic rice G2-6 indicated that the 5'-terminal of T-DNA was inserted into chromosome 8 of the rice genome. An event-specific PCR test system was established and the limit of detection of the primers reached five copies. Overall, the G2-EPSPS gene significantly improved glyphosate-tolerance in transgenic rice; furthermore, it is a useful candidate gene for the future development of commercial transgenic rice.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.pone.0110808URLPMID:25354084 [本文引用: 1]
Trace quantities of contaminating DNA are widespread in the laboratory environment, but their presence has received little attention in the context of high throughput sequencing. This issue is highlighted by recent works that have rested controversial claims upon sequencing data that appear to support the presence of unexpected exogenous species. I used reads that preferentially aligned to alternate genomes to infer the distribution of potential contaminant species in a set of independent sequencing experiments. I confirmed that dilute samples are more exposed to contaminating DNA, and, focusing on four single-cell sequencing experiments, found that these contaminants appear to originate from a wide diversity of clades. Although negative control libraries prepared from 'blank' samples recovered the highest-frequency contaminants, low-frequency contaminants, which appeared to make heterogeneous contributions to samples prepared in parallel within a single experiment, were not well controlled for. I used these results to show that, despite heavy replication and plausible controls, contamination can explain all of the observations used to support a recent claim that complete genes pass from food to human blood. Contamination must be considered a potential source of signals of exogenous species in sequencing data, even if these signals are replicated in independent experiments, vary across conditions, or indicate a species which seems a priori unlikely to contaminate. Negative control libraries processed in parallel are essential to control for contaminant DNAs, but their limited ability to recover low-frequency contaminants must be recognized.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}