 ,1, 郭军1, 刘成1, 李豪圣1, 宋健民1, 刘爱峰1, 曹新有1, 程敦公1, 李法计1, 何中虎2, 夏先春,2,*, 刘建军,1,*
,1, 郭军1, 刘成1, 李豪圣1, 宋健民1, 刘爱峰1, 曹新有1, 程敦公1, 李法计1, 何中虎2, 夏先春,2,*, 刘建军,1,*Functional analysis of Lcye gene involved in the carotenoid synthesis in common wheat
ZHAI Sheng-Nan,1, GUO Jun1, LIU Cheng1, LI Hao-Sheng1, SONG Jian-Min1, LIU Ai-Feng1, CAO Xin-You1, CHENG Dun-Gong1, LI Fa-Ji1, HE Zhong-Hu2, XIA Xian-Chun,2,*, LIU Jian-Jun,1,*通讯作者:
收稿日期:2020-02-18接受日期:2020-06-2网络出版日期:2020-06-12
| 基金资助: |
Received:2020-02-18Accepted:2020-06-2Online:2020-06-12
| Fund supported: |
作者简介 About authors
E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1755KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
翟胜男, 郭军, 刘成, 李豪圣, 宋健民, 刘爱峰, 曹新有, 程敦公, 李法计, 何中虎, 夏先春, 刘建军. 小麦类胡萝卜素合成途径关键基因Lcye功能分析[J]. 作物学报, 2020, 46(10): 1485-1495. doi:10.3724/SP.J.1006.2020.01013
ZHAI Sheng-Nan, GUO Jun, LIU Cheng, LI Hao-Sheng, SONG Jian-Min, LIU Ai-Feng, CAO Xin-You, CHENG Dun-Gong, LI Fa-Ji, HE Zhong-Hu, XIA Xian-Chun, LIU Jian-Jun.
面粉色泽是评价小麦粉品质的重要感官指标和市场指标。黄色素是小麦籽粒中最主要的天然色素, 是面粉及其制品黄度形成的主要原因。籽粒黄色素含量与面粉、面团黄度以及面包、面条颜色显著相关, 相关系数分别高达0.8~0.9和0.69~0.76 [1,2,3,4]。中式面制品, 如面条、馒头、包子、饺子等对面粉的白度要求比较高。
类胡萝卜素是构成黄色素的主要组分。类胡萝卜素, 尤其是β-胡萝卜素, 具有抗氧化、抗癌、维生素A原、预防眼睛老年性黄斑病变、延缓衰老、提高免疫力等重要生理保健功能。人类和动物不能自身合成类胡萝卜素, 因此必须从外界摄取[5,6]。近年来, 随着人们营养和保健意识的增强, 提高小麦籽粒类胡萝卜素含量、培育亮黄色的面粉和面制品小麦品种, 逐渐成为新的育种目标。
植物类胡萝卜素生物合成涉及一个复杂的基因调控网络[6,7]。番茄红素环化是类胡萝卜素合成途径的重要分支点。植物体内普遍存在ε-番茄红素环化酶(lycopene epsilon cyclase, LCYE)和β-番茄红素环化酶(lycopene beta cyclase, LCYB)两种番茄红素环化酶。Howitt等[8]克隆了普通小麦Lcye基因, 发现其与3B染色体上黄色素含量QTL位点共分离, 证实Lcye基因是影响籽粒黄色素含量的关键基因。董长海[9]克隆了普通小麦3B和3D染色体上的Lcye基因全长, 并针对B基因组序列差异开发了显性标记YP3B-1。Crawford和Francki[10]克隆了Lcye-3A基因, 并根据序列差异开发了功能标记e-LCY3A-3。综上所述, 目前对小麦Lcye基因研究局限于QTL定位、基因克隆和分子标记开发, 其功能和遗传调控机制尚不明确, 严重影响和制约了小麦面粉及其制品颜色性状遗传改良的育种进程。因此, 加强Lcye基因功能及其遗传调控机制研究, 有助于进一步了解小麦籽粒黄色素含量形成的分子机制, 为培育符合市场需求的小麦新品种奠定理论基础。
定向诱导基因组局部突变技术(targeting induced local lesions in genomes, TILLING)是一种将诱发产生高频率点突变的化学诱变方法与PCR筛选和高通量检测方法有效结合, 快速高效检测目标区域点突变, 通过对其表型鉴定分析, 获得基因功能的反向遗传学研究方法[11,12]。与转基因和分子标记辅助选择等分子育种技术相比, TILLING技术还具有诱变育种稳定快、仅改变少数目标性状、无需进行繁琐耗时的转基因及杂交、回交等优点, 是一种高效定向的分子育种技术[13,14]。随着测序技术及作物基因组学的迅速发展, TILLING技术将在小麦基因功能分析、遗传调控及重要农艺、品质性状遗传改良中发挥重要作用[15,16,17]。
本研究应用TILLING技术筛选甲基磺酸乙酯(ethyl methane sulfonate, EMS)诱变群体, 根据小麦Lcye基因序列设计特异引物, 通过非变性聚丙烯酰胺凝胶电泳技术检测突变位点, 获得不同等位基因变异的突变植株, 对不同等位基因的遗传群体与表型进行分析, 鉴定各突变位点对LCYE功能的影响, 揭示小麦类胡萝卜素合成途径关键基因Lcye的功能和遗传调控机制, 为面制品颜色性状遗传改良提供理论基础和种质资源。
1 材料与方法
1.1 EMS诱变群体构建
EMS诱变群体构建参照Slade等[18]方法。首先筛选一批纯合稳定、大小一致、籽粒饱满的济麦20和济麦22小麦种子, 利用1.2%浓度的EMS进行诱变处理, 产生一系列的点突变; 将处理后的种子温室种植获得突变群体M1代; M1代种子大田种植获得2491份M2代植株(1251份济麦20, 1240份济麦22)。1.2 应用TILLING技术筛选EMS突变体库
EMS突变库筛选参照Till等[19]方法, 具体步骤如下: (1) 突变体DNA池构建: 应用CTAB法[20]分别提取每个M2突变体植株的基因组DNA, 将其存放于96孔板中。利用NanoDrop-2000超微量分光光度仪(thermo scientific)测定DNA的浓度和质量。按8个样品一组将DNA进行等量混合, 构建8倍DNA混合池, 4℃保存备用。(2) Lcye特异性引物设计: 基于小麦Lcye同源基因序列差异, 设计A、B和D基因组特异性引物。利用中国春缺体-四体材料及PCR产物测序方法, 进行引物基因组特异性验证; 应用CODDLE (codons optimized to discover deleterious lesions, http://www.proweb.org/coddle/)软件分析扩增区域是否对基因功能起重要作用。最终4对Lcye特异性引物, 用于突变体筛选(表1)。(3) PCR扩增与异源双链生成: 以DNA混合池为模板进行PCR扩增, 反应体系: DNA 50 ng, PreMix 7.5 μL, 10 μmol L?1上、下游引物各1 μL, ddH2O补足至15 μL; 反应程序: 首先95℃变性5 min; 然后95℃变性30 s, 退火温度从66℃开始, 每个循环降低0.3℃, 退火45 s, 72℃延伸1.5 min, 重复35个循环; 72℃延伸10 min; 然后99℃ 10 min, 85℃1 min, 退火温度从85℃开始, 每个循环降低0.5℃, 退火时间30 s, 共99个循环; 最后16℃保温备用。如果DNA混合池样本目标片段中含有突变位点, 扩增产物经过反复变性、复性将形成野生型和突变体扩增片段的异源双链(即含有错配碱基)。(4) CEL I酶切: 用特异性识别并切割错配碱基的核酸内切酶CEL I剪切异源双链核酸分子。酶切反应体系: 10×消化缓冲液2 μL, CEL I 1 μL, 异源双链DNA 15 μL, ddH2O补足至20 μL。45℃反应20 min, 随后加入EDTA (0.25 mol L-1) 5 μL终止酶切反应。(5) 非变性聚丙烯酰胺凝胶电泳检测: 利用非变性聚丙烯酰胺凝胶电泳技术检测CEL I酶切片段, 筛选获得含有突变位点的阳性DNA混合池。对阳性DNA混合池中的每个样本DNA逐一与野生型DNA进行等量混合, 重复上述步骤, 筛选获得阳性突变体单株。(6) 突变样本克隆测序: 对突变个体PCR产物进行克隆测序, 鉴定突变类型和位置。Table 1
表1
表1利用TILLING技术筛选Lcye突变体的引物信息
Table 1
| 基因 Gene | 名称 Name | 上游引物序列 Forward primer (5′-3′) | 下游引物序列 Reverse primer (5′-3′) | 扩增长度 Length (bp) |
|---|---|---|---|---|
| Lcye-A1 | A3F-A7R | CCACAGTAGCAAAAATTAGTCA | TGCTACATTTCACAGTGGTGAA | 1450 |
| Lcye-A1 | A8F-A9R | GGTTGAAAGATATCCGTACAAC | TTTGGGTAACCGGAAAAAGGTT | 978 |
| Lcye-B1 | B4F-B6R | CACCAACCCTGCACAAAGTGCC | GGAATATAAGACCACTCCTGAG | 578 |
| Lcye-D1 | D2F-D5R | GCTGAGAAGGTACATTCTATCA | TTGAACTGGTGCACAAACAACA | 437 |
新窗口打开|下载CSV
1.3 突变位点对蛋白功能影响预测
利用PARSESNP (project aligned related sequen ces and evaluate SNPs, http://www.proweb.org/pars esnp/)软件分析突变体植株DNA序列的突变类型, 预测突变位点对LCYE功能是否造成影响。当PSSM值大于10和SIFT (sorting intolerant from tolerant)值小于0.05的氨基酸改变被认为可能对蛋白质功能造成重要影响[21,22]。1.4 突变位点功能分析
为了减小其他突变背景影响, 将含有Lcye错义突变位点的纯合M3植株与野生型植株进行杂交, 构建F2代群体, 用于分析突变位点对基因表达水平及蛋白质功能的影响。F2群体于2014—2015年度种植于北京, 行长2 m, 行距25 cm, 每行20株, 每个F2群体种20行, 田间管理采用常规方法。利用克隆测序的方法, 鉴定F2群体中每个株系Lcye基因型(纯合突变型、杂合突变型和野生型)。每种基因型各选取10个生长发育进程一致的生物学重复, 记录开花期, 分别采集花后7、14、21和28 d籽粒, 立即置于液氮中, -80℃保存, 用于RNA提取和Lcye基因表达水平分析。单株收获, 成熟籽粒-20℃保存, 用于黄色素含量测定。
利用实时定量PCR (quantitative real-time PCR, qRT-PCR)技术, 测定F2群体中纯合突变型、杂合突变型和野生型植株籽粒不同发育时期(花后7、14、21和28 d) Lcye基因及其各同源基因表达量, 分析突变位点对Lcye基因表达水平的影响。具体步骤为: 应用RNAprep Pure植物总RNA提取试剂盒(TIANGEN Biotech, 中国北京)分别提取花后7、14、21和28 d籽粒总RNA, 纯合突变型、杂合突变型和野生型植株各3个生物学重复; 利用PrimeScript RT Reagent试剂盒(TaKaRa Bio Inc., Otsu, Japan)将其反转录成cDNA, 置于-20℃保存备用; 基于小麦Lcye同源基因cDNA序列间的保守性和差异性, 设计Lcye基因的保守性引物和A、B、D基因组特异性引物(表2), 对其进行溶解曲线分析和qRT-PCR产物克隆测序, 验证引物保守性和特异性, 普通小麦β-actin基因(AB181991)作为内参基因; 反应体系: LightCycler FastStart DNA Master SYBR Green (Roche Applied Sciences, USA) 10 μL, 上下游引物0.5 μmol L?1, cDNA 50 ng, ddH2O补充至20 μL。反应程序: 95℃ 10 min; 95℃ 15 s, 60℃ 20 s和72℃ 20 s, 共40个循环; 应用公式2?ΔΔCT计算目标基因相对表达量[23]。首先, 利用同一样本中β-actin基因转录水平来校正目标基因相对表达水平; 其次将野生型植株花后28 d籽粒目标基因相对表达量设为1, 计算不同基因型植株的籽粒不同发育时期目标基因的相对表达量。每个样本3次技术重复, 基因相对表达量用平均值±标准误差(standard error, SE)表示。
Table 2
表2
表2Lcye基因的qRT-PCR引物信息
Table 2
| 基因Gene | 名称Name | 序列Sequence (5'-3') |
|---|---|---|
| Lcye-all | Lcye-all-F2 | TGACCACYGAATATCCAGTTGC |
| Lcye-all-R6 | AGTTTTCTTTGAGGAAACATGC | |
| Lcye-A1 | Lcye-A1-F7 | GTTGCTGAGAAGATGCAACGAT |
| Lcye-A1-R7 | CAAAGTATCTTGCGGTCCCTTT | |
| Lcye-B1 | Lcye-B1-F3 | ATCTCCAGATGGACATCGAGTG |
| Lcye-B1-R3 | TCCAACCTCATACTCTAGAAGT | |
| Lcye-D1 | Lcye-D1-F3 | TTGGCCCTGATCTTCCATTC |
| Lcye-D1-R1 | ATATACTACTCGATGTCCATCA | |
| β-actin | Actin-F | CTGATCGCATGAGCAAAGAG |
| Actin-R | CCACCGATCCAGACACTGTA |
新窗口打开|下载CSV
测定F2群体中纯合突变型、杂合突变型与野生型植株籽粒黄色素含量, 分析突变位点对LCYE蛋白功能的影响。每种基因型测定5个单株, 平均值作为该基因型的黄色素含量。籽粒黄色素含量的测定参照AACC方法14-50, 稍作改动。称取1 g全麦粉, 用水饱和正丁醇溶液(5∶1)振荡提取1 h; 2823 × g离心10 min。应用分光光度计测定上清液在436.5 nm处吸光值, 计算黄色素含量。每个样本3次技术重复, 黄色素含量用平均值±SE表示。
1.5 LCYE功能结构域预测
利用NCBI (national center for biotechnology information, http://www.ncbi.nlm.nih.gov/)数据库, 获得目前已知的27个物种Lcye基因的cDNA序列(附表1), 利用MEME Suite 5.1.0 (http://meme-suite. org/)预测LCYE功能结构域, 分析各突变位点在结构域中的分布情况。Supplementary table 1
附表1
附表127个物种Lcye基因cDNA序列信息
Supplementary table 1
| 种名 Species name | GenBank登录号 GenBank accession number | 种名 Species name | GenBank登录号 GenBank accession number |
|---|---|---|---|
| 拟南芥Arabidopsis thaliana | NM_125085 | 小立碗藓Physcomitrella patens | XM_001753846 |
| 二穗短柄草Brachypodium distachyon | XM_003569209 | 碧桃Prunus persica | XM_007203578 |
| 大白菜Brassica rapa | XM_009133907 | 蓖麻Ricinus communis | XM_002514090 |
| 荠菜Capsella rubella | XM_006280236 | 谷子Setaria italica | XM_004969360 |
| 莱茵衣藻Chlamydomonas reinhardtii | XM_001696477 | 番茄Solanum lycopersicum | EU533951 |
| 柑橘Citrus sinensis | AY533827 | 马铃薯Solanum tuberosum | XM_006353482 |
| 黄瓜Cucumis sativus | XM_004157912 | 高粱Sorghum bicolor | XM_002455793 |
| 草莓Fragaria vesca | XM_004287534 | 可可Theobroma cacao | XM_007012707 |
| 大豆Glycine max | XM_003533727 | 普通小麦Triticum aestivum | EU649785 |
| 大麦Hordeum vulgare | AK371513 | 圆锥小麦Triticum turgidum | GAKM01004311 |
| 亚麻Linum usitatissimum | KC565894 | 乌拉尔图小麦Triticum urartu | GAKL01018490 |
| 苹果Malus domestica | XM_008389970 | 葡萄Vitis vinifera | JQ319637 |
| 蒺藜苜蓿Medicago truncatula | XM_003595195 | 玉米Zea mays | EU924262 |
| 水稻Oryza sativa | NM_001049945 |
新窗口打开|下载CSV
1.6 统计分析
应用Student’s t检验对F2群体中纯合突变型、杂合突变型和野生型植株籽粒Lcye基因表达差异和黄色素含量进行显著性分析。Lcye基因在EMS诱变群体中的突变密度=点突变数/检测总碱基数。2 结果与分析
2.1 EMS突变体库筛选
应用TILLING技术, 在2491份M2代EMS诱变群体中共检测到21个Lcye基因的突变植株(表3)。采用非变性聚丙烯酰胺凝胶电泳技术分离CEL I酶切产物, 扩增片段150 bp内的错配超出检测范围, 将无法被检测到, 因此推测Lcye基因在该EMS诱变群体中的突变密度为1/266.1 kb。Table 3
表3
表3利用TILLING技术筛选获得Lcye突变体信息
Table 3
| 基因 Gene | 突变体编号 Number of M2 plant | 外显子/内含子 Exon/intron | 核苷酸改变 Nucleotide change | 密码子改变 Codon change | 氨基酸改变 Amino acid change | 基因型 Zygosity |
|---|---|---|---|---|---|---|
| Lcye-A1 | M091034 | Intron | C1184T | Hom | ||
| M091686 | Intron | C1243T | Hom | |||
| M091772 | Intron | C1418T | Hom | |||
| M092043 | Intron | C1478T | Hom | |||
| M092852 | Intron | C3222T | Hom | |||
| M090431 | Intron | C858T | Het | |||
| M090631 | Intron | T1461C | Het | |||
| M090897 | Intron | G1068A | Hom | |||
| M091996 | Intron | G1575A | Hom | |||
| M090147 | Intron | G3073A | Hom | |||
| M091648 | Exon | G3284A | GGA→GAA | G392E | Hom | |
| M090201 | Exon | G3306A | TTA→TTG | L399= | Hom | |
| Lcye-B1 | M091626 | Intron | G2406A | Hom | ||
| Lcye-D1 | M092404 | Intron | C2014T | Het | ||
| M091884 | Intron | C2017T | Het | |||
| M090945 | Exon | C2086T | TAC→TAT | Y214= | Hom | |
| M092089 | Exon | C2087T | CTC→TTC | L215F | Hom | |
| M091328 | Exon | C2121T | CCT→CTT | P226L | Het | |
| M090815 | Exon | C2202T | TCT→TTT | S253F | Het | |
| M092230 | Exon | G2195A | GCA→ACA | A251T | Hom | |
| M091075 | Exon | G2262A | GGT→GAT | G273D | Hom |
新窗口打开|下载CSV
突变体克隆测序和序列分析显示, 核苷酸从C到T的突变频率为57.1%, G到A的突变频率为38.1%, 还检测到1个T到C的特异突变位点(表3)。根据突变位置分类, 8个位于外显子区, 13个分布于内含子区(图1)。外显子区域的点突变又分为6个错义突变和2个同义突变(表3)。
图1
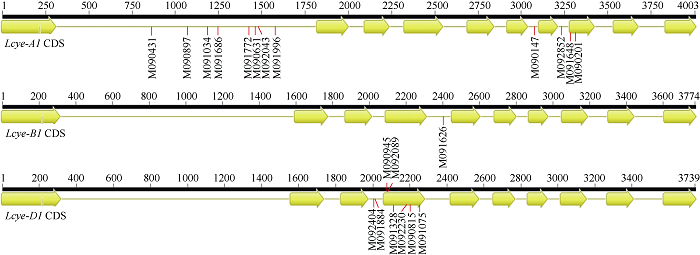 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Lcye突变位点分布图
黄色箭头代表外显子; 连线代表内含子。
Fig. 1Distribution of mutation sites in Lcye
Exons are represented by yellow arrows and introns by connecting lines.
2.2 突变位点对蛋白质功能影响预测
PARSESNP软件分析结果显示, 错义突变体M090815 (C2202T)和M091648 (G3284A)的PSSM值分别为26.9和27.7, SIFT值均为0, 推测这些突变位点可能对LCYE蛋白功能产生严重影响(表4)。Table 4
表4
表4突变位点对蛋白功能影响严重度预测
Table 4
| 基因 Gene | 突变体编号 Number of M2 plant | 核苷酸改变 Nucleotide change | 氨基酸改变 Amino acid change | PSSM值 PSSM difference | SIFT值 SIFT score |
|---|---|---|---|---|---|
| Lcye-D1 | M090815 | C2202T | S253F | 26.9 | 0 |
| Lcye-A1 | M091648 | G3284A | G392E | 27.7 | 0 |
新窗口打开|下载CSV
2.3 突变体Lcye基因表达和黄色素含量分析
将6个纯合错义突变体与野生型植株杂交, 构建F2代群体, 分析突变位点对Lcye基因表达水平和籽粒黄色素含量的影响。结果表明, 6个F2群体中, 仅M090815 (C2202T)突变位点显著降低了Lcye基因表达和黄色素含量(图2和图3), 表明该突变位点对LCYE蛋白功能产生重要影响。图2
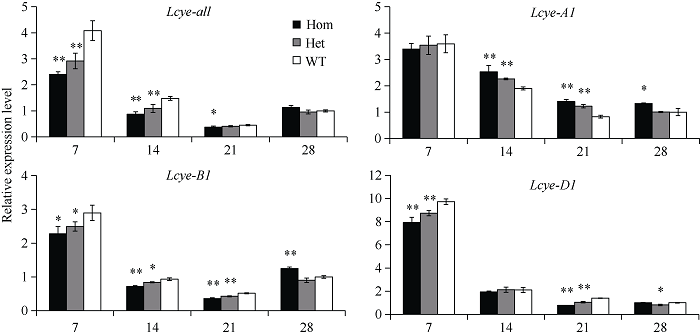 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2突变体M090815的F2群体中不同基因型植株籽粒Lcye及其同源基因的相对表达量
* 和**分别代表0.05和0.01显著水平的差异。Hom: 纯合突变型; Het: 杂合突变型; WT: 野生型。
Fig. 2The relative expression analysis of Lcye and its homoeologs in grains of three genotypes in F2 population derived from the homozygous M090815 mutant crossed with the wild-type plant during grain development at 7, 14, 21, and 28 days post anthesis
* P < 0.05, ** P < 0.01. Hom: homozygous mutants; Het: heterozygous mutants; WT: wild-type genotypes.
图3
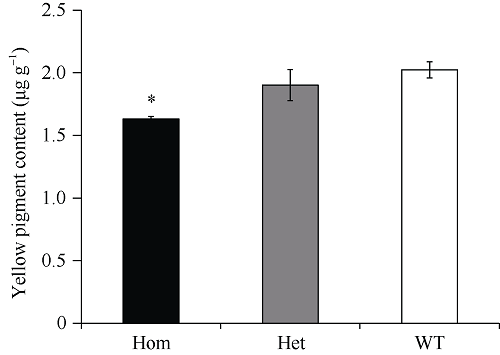 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3突变体M090815的F2群体中不同基因型植株籽粒黄色素含量
* 代表0.05显著水平的差异。Hom: 纯合突变型; Het: 杂合突变型; WT: 野生型。
Fig. 3Yellow pigment content of different genotypes in F2 populations derived from homozygous M090815 mutant crossed with wild-type plant
* P < 0.05. Hom: homozygous mutants; Het: heterozygous mutants; WT: wild-type genotypes.
突变体M090815构建的F2群体中, 不同基因型植株籽粒Lcye及其同源基因表达水平如图2所示。花后7~21 d, 纯合突变型植株籽粒Lcye基因总表达量降低至野生型的59.0%~83.0%, 杂合型降低至71.5%~91.1%, 花后28 d各基因型表达差异不显著。除了Lcye-B1纯合突变型在花后28 d表达量显著高于野生型外, 花后各时期Lcye-B1和Lcye-D1基因表达显示相似的下降趋势, 纯合突变型植株表达量降低至野生型的70.0%~78.7%和55.5%~92.0%, 杂合型降低至83.1%~90.0%和75.2%~90.0%。Lcye-A1在花后7 d, 各基因型表达量差异不显著, 花后14~28 d, 表现出补偿效应, 纯合突变型比野生型表达量高33.4%~70.0%, 杂合型高0.7%~ 48.1%。相应地, 在突变体M090815构建的F2群体中, 纯合突变型植株成熟籽粒黄色素含量显著低于杂合突变型和野生型植株(图3, 1.63 vs. 1.90和2.02 μg g?1)。
2.4 LCYE功能结构域预测
基于拟南芥(Arabidopsis thaliana)、水稻(Oryza sativa)、大麦(Hordeum vulgare)等27个物种已知的Lcye基因cDNA序列(附表1), 利用MEME Suite 5.1.0预测LCYE功能结构域, 共检测到3个结构域(图4)。6个错义突变中, M090815 (C2202T)和M092230 (G2195A)突变位点恰好位于结构域1内, 且M090815突变位点在27个物种中非常保守, M092230突变位点存在G、A和U三种碱基变异。图4
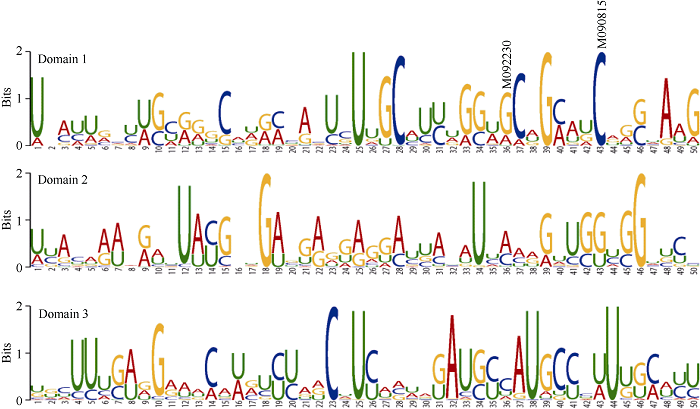 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4Lcye功能结构域预测
Fig. 4Functional domains predication of Lcye by MEME
3 讨论
3.1 LCYE调控小麦籽粒类胡萝卜素合成
小麦籽粒类胡萝卜素影响面制品营养品质和表观色泽。番茄红素环化是类胡萝卜素合成途径中一个重要的分节点, 番茄红素在LCYB催化下, 经β-β途径生成β-胡萝卜素、玉米黄质、花药黄质和堇菜黄质等叶黄素类物质; 在LCYE和LCYB共同催化下, 经β-ε途径生成α-胡萝卜素和叶黄体素。类胡萝卜素含量和组成在小麦籽粒发育过程中是动态变化的, 具有复杂的遗传调控网络[8]。β-β途径在籽粒发育早期占有较高表达水平, 随着籽粒生长发育表达量和所占比例逐渐下降, 成熟籽粒几乎检测不到玉米黄质、环氧玉米黄质和堇菜黄质等类胡萝卜素。相反, β-ε途径在整个籽粒发育过程中稳定表达, 叶黄体素成为构成成熟籽粒类胡萝卜素的主要成分[8]。通过调节LCYB和LCYE的相对活性和相对含量可以决定番茄红素转化为α-胡萝卜素和β-胡萝卜素的比例。在番茄Delta突变体中, Lcye转录水平提高, 果实内δ-胡萝卜素含量大量增多; 而在番茄Beta突变体中, Lcyb过表达, 果实中累积大量的β-胡萝卜素[24]。对比两种突变体的差异进一步证实番茄果实中类胡萝卜素的积累主要是相关基因在转录水平上差异表达的结果[25]。
Richaud等[26]应用TILLING技术, 筛选硬粒小麦Lcye突变体, W437*(Lcye-A1)突变位点显著增加突变体叶片中β-胡萝卜素和类胡萝卜素的含量, 但对籽粒中类胡萝卜素含量无显著影响。本研究利用TILLING技术筛选普通小麦EMS突变体库, 获得Lcye基因一系列等位变异, 研究其对基因表达和籽粒黄色素含量的影响, 为基因功能研究和面制品颜色性状改良奠定理论基础和提供重要种质资源。
3.2 Lcye突变体筛选
TILLING技术有效结合了高频率点突变与现代分析检测技术, 仅需要较小的突变群体便可快速有效地筛选到一系列目标基因点突变, 逐渐成为植物功能基因组学、作物遗传育种以及自然资源遗传多样性评估等研究的重要手段[27,28]。目前, 该技术已广泛应用于水稻[29]、玉米[30]、小麦[31,32]、大麦[33]、高粱[34]等20多种作物。研究表明小麦基因突变频率远远高于拟南芥、水稻和玉米等植物, TILLING技术在小麦等麦类作物中的应用潜力更为巨大、前景更为光明。对于小麦等基因组复杂的多倍体物种, 基因多拷贝是限制TILLING技术高效应用的一个瓶颈。设计特异性引物至关重要, 否则会影响检测效率。小麦Lcye基因A、B和D同源基因间序列相似性非常高(89.1%~97.0%), 设计特异性引物非常困难。结合CODDLE程序给出的EMS诱导植株产生有害突变的可能范围, 最终筛选到4对引物用于Lcye突变体检测(表1)。
酶切产物的检测方法有多种, 一般采用高效液相色谱技术、聚丙烯酰胺凝胶电泳、琼脂糖凝胶电泳以及双色红外荧光检测技术[35,36,37,38]。本研究采用非变性聚丙烯酰胺凝胶电泳检测技术, 既不使用荧光标记引物, 又不使用昂贵的变性凝胶电泳成像系统, 有效地简化了实验流程, 降低了实验成本, 在一定程度上扩大TILLING的应用范围和工作效率。共筛选到21个Lcye突变位点, 推测Lcye在EMS诱变群体中的突变频率为(1/266.1 kb), 远低于Uauy等[37]应用非变性聚丙烯酰胺凝胶技术检测到的六倍体小麦突变频率(1/38 kb)。这可能是由于基因目标区域G/C含量不同或者非变性聚丙烯酰胺凝胶电泳检测技术对8样本DNA混池的检测灵敏度较低所致。
3.3 影响LCYE功能的重要调控位点
基于拟南芥、水稻、大麦等27个物种Lcye的cDNA序列, 利用MEME预测LCYE功能结构域, 结果显示M090815 (C2202T)和M092230 (G2195A)突变位点恰好位于结构域1内(图4)。M092230 (G2195A)突变位点在自然界物种进化中存在G、A和U三种碱基变异, 因此推测G到A核苷酸突变对LCYE功能未产生影响。而M090815 (C2202T)突变位点在27个物种中高度保守, 推测该位点对LCYE功能具有重要影响。PARSESNP软件预测显示M090815 (C2202T)和M091648 (G3284A)突变位点可能对蛋白功能产生严重影响(表4), 进一步表明M090815 (C2202T)突变位点对LCYE蛋白功能的重要性。M090815构建的F2群体中, 与野生型植株相比, 纯合突变型植株籽粒黄色素含量显著下降(图3), 证实M090815 (C2202T)突变位点对LCYE功能具有重要影响。然而在M091648构建的F2群体中纯合突变型与野生型植株相比籽粒黄色素含量差异并不显著。这种不一致性可能是由于PARSESN软件是基于序列同源性及氨基酸物理特性进行突变位点对蛋白功能影响严重度的预测, 而有些位点可能并不参与蛋白功能调控[21,22]。对M090815构建的F2群体进行Lcye及其同源基因表达水平分析显示, 与野生型植株相比, 纯合突变型植株籽粒Lcye基因总表达量显著降低(图2)。籽粒发育各时期, 除Lcye-B1在纯合突变型植株中表达水平在花后28 d高于野生型外, Lcye-B1和Lcye-D1基因表达水平均呈下降趋势。而Lcye-A1在花后7 d, 各基因型植株表达差异不显著, 花后14~28 d, 表现出补偿效应。推测Lcye-B1和Lcye-D1基因表达受协同调控, 而Lcye-A1表达调控机制可能与之不同。序列分析发现, Lcye-A1与Lcye-B1和Lcye-D1的序列相似性分别为89.1%和89.2%, 而Lcye-B1和Lcye-D1基因序列相似性高达97.0%, 为上述推测提供间接支持。鉴于上述结果只通过一个F2群体获得, 因此仍需进一步研究。
3.4 分子育种
种质资源缺乏成为小麦面粉及其制品颜色改良的“瓶颈”, 严重影响我国小麦品种面制品颜色性状的遗传改良。EMS诱变可以创造大量的等位变异, 且在后代稳定遗传, 由于不涉及转基因操作, 获得的优异突变体可以直接用于育种实践[39,40]。在我国, 面制品以蒸煮为主, 细腻洁白的面粉备受消费者青睐。本研究采用TILLING技术筛选获得的显著降低籽粒黄色素含量的M090815突变体, 该突变体农艺性状与野生型济麦20无显著差异, 可作为面粉颜色遗传改良的重要种质资源。此外, 济麦20为高产优质面包、面条兼用型强筋小麦; 济麦22为超高产稳产广适多抗小麦品种, 两个均为大面积推广种植的优良品种, 综合性状良好。以上述两个品种作诱变基础材料创制的优异突变体, 更适宜于用作杂交亲本, 以期快速培育出高产优质小麦新品种。竞争性等位基因特异性PCR (Kompetitive Allele Specific PCR, KASP)是基于引物末端碱基的特异性匹配对SNP进行双等位基因分型技术, 具有高通量、准确性高、稳定性好和检测成本低等优点, 是目前广泛使用的SNP分型方法[41,42]。因此, 在后续工作中针对M090815突变体Lcye-D1基因C2202T的SNP变异位点开发KSAP标记, 以期为利用M090815突变体对面制品颜色性状进行高效、可靠的分子标记辅助育种提供技术支撑。
4 结论
本研究以济麦20和济麦22的EMS诱变群体为材料, 利用TILLING技术筛选小麦类胡萝卜素合成关键基因Lcye的突变体, 共获得21个Lcye基因的点突变。生物信息学分析、Lcye基因表达水平和黄色素含量测定均显示, M090815 (C2202T)突变位点对LCYE蛋白功能具有重要影响。M090815突变位点显著降低Lcye基因表达水平, 且Lcye-B和Lcye-D基因表达降低趋势相似, 而Lcye-A在花后14~28 d表现出补偿效应。本研究获得的突变体不仅为开展Lcye基因功能研究提供了良好的试验材料, 也为面制品颜色性状遗传改良提供了丰富的种质资源。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1071/AR01048URL [本文引用: 1]
DOI:10.1021/jf030404lURLPMID:14664553 [本文引用: 1]

DOI:10.1021/jf040351nURLPMID:15796565 [本文引用: 1]
Color is an important parameter involved in the definition of semolina and pasta quality. This character is mainly due to natural pigments (carotenoids) that are present at different levels in cereals and cereal products, due to botanical origin, growing conditions, distribution in the kernel, and technological processes. In food industries, color measurements are usually performed by means of automatic instruments that are rapid and safe, as alternatives to the chemical extraction methods. In this study, automatic measurements (CIE, color-space system L, a, b), water-saturated butanol (WSB), and HPLC determinations have been applied to evaluate the carotenoid content in whole meals and respective semolina samples produced from wheat cultivated in the years 2001 and 2002. In whole meals, total carotenoids, determined by HPLC, were about 3.0 microg/g (2001) and 3.5 microg/g (2002) calculated on dry weight (dw) and about 3.0 and 3.2 microg/g dw in corresponding semolina samples. The b values for the same period were 19.78 and 15.75, respectively, in raw materials and 20.03-21.67 in semolina. Results have confirmed lutein and beta-carotene as the main components mainly responsible for the yellow color in wheat grains. The ability of the index b to express natural dyeing was dependent on sample characteristics as demonstrated by the relationships found between this index and pigments, although the best correlation resulted between HPLC and WSB.
DOI:10.1007/s11033-009-9922-7URL [本文引用: 1]
TaPDS and TaZDS were cloned from wheat (Triticum aestivum cv. Chinese Spring) respectively, using the rapid amplification of cDNA ends (RACE) approach. The cDNA of TaPDS sequence was 2076bp long, containing a 1731bp open reading frame (ORF) which deduced protein having 576 amino acid residues with predicted molecular mass of 64.3kDa and having a putative transit sequence for plastid targeting in the N-terminal region. While the cDNA sequence of TaZDS was 2150bp long, contained an ORF sequence of 1707bp, and encoded a putative protein of 568 amino acid residues with an estimated molecular mass of 62.5kDa. Phylogenetic analysis demonstrated that TaPDS and TaZDS showed high homology with other PDSs and ZDSs in higher plant species, respectively. Moreover, sequences analysis also showed a high degree of conservation among plant PDSs and ZDSs. The deduced TaPDS and TaZDS protein both have the dinucleotide binding domain and conserved regions characteristic of other carotene desaturases. Analysis of the expression pattern of wheat TaPDS and TaZDS in different tissues revealed that the transcripts levels were higher in leaves and flowers petals, followed by in inflorescences, and were nearly absent in the roots and seeds. Southern analysis of genomic DNA indicated that the wheat TaPDS and TaZDS probably belong to a low-copy-number gene family.]]>
DOI:10.3724/SP.J.1006.2016.00706URL [本文引用: 1]
(ultra performance liquid chromatography, UPLC)测定其组分的技术体系。研究表明,用含0.1% BHT的正己烷∶丙酮(80∶20, v/v)混合液作为提取液,结合恒温振荡法(300转 min–1, 35℃, 振荡1 h)的提取效果最好。以YMC C30胡萝卜素分析专用色谱柱(高100 mm, 直径4.6 mm, 粒径3 µm)洗脱,乙腈∶甲醇∶水∶三乙胺(81∶14∶5∶0.05, v/v/v/v)为流动相A,甲醇∶乙酸乙酯∶三乙胺(68∶32∶0.05, v/v/v)为流动相B,流速0.4 mL min–1,柱温35℃,分离时间25 min;以二极管阵列检测器(photodiode array, PDA)在450 nm波长下,能有效检测叶黄素、玉米黄质和β-胡萝卜素3种组分。该技术体系可作为普通小麦类胡萝卜素组分及营养品质研究的有效方法。]]>
[本文引用: 1]
DOI:10.1016/j.tplants.2010.02.003URLPMID:20303820 [本文引用: 2]
Carotenoids are a diverse group of colourful pigments naturally found in plants, algae, fungi and bacteria. They play essential roles in development, photosynthesis, root-mycorrhizal interactions and the production of phytohormones, such as abscisic acid and strigolactone. Carotenoid biosynthesis is regulated throughout the life cycle of a plant with dynamic changes in composition matched to prevailing developmental requirements and in response to external environmental stimuli. There are key regulatory nodes in the pathway that control the flux of metabolites into the pathway and alter flux through the pathway. The molecular nature of the mechanisms regulating carotenoid biosynthesis, including evidence for metabolite feedback, transcription and epigenetic control as well as their accumulation, storage and degradation will be the focus of this review.
DOI:10.1016/j.abb.2010.07.028URLPMID:20688043 [本文引用: 1]
Carotenoids fulfill many processes that are essential for normal growth and development in plants, but they are also responsible for the breathtaking variety of red-to-yellow colors we see in flowers and fruits. Although such visual diversity helps to attract pollinators and encourages herbivores to distribute seeds, humans also benefit from the aesthetic properties of flowers and an entire floriculture industry has developed on the basis that new and attractive varieties can be produced. Over the last decade, much has been learned about the impact of carotenoid metabolism on flower color development and the molecular basis of flower color. A number of different regulatory mechanisms have been described ranging from the transcriptional regulation of genes involved in carotenoid synthesis to the control of carotenoid storage in sink organs. This means we can now explain many of the natural colorful varieties we see around us and also engineer plants to produce flowers with novel and exciting varieties that are not provided by nature.
DOI:10.1007/s10142-009-0121-3URL [本文引用: 3]
Endosperm carotenoid content in wheat is a primary determinant of flour colour and this affects both the nutritional value of the grain and its utility for different applications. Utilising wheat rice synteny two genes, ε-cyclase (ε-LCY) and phytoene synthase (Psy-A1), were identified as candidate genes for two of the QTL affecting lutein content in wheat endosperm. Analysis of the sequence changes in ε-LCY and Psy-A1 revealed possible causal mechanisms for both QTL. A point mutation in ε−LCY results in the substitution of a conserved amino acid in the high lutein allele. This substitution has been observed in high lutein-accumulating species from the Gentiales order. In Psy-A1, a sequence duplication at the end of exon 2 creates a new splice site and causes alternative splicing of the transcript and activation of a cryptic exon, resulting in four different transcripts: a wild-type transcript, two transcripts with early terminations and a transcript that would produce an in-frame, albeit longer protein. Only the wild-type splice variant produced an enzymatically active protein and its mRNA abundance was reduced by titration with the other splice variants. This reduction in wild-type mRNA is argued to result in a reduction in PSY protein and thus carotenoid content in wheat.]]>
河北农业大学硕士学位论文,
[本文引用: 1]
MS Thesis of Agricultural University of Hebei, Baoding, Hebei,
[本文引用: 1]
DOI:10.1007/s11032-012-9812-xURL [本文引用: 1]
In: Grotewold E ed.
[本文引用: 1]
DOI:10.11869/j.issn.100-8551.2014.02.0224URL [本文引用: 1]
TILLING技术(Targeting Induced Local Lesions IN Genomes)经过十多年的快速发展,已经应用到多种植物点突变高通量筛选与基因功能研究中,检测方法也逐渐趋于多样化,直接测序、毛细管电泳、高分辨熔解曲线等检测方法均得到了较多的应用。与此同时EcoTILLING、DeTILLING及iTILLING等相关技术的发展扩大了TILLING技术的应用范围。本文着重介绍了目前TILLING分析中用到的不同检测方法及相关生物信息学工具,详细评述了TILLING技术在模式植物、主要粮食作物、油料作物、蔬菜等其它作物中的应用与研究进展。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.7606/j.issn.1009-1041.2013.05.036URL [本文引用: 1]
Tilling(Targeting induced local lesions in genomes)技术是近年来兴起的利用化学诱变或者物理诱变方法产生一系列的点突变,经过PCR快速筛选突变体,进而验证基因功能的方法。近年来,Tilling技术被用于多种生物的研究中。对小麦这种基因组庞大、遗传背景复杂的作物来说,Tilling技术在分析基因功能、挖掘重要基因资源和改良作物品质方面比传统的转基因方法有更大的优势。然而,Tilling技术在小麦中的应用还存在筛选工作量大等问题。本文介绍了Tilling技术的操作流程、问题改进及在小麦基因功能研究和品质改良方面的研究现状。
[本文引用: 1]
DOI:10.7606/j.issn.1009-1041.2010.01.035URL [本文引用: 1]
TILLING(Targeting induced local lesions in genomes,定向诱导基因组局部突变技术)是一种高通量的等位变异创制和突变体快速鉴定技术,其实质是将传统的化学诱变方法和突变的高效筛选有效结合的反向遗传学研究方法。其技术原理是将传统的酶切技术与PCR技术相结合后采用红外双色荧光系统进行结果鉴定,从而筛选出相应的突变体。传统的TILLING技术主要用于筛选由人工诱导产生的突变体。Ecotilling技术由TILLING技术延伸而来,主要用于鉴定自然界中已经存在的突变体,其与传统的TILLING技术的区别主要为构建DNA池时略有差异。随着该项技术在拟南芥等模式植物中的成功应用,越来越多的人开始将其用于基因组较大的植物之中。本文对近年来TILLING技术在麦类作物中的应用进行了分析,并通过比较不同植物突变体库中的突变频率发现,经EMS处理的小麦等麦类作物突变体库中的突变频率显著高于其他植物,因此相信,TILLING技术将会作为一种常规手段在麦类作物尤其是普通小麦改良中得到越来越广泛的应用。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12870-016-0916-zURLPMID:27769185 [本文引用: 1]
BACKGROUND: Phytoene synthase 1 (PSY1) is the most important regulatory enzyme in carotenoid biosynthesis, whereas its function is hardly known in common wheat. The aims of the present study were to investigate Psy1 function and genetic regulation using reverse genetics approaches. RESULTS: Transcript levels of Psy1 in RNAi transgenic lines were decreased by 54-76 % and yellow pigment content (YPC) was reduced by 26-35 % compared with controls, confirming the impact of Psy1 on carotenoid accumulation. A series of candidate genes involved in secondary metabolic pathways and core metabolic processes responded to Psy1 down-regulation. The aspartate rich domain (DXXXD) was important for PSY1 function, and conserved nucleotides adjacent to the domain influenced YPC by regulating gene expression, enzyme activity or alternative splicing. Compensatory responses analysis indicated that three Psy1 homoeologs may be coordinately regulated under normal conditions, but separately regulated under stress. The period 14 days post anthesis (DPA) was found to be a key regulation node during grain development. CONCLUSION: The findings define key aspects of flour color regulation in wheat and facilitate the genetic improvement of wheat quality targeting color/nutritional specifications required for specific end products.
DOI:10.1038/nbt1043URLPMID:15580263 [本文引用: 1]
We report the use of TILLING (targeting induced local lesions in genomes), a reverse genetic, nontransgenic method, to improve a quality trait in a polyploid crop plant. Waxy starches, composed mostly of amylopectin, have unique physiochemical properties. Wheat with only one or two functional waxy genes (granule-bound starch synthase I, or GBSSI) produces starch with intermediate levels of amylopectin. We have identified 246 alleles of the waxy genes by TILLING each homoeolog in 1,920 allohexaploid and allotetraploid wheat individuals. These alleles encode waxy enzymes ranging in activity from near wild type to null, and they represent more genetic diversity than had been described in the preceding 25 years. A line of bread wheat containing homozygous mutations in two waxy homoeologs created through TILLING and a preexisting deletion of the third waxy homoeolog displays a near-null waxy phenotype. This approach to creating and identifying genetic variation shows potential as a tool for crop improvement.
DOI:10.1038/nprot.2006.329URLPMID:17406493 [本文引用: 1]
We describe Targeting-Induced Local Lesions IN Genomes (TILLING), a reverse-genetic strategy for the discovery and mapping of induced mutations. TILLING is suitable for essentially any organism that can be mutagenized. The TILLING procedure has also been adapted for the discovery and cataloguing of natural polymorphisms, a method called Ecotilling. To discover nucleotide changes within a particular gene, PCR is performed with gene-specific primers that are end-labeled with fluorescent molecules. After PCR, samples are denatured and annealed to form heteroduplexes between polymorphic DNA strands. Mismatched base pairs in these heteroduplexes are cleaved by digestion with a single-strand specific nuclease. The resulting products are size-fractionated using denaturing polyacrylamide gel electrophoresis and visualized by fluorescence detection. The migration of cleaved products indicates the approximate location of nucleotide polymorphisms. Throughput is increased and costs are reduced by sample pooling, multi-well liquid handling and automated gel band mapping. Once genomic DNA samples have been obtained, pooled and arrayed, thousands of samples can be screened daily.
[本文引用: 1]
DOI:10.1093/nar/gkg509URLPMID:12824425 [本文引用: 2]
Single nucleotide polymorphism (SNP) studies and random mutagenesis projects identify amino acid substitutions in protein-coding regions. Each substitution has the potential to affect protein function. SIFT (Sorting Intolerant From Tolerant) is a program that predicts whether an amino acid substitution affects protein function so that users can prioritize substitutions for further study. We have shown that SIFT can distinguish between functionally neutral and deleterious amino acid changes in mutagenesis studies and on human polymorphisms. SIFT is available at http://blocks.fhcrc.org/sift/SIFT.html.
DOI:10.1093/nar/gkg574URLPMID:12824424 [本文引用: 2]
PARSESNP is a tool for the display and analysis of polymorphisms in genes. Using a reference DNA sequence, an exon/intron position model and a list of polymorphisms, it determines the effects of these polymorphisms on the expressed gene product, as well as the changes in restriction enzyme recognition sites. It shows the locations and effects of the polymorphisms in summary on a stylized graphic and in detail on a display of the protein sequence aligned with the DNA sequence. The addition of a homology model, in the form of an alignment of related protein sequences, allows for prediction of the severity of missense changes. PARSESNP is available on the World Wide Web at http://www.proweb.org/parsesnp/.
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1021/jf991020rURLPMID:10995282 [本文引用: 1]
Changes in the biosynthesis of individual carotenoid pigments have been investigated during fruit ripening of five cultivars of red pepper (Capsicum annuum L.): Mana, Numex, Belrubi, Delfin, and Negral (a chlorophyll-retaining mutant when ripe). The study was carried out throughout the ripening process, and with special emphasis on the ripe stage, to discover possible differences between cultivars and to characterize these by their carotenoid pattern and content for selecting the best varieties for breeding programs. Ripening fruit of the five cultivars showed the typical and characteristic pattern of carotenoid biosynthesis for the Capsicum genus. In the five cultivars, lutein and neoxanthin, both characteristic chloroplast pigments, decreased in concentration with ripening and eventually disappeared. beta-Carotene, antheraxanthin, and violaxanthin increased in concentration, and other pigments were biosynthesized de novo: zeaxanthin, beta-cryptoxanthin, capsanthin, capsorubin, capsanthin-5,6-epoxide, and cucurbitaxanthin A. A pool of zeaxanthin stands out of the rest of pigment during ripening, which reveals the importance of this pigment as a branching point in the carotenoid biosynthesis in Capsicum. Quantitatively, Negral cultivar showed the highest increase in total carotenoid content (48. 39-fold), followed by Mana and Delfin with 38.03- and 36.8-fold, respectively, and by Belrubi and Numex with 28.03- and 23.48-fold, respectively. In all the red varieties, there was an inverse relationship between total carotenoid content and the red to yellow isochromic pigment fraction ratio (R/Y) and the capsanthin-to-zeaxanthin ratio (Caps/Zeax). This seems to be related to the carotenogenic capacity of the cultivar, and thus selection and breeding should not only seek a higher total carotenoid content but also attempt to increase these ratios. In the present study, the cultivar Mana had the highest total carotenoid content (13 208 mg/kg dwt), but the lowest R/Y (1.25) and Caps/Zeax (3.38) ratios, which are therefore the parameters to improve. The cultivar Negral had a high carotenoid content (8797 mg/kg dwt) and high R/Y and Caps/Zeax ratios and could be used for transfer of these characters in direct crosses with the cultivar Mana. The cultivar Numex had the highest Caps/Zeax ratio (7.17) and is thus an ideal progenitor for this character.
DOI:10.1073/pnas.190177497URLPMID:10995464 [本文引用: 1]
Carotenoid pigments in plants fulfill indispensable functions in photosynthesis. Carotenoids that accumulate as secondary metabolites in chromoplasts provide distinct coloration to flowers and fruits. In this work we investigated the genetic mechanisms that regulate accumulation of carotenoids as secondary metabolites during ripening of tomato fruits. We analyzed two mutations that affect fruit pigmentation in tomato (Lycopersicon esculentum): Beta (B), a single dominant gene that increases beta-carotene in the fruit, and old-gold (og), a recessive mutation that abolishes beta-carotene and increases lycopene. Using a map-based cloning approach we cloned the genes B and og. Molecular analysis revealed that B encodes a novel type of lycopene beta-cyclase, an enzyme that converts lycopene to beta-carotene. The amino acid sequence of B is similar to capsanthin-capsorubin synthase, an enzyme that produces red xanthophylls in fruits of pepper (Capsicum annum). Our results prove that beta-carotene is synthesized de novo during tomato fruit development by the B lycopene cyclase. In wild-type tomatoes B is expressed at low levels during the breaker stage of ripening, whereas in the Beta mutant its transcription is dramatically increased. Null mutations in the gene B are responsible for the phenotype in og, indicating that og is an allele of B. These results confirm that developmentally regulated transcription is the major mechanism that governs lycopene accumulation in ripening fruits. The cloned B genes can be used in various genetic manipulations toward altering pigmentation and enhancing nutritional value of plant foods.
DOI:10.1371/journal.pone.0208948URLPMID:30532162 [本文引用: 1]
Increasing beta-carotene (a vitamin A precursor) content in Triticum turgidum L. ssp. durum (durum wheat) grains is important to improve pasta nutritional quality. Studies in other species show that altering the expression of LCYE genes increases the flux towards the beta-beta branch, accumulating higher beta-carotene levels. Durum wheat is a tetraploid species that has two LCYE genes (LCYE-A and LCYE-B) associated to the A and B genomes. The objective of this work was to produce durum wheat LCYE mutants through EMS to potentially increase beta-carotene content. The LCYE point mutations created with EMS were identified using a Kronos TILLING (Targeting Induced Local Lesion IN Genomes) mutant population. Specific primers that amplified exons 3 through 10 of the LCYE genes were designed and validated. To simplify the TILLING procedure, fragments were digested with CJE (Celery Juice Extract) and visualized on 2% agarose gels. 6X mutant pools were identified, which showed cleavage products and then made into 2X pools to identify mutant individuals. LCYE mutants were then sequenced and evaluated with BLOSUM62, SIFT and PSSM algorithms. Mutants with substitutions W437*, P334L and G368R in LCYE-A and P405L, G352R and T393I in LCYE-B predicted to affect protein function were selected. Substitution W437* increased beta-carotene in 75% and overall total carotenoids content in leaves of the mutant 2426 (A1 mutant line), but no significant differences relative to the control were found in grains through HPLC. Finally, the increased levels of beta-carotene on leaves have potential applications to improving plant resistance under contaminated environmental conditions.
DOI:10.1016/j.pbi.2005.01.004URLPMID:15753003 [本文引用: 1]
TILLING (Targeting Induced Local Lesions IN Genomes) is a powerful reverse genetic technique that employs a mismatch-specific endonuclease to detect induced or natural DNA polymorphisms. Its advantages over other reverse genetic techniques include its applicability to virtually any organism, its facility for high-throughput and its independence of genome size, reproductive system or generation time. TILLING is currently being used for the detection of both induced and natural variation in several plant species.
In: Varshney R, Pandey M, Chitikineni A, eds. Plant Genetics and Molecular Biology.
URL [本文引用: 1]
DOI:10.1186/1471-2229-7-19URLPMID:17428339 [本文引用: 1]
BACKGROUND: Rice is both a food source for a majority of the world's population and an important model system. Available functional genomics resources include targeted insertion mutagenesis and transgenic tools. While these can be powerful, a non-transgenic, unbiased targeted mutagenesis method that can generate a range of allele types would add considerably to the analysis of the rice genome. TILLING (Targeting Induced Local Lesions in Genomes), a general reverse genetic technique that combines traditional mutagenesis with high throughput methods for mutation discovery, is such a method. RESULTS: To apply TILLING to rice, we developed two mutagenized rice populations. One population was developed by treatment with the chemical mutagen ethyl methanesulphonate (EMS), and the other with a combination of sodium azide plus methyl-nitrosourea (Az-MNU). To find induced mutations, target regions of 0.7-1.5 kilobases were PCR amplified using gene specific primers labeled with fluorescent dyes. Heteroduplexes were formed through denaturation and annealing of PCR products, mismatches digested with a crude preparation of CEL I nuclease and cleaved fragments visualized using denaturing polyacrylamide gel electrophoresis. In 10 target genes screened, we identified 27 nucleotide changes in the EMS-treated population and 30 in the Az-MNU population. CONCLUSION: We estimate that the density of induced mutations is two- to threefold higher than previously reported rice populations (about 1/300 kb). By comparison to other plants used in public TILLING services, we conclude that the populations described here would be suitable for use in a large scale TILLING project.
DOI:10.1186/1471-2229-4-12URLPMID:15282033 [本文引用: 1]
BACKGROUND: Going from a gene sequence to its function in the context of a whole organism requires a strategy for targeting mutations, referred to as reverse genetics. Reverse genetics is highly desirable in the modern genomics era; however, the most powerful methods are generally restricted to a few model organisms. Previously, we introduced a reverse-genetic strategy with the potential for general applicability to organisms that lack well-developed genetic tools. Our TILLING (Targeting Induced Local Lesions IN Genomes) method uses chemical mutagenesis followed by screening for single-base changes to discover induced mutations that alter protein function. TILLING was shown to be an effective reverse genetic strategy by the establishment of a high-throughput TILLING facility and the delivery of thousands of point mutations in hundreds of Arabidopsis genes to members of the plant biology community. RESULTS: We demonstrate that high-throughput TILLING is applicable to maize, an important crop plant with a large genome but with limited reverse-genetic resources currently available. We screened pools of DNA samples for mutations in 1-kb segments from 11 different genes, obtaining 17 independent induced mutations from a population of 750 pollen-mutagenized maize plants. One of the genes targeted was the DMT102 chromomethylase gene, for which we obtained an allelic series of three missense mutations that are predicted to be strongly deleterious. CONCLUSIONS: Our findings indicate that TILLING is a broadly applicable and efficient reverse-genetic strategy. We are establishing a public TILLING service for maize modeled on the existing Arabidopsis TILLING Project.
DOI:10.1111/pbi.12631URLPMID:27565953 [本文引用: 1]
Wheat is one of the most widely grown cereal crops in the world and is an important food grain source for humans. However, wheat yields can be reduced by many abiotic and biotic stress factors, including powdery mildew disease caused by Blumeria graminis f.sp. tritici (Bgt). Generating resistant varieties is thus a major effort in plant breeding. Here, we took advantage of the non-transgenic Targeting Induced Lesions IN Genomes (TILLING) technology to select partial loss-of-function alleles of TaMlo, the orthologue of the barley Mlo (Mildew resistance locus o) gene. Natural and induced loss-of-function alleles (mlo) of barley Mlo are known to confer durable broad-spectrum powdery mildew resistance, typically at the expense of pleiotropic phenotypes such as premature leaf senescence. We identified 16 missense mutations in the three wheat TaMlo homoeologues, TaMlo-A1, TaMlo-B1 and TaMlo-D1 that each lead to single amino acid exchanges. Using transient gene expression assays in barley single cells, we functionally analysed the different missense mutants and identified the most promising candidates affecting powdery mildew susceptibility. By stacking of selected mutant alleles we generated four independent lines with non-conservative mutations in each of the three TaMlo homoeologues. Homozygous triple mutant lines and surprisingly also some of the homozygous double mutant lines showed enhanced, yet incomplete, Bgt resistance without the occurrence of discernible pleiotropic phenotypes. These lines thus represent an important step towards the production of commercial non-transgenic, powdery mildew-resistant bread wheat varieties.
DOI:10.1007/s10681-017-2093-zURL [本文引用: 1]
DOI:10.1186/1756-0500-2-258URLPMID:20017921 [本文引用: 1]
BACKGROUND: The economic importance of cereals such as barley, and the demand for improved yield and quality require a better understanding of the genetic components that modulate biologically and commercially relevant traits. While Arabidopsis thaliana is the premiere model plant system, the spectrum of its traits cannot address all of the fundamental questions of crop plant development. Unlike Arabidopsis, barley is both a crop and a model system for scientific research, and it is increasingly being used for genetic and molecular investigations into the conserved biological processes of cereals. A common challenge in genetic studies in plants with large genomes arises from the very time-consuming work of associating mutant phenotypes with gene sequence information, especially if insertion mutagenesis is not routine, as in barley. Reverse genetics based on chemical mutagenesis represents the best solution to this obstacle. FINDINGS: In barley, we generated a new TILLING (Targeting Local Lesions IN Genomes) resource comprising 10,279 M(2 )mutants in the two-rowed malting cultivar 'Barke,' which has been used in the generation of other genomic resources in barley (~150,000 ESTs, DH mapping population). The value of this new resource was tested using selected candidate genes. An average frequency of approximately one mutation per 0.5 Mb was determined by screening ten fragments of six different genes. The ethyl methanesulphonate (EMS)mutagenesis efficiency was studied by recording and relating the mutagenesis-dependent effects found in the three mutant generations (M(1)-M(3)). A detailed analysis was performed for the homeodomain-leucine-zipper (HD-ZIP) gene HvHox1. Thirty-one mutations were identified by screening a 1,270-bp fragment in 7,348 M(2 )lines. Three of the newly identified mutants exhibited either a six-rowed or an intermedium-spike phenotype, and one mutant displayed a significantly altered spikelet morphology compared to that of the 'Barke' wild type. Our results indicate a bias in the frequency of independent functional mutations at specific base pair (bp) positions within the gene HvHox1. CONCLUSIONS: A new TILLING population was developed as a resource for high-throughput gene discovery in an alternative barley germplasm. Pilot screening demonstrated a similar or even slightly higher mutation frequency when compared to previously published barley TILLING populations that should allow for the identification of diverse allelic variation. Partial phenotypic evaluation of the M(2 )and M(3 )generations has revealed the presence of a wide spectrum of morphological diversity that highlights the great potential of this resource for use in forward genetic screens. Altogether, our study shows the efficiency of screening and the applicability of the new TILLING population for genetic studies in the barley crop model system.
DOI:10.1111/tpj.13161URLPMID:26959378 [本文引用: 1]
Screening large populations for carriers of known or de novo rare single nucleotide polymorphisms (SNPs) is required both in Targeting induced local lesions in genomes (TILLING) experiments in plants and in screening of human populations. We previously suggested an approach that combines the mathematical field of compressed sensing with next-generation sequencing to allow such large-scale screening. Based on pooled measurements, this method identifies multiple carriers of heterozygous or homozygous rare alleles while using only a small fraction of resources. Its rigorous mathematical foundations allow scalable and robust detection, and provide error correction and resilience to experimental noise. Here we present a large-scale experimental demonstration of our computational approach, in which we targeted a TILLING population of 1024 Sorghum bicolor lines to detect carriers of de novo SNPs whose frequency was less than 0.1%, using only 48 pools. Subsequent validation confirmed that all detected lines were indeed carriers of the predicted mutations. This novel approach provides a highly cost-effective and robust tool for biologists and breeders to allow identification of novel alleles and subsequent functional analysis.
DOI:10.1104/pp.126.2.480URLPMID:11402178 [本文引用: 1]
DOI:10.3835/plantgenome2008.10.0012URL [本文引用: 1]
DOI:10.1186/1471-2229-9-115URLPMID:19712486 [本文引用: 2]
BACKGROUND: Wheat (Triticum ssp.) is an important food source for humans in many regions around the world. However, the ability to understand and modify gene function for crop improvement is hindered by the lack of available genomic resources. TILLING is a powerful reverse genetics approach that combines chemical mutagenesis with a high-throughput screen for mutations. Wheat is specially well-suited for TILLING due to the high mutation densities tolerated by polyploids, which allow for very efficient screens. Despite this, few TILLING populations are currently available. In addition, current TILLING screening protocols require high-throughput genotyping platforms, limiting their use. RESULTS: We developed mutant populations of pasta and common wheat and organized them for TILLING. To simplify and decrease costs, we developed a non-denaturing polyacrylamide gel set-up that uses ethidium bromide to detect fragments generated by crude celery juice extract digestion of heteroduplexes. This detection method had similar sensitivity as traditional LI-COR screens, suggesting that it represents a valid alternative. We developed genome-specific primers to circumvent the presence of multiple homoeologous copies of our target genes. Each mutant library was characterized by TILLING multiple genes, revealing high mutation densities in both the hexaploid (~1/38 kb) and tetraploid (~1/51 kb) populations for 50% GC targets. These mutation frequencies predict that screening 1,536 lines for an effective target region of 1.3 kb with 50% GC content will result in ~52 hexaploid and ~39 tetraploid mutant alleles. This implies a high probability of obtaining knock-out alleles (P = 0.91 for hexaploid, P = 0.84 for tetraploid), in addition to multiple missense mutations. In total, we identified over 275 novel alleles in eleven targeted gene/genome combinations in hexaploid and tetraploid wheat and have validated the presence of a subset of them in our seed stock. CONCLUSION: We have generated reverse genetics TILLING resources for pasta and bread wheat and achieved a high mutation density in both populations. We also developed a modified screening method that will lower barriers to adopt this promising technology. We hope that the use of this reverse genetics resource will enable more researchers to pursue wheat functional genomics and provide novel allelic diversity for wheat improvement.
DOI:10.1186/s12863-016-0350-0URLPMID:26884094 [本文引用: 1]
BACKGROUND: Durum wheat (Triticum turgidum L.) is a cereal crop widely grown in the Mediterranean regions; the amber grain is mainly used for the production of pasta, couscous and typical breads. Single nucleotide polymorphism (SNP) detection technologies and high-throughput mutation induction represent a new challenge in wheat breeding to identify allelic variation in large populations. The TILLING strategy makes use of traditional chemical mutagenesis followed by screening for single base mismatches to identify novel mutant loci. Although TILLING has been combined to several sensitive pre-screening methods for SNP analysis, most rely on expensive equipment. Recently, a new low cost and time saving DHPLC protocol has been used in molecular human diagnostic to detect unknown mutations. RESULTS: In this work, we developed a new durum wheat TILLING population (cv. Marco Aurelio) using 0.70-0.85% ethyl methane sulfonate (EMS). To investigate the efficiency of the mutagenic treatments, a pilot screening was carried out on 1,140 mutant lines focusing on two target genes (Lycopene epsilon-cyclase, epsilon-LCY, and Lycopene beta-cyclase, beta-LCY) involved in carotenoid metabolism in wheat grains. We simplify the heteroduplex detection by two low cost methods: the enzymatic cleavage (CelI)/agarose gel technique and the denaturing high-performance liquid chromatography (DHPLC). The CelI/agarose gel approach allowed us to identify 31 mutations, whereas the DHPLC procedure detected a total of 46 mutations for both genes. All detected mutations were confirmed by direct sequencing. The estimated overall mutation frequency for the pilot assay by the DHPLC methodology resulted to be of 1/77 kb, representing a high probability to detect interesting mutations in the target genes. CONCLUSION: We demonstrated the applicability and efficiency of a new strategy for the detection of induced variability. We produced and characterized a new durum wheat TILLING population useful for a better understanding of key gene functions. The availability of this tool together with TILLING technique will expand the polymorphisms in candidate genes of agronomically important traits in wheat.
DOI:10.1186/1471-2229-12-69URLPMID:22584013 [本文引用: 1]
BACKGROUND: Wheat (Triticum spp.) is an important source of food worldwide and the focus of considerable efforts to identify new combinations of genetic diversity for crop improvement. In particular, wheat starch composition is a major target for changes that could benefit human health. Starches with increased levels of amylose are of interest because of the correlation between higher amylose content and elevated levels of resistant starch, which has been shown to have beneficial effects on health for combating obesity and diabetes. TILLING (Targeting Induced Local Lesions in Genomes) is a means to identify novel genetic variation without the need for direct selection of phenotypes. RESULTS: Using TILLING to identify novel genetic variation in each of the A and B genomes in tetraploid durum wheat and the A, B and D genomes in hexaploid bread wheat, we have identified mutations in the form of single nucleotide polymorphisms (SNPs) in starch branching enzyme IIa genes (SBEIIa). Combining these new alleles of SBEIIa through breeding resulted in the development of high amylose durum and bread wheat varieties containing 47-55% amylose and having elevated resistant starch levels compared to wild-type wheat. High amylose lines also had reduced expression of SBEIIa RNA, changes in starch granule morphology and altered starch granule protein profiles as evaluated by mass spectrometry. CONCLUSIONS: We report the use of TILLING to develop new traits in crops with complex genomes without the use of transgenic modifications. Combined mutations in SBEIIa in durum and bread wheat varieties resulted in lines with significantly increased amylose and resistant starch contents.
DOI:10.3724/SP.J.1006.2011.00202URL [本文引用: 1]
近10年我国小麦育种研究在3个方面取得新进展:育成一批高产优质多抗新品种,周8425B、鲁麦14和普通小麦-簇毛麦6VS/6AL易位系在全国小麦育种中发挥了重要作用,育种技术研究也取得重要进展。但育种工作也存在4个主要问题。从育种角度评述分子标记辅助育种中连锁标记和功能标记的研究现状和存在的主要问题,并提出今后的重点领域。概括小麦品质研究中与育种密切相关的实用技术和方法,即面包、面条和饼干品质育种中的品质评价方法和选择指标,建议今后加强5个方面的工作。对未来小麦育种4个重要问题做了分析,提出国内进一步加强高产潜力研究的初步设想,建议加大持久抗性的研究力度,重视抗旱、抗热及适应性等与气候变化相关性状的研究,还分析了种业商业化等问题。
[本文引用: 1]
DOI:10.1007/s11032-013-9917-xURL [本文引用: 1]
DOI:10.1155/2017/6572969URLPMID:28630621 [本文引用: 1]
Seed composition is one of the most important determinants of the economic values in soybean. The quality and quantity of different seed components, such as oil, protein, and carbohydrates, are crucial ingredients in food, feed, and numerous industrial products. Soybean researchers have successfully developed and utilized a diverse set of molecular markers for seed trait improvement in soybean breeding programs. It is imperative to design and develop molecular assays that are accurate, robust, high-throughput, cost-effective, and available on a common genotyping platform. In the present study, we developed and validated KASP (Kompetitive allele-specific polymerase chain reaction) genotyping assays based on previously known functional mutant alleles for the seed composition traits, including fatty acids, oligosaccharides, trypsin inhibitor, and lipoxygenase. These assays were validated on mutant sources as well as mapping populations and precisely distinguish the homozygotes and heterozygotes of the mutant genes. With the obvious advantages, newly developed KASP assays in this study can substitute the genotyping assays that were previously developed for marker-assisted selection (MAS). The functional gene-based assay resource developed using common genotyping platform will be helpful to accelerate efforts to improve soybean seed composition traits.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}