 ,1,**, 纪志远,1,**, 郝巍1, 唐永超1, 韦叶娜1,2, 胡运高2, 赵开军1, 王春连,1,*
,1,**, 纪志远,1,**, 郝巍1, 唐永超1, 韦叶娜1,2, 胡运高2, 赵开军1, 王春连,1,*Molecular identification of rice bacterial blight susceptible gene Xig1 and creation of disease resistant resources
ZHENG Kai-Li,1,**, JI Zhi-Yuan,1,**, HAO Wei1, TANG Yong-Chao1, WEI Ye-Na1,2, HU Yun-Gao2, ZHAO Kai-Jun1, WANG Chun-Lian,1,*通讯作者:
收稿日期:2020-02-27接受日期:2020-04-15网络出版日期:2020-09-12
| 基金资助: |
Received:2020-02-27Accepted:2020-04-15Online:2020-09-12
| Fund supported: |
作者简介 About authors
郑凯丽, E-mail:
纪志远, E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2288KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郑凯丽, 纪志远, 郝巍, 唐永超, 韦叶娜, 胡运高, 赵开军, 王春连. 水稻白叶枯病感病相关基因Xig1的分子鉴定及抗病资源创制[J]. 作物学报, 2020, 46(9): 1332-1339. doi:10.3724/SP.J.1006.2020.02013
ZHENG Kai-Li, JI Zhi-Yuan, HAO Wei, TANG Yong-Chao, WEI Ye-Na, HU Yun-Gao, ZHAO Kai-Jun, WANG Chun-Lian.
水稻(Oryza sativa L.)在世界粮食作物中占有举足轻重的地位, 水稻的产量与品质关乎到世界粮食安全与人口的可持续发展。而由稻黄单胞杆菌水稻致病变种Xoo (Xanthomonas oryzae pv. oryzae)引起的白叶枯病是世界水稻生产中最严重的细菌病害之一, 是限制水稻高产稳产的重要因子。一旦水稻白叶枯病发生会引起水稻不同程度的减产, 发生严重时可减产50%以上甚至绝收[1]。Xoo主要是通过水稻叶片的伤口或水孔侵入, 在维管束内生长并大量繁殖, 导致维管束阻塞, 严重阻碍植物生长所需的各类营养元素的运输, 进而导致病害发生。
Xoo能成功侵染水稻并从其体内攫取养分, 主要依赖于各类蛋白系统(如III型分泌系统)分泌的多种毒性因子。毒性因子不仅需要成功抑制住寄主的防御反应[2], 同时还要操纵感病基因来增强寄主感病性。SWEET家族基因是植物病原黄单胞菌的重要感病靶标, Xoo通过III型分泌系统(T3SS)将效应子TALE (transcription activator-like effector)转运到水稻细胞内, TALE蛋白靶向到SWEET基因的启动子区的EBE (effector binding element)序列, 吸引寄主的其他蛋白形成转录复合体, 激活靶基因的转录表达[3,4]。SWEET基因的表达使寄主的糖类转运更利于病原菌的攫取。主效毒性因子PthXo1和PthXo2就分别靶向OsSWEET11 (Os8N3/Xa13)和OsSWEET13 (Os12N3/Xa25)[5,6,7], 某些感病基因位点甚至被多个TALE所操控激活, 如OsSWEET14 (Os11N3)可以被4个TAL效应子AvrXa7、PthXo3、TalC和TalF绑定激活[7,8,9]。Streubel等[9]分析发现白叶枯病菌小种PXO99A (P6)中Tal7b和Tal8b蛋白可能作用于OsSWEET15。还有一些潜在的感病基因未有互作的毒性因子发现, Li等[10]根据日本晴中OsSWEET12的启动子序列, 人工设计dTALE并成功诱导该基因表达, 最终使宿主水稻呈现感病, 证明OsSWEET12是一个潜在的感病靶点。自然变异或人工突变造成EBE序列改变可以阻断TALE的作用并产生抗性, 例如OsSWEET11和OsSWEET13的抗病等位基因xa13和xa25[9,11]。
目前获得抗源的途径有很多, 如从野生稻、地方特色品种中寻找基因资源、利用物理或化学诱变技术创制抗病材料、利用外源基因(T-DNA)随机突变获得抗病材料等技术方法, 但这些方法存在耗费时间长或突变效率低等缺点。最近基因编辑技术快速发展, 尤其是CRISPR/Cas9技术发明后, 使通过编辑感病基因创制优异资源并服务于育种利用变的更便捷。利用该技术已成功对水稻白叶枯病感病基因SWEET13进行敲除并使水稻获得了抗性[6], 靶向修饰稻瘟病负调控基因OsERF922后提高了水稻对稻瘟病的抗病能力[12]。通过编辑感病品种IR24中的Pong2-1和Pong11-1基因, 创制出抗白叶枯病水稻材料[13]。尤其是近期2个实验室相继报道通过同时突变3个易感基因OsSWEET11、OsSWEET13和OsSWET14的TALE效应子的特异性结合位点(EBE), 最终获得了广谱抗白叶枯病的水稻新种质[14,15]。这些研究结果表明, 通过修饰感病水稻品种创制抗病资源是有效途径之一。
本实验室前期通过转录组测序筛选到一个在感病水稻品种IR24中高度表达, 而在抗病野生稻基因导入系W6023 (IR24为母本)中不表达的基因Os11g39100, 我们将其命名为Xig1 (Xoo-induced- gene 1), 并推测该基因可能与寄主感病相关。本文报道关于Xig1的功能分析和抗病材料创制的研究工作。
1 材料与方法
1.1 水稻材料与生长条件
广谱感白叶枯病籼稻品种IR24、野生稻基因导入系W6023 (以IR24为母本, 普通野生稻编号03-66为父本杂交, 经自交、回交至BC5F6代)和基因编辑不同世代水稻材料。每年5月至10月种植于中国农业科学院作物科学研究所顺义试验基地, 每年11月至翌年4月种植于中国农业科学院作物科学研究所温室(温度范围在25~32°C, 自然光照条件)。所有材料常规水肥料管理。1.2 病原菌及生长条件
大肠杆菌TOP10和农杆菌EHA105在LB培养基(胰蛋白胨15 g L-1; 酵母提取物5 g L-1; 氯化钠5 g L-1; 琼脂15 g L-1; pH 6.8~7.0)上生长, 培养温度分别为37°C和28°C。构建的质粒载体用热激的方法转入大肠杆菌, 用电击的方法转入农杆菌, 在含有相应抗生素的培养基上生长。水稻白叶枯病菌P6在PSA培养基(多聚蛋白胨10 g L-1; 蔗糖10 g L-1; 谷氨酸1 g L-1; 琼脂15 g L-1; pH 6.8~7.0)上生长, 30℃培养2 d, 用无菌水将菌洗脱、混匀, 菌液浓度调到OD600为1.0用于水稻接种。1.3 水稻白叶枯病菌接种与鉴定
水稻材料长至五叶一心时, 用注射器(去掉针头)进行注射接种, 接种时从水稻叶片背面缓缓将菌液推进叶片组织内, 取注射0 h (null)、12 h、24 h、36 h、48 h的叶片用于表达分析。在水稻成株期用剪刀蘸取菌液进行剪叶接种, 分别在7 d、15 d量取病斑长度, 每个株系测量10株, 每株选择3片叶进行测量, 选取平均值, 并进行方差分析和多重比较。1.4 RNA提取与实时荧光定量PCR (Quantitative Real-time PCR, qRT-PCR)
对注射不同时间点的叶片采用TRIzol法提取总RNA。利用Dnase I (TAKARA公司)去除基因组DNA污染。取2 μg总RNA在20 μL反应体系中进行反转录PCR获取第1链cDNA (试剂盒购自天根生化科技(北京)有限公司), 将得到的 cDNA经5倍稀释之后作为qRT-PCR模板DNA。用ABI 7500 Real-time PCR System分析, 水稻泛素基因OsUbi (LOC_Os03g13170)做为标准化内参, 基因引物OsUbiF: 5′-GCTCCGTGGCGGTATCAT-3′, OsUbiR: 5′-CGGCAGTTGACAGCCCTAG-3′。qRT-PCR扩增采用SYBR Premix ExTaq II试剂盒(大连宝生物公司)。20 μL反应体系含2 μL cDNA、0.8 μL引物、10 μL SYBR Green PCR Master Mix和7.2 μL ddH2O。PCR 程序为95℃预变性2 min; 95℃ 15 s, 60℃ 30 s, 40个循环。每个样品设置3个重复。相对表达量分析参考Livak等[16]报道的2-ΔΔCt法。1.5 水稻基因组DNA提取及PCR检测
本实验采用McCouch等[17]方法提取供体及基因编辑材料基因组DNA, 用于PCR 扩增。PCR总反应体积25 μL, 2×PCR Mix 12.5 μL, 正向引物0.4 μL, 反向引物 0.4 μL, DNA 1.0 μL, 加ddH2O补足。PCR程序为94℃预变性3 min; 94℃变性 30 s、55℃退火 30 s、72℃延伸 50 s, 35个循环; 72℃延伸 5 min。在0.8%琼脂糖凝胶上电泳检测 PCR扩增产物。1.6 基因信息分析、引物设计
根据日本晴基因组注释信息(1.7 瞬时表达载体的构建和原生质体的转化
分别扩增Xig1与黄色荧光蛋白基因yfp片段, 利用诺唯赞公司重组酶将融合片段构建到携带有35S启动子的pUC19载体, 测序正确后, 大量提取质粒DNA。原生质体的制备与转化参照Wang等[18]方法。经25℃、16 h暗培养, 使用激光共聚焦显微镜观察融合蛋白在原生质体细胞中的分布。1.8 CRISPR表达载体构建及遗传转化
根据Xig1基因序列, 在基因内部设计三靶点突变体, CRISPR载体pYLCRISPR/Cas9Pubi-H由华南农业大学刘耀光院士实验室提供。靶点1 (S1)的接头引物为XIG1F1/R1: 5′-GGCAATGCGCGGAAC AGCAGCAC/AAACGTGCTGCTGTTCCGCGCAT- 3′; 靶点2 (S2)的接头引物为XIG1F2/R2: 5′-GCC GCCATGAACTCGGTAATGCG/AAACCGCATTACCGAGTTCATGG-3′; 靶点3 (S3)的接头引物为XIG 1F3/R3: 5′-TCAGCGTCGCGCTCCTTAAGCGG/AA ACCCGCTTAAGGAGCGCGACG-3′。载体构建具体过程参考Ma等[19]方法。载体构建完成后, 通过农杆菌介导法转化到籼稻品种IR24中。根据Cas9基因序列, 设计了一对特异引物Cas9pF: 5′-TTCGACC AGTCCAAGAACGG/Cas9pR: CTTGACCTTGGTGA GCTCGT-3′, 用以检测转基因阳性植株。2 结果与分析
2.1 Xig1基因表达分析
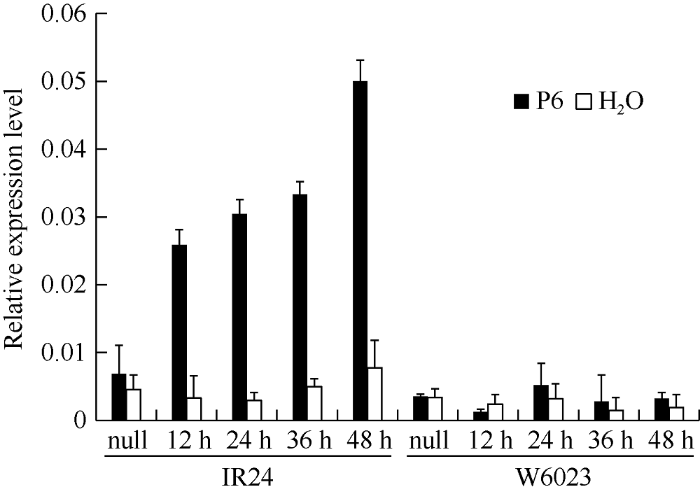
本实验室前期用白叶枯病强致病菌P6注射接种IR24和W6023。取注射不同时间点的叶片进行转录组测序(RNA-Seq)分析, 发掘到一个在IR24中表达量显著高于W6023的基因, 推测此基因可能与感病相关, 暂命名为Xig1。为了进一步验证转录组测序结果, 依据日本晴基因组信息设计qRT-PCR引物XIG1QF/ R, 对用P6和H2O注射接种IR24和W6023后不同时间点的Xig1基因的表达进行分析(图1)。IR24在接种前后Xig1基因的表达量有明显差异, 并且随着注射时间的延长, Xig1基因的表达量逐渐升高, 并受病原菌的诱导表达。而W6023接种与未接种比较, Xig1基因表达没有明显变化。用无菌水注射IR24和W6023, Xig1基因的相对表达量均无明显差异。结果表明: qRT-PCR的结果与实验室前期RNA-Seq的结果一致, 而且Xig1基因受病原菌诱导表达, 推测此基因是一个水稻白叶枯病感病相关基因。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1接种P6后IR24和W6023中Xig1基因相对表达量
Fig. 1Relative expression level of Xig1 in IR24 and W6023 under the inoculation of P6
2.2 Xig1基因的克隆及生物信息学分析
根据转录组测序获得的Xig1基因的cDNA序列及Rice Genome Annotation Project注释信息, 序列比对结果分析发现, Xig1 (Os11g39100)位于水稻11号染色体上, 功能未知。根据参考序列, 设计引物XIG1F和XIG1R4, 以IR24和W6023为模板, 扩增该基因的全长。测序后分析该基因全长612 bp, 没有内含子, 包含一个完整开放阅读框, 编码203个氨基酸。Bioedit软件比对Xig1基因在IR24和W6023中核酸和氨基酸序列的差异, 分析结果表明在编码序列+27、+95、+245处存在3个SNP的差异, 氨基酸序列在第32个和第82个氨基酸上存在差异, 核酸序列在+27上的差异在氨基酸上没有差异。但IR24和W6023在Xig1启动子区域存在较大差别, 我们对两者起始密码子上游500 bp区间作了比较分析。利用PlantCARE网站(
Table 1
表1
表1Xig1IR24基因启动子区内顺式元件特征分析
Table 1
| 顺式作用元件 Cis-acting element | 核心序列 Core sequence | 功能 Function | 数量 Amount |
|---|---|---|---|
| GARE-motif | AAACAGA | 赤霉素反应元件Gibberellin response-related elements | 1 |
| GT1-motif | GGTTAA | 光响应元件Light response elements | 2 |
| MBS | CGGTCA, CAACTG | MYB结合位点、参与干旱诱导 MYB binding sites involved in drought-inducibility | 4 |
| Skn-1motif | GTCAT | 胚乳表达所需元件Elements related to endosperm expression | 4 |
| TA-rich region | TATATATATATATATATATATA | 增强子Enhancer | 1 |
| TATA-box | TATA, TTA, TAATA | 核心启动子元件Core promoter element | 21 |
| TC-rich repeats | GTTTTCTTAC, ATTTTCTCCA | 防御和应激反应元件 Elements involved in defense and stress responsiveness | 2 |
| TGA-element | AACGAC | 生长素应答元件Auxin response elements | 1 |
新窗口打开|下载CSV
2.3 XIG1的亚细胞定位分析
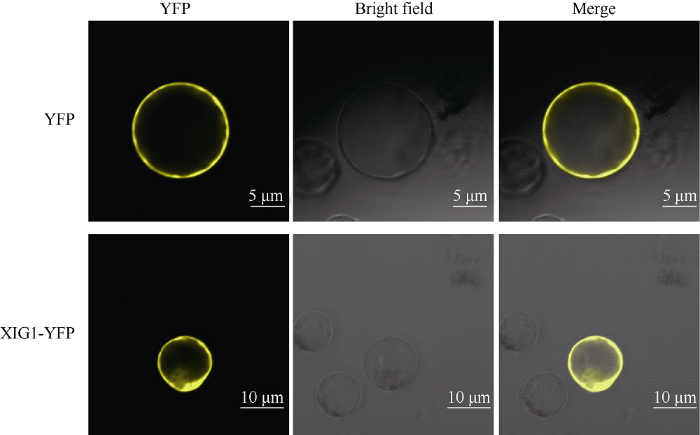
将构建好的重组质粒转化到水稻原生质体, 用LSM 700激光共聚焦显微镜观察XIG1::YFP在细胞中的定位情况。XIG1::YFP融合蛋白的荧光发光结果(图2), 融合蛋白出现在细胞质部位。初步判断XIG定位于细胞质。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2YFP标记的XIG1::YFP融合蛋白的亚细胞定位结果
YFP: 黄色荧光; Bright field: 可见光; Merge: 黄色荧光与可见光叠加。
Fig. 2Subcellular localization of XIG1::YFP in rice protoplast
YFP: yellow fluorescent signal; Merge: YFP and bright field images were merged.
2.4 利用CRISPR/Cas9技术创制Xig1基因编辑突变体
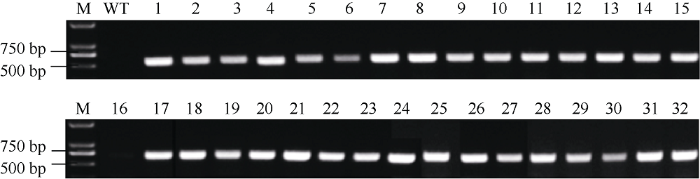
为了研究Xig1基因是否与水稻的抗感病性有关, 本研究利用CRISPR/Cas9系统对Xig1基因进行靶向编辑。通过农杆菌转化法将构建好的载体(pC-Xig1)转化到籼稻品种IR24中, 获得32株T0代转化体。使用引物Cas9特异性引物Cas9pF/Cas9pR检测T0代植株, 受体IR24为阴性对照, 检测结果显示31株T0代转基因植株呈阳性(图3)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3T0代转基因材料的分子检测
M: 分子量标记; WT: IR24 (阴性对照); 1~32: T0代转基因植株。
Fig. 3Molecular detection of T0 transgenic plants
M: marker; WT: IR24 (negative control); 1-32: T0 transgenic plants.
使用Xig1基因的特异引物XIG1F/XIG1R4分别检测T0、T1和T2代植株, 回收PCR产物并测序。序列结果与野生型进行比对分析发现, 在31个T0代Cas9阳性植株中有28个发生了靶点编辑, 编辑概率为90.3%。T0和T1代的靶点序列都分别出现了纯合型、杂合型、双等位型和嵌合型4种不同类型的编辑情况。经过自交在T2代筛选到17个纯合的编辑株系, 编辑类型有单碱基插入和不同类型的碱基缺失, 具体编辑情况如图4。从图中看出, S1中11个为单碱基插入, 6个不同碱基数的缺失; S2中只有4个为单碱基插入, 13个不同碱基数的缺失, 并且缺失数目较多; S3中有12个为单碱基插入, 5个不同碱基数的缺失。其中Xig1-5和Xig1-10在S2和S3之间有大片段的缺失。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图417个T2代基因编辑纯合株系靶点序列
大写加粗字母为PAM核苷酸序列; 下画线序列为靶点序列; 红色破折号表示核苷酸缺失; 红色小写字母表示插入的核苷酸; 斜体大写字母为起始密码子。序列右侧的数字代表各靶位点突变类型及突变所涉及的核苷酸数。“-”和“+”分别表示缺失和插入所指示的核苷酸数。100 bp为此序列中未显示的一段碱基序列。
Fig. 4The target sequences of the 17 homozygous Xig1-edited lines of T2
Capitalized bold letters are PAM sequences; underlined sequences are target sequences; red dashes indicate nucleotide deletions; red lowercase letters indicate inserted nucleotides; italic capital letters are initiation codons. The numbers on the right side of the sequences represent the types of mutation at each target site and the number of nucleotides involved in the mutation. “-” and “+” indicate the number of deletion and insertion nucleotides, respectively. The 100 bp are the bases not shown in this sequence.
2.5 Xig1基因编辑株系白叶枯病抗性检测
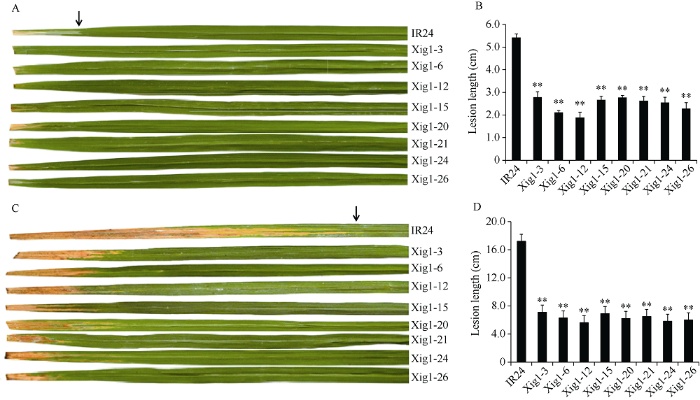
为了研究Xig1是否与寄主感病性相关, 我们选取8个无T-DNA 残留的T2代Xig1突变株系进行水稻白叶枯病菌接种鉴定。采用P6菌株对成株期植株进行剪叶接种, 接种7 d后测量叶片的病斑长度, 每个株系测量10株, 每株选择3片叶进行测量。野生型IR24的平均病斑长度为5.5 cm左右, 编辑株系的平均病斑长度在2.0~3.0 cm (图5-A, B)。接种14 d后再次测量病斑长度, 野生型IR24的平均病斑长度延伸到17 cm左右, 编辑株系的病斑长度在6~8 cm (图5-C, D)。鉴定结果表明, 在白叶枯病发生前期和后期, 基因编辑植株与野生型IR24相比病斑长度均明显缩短, 方差分析达到了极显著, Xig1突变后水稻白叶枯病的抗性明显提高。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图58个T2转基因抗病株系抗病反应
A, C: 分别为接种7 d和14 d时8个转基因株系与对照IR24病斑长度症状图。B, D: 分别为接种7 d和14 d时8个转基因株系与对照IR24病斑长度柱形图。**代表极显著差异(P < 0.01)。箭头: 接种的IR24叶片。
Fig. 5Disease response to Xoo strain P6 of 8 T2 transgenic lines
A, C: Disease reactions of 7 days and 14 days after inoculation in eight transgenic lines and IR24. B, D: Lesion length of 7 days and 14 days after inoculation with PXO99A in eight transgenic lines compared with IR24. ** denotes significantly different (P < 0.01). Arrows indicates the inoculated leaves of IR24.
2.6 Xig1基因编辑株系主要农艺性状考察
对Xig1基因编辑后的8个T3纯合株系进行农艺性状考察, 包括株高、分蘖数、结实率和千粒重, 每个株系考察10个植株。考察结果见表2, 8个基因编辑株系的株高、分蘖数、结实率和千粒重, 取平均值, 并作方差分析, 采用Duncan氏测验进行株系间多重比较, 结果显示8个基因编辑株系的主要农艺性状与IR24相比均无显著性差异。由此说明, Xig1在基因编辑后对水稻的主要农艺性状无显著影响。Table 2
表2
表28个基因编辑株系的农艺性状分析
Table 2
| 基因编辑株系 Lines | 株高 Plant height (cm) | 分蘖数 Number of tillers | 穗粒数 Number of grains per panicle | 结实率 Seed-setting rate (%) | 千粒重 Thousand-seed weight (g) | 籽粒产量 Grain yield (g) |
|---|---|---|---|---|---|---|
| IR24 | 87.2±2.1 a | 10.8±1.3 a | 112.7±12.6 a | 82.9±1.9 a | 24.8±0.8 a | 24.5±2.9 a |
| Xig1-3 | 87.1±1.6 a | 11.9±1.7 a | 110.9±9.9 a | 80.2±1.4 a | 24.4±1.0 a | 26.0±4.5 a |
| Xig1-6 | 87.8±2.6 a | 11.8±1.9 a | 108.8±5.5 a | 80.0±1.7 a | 25.9±0.9 a | 26.7±2.6 a |
| Xig1-12 | 86.7±2.5 a | 10.6±0.9 a | 114.5±10.9 a | 81.2±1.5 a | 24.6±0.7 a | 24.9±6.0 a |
| Xig1-15 | 86.5±1.5 a | 10.5±1.7 a | 114.6±11.3 a | 83.6±0.4 a | 24.9±1.8 a | 25.2±2.5 a |
| Xig1-20 | 86.7±1.5 a | 11.3±1.6 a | 116.7±8.9 a | 84.1±1.1 a | 25.1±1.2 a | 28.0±3.7 a |
| Xig1-21 | 87.6±2.3 a | 10.4±1.5 a | 123.0±10.0 a | 82.7±2.2 a | 25.9±1.3 a | 27.2±2.0 a |
| Xig1-24 | 88.2±1.9 a | 13.2±3.2 a | 97.4±4.3 a | 83.2±3.3 a | 25.2±0.5 a | 26.8±2.1 a |
| Xig1-26 | 88.4±1.7 a | 10.2±2.2 a | 118.0±12.9 a | 84.2±3.3 a | 25.5±0.8 a | 25.6±3.1 a |
新窗口打开|下载CSV
3 讨论
植物-病原菌的协同进化是复杂而又漫长的过程, 微生物具备一定适应能力并在寄主上定殖是成为病原物的第一步, 适应性病原物(adapted pathogens)同时还需要克服寄主长期进化形成的抗病机制才能最终攫取到养分。因此, 在自然和寄主等压力的作用下, 病原物需要不断改变致病性(pathogencity)来适应各类变化, 如产生新的毒性因子[20]。干扰或操控寄主感病基因是病原物常用的策略之一: 除SWEET家族基因外, Xoo操控的靶标基因还有转录因子类基因OsTFX1和OsTFⅡAγ1[21]; 水稻细菌性条斑病菌(Xanthomonas oryzae pv. oryzicola, Xoc)的感病靶标基因有硫转运蛋白基因OsSULTR3;6[22]和bHLH转录因子家族基因(Os02g47660和Os03g07540)[23]等。寄主感病基因的突变可以获得广谱和持久的抗病性, 例如OsTFⅡAγ5 (Xa5)是TALE激活靶标时转录复合体的必要组分, 当Xa5的第39位缬氨酸突变为谷氨酸后(xa5), TALE操控感病基因的诱导表达水平下降, 水稻获得广谱、持久的抗白叶枯病能力[24]; Xa13 (OsSWEET11)启动子的自然突变产生xa13, 寄主获得了对菲律宾多个小种的抗性[7]。
本研究前期通过转录组测序发现, 水稻参考基因组注释中功能未知的Os11g39100 (Xig1)基因在感病亲本IR24中表达, 在抗病野生稻导入系W6023中不表达。Xig1IR24的表达受病原菌诱导, 表达量随诱导时间延长而增高, 而Xig1在W6023中几乎不表达, 推测该基因可能是一个感病基因。比对IR24和W6023来源的Xig1, 该基因在抗感品种中的全长均为612 bp, 仅含1个外显子, 在核酸水平上存在3个单碱基的差异位点, 在蛋白质水平上存在2个氨基酸差异位点。本研究从IR24中获得了3 kb的启动子序列, Xig1IR24的启动子和编码区序列与籼稻品种蜀恢498 (http://www.mbkbase.org/R498/)的完全一致。在此范围内只成功获得Xig1W6023的启动子500 bp序列, 通过测序发现IR24和W6023在Xig1启动子区域存在较大差别, 推测野生稻中Xig1的启动子存在变异, 启动子变异可能是抗感品种中基因差异表达的主要原因。
为研究Xig1抗感病功能, 我们利用CRISPR/ Cas9系统构建了IR24的Xig1突变体株系, 获得T0代转基因阳性植株31株。对基因编辑植株自交后代株系进行分子检测, 得到了17个无T-DNA插入的纯合株系。从纯合株系中选择部分代表进行田间抗病性分析, 接种后7 d和14 d的调查结果显示, Xig1突变后植株病斑长度较野生型IR24相比都有所缩短, 证明Xig1对水稻感病性起重要贡献。农艺性状考察结果显示, 编辑株系在主要的农艺性状上与野生型IR24无显著差异。综上所述, Xig1可能是Xoo操控的感病靶标或水稻感病通路中的关键基因, 利用基因编辑技术对Xig1的突变可以提高水稻的抗白叶枯病能力, 本研究获得的Xig1基因定点突变株系是新的水稻抗白叶枯病种质资源。
4 结论
Xig1是新发现的水稻白叶枯病的感病基因, 受病原菌诱导表达。Xig1等位基因序列差异在抗感材料中主要集中在启动子区, 亚细胞定位结果显示XIG1定位于细胞质中。利用CRISPR/Cas9系统对Xig1靶点进行基因组编辑, 获得8个Xig1基因定点突变的纯合株系, 接种鉴定发现对白叶枯病的抗性得到明显提高, 而农艺性状无显著差异, 为水稻抗病育种提供了新的种质资源。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1146/annurev-phyto-102313-045926URLPMID:24906128 [本文引用: 1]

Rice feeds more than half of the world's population. Rice blast, caused by the fungal pathogen Magnaporthe oryzae, and bacterial blight, caused by the bacterial pathogen Xanthomonas oryzae pv. oryzae, are major constraints to rice production worldwide. Genome sequencing and extensive molecular analysis has led to the identification of many new pathogen-associated molecular patterns (PAMPs) and avirulence and virulence effectors in both pathogens, as well as effector targets and receptors in the rice host. Characterization of these effectors, host targets, and resistance genes has provided new insight into innate immunity in plants. Some of the new findings, such as the binding activity of X. oryzae transcriptional activator-like (TAL) effectors to specific rice genomic sequences, are being used for the development of effective disease control methods and genome modification tools. This review summarizes the recent progress toward understanding the recognition and signaling events that govern rice innate immunity.
DOI:10.1016/j.pbi.2012.03.004URLPMID:22465133 [本文引用: 1]
Effectors secreted by the bacterial type III system play a central role in the interaction between Gram-negative bacterial pathogens and their host plants. Recent advances in the effector studies have helped cementing several key concepts concerning bacterial pathogenesis, plant immunity, and plant-pathogen co-evolution. Type III effectors use a variety of biochemical mechanisms to target specific host proteins or DNA for pathogenesis. The identifications of their host targets led to the identification of novel components of plant innate immune system. Key modules of plant immune signaling pathways such as immune receptor complexes and MAPK cascades have emerged as a major battle ground for host-pathogen adaptation. These modules are attacked by multiple type III effectors, and some components of these modules have evolved to actively sense the effectors and trigger immunity.
URLPMID:17962565 [本文引用: 1]
DOI:10.1126/science.1144958URLPMID:17962564 [本文引用: 1]
Plant disease resistance (R) proteins recognize matching pathogen avirulence proteins. Alleles of the pepper R gene Bs3 mediate recognition of the Xanthomonas campestris pv. vesicatoria (Xcv) type III effector protein AvrBs3 and its deletion derivative AvrBs3Deltarep16. Pepper Bs3 and its allelic variant Bs3-E encode flavin monooxygenases with a previously unknown structure and are transcriptionally activated by the Xcv effector proteins AvrBs3 and AvrBs3Deltarep16, respectively. We found that recognition specificity resides in the Bs3 and Bs3-E promoters and is determined by binding of AvrBs3 or AvrBs3Deltarep16 to a defined promoter region. Our data suggest a recognition mechanism in which the Avr protein binds and activates the promoter of the cognate R gene.
DOI:10.1073/pnas.0604088103URLPMID:16798873 [本文引用: 1]
Many bacterial diseases of plants depend on the interaction of type III effector genes of the pathogen and disease-susceptibility genes of the host. The host susceptibility genes are largely unknown. Here, we show that expression of the rice gene Os8N3, a member of the MtN3 gene family from plants and animals, is elevated upon infection by Xanthomonas oryzae pv. oryzae strain PXO99(A) and depends on the type III effector gene pthXo1. Os8N3 resides near xa13, and PXO99(A) failed to induce Os8N3 in rice lines with xa13. Silencing of Os8N3 by inhibitory RNA produced plants that were resistant to infection by strain PXO99(A) yet remained susceptible to other strains of the pathogen. The effector gene avrXa7 from strain PXO86 enabled PXO99(A) compatibility on either xa13- or Os8N3-silenced plants. The findings indicate that Os8N3 is a host susceptibility gene for bacterial blight targeted by the type III effector PthXo1. The results support the hypothesis that X. oryzae pv. oryzae commandeers the regulation of otherwise developmentally regulated host genes to induce a state of disease susceptibility. Furthermore, the results support a model in which the pathogen induces disease susceptibility in a gene-for-gene manner.
DOI:10.1111/tpj.12838URLPMID:25824104 [本文引用: 2]
Bacterial blight of rice is caused by the gamma-proteobacterium Xanthomonas oryzae pv. oryzae, which utilizes a group of type III TAL (transcription activator-like) effectors to induce host gene expression and condition host susceptibility. Five SWEET genes are functionally redundant to support bacterial disease, but only two were experimentally proven targets of natural TAL effectors. Here, we report the identification of the sucrose transporter gene OsSWEET13 as the disease-susceptibility gene for PthXo2 and the existence of cryptic recessive resistance to PthXo2-dependent X. oryzae pv. oryzae due to promoter variations of OsSWEET13 in japonica rice. PthXo2-containing strains induce OsSWEET13 in indica rice IR24 due to the presence of an unpredicted and undescribed effector binding site not present in the alleles in japonica rice Nipponbare and Kitaake. The specificity of effector-associated gene induction and disease susceptibility is attributable to a single nucleotide polymorphism (SNP), which is also found in a polymorphic allele of OsSWEET13 known as the recessive resistance gene xa25 from the rice cultivar Minghui 63. The mutation of OsSWEET13 with CRISPR/Cas9 technology further corroborates the requirement of OsSWEET13 expression for the state of PthXo2-dependent disease susceptibility to X. oryzae pv. oryzae. Gene profiling of a collection of 104 strains revealed OsSWEET13 induction by 42 isolates of X. oryzae pv. oryzae. Heterologous expression of OsSWEET13 in Nicotiana benthamiana leaf cells elevates sucrose concentrations in the apoplasm. The results corroborate a model whereby X. oryzae pv. oryzae enhances the release of sucrose from host cells in order to exploit the host resources.
DOI:10.1105/tpc.110.078964URLPMID:21098734 [本文引用: 3]
The rice (Oryza sativa) gene xa13 is a recessive resistance allele of Os-8N3, a member of the NODULIN3 (N3) gene family, located on rice chromosome 8. Os-8N3 is a susceptibility (S) gene for Xanthomonas oryzae pv oryzae, the causal agent of bacterial blight, and the recessive allele is defeated by strains of the pathogen producing any one of the type III effectors AvrXa7, PthXo2, or PthXo3, which are all members of the transcription activator-like (TAL) effector family. Both AvrXa7 and PthXo3 induce the expression of a second member of the N3 gene family, here named Os-11N3. Insertional mutagenesis or RNA-mediated silencing of Os-11N3 resulted in plants with loss of susceptibility specifically to strains of X. oryzae pv oryzae dependent on AvrXa7 or PthXo3 for virulence. We further show that AvrXa7 drives expression of Os-11N3 and that AvrXa7 interacts and binds specifically to an effector binding element within the Os-11N3 promoter, lending support to the predictive models for TAL effector binding specificity. The result indicates that variations in the TAL effector repetitive domains are driven by selection to overcome both dominant and recessive forms of resistance to bacterial blight in rice. The finding that Os-8N3 and Os-11N3 encode closely related proteins also provides evidence that N3 proteins have a specific function in facilitating bacterial blight disease.
DOI:10.1094/MPMI-11-10-0254URLPMID:21679014 [本文引用: 1]
African strains of Xanthomonas oryzae pv. oryzae contain fewer TAL effectors than Asian strains, and their contribution to pathogenicity is unknown. Systematic mutagenesis of tal genes was used to decipher the contribution of each of the eight TAL effector paralogs to pathogenicity of African X. oryzae pv. oryzae BAI3. A strain mutated in talC was severely affected in the production of disease symptoms. Analysis of growth in planta upon leaf-clip inoculation showed that mutant bacteria multiplied only at the site of inoculation at the apex of the leaf, suggesting a requirement for talC during colonization of vascular tissues. Such tissue-specific effect of a tal mutant is a novel phenotype, which has not yet been characterized in other xanthomonads. Microarray experiments comparing the host response of rice leaves challenged with BAI3(R) vs. BAI3(R)DeltatalC were performed to identify genes targeted by TalC. A total of 120 upregulated and 21 downregulated genes were identified, among them Os11N3, which is a member of the MtN3/saliva family. Based on semiquantitative reverse transcription-polymerase chain reaction and beta-glucuronidase reporter assays, we show that Os11N3 is directly upregulated by TalC and identify a TalC DNA target box within the Os11N3 upstream sequence.
DOI:10.1111/nph.12411URL [本文引用: 3]
Bacterial plant-pathogenic Xanthomonas strains translocate transcription activator-like (TAL) effectors into plant cells to function as specific transcription factors. Only a few plant target genes of TAL effectors have been identified, so far. Three plant SWEET genes encoding putative sugar transporters are known to be induced by TAL effectors from rice-pathogenic Xanthomonas oryzae pv. oryzae (Xoo). We predict and validate that expression of OsSWEET14 is induced by a novel TAL effector, Tal5, from an African Xoo strain. Artificial TAL effectors (ArtTALs) were constructed to individually target 20 SWEET orthologs in rice. They were used as designer virulence factors to study which rice SWEET genes can support Xoo virulence. The Tal5 target box differs from those of the already known TAL effectors TalC, AvrXa7 and PthXo3, which also induce expression of OsSWEET14, suggesting evolutionary convergence on key targets. ArtTALs efficiently complemented an Xoo talC mutant, demonstrating that specific induction of OsSWEET14 is the key target of TalC. ArtTALs that specifically target individual members of the rice SWEET family revealed three known and two novel SWEET genes to support bacterial virulence. Our results demonstrate that five phylogenetically close SWEET proteins, which presumably act as sucrose transporters, can support Xoo virulence.
URLPMID:23430045 [本文引用: 1]
URLPMID:12750790 [本文引用: 1]
URLPMID:27116122 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.molp.2019.08.006URLPMID:31493565 [本文引用: 1]
Xanthomonas oryzae pv. oryzae (Xoo), the causal agent of bacterial blight of rice, employs the transcription activator-like effectors (TALEs) to induce the expression of the OsSWEET family of putative sugar transporter genes, which function in conferring disease susceptibility (S) in rice plants. To engineer broad-spectrum bacterial blight resistance, we used CRISPR/Cas9-mediated gene editing to disrupt the TALE-binding elements (EBEs) of two S genes, OsSWEET11 and OsSWEET14, in rice cv. Kitaake, which harbors the recessive resistance allele of Xa25/OsSWEET13. The engineered rice line MS14K exhibited broad-spectrum resistance to most Xoo strains with a few exceptions, suggesting that the compatible strains may contain new TALEs. We identified two PthXo2-like TALEs, Tal5LN18 and Tal7PXO61, as major virulence factors in the compatible Xoo strains LN18 and PXO61, respectively, and found that Xoo encodes at least five types of PthXo2-like effectors. Given that PthXo2/PthXo2.1 target OsSWEET13 for transcriptional activation, the genomes of 3000 rice varieties were analyzed for EBE variationsin the OsSWEET13 promoter, and 10 Xa25-like haplotypes were identified. We found that Tal5LN18 and Tal7PXO61 bind slightly different EBE sequences in the OsSWEET13 promoter to activate its expression. CRISPR/Cas9 technology was then used to generate InDels in the EBE of the OsSWEET13 promoter in MS14K to creat a new germplasm with three edited OsSWEET EBEs and broad-spectrum resistance against all Xoo strains tested. Collectively, our findings illustrate how to disarm TALE-S co-evolved loci to generate broad-spectrum resistance through the loss of effector-triggered susceptibility in plants.
DOI:10.1038/s41587-019-0267-zURLPMID:31659337 [本文引用: 1]
Bacterial blight of rice is an important disease in Asia and Africa. The pathogen, Xanthomonas oryzae pv. oryzae (Xoo), secretes one or more of six known transcription-activator-like effectors (TALes) that bind specific promoter sequences and induce, at minimum, one of the three host sucrose transporter genes SWEET11, SWEET13 and SWEET14, the expression of which is required for disease susceptibility. We used CRISPR-Cas9-mediated genome editing to introduce mutations in all three SWEET gene promoters. Editing was further informed by sequence analyses of TALe genes in 63 Xoo strains, which revealed multiple TALe variants for SWEET13 alleles. Mutations were also created in SWEET14, which is also targeted by two TALes from an African Xoo lineage. A total of five promoter mutations were simultaneously introduced into the rice line Kitaake and the elite mega varieties IR64 and Ciherang-Sub1. Paddy trials showed that genome-edited SWEET promoters endow rice lines with robust, broad-spectrum resistance.
URLPMID:11846609 [本文引用: 1]
DOI:10.1007/BF00273666URLPMID:24232389 [本文引用: 1]
We report the construction of an RFLP genetic map of rice (Oryza sativa) chromosomes. The map is comprised of 135 loci corresponding to clones selected from a PstI genomic library. This molecular map covers 1,389 cM of the rice genome and exceeds the current classical maps by more than 20%. The map was generated from F2 segregation data (50 individuals) from a cross between an indica and javanica rice cultivar. Primary trisomics were used to assign linkage groups to each of the 12 rice chromosomes. Seventy-eight percent of the clones assayed revealed RFLPs between the two parental cultivars, indicating that rice contains a significant amount of RFLP variation. Strong correlations between size of hybridizing restriction fragments and level of polymorphism indicate that a significant proportion of the RFLPs in rice are generated by insertions/delections. This conclusion is supported by the occurrence of null alleles for some clones (presumably created by insertion or deletion events). One clone, RG229, hybridized to sequences in both the indica and javanica genomes, which have apparently transposed since the divergence of the two cultivars from their last common ancestor, providing evidence for sequence movement in rice. As a by product of this mapping project, we have discovered that rice DNA is less C-methylated than tomato or maize DNA. Our results also suggest the notion that a large fraction of the rice genome (approximately 50%) is single copy.
[本文引用: 1]
URLPMID:25917172 [本文引用: 1]
DOI:10.1016/j.molp.2014.10.010URLPMID:25616388 [本文引用: 1]
The majority of plant disease resistance (R) genes encode proteins that share common structural features. However, the transcription activator-like effector (TALE)-associated executor type R genes show no considerable sequence homology to any known R genes. We adopted a map-based cloning approach and TALE-based technology to isolate and characterize Xa23, a new executor R gene derived from wild rice (Oryza rufipogon) that confers an extremely broad spectrum of resistance to bacterial blight caused by Xanthomonas oryzae pv. oryzae (Xoo). Xa23 encodes a 113 amino acid protein that shares 50% identity with the known executor R protein XA10. The predicted transmembrane helices in XA23 also overlap with those of XA10. Unlike Xa10, however, Xa23 transcription is specifically activated by AvrXa23, a TALE present in all examined Xoo field isolates. Moreover, the susceptible xa23 allele has an identical open reading frame of Xa23 but differs in promoter region by lacking the TALE binding element (EBE) for AvrXa23. XA23 can trigger a strong hypersensitive response in rice, tobacco, and tomato. Our results provide the first evidence that plant genomes have an executor R gene family of which members execute their function and spectrum of disease resistance by recognizing the cognate TALEs in the pathogen.
URLPMID:17563377 [本文引用: 1]
DOI:10.1371/journal.pone.0235898URLPMID:32833999 [本文引用: 1]
Myo/Nog cells were discovered in the chick embryo epiblast. Their expression of MyoD reflects a commitment to the skeletal muscle lineage and capacity to differentiate into myofibroblasts. Release of Noggin by Myo/Nog cells is essential for normal morphogenesis. Myo/Nog cells rapidly respond to wounding in the skin and eyes. In this report, we present evidence suggesting that Myo/Nog cells phagocytose tattoo ink in tissue sections of human skin and engulf cell corpses in cultures of anterior human lens tissue and magnetic beads injected into the anterior chamber of mice in vivo. Myo/Nog cells are distinct from macrophages in the skin and eyes indicated by the absence of labeling with an antibody to ionized calcium binding adaptor molecule 1. In addition to their primary roles as regulators of BMP signaling and progenitors of myofibroblasts, Myo/Nog cells behave as nonprofessional phagocytes defined as cells whose primary functions are unrelated to phagocytosis but are capable of engulfment.
DOI:10.1104/pp.106.080580URLPMID:16896230 [本文引用: 1]
The basic/helix-loop-helix (bHLH) transcription factors and their homologs form a large family in plant and animal genomes. They are known to play important roles in the specification of tissue types in animals. On the other hand, few plant bHLH proteins have been studied functionally. Recent completion of whole genome sequences of model plants Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa) allows genome-wide analysis and comparison of the bHLH family in flowering plants. We have identified 167 bHLH genes in the rice genome, and their phylogenetic analysis indicates that they form well-supported clades, which are defined as subfamilies. In addition, sequence analysis of potential DNA-binding activity, the sequence motifs outside the bHLH domain, and the conservation of intron/exon structural patterns further support the evolutionary relationships among these proteins. The genome distribution of rice bHLH genes strongly supports the hypothesis that genome-wide and tandem duplication contributed to the expansion of the bHLH gene family, consistent with the birth-and-death theory of gene family evolution. Bioinformatics analysis suggests that rice bHLH proteins can potentially participate in a variety of combinatorial interactions, endowing them with the capacity to regulate a multitude of transcriptional programs. In addition, similar expression patterns suggest functional conservation between some rice bHLH genes and their close Arabidopsis homologs.
DOI:10.1007/s00438-005-0091-7URLPMID:16614777 [本文引用: 1]
The recessive gene xa5 for resistance to bacterial blight resistance of rice is located on chromosome 5, and evidence based on genetic recombination has been shown to encode a small subunit of the basal transcription factor IIA (Iyer and McCouch in MPMI 17(12):1348-1354, 2004). However, xa5 has not been demonstrated by a complementation test. In this study, we introduced the dominant allele Xa5 into a homozygous xa5-line, which was developed from a cross between IRBB5 (an indica variety with xa5) and Nipponbare (a japonica variety with Xa5). Transformation of Xa5 and subsequent segregation analysis confirmed that xa5 is a V39E substitution variant of the gene for TFIIAgamma on chromosome 5 (TFIIAgamma5 or Xa5). The rice has an addition gene for TFIIAgamma exists on chromosome 1 (TFIIAgamma1). Analysis of the expression patterns of Xa5 (TFIIAgamma5)/xa5 and TFIIAgamma1 revealed that both the genes are constitutively expressed in different rice organs. However, no expression of TFIIAgamma1 could be detected in the panicle by reverse transcriptase-polymerase chain reaction. To compare the structural difference between the Xa5/xa5 and TFIIAgamma1 proteins, 3-D structures were predicted using computer-aided modeling techniques. The modeled structures of Xa5 (xa5) and TFIIAgamma1 fit well with the structure of TFIIA small subunit from human, suggesting that they may all act as a small subunit of TFIIA. The E39V substitution in the xa5 protein occurs in the alpha-helix domain, a supposed conservative substitutable site, which should not affect the basal transcription function of TFIIAgamma. The structural analysis indicates that xa5 and Xa5 potentially retain their basic transcription factor function, which, in turn, may mediate the novel pathway for bacterial blight resistance and susceptibility, respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}