 ,1,2,**, 霍强,1,2,**, 高玉敏1,2, 李阳阳1,2, 谢玲1,2, 魏丽娟1,2, 刘列钊1,2, 卢坤1,2, 李加纳,1,2,*
,1,2,**, 霍强,1,2,**, 高玉敏1,2, 李阳阳1,2, 谢玲1,2, 魏丽娟1,2, 刘列钊1,2, 卢坤1,2, 李加纳,1,2,*Selection of candidate genes for chlorophyll content in leaves of Brassica napus using genome-wide association analysis
JIAN Hong-Ju,1,2,**, HUO Qiang,1,2,**, GAO Yu-Min1,2, LI Yang-Yang1,2, XIE Ling1,2, WEI Li-Juan1,2, LIU Lie-Zhao1,2, LU Kun1,2, LI Jia-Na,1,2,*通讯作者:
收稿日期:2020-01-12接受日期:2020-04-15网络出版日期:2020-04-27
| 基金资助: |
Received:2020-01-12Accepted:2020-04-15Online:2020-04-27
| Fund supported: |
作者简介 About authors
荐红举, E-mail:
霍强, E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2958KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
荐红举, 霍强, 高玉敏, 李阳阳, 谢玲, 魏丽娟, 刘列钊, 卢坤, 李加纳. 用全基因组关联分析筛选甘蓝型油菜叶片叶绿素含量候选基因[J]. 作物学报, 2020, 46(10): 1557-1565. doi:10.3724/SP.J.1006.2020.04007
JIAN Hong-Ju, HUO Qiang, GAO Yu-Min, LI Yang-Yang, XIE Ling, WEI Li-Juan, LIU Lie-Zhao, LU Kun, LI Jia-Na.
油菜是中国四大油料作物之一, 也是产油效率最高的油料作物, 而菜籽作为我国食用植物油的最大来源, 自给率严重不足, 因此, 提高油菜产量和含油量是油菜育种的迫切目标。叶绿素是光合作用的重要色素, 筛选并研究控制油菜叶绿素含量的基因对提高油菜产量具有重要意义。
叶绿素在光合作用中能够吸收光能并参与能量传递[1]。在生物学和遗传学上已经对叶绿素代谢进行了广泛的研究[2,3,4]。叶绿素的生物合成首先是叶绿素前体L-谷氨酰tRNA (ALA)到原卟啉IX的合成[4,5], 然后通过ALA脱水酶(ALAD)将ALA的2个分子缩合成吡咯分子, 即胆色素原。再通过酶的连续转化和原卟啉IX的金属螯合反应来划分通路, 从而指导最终产物叶绿素和血红素的形成[4]。在黑暗中, 叶绿素的生物合成分支在中间原叶绿素酸酯(Pchlide)被阻断, 因为原叶绿素酸酯向叶绿素的转化是由依赖光的NADPH催化的[6]。然而, 过量的原叶绿素酸酯游离和其他吡啶中间体在黑暗中积累可能在光照射下产生活性氧(ROS), 从而导致子叶光漂白甚至细胞死亡[7]。因此, 叶绿素的生物合成在植物生长发育过程中扮演着重要的角色。叶绿素生物合成相关基因的表达差异造成了叶绿素含量的不同。
叶片是植物光合作用的主要场所, 也是植物源到库的重要源场所。而植物叶色突变体的叶色主要表现在苗期。甘蓝型油菜中的叶色黄化突变体BnaC.ygl是由于血红素加氧酶(HO1)的活性降低导致血红素的积累, 从而抑制了叶绿素合成前体ALA的合成, 造成叶绿素生物合成受到抑制导致叶色变黄[8]; 玉米中5个黄化突变体是由于ZmPTAC2、ZmMurE、ZmPTAC10、ZmPTAC12和ZmPRIN2缺失所引起, 这5个基因与磷酸烯醇式丙酮酸(PEP)合成相关, 相关基因的缺失导致细胞中含有较少的质体核糖体和光合复合物, 表明PEP在叶绿素生物合成中起到关键作用[9]。植物光合作用可以通过叶绿素将CO2和H2O转化为可储存的有机物, 因此叶绿素是植物有机物积累的重要物质。通过降低叶绿素降解的时间可以增加产量[10]。另外, 叶片中叶绿素的含量也与作物粒饱满程度、有机物积累及单株产量显著相关[11]。
近年来在高粱[12]、玉米[13]、小麦[14]、水稻[15]、大豆[16]等植物中进行了叶绿素相关研究, 但关于甘蓝型油菜叶片叶绿素含量相关基因鉴定的报道较少[17,18]。Ge等[19]利用白菜一个F2:3群体2年的叶绿素a和叶绿素b含量数据进行QTL定位, 一共检测到10个QTL, 单个QTL解释表型率为7%~17%。Wang等[17]利用一个甘蓝型油菜叶绿素缺失突变体cde1和中双11构建的F2群体及620个F2:3群体将CDE1基因定位在一个长度为311 kb区间内, 该定位区间包括22个候选基因; Qian等[18]探究了甘蓝型油菜中bnnye1-A01的缺失对光系统(LHC)相关基因表达及光合反应中心蛋白PSI和PSII表达的影响以及对籽粒油脂积累的影响, 同时也发现携带bnnye1-A01基因也会影响株高。
随着高通量测序技术的成熟以及基因型分型成本的降低, 全基因组关联分析(genome-wide association)已经成为植物基因探究以及复杂性状解析的重要方法[20]。全基因组关联分析已经在棉花[21,22]、杨树[23]、大豆[24]等植物中广泛应用。本研究对588份甘蓝型油菜自然群体的叶片进行2年叶绿素含量测定并进行全基因组关联分析, 筛选出控制甘蓝型油菜叶片叶绿素含量相关的候选基因, 为油菜叶绿素含量的遗传改良奠定基础。
1 材料方法
1.1 试验材料
用于关联分析的588份甘蓝型油菜是由全球各地征集的油菜品种组成, 其中大部分来自中国重庆、湖北、湖南、江苏、陕西等地, 部分来自加拿大、德国、瑞典、丹麦、澳大利亚等国家。所有材料由重庆市西南大学油菜工程中心提供。1.2 田间试验与叶绿素测定
该群体于2017—2018年, 2018—2019年连续种植于重庆市北碚区歇马镇油菜基地。采用随机区组设计, 2个重复, 以育苗移栽方式每小区种植3行, 每行15株, 行距40 cm, 株距20 cm。按照常规生产方式进行田间管理。在植株苗期用SPAD-502Plus叶绿素仪测定完全伸展的叶片中间部位叶绿素含量, 对每个品种测定5株。利用IBM SPSS Statistics 19.0统计分析软件进行描述性统计分析并制作正态分布图。1.3 全基因组关联分析的方法
参考文献进行基因型数据测定与分析、群体结构、亲缘关系分析与连锁不平衡分析[25]。全基因组关联分析采用naive、Q、PCA、K、Q+K和PCA+K六种统计模型来评估群体结构和亲缘关系的影响(Q: 群体结构; PCA: 主成分; K: 亲缘关系), 进行表型和SNP关联分析。运用TASSEL 5.0软件进行这6种模型的关联分析, 其中使用一般线性模型(GLM)分析naive、Q和PCA模型, 使用混合线性模型(MLM)分析K、Q+K和PCA+K模型。利用SAS软件检测这6种模型的运算结果, 对其lg(P)的观测值和lg(P)期望值绘制Quantile-quantile散点图, 通过QQ图比较后确定最佳模型, 在最佳模型下进行叶片叶绿素含量的全基因组关联分析, 利用R软件作Manhattan图。本试验使用的SNP数据为385,692个, P值小于阈值(1/385,692 = 2.593E-06)的位点为显著关联位点。1.4 候选基因的挖掘
根据甘蓝型油菜“Darmor-bzh”参考基因组[26], 提取分析得到的叶片叶绿素含量显著关联的SNP标记各位点500 kb内的基因[27]; 将需要功能分析的基因序列与拟南芥所有基因序列进行BLASTn比对, e-value阈值为1e-10, 并以同源性最高的拟南芥基因作为待分析基因进行功能注释来筛选与叶绿素含量相关的候选基因。1.5 候选基因验证
对叶绿素含量极端材料在苗期取3个生物学重复的叶片并提取RNA。根据前期所取材料的叶片提取的RNA, 使用TaKaRa公司的PrimeScript RT reagent Kit with gDNA Eraser反转录合成cDNA, 用于qRT-PCR验证。以油菜UBC21作为内参基因, 使用TaKaRa公司的SYBR Premix Ex Taq II试剂盒进行qRT-PCR验证候选基因的相对表达量。设置每个样品3次技术重复。在BIO-RAD定量PCR仪上进行qRT-PCR扩增。在网站http://biodb.swu.edu.cn/ qprimerdb上查找设计候选基因的qRT-PCR引物(表1), 由上海生工生物工程有限公司合成。Table 1
表1
表1候选基因引物
Table 1
| 基因Gene | 引物序列Primer sequence (5°-3°) |
|---|---|
| BnaA04g00600D | F: AAATGTCGTTGAGGAATTACGC; R: TGAATGATATACGTCAGCACCA |
| BnaC09g54390D | F: AAGGAGCTAACACAATGACTGA; R: GGTATCATCACAGGTAACCCAT |
| BnaC03g18820D | F: AATCATGTTTCCATTAGCCACG; R: CATTACCACCGTTGAAAACCAA |
| BnaC06g24160D | F: AATGTTGCGCATTATCTTCCTC; R: TGTTTGTAGTGTTTCCAGGAGT |
| BnaC07g39280D | F: AGAAAACGATGCTTATGCTGAC; R: CATAGCTGCAGAGATATCGCTA |
| BnaA04g00500D | F: AGCTCATATACATTGTGGAGCA; R: TGCTGCTAGATTGGTCTAAGAG |
| BnaA02g24670D | F: CTAATCGAATCAAGCCCTGTTG; R: GGTGAAAACGCAAAGAAATTGG |
| BnaC02g01240D | F: CTCAAGGAGCTATTGGAATTGC; R: TATCCAGCAATAGGTGTACGAC |
| BnaC03g20950D | F: CTTTATGGTGTTGGCCAAGTAC; R: AACTTTACCACGCCGTATTTTC |
| BnaC05g38600D | F: GACTCTCTGAAGATACTGAGGC; R: ACTCTTGATCACCGTATACGTC |
| BnaA10g02050D | F: GAGCTGGAACAATAACTCCTCT; R: CACAGTTTCTTTGCCTTCTCTG |
| BnaC05g10770D | F: GAGTGTTCCTGATTGTATGCAC; R: AGATGTGCCAATCCAAAAGAAC |
| BnaC04g01850D | F: GATAGGAACGGTGATGGTTTTG; R: TTCCGTCACGAAATTAAAACCC |
| BnaC02g25030D | F: GTAAATGTGCCTGATGAGGTTG; R: TGAACAATCTCTTCGTCAAGGA |
| BnaA06g06000D | F: GTCGCTGCTTAAACAGTTACAA; R: GAAACCCAACAAAGAGTGAACA |
| BnaA05g01720D | F: GTTCACAACGTCAATGATCACA; R: GTGGCCCAAGAATCAAAGATTT |
| BnaC08g27000D | F: GTTTCTGAACGAACAGGTTGAA; R: ATTTAAGCAGCAGCAGAAGTTC |
| BnaC09g05970D | F: TTACTAGCTTGGCTCACACTAC; R: CGTTCTCATCAACAACACTCAG |
| BnaC09g05970D | F: TTACTAGCTTGGCTCACACTAC; R: CGTTCTCATCAACAACACTCAG |
| BnaA01g20460D | F: TTATCAAGCGTGCGAAGTAAAG; R: CGCGCTGATCTTAACCTAGTTA |
| BnaC08g27310D | F: TTTGCTACCTGTTGAAAACGAC; R: TTATTCAACTGTGACAAAGCCG |
新窗口打开|下载CSV
2 结果与分析
2.1 叶绿素含量表型分析
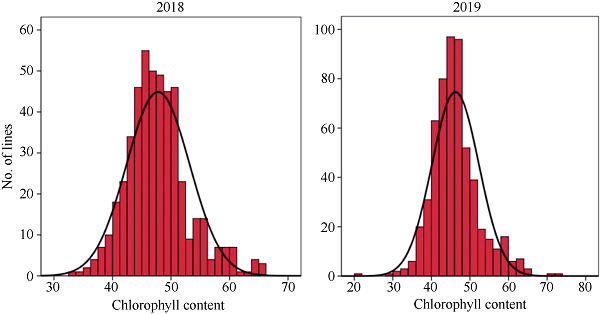
自然群体中, 叶绿素含量在2018年和2019年的变异系数分别为11.27%和12.73%, 2年间的变异系数较为稳定(表2)。自然群体2年叶绿素含量表现出连续正态分布(图1), 说明该性状符合由多基因控制的数量性状特点, 适合全基因组关联分析。Table 2
表2
表2甘蓝型油菜叶绿素含量的表型数据统计分析
Table 2
| 性状 Trait | 环境 Environment | 均值±标准差 Mean ± SD | 范围 Range | 变异系数 CV (%) |
|---|---|---|---|---|
| 叶绿素含量 | 2018 | 47.79±5.39 | 33.44-65.82 | 11.27 |
| Chlorophyll content | 2019 | 46.37±5.90 | 32.92-72.04 | 12.73 |
新窗口打开|下载CSV
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1甘蓝型油菜叶片叶绿素含量的频次分布
Fig. 1Frequency distribution of chlorophyll content in leaves of B. napus
2.2 甘蓝型油菜叶片叶绿素含量全基因组关联分析
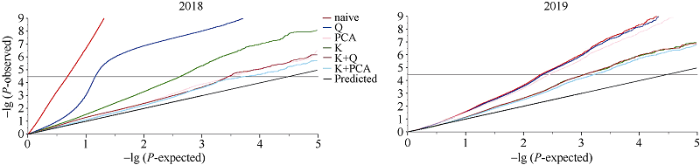
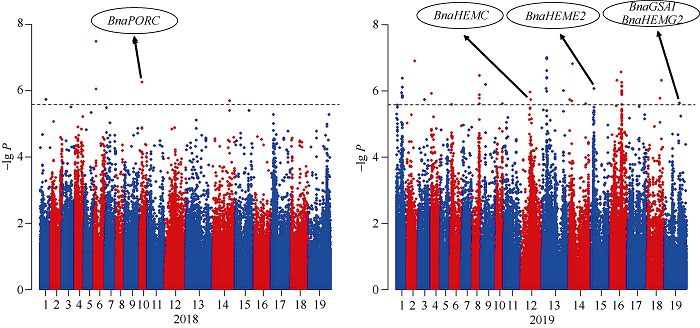
对叶绿素含量关联分析6种模型下的结果绘制Quantile-quantile散点图(图2), 由图2可知, 最佳模型为P+K模型。在最佳模型下, 利用385,692个SNPs, 以P值小于阈值(1/385,692 = 2.593E-06)确定显著关联SNP位点(表3), 并绘制Manhattan图(图3)。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2甘蓝型油菜叶绿素含量在各模型中的QQ图
Fig. 2Quantile-quantile plot for six models of chlorophyll contents
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3甘蓝型油菜叶绿素含量在最佳模型中的曼哈顿图
Fig. 3Manhattan plots of association analysis using the optimal model of chlorophyll contents
Table 3
表3
表3最佳模型下叶绿素含量显著位点表
Table 3
| 环境Environment | 位点 SNP | 染色体 Chr. | 位置 Position | 阈值 lg (P-value) | 贡献率 R2 (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 2018 | S1_12845378 | A03 | 12,845,378 | 5.73 | 5.51 | ||||
| S6_3493805 | A06 | 3,493,805 | 7.49 | 7.89 | |||||
| S6_3812220 | A06 | 3,812,220 | 6.05 | 6.10 | |||||
| S10_4969863 | A10 | 4,969,863 | 6.25 | 6.23 | |||||
| S14_37741561 | C04 | 37,741,561 | 5.69 | 5.67 | |||||
| 2019 | S1_12688640 | A01 | 12,688,640 | 6.39 | 8.42 | ||||
| S1_12540615 | A01 | 12,540,615 | 6.10 | 8.24 | |||||
| S1_10570293 | A01 | 10,570,293 | 5.93 | 8.51 | |||||
| S1_12628926 | A01 | 12,628,926 | 5.86 | 8.28 | |||||
| S1_12540545 | A01 | 12,540,545 | 5.84 | 7.67 | |||||
| S1_12540622 | A01 | 12,540,622 | 5.83 | 7.66 | |||||
| 环境Environment | 位点 SNP | 染色体 Chr. | 位置 Position | 域值 lg (P-value) | 贡献率 R2 (%) | ||||
| 2019 | S1_12628563 | A01 | 12,628,563 | 5.79 | 9.76 | ||||
| S1_12568566 | A01 | 12,568,566 | 5.78 | 7.55 | |||||
| S2_17839296 | A02 | 17,839,296 | 6.91 | 9.73 | |||||
| S3_13514793 | A03 | 13,514,793 | 5.73 | 7.72 | |||||
| S4_547942 | A04 | 547,942 | 5.93 | 8.08 | |||||
| S8_16108365 | A08 | 16,108,365 | 6.46 | 8.44 | |||||
| S8_16192480 | A08 | 16,192,480 | 5.88 | 7.80 | |||||
| S8_16181282 | A08 | 16,181,282 | 5.78 | 7.85 | |||||
| S8_16103637 | A08 | 16,103,637 | 5.61 | 7.97 | |||||
| S8_16181355 | A08 | 16,181,355 | 5.60 | 7.29 | |||||
| S9_10532790 | A09 | 10,532,790 | 6.18 | 8.70 | |||||
| S10_14050715 | A10 | 14,050,715 | 5.61 | 7.72 | |||||
| S12_20495602 | C02 | 20,495,602 | 5.95 | 9.35 | |||||
| S12_22225349 | C02 | 22,225,349 | 5.74 | 8.76 | |||||
| S13_11413088 | C03 | 11,413,088 | 7.01 | 10.34 | |||||
| S13_11423629 | C03 | 11,423,629 | 6.95 | 9.31 | |||||
| S13_11415175 | C03 | 11,415,175 | 6.61 | 8.92 | |||||
| S13_11433444 | C03 | 11,433,444 | 6.39 | 8.88 | |||||
| S13_11433450 | C03 | 11,433,450 | 6.38 | 8.87 | |||||
| S13_9816561 | C03 | 9,816,561 | 6.06 | 8.70 | |||||
| S13_48351338 | C03 | 48,351,338 | 6.00 | 8.06 | |||||
| S13_11249216 | C03 | 11,249,216 | 5.97 | 8.22 | |||||
| S14_7018412 | C04 | 7,018,412 | 6.81 | 9.39 | |||||
| S14_1487936 | C04 | 1,487,936 | 5.73 | 7.96 | |||||
| S14_5798300 | C04 | 5,798,300 | 5.68 | 8.45 | |||||
| S14_36521995 | C04 | 36,521,995 | 5.60 | 7.95 | |||||
| S15_6002527 | C05 | 6,002,527 | 6.06 | 8.65 | |||||
| S16_24042352 | C06 | 24,042,352 | 6.55 | 9.94 | |||||
| S16_14397036 | C06 | 14,397,036 | 6.31 | 8.84 | |||||
| S16_25797577 | C06 | 25,797,577 | 6.25 | 8.34 | |||||
| S16_25797598 | C06 | 25,797,598 | 6.14 | 8.15 | |||||
| S16_25233735 | C06 | 25,233,735 | 6.14 | 8.09 | |||||
| S16_25797558 | C06 | 25,797,558 | 6.00 | 8.46 | |||||
| S16_25234451 | C06 | 25,234,451 | 5.97 | 8.45 | |||||
| S16_25234262 | C06 | 25,234,262 | 5.92 | 8.11 | |||||
| S16_25797570 | C06 | 25,797,570 | 5.89 | 8.06 | |||||
| S16_25234230 | C06 | 25,234,230 | 5.88 | 8.31 | |||||
| S18_31711591 | C08 | 31,711,591 | 6.32 | 8.85 | |||||
| S18_28228959 | C08 | 28,228,959 | 5.77 | 8.36 | |||||
| S19_32818852 | C09 | 32,818,852 | 5.62 | 8.15 | |||||
新窗口打开|下载CSV
2018年检测到5个SNP位点, 分别位于A01 (1个)、A06 (2个)、A10 (1个)和C04 (1个)染色体, 它们的贡献率为5.51%~7.89%, 其中S6_3493805位点贡献率最大; 2019年检测到46个SNP位点, 分别位于A01 (8个)、A02 (1个)、A03 (1个)、A04 (1个)、A08 (5个)、A09 (1个)、A10 (1个)、C02 (2个)、C03 (8个)、C04 (4个)、C05 (1个)、C06 (10个)、C08 (2个)和C09 (1个)染色体, 贡献率为7.29%~10.34%, 其中S13_11413088位点贡献率最大。
2.3 甘蓝型油菜叶片叶绿素含量候选基因筛选
初步筛选出23个与油菜叶片叶绿素含量显著关联的候选基因, 与拟南芥基因序列进行Blastn比对, 结合前人已报道的拟南芥同源基因功能, 筛选可能在本研究中发挥作用的候选基因(表4)。Table 4
表4
表4叶绿素含量候选基因
Table 4
| 基因名字 Gene name | 物理位置 Physical position | 拟南芥同源基因 Homologs in A. thaliana | 功能注释 Functional annotation |
|---|---|---|---|
| BnaA01g20460D | A01:12331065-1233745 | AT3G48740 | Nodulin MtN3 family protein |
| BnaA02g24670D | A02:17989410-17990090 | AT5G46780 | VQ motif-containing protein |
| BnaA04g00500D | A04:365464-366530 | AT3G61890 | Homeobox 12 (HB-12) |
| BnaA04g00600D | A04:460722-461017 | AT3G61640 | Arabinogalactan protein 20 (AGP20) |
| BnaA05g01720D | A05:984996-986020 | AT2G40490 | HEME2 |
| BnaA06g06000D | A06:3306811-3307795 | AT1G10200 | WLIM1 |
| BnaA10g02050D | A10:1025298-1027102 | AT1G03630 | Protochlorophyllide oxidoreductase C (POR C) |
| BnaC02g01240D | C02:511615-513517 | AT5G08280 | Hydroxymethylbilane synthase (HEMC) |
| BnaC02g25030D | C02:22402218-22403498 | AT1G07440 | NAD(P)-binding Rossmann-fold superfamily protein |
| BnaC03g18820D | C03:9634933-9635479 | AT2G33830 | Dormancy/auxin associated family protein |
| BnaC03g20950D | C03:11192271-11192876 | AT2G37970 | SOUL-1 |
| BnaC04g01850D | C04:1467437-1468078 | AT2G41410 | Calcium-binding EF-hand family protein |
| BnaC05g10770D | C05:6194068-6195531 | AT1G14480 | Ankyrin repeat family protein |
| 基因名字 Gene name | 物理位置 Physical position | 拟南芥同源基因 Homologs in A. thaliana | 功能注释 Functional annotation |
| BnaC05g38600D | C05:37280225-37281849 | AT3G14930 | HEME1 |
| BnaC06g24160D | C06:25958767-25959390 | AT1G72610 | Germin-like protein 1 (GER1) |
| BnaC07g39280D | C07:40238482-40239858 | AT4G25080 | Magnesium-protoporphyrin IX methyltransferase (CH LM) |
| BnaC08g27000D | C08:28191454-28192970 | AT3G56090 | Ferritin 3 (FER3) |
| BnaC08g27310D | C08:28357417-28358091 | AT3G56360 | Unknown protein |
| BnaC09g05970D | C09:3640560-3642915 | AT5G63570 | Glutamate-1-semialdehyde-2,1-aminomutase (GSA1) |
| BnaC03g18980D | chrC03:9831772-9832572 | AT2G34430 | Light-harvesting chlorophyll-protein complex II subunit B1 (LHB1B1) |
| BnaC02g25060D | chrC02:22446772-22448037 | AT1G78600 | Light-regulated zinc finger protein 1 (LZF1) |
| BnaA04g00660D | chrA04:488057-489279 | AT3G61470 | Photosystem I light harvesting complex gene 2 (LHCA2) |
| BnaA08g22200D | chrA08:16221799-16222764 | AT1G19150 | Photosystem I light harvesting complex gene 6 (LHCA6) |
新窗口打开|下载CSV
2.4 候选基因的验证
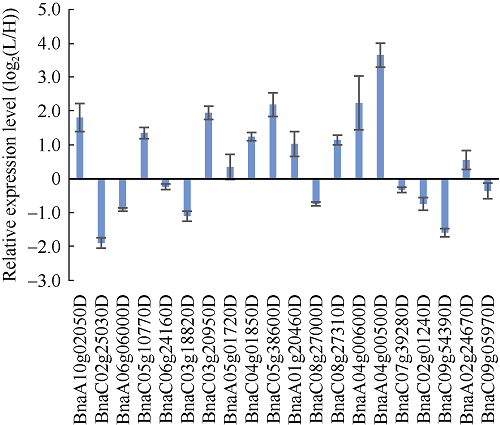
挑选20个候选基因进行荧光定量验证, 检测其在2类材料中的表达情况表明, 相比叶绿素含量高的材料, 在叶绿素含量低的材料中, 11个基因上调表达, 9个基因下调表达(图4)。上调基因包括BnaC05g38600D, 其编码HEME1蛋白, 下调基因包括BnaC09g54390D, 其编码HEMG2, 这些基因均参与叶绿素合成途径。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4甘蓝型油菜叶片叶绿素含量候选基因定量验证
Fig. 4Expression profiles of chlorophyll content candidate genes in leaves of B. napus using qRT-PCR
3 讨论
利用数量性状座位(quantitative trait loci, QTL)或者全基因组关联分析可以快速鉴定与性状关联的分子标记和基因, 是非常有效的定位候选基因的方法。定位甘蓝型油菜重要农艺性状QTL已经在产量性状如千粒重[27]、角果长度[28]和农艺性状如开花期[29]、株高[30]等方面取得重要进展。利用GWAS定位甘蓝型油菜复杂的数量性状同样取得重要进展[31,32,33]。但是, 影响甘蓝型油菜叶片叶绿素含量相关QTL或者候选基因的研究相对较少, 仅有1篇通过突变体鉴定到的一个候选基因BnaC.YGL[8]。本研究中, 利用588份全球搜集的优质种质资源, 具有较为丰富的表型变异, 叶绿素含量变异范围广(表2); 课题组前期已经对该资源材料进行重测序, 挖掘出385,692个高质量的SNP位点[25], 这些为通过GWAS分析筛选叶绿素含量显著关联位点奠定基础。植物合成的叶绿素含量不仅受到遗传因素的调控, 也会受到外界环境的影响, 如外界光照、温度和营养[34]。而当植物叶绿素含量发生变化, 细胞内叶绿素的超微结构、叶片生理生化特征以及叶色调控机制也都会发生改变[17]。油菜叶片叶绿素含量受年度间水肥条件和光温条件差异影响, 基因型与环境互作较大, 不同环境下有不同的基因群发挥作用, 进而造成关联位点的差异。通过测定甘蓝型油菜苗期叶片叶绿素含量, 2018和2019年分别筛选出与叶绿素显著关联的SNP位点5个和46个。通过对SNP位点上下游各500 kb范围内候选基因的筛选, 共得到2022个候选基因, 其中, BnaA10g02050D (BnaPOR C)、BnaC02g01240D (BnaHEMC)、Bna C05g38600D (BnaHEME2)和BnaC09g54390D (BnaHEMG2) 4个基因的同源基因在拟南芥中参与叶绿素合成途径(表4)。这些将作为重要候选基因进一步分析。
另外, 4个基因参与叶绿体发育和光合作用过程, 如BnaC03g18980D, 编码叶绿素a/b结合蛋白(LIGHT-HARVESTING CHLOROPHYLL-PROTEIN COMPLEX II SUBUNIT B1, LHB1B1), 该蛋白定位在叶绿体膜、类囊体膜上参与光捕获, 响应光刺激[35]。BnaA04g00660D和BnaA08g22200D分别编码光合系统I的光捕获复合体基因2 (PHOTOSYSTEM I LIGHT HARVESTING COMP LEX GENE 2, LHCA2)和LHCA6, 都定位在叶绿体类囊体膜, 参与光捕获过程[36,37]。BnaC02g25060D编码光调控的锌指蛋白1 (light-regulated zinc finger protein 1, LZF1), 又名B-box domain protein 22 (BBX22和DBB3), 其定位在细胞核, 参与含花青素的复合生物合成过程、叶绿素生物合成过程、叶绿体组织等调控过程[38,39]。
4 结论
2年分别鉴定到5个和46个显著关联的SNP标记, SNP位点上下游500 kb区间内共筛选出23个与叶绿素合成途径相关的候选基因, 并进行了定量验证, 本研究为油菜后续叶片叶绿素含量的遗传改良奠定了基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1105/tpc.7.7.1039URLPMID:12242396 [本文引用: 1]
DOI:10.1007/s11103-004-2331-3URL [本文引用: 1]

Chlorophyll (Chl) has unique and essential roles in photosynthetic light-harvesting and energy transduction, but its biosynthesis, accumulation and degradation is also associated with chloroplast development, photomorphogenesis and chloroplast-nuclear signaling. Biochemical analyses of the enzymatic steps paved the way to the identification of their encoding genes. Thus, important progress has been made in the recent elucidation of almost all genes involved in Chl biosynthesis and breakdown. In addition, analysis of mutants mainly in Arabidopsis, genetically engineered plants and the application of photo-reactive herbicides contributed to the genetic and regulatory characterization of the formation and breakdown of Chl. This review highlights recent progress in Chl metabolism indicating highly regulated pathways from the synthesis of precursors to Chl and its degradation to intermediates, which are not longer photochemically active.]]>
DOI:10.1016/j.pbi.2006.03.011URLPMID:16603411 [本文引用: 1]
Since the 1970s, researchers have proposed several regulatory pathways governing chlorophyll metabolism, but only recently have the underlying molecular mechanisms been elucidated. The recent data indicate that such regulatory systems are more complex than originally anticipated. For instance, the pathways involve a series of protein-protein interactions, including complex formation, the dual localization of enzymes within chloroplasts, and a novel protein degradation mechanism that is triggered by pigments. Furthermore, several lines of evidence suggest that chlorophyll metabolism might not only significantly impact the assembly of photosynthetic machineries but also influence processes such as programmed cell death, the 'stay-green' phenomenon, and chloroplast-nucleus communication.
DOI:10.1146/annurev.arplant.57.032905.105448URLPMID:17227226 [本文引用: 3]
Tetrapyrroles play vital roles in various biological processes, including photosynthesis and respiration. Higher plants contain four classes of tetrapyrroles, namely, chlorophyll, heme, siroheme, and phytochromobilin. All of the tetrapyrroles are derived from a common biosynthetic pathway. Here we review recent progress in the research of tetrapyrrole biosynthesis from a cellular biological view. The progress consists of biochemical, structural, and genetic analyses, which contribute to our understanding of how the flow and the synthesis of tetrapyrrole molecules are regulated and how the potentially toxic intermediates of tetrapyrrole synthesis are maintained at low levels. We also describe interactions of tetrapyrrole biosynthesis and other cellular processes including the stay-green events, the cell-death program, and the plastid-to-nucleus signal transduction. Finally, we present several reports on attempts for agricultural and horticultural applications in which the tetrapyrrole biosynthesis pathway was genetically modified.
DOI:10.1016/j.tplants.2010.05.012URLPMID:20598625 [本文引用: 1]
Tetrapyrroles such as chlorophyll and heme are co-factors for essential proteins involved in a wide variety of crucial cellular functions. Nearly 2% of the proteins encoded by the Arabidopsis thaliana genome are thought to bind tetrapyrroles, demonstrating their central role in plant metabolism. Although the enzymes required for tetrapyrrole biosynthesis are well characterized, there are still major questions about the regulation of the pathway, and the transport of tetrapyrroles within cells. These issues are important, as misregulation of tetrapyrrole metabolism can lead to severe photo-oxidative stress, and because tetrapyrroles have been implicated in signaling pathways coordinating interactions between plant organelles. In this review, we discuss the cell biology of tetrapyrrole metabolism and its implications for tetrapyrroles as signaling molecules.
DOI:10.1007/s11120-010-9568-2URL [本文引用: 1]
Chloroplast development is usually regarded as proceeding from proplastids. However, direct or indirect conversion pathways have been described in the literature, the latter involving the etioplast or the etio-chloroplast stages. Etioplasts are characterized by the absence of chlorophylls (Chl-s) and the presence of a unique inner membrane network, the prolamellar body (PLB), whereas etio-chloroplasts contain Chl-s and small PLBs interconnected with chloroplast thylakoids. As etioplast development requires growth in darkness for several days, this stage is generally regarded as a nonnatural pathway of chloroplast development occurring only under laboratory conditions. In this article, we have reviewed the data in favor of the involvement of etioplasts and etio-chloroplasts as intermediary stage(s) in chloroplast formation under natural conditions, the molecular aspects of PLB formation and we propose a dynamic model for its regulation.
DOI:10.1073/pnas.0803950105URLPMID:18723681 [本文引用: 1]
A homology model of NADPH:protochlorophyllide (Pchlide) oxidoreductase A (POR; E.C. 1.3.33.1) of barley is developed and verified by site-directed mutagenesis. PORA is considered a globular protein consisting of nine alpha-helices and seven beta-strands. The model predicts the presence of two functionally distinctive Pchlide binding sites where the pigment is coordinated by cysteine residues. The pigment bound to the first, high-affinity Pchlide binding site is used for the formation of the photoactive state of the enzyme. The pigment bound to the second, low-affinity Pchlide binding site is involved in the PORA:PORB interaction, allowing for resonance energy transfer between the neighboring PORs in the complex. In the in vitro reconstituted light-harvesting POR:Pchlide complex (LHPP), light absorbed by PORA-bound Pchlide b is transferred to PORB-bound Pchlide a. That induces the conversion of Pchlide a to chlorophyllide (Chlide) a. This energy transfer eliminates the possibility of Pchlide b photoreduction and prevents that excited triplet states of either Pchlides a or b accumulate and provoke singlet oxygen production. Together, our results provide a photoprotective role of PORA during greening.
DOI:10.1007/s11103-017-0583-yURLPMID:28108964 [本文引用: 2]
We previously described a Brassica napus chlorophyll-deficient mutant (ygl) with yellow-green seedling leaves and mapped the related gene, BnaC.YGL, to a 0.35 cM region. However, the molecular mechanisms involved in this chlorophyll defect are still unknown. In this study, the BnaC07.HO1 gene (equivalent to BnaC.YGL) was isolated by the candidate gene approach, and its function was confirmed by genetic complementation. Comparative sequencing analysis suggested that BnaC07.HO1 was lost in the mutant, while a long noncoding-RNA was inserted into the promoter of the homologous gene BnaA07.HO1. This insert was widely present in B. napus cultivars and down-regulated BnaA07.HO1 expression. BnaC07.HO1 was highly expressed in the seedling leaves and encoded heme oxygenase 1, which was localized in the chloroplast. Biochemical analysis showed that BnaC07.HO1 can catalyze heme conversion to form biliverdin IXalpha. RNA-seq analysis revealed that the loss of BnaC07.HO1 impaired tetrapyrrole metabolism, especially chlorophyll biosynthesis. According, the levels of chlorophyll intermediates were reduced in the ygl mutant. In addition, gene expression in multiple pathways was affected in ygl. These findings provide molecular evidences for the basis of the yellow-green leaf phenotype and further insights into the crucial role of HO1 in B. napus.
DOI:10.1104/pp.113.228726URL [本文引用: 1]
Chloroplast transcription in land plants relies on collaboration between a plastid-encoded RNA polymerase (PEP) of cyanobacterial ancestry and a nucleus-encoded RNA polymerase of phage ancestry. PEP associates with additional proteins that are unrelated to bacterial transcription factors, many of which have been shown to be important for PEP activity in Arabidopsis (Arabidopsis thaliana). However, the biochemical roles of these PEP-associated proteins are not known. We describe phenotypes conditioned by transposon insertions in genes encoding the maize (Zea mays) orthologs of five such proteins: ZmPTAC2, ZmMurE, ZmPTAC10, ZmPTAC12, and ZmPRIN2. These mutants have similar ivory/virescent pigmentation and similar reductions in plastid ribosomes and photosynthetic complexes. RNA gel-blot and microarray hybridizations revealed numerous changes in plastid transcript populations, many of which resemble those reported for the orthologous mutants in Arabidopsis. However, unanticipated reductions in the abundance of numerous transfer RNAs (tRNAs) dominated the microarray data and were validated on RNA gel blots. The magnitude of the deficiencies for several tRNAs was similar to that of the most severely affected messenger RNAs, with the loss of trnL-UAA being particularly severe. These findings suggest that PEP and its associated proteins are critical for the robust transcription of numerous plastid tRNAs and that this function is essential for the prodigious translation of plastid-encoded proteins that is required during the installation of the photosynthetic apparatus.
DOI:10.1556/CRC.37.2009.4.3URL [本文引用: 1]
DOI:10.1111/jac.12045URL [本文引用: 1]
Heat stress has become an increasingly important factor in limiting wheat yields. In northern China, high temperature (>30 degrees C) during the grain filling is one of the major constraints in increasing wheat productivity. We used two winter wheat (Triticum aestivum L.) cultivars with different sensitivities to heat stress (Jimai 22 'JM22', low sensitivity and Xinmai 26 'XM26', high sensitivity) to study the various aspects of photosynthetic characteristics during the grain filling stage under heat stress. The results showed that photosynthesis rates (P-n) in flag leaves of XM26 decreased faster than in JM22 under heat stress during the grain-filling stage. P-n decreased more rapidly under heat stress than without stress, by up to 69.9% and 59.3%, respectively, at 10days following heat stress (10 DAS). This decline of P-n was not caused by heat-induced stomatal limitation, but rather by a decline in Rubisco activity and a functional drop in photosystem II (PSII). After heat stress, the grain yield of JM22 decreased by 6.41%, but XM26 decreased by 11.43%, when compared with their respective controls. Heat stress also caused an alteration of mesophyll cell ultrastructure. Injury caused by heat stress to organelles in XM26 was more severe than JM22. Moreover, the JM22 cultivar showed some self-repair capacity following heat stress injury. These results indicate that declines in photosynthetic performance caused by heat stress were cultivar-dependent. Compared with XM26, the JM22 cultivar had superior heat stability in terms of PSII function and carboxylation activity, both of which are susceptible to heat stress.
DOI:10.1007/s10681-009-0108-0URL [本文引用: 1]
2F3/BC1F4 generations) of four stable stay-green QTLs (StgB, Stg1, Stg3 and Stg4) from donor parent B35 to senescent variety R 16. The majority of the introgression lines had higher leaf chlorophyll levels at flowering (a distinctive trait of the donor parent) and a greater percentage green leaf area during the latter part of grain filling, than did R 16, indicating that the stay-green QTLs were expressed phenotypically in the R 16 background. None of the QTL introgression lines achieved the same level of stay-green as B35, however. Maintenance of a greater relative green leaf area during the latter half of grain filling was related to a greater relative grain yield in two of three post-flowering moisture deficit environments in which the materials were evaluated (r 2=0.34 in 2004–2005 and r 2=0.76 in 2005–2006), as was a direct measure of leaf chlorophyll in one of the post-flowering stress environments in which this was measured (r 2=0.42, P<0.05). Thus the study provided useful evidence that the marker-assisted backcross transfer of stay-green QTLs from B35 into an adapted, but senescent background has the potential to enhance tolerance of post-flowering drought stress in sorghum.]]>
DOI:10.1007/s11032-011-9615-5URL [本文引用: 1]
DOI:10.1007/s11032-013-9862-8URLPMID:23794940 [本文引用: 1]
Relatively little is known of the genetic control of chlorophyll fluorescence (CF) and pigment traits important in determining efficiency of photosynthesis in wheat and its association with biomass productivity. A doubled haploid population of 94 lines from the wheat cross Chinese Spring x SQ1 was trialled under optimum glasshouse conditions for 4 years to identify quantitative trait loci (QTL) for CF traits including, for the first time in wheat, JIP-test parameters per excited cross section (CSm): ABS/CSm, DIo/CSm, TRo/CSm, RC/CSm and ETo/CSm, key parameters determining efficiency of the photosynthetic apparatus, as well as chlorophyll and carotenoid contents to establish associations with biomass and grain yield. The existing genetic map was extended to 920 loci by adding Diversity Arrays Technology markers. Markers and selected genes for photosynthetic light reactions, pigment metabolism and biomass accumulation were located to chromosome deletion bins. Across all CF traits and years, 116 QTL for CF were located on all chromosomes except 7B, and 39 QTL were identified for pigments on the majority of chromosomes, excluding 1A, 2A, 4A, 3B, 5B, 1D, 2D, 5D, 6D and 7D. Thirty QTL for plant productivity traits were mapped on chromosomes 3A, 5A, 6A, 7A, 1B, 2B, 4B, 6B, 7B, 3D and 4D. A region on chromosome 6B was identified where 14 QTL for CF parameters coincided with QTL for chlorophyll content and grain weight per ear. Thirty-five QTL regions were coincident with candidate genes. The environment was shown to dominate in determining expression of genes for those traits.
DOI:10.1093/jhered/ess041URL [本文引用: 1]
The chlorophyll content is one of the most important traits selected by breeders, and it is controlled by quantitative trait loci (QTLs) derived from natural variations in rice. We analyzed the QTL controlling chlorophyll content by using 94 RILs derived from a cross between 2 japonica rice cultivars, Lijiangxintuanheigu (LTH) and Shennong265 (SN265). Twenty-two QTLs controlling chlorophyll content at tillering stage, heading stage, and maturity stage were detected, respectively. Among them, Rice cv. LTH had a positive allele only at 1 locus (qCTH4) on chromosome 4. Further analysis indicated that the genetic effect of qCTH4 was the net effects within the period from tillering to heading. The QTL qCTH4 controlling chlorophyll content from tillering to heading locates between RM255 and RM349 on chromosome 4 with a LOD score 19.41, and the QTL qCTH4 explains 61.42% of phenotypic variation. In order to eliminate the influence of other QTLs, 1 single residual heterozygous plant, RH-qCTH4, was selected based on the genotypes of 114 Simple Sequence Repeat (SSR) markers. Using the segregating population derived from RH-qCTH4 by self-crossing, this region was narrowed down to an interval between RM3276 and RM17494 in an approximately 771 kb target region. These results are useful for map-based cloning of qCTH4 and for marker-assisted selection of high photosynthetic efficiency variety.
DOI:10.1186/s12870-016-0861-xURLPMID:27488358 [本文引用: 1]
BACKGROUND: Chlorophyll is a major component of chloroplasts and a better understanding of the genetic basis of chlorophyll in soybean [Glycine max (L.) Merr.] might contribute to improving photosynthetic capacity and yield in regions with adverse environmental conditions. A collection of 332 diverse soybean genotypes were grown in 2 years (2009 and 2010) and chlorophyll a (eChl_A), chlorophyll b (eChl_B), and total chlorophyll (eChl_T) content as well as chlorophyll a/b ratio (eChl_R) in leaf tissues were determined by extraction and spectrometric determination. Total chlorophyll was also derived from canopy spectral reflectance measurements using a model of wavelet transformed spectra (tChl_T) as well as with a spectral reflectance index (iChl_T). RESULTS: A genome-wide associating mapping approach was employed using 31,253 single nucleotide polymorphisms (SNPs) to identify loci associated with the extract based eChl_A, eChl_B, eChl_R and eChl_T measurements and the two canopy spectral reflectance-based methods (tChl_T and iChl_T). A total of 23 (14 loci), 15 (7 loci) and 14 SNPs (10 loci) showed significant association with eChl_A, eChl_B and eChl_R respectively. A total of 52 unique SNPs were significantly associated with total chlorophyll content based on at least one of the three approaches (eChl_T, tChl_T and iChl_T) and likely tagged 27 putative loci for total chlorophyll content, four of which were indicated by all three approaches. CONCLUSIONS: Results presented here show that markers for chlorophyll traits can be identified in soybean using both extract-based and canopy spectral reflectance-based phenotypes, and confirm that high-throughput phenotyping-amenable canopy spectral reflectance measurements can be used for association mapping.
DOI:10.1038/srep31419URLPMID:27506952 [本文引用: 3]
Leaf colour regulation is important in photosynthesis and dry material production. Most of the reported chlorophyll-deficient loci are recessive. The dominant locus is rarely reported, although it may be more important than the recessive locus in the regulation of photosynthesis efficiency. During the present study, we mapped a chlorophyll-deficient dominant locus (CDE1) from the ethyl methanesulfonate-mutagenized Brassica napus line NJ7982. Using an F2 population derived from the chlorophyll-deficient mutant (cde1) and the canola variety 'zhongshuang11', a high-density linkage map was constructed, consisting of 19 linkage groups with 2,878 bins containing 13,347 SNP markers, with a total linkage map length of 1,968.6 cM. Next, the CDE1 locus was mapped in a 0.9-cM interval of chromosome C08 of B. napus, co-segregating with nine SNP markers. In the following fine-mapping of the gene using the inherited F2:3 populations of 620 individuals, the locus was identified in an interval with a length of 311 kb. A bioinformatics analysis revealed that the mapping interval contained 22 genes. These results produced a good foundation for continued research on the dominant locus involved in chlorophyll content regulation.
DOI:10.1016/j.molp.2016.10.017URLPMID:27825945 [本文引用: 2]
Chlorophyll levels provide important information about plant growth and physiological plasticity in response to changing environments. The stay-green gene NON-YELLOWING 1 (NYE1) is believed to regulate chlorophyll degradation during senescence, concomitantly affecting the disassembly of the light-harvesting complex and hence indirectly influencing photosynthesis. We identified Brassica napus accessions carrying an NYE1 deletion associated with increased chlorophyll content, and with upregulated expression of light-harvesting complex and photosynthetic reaction center (PSI and PSII) genes. Comparative analysis of the seed oil content of accessions with related genetic backgrounds revealed that the B. napus NYE1 gene deletion (bnnye1) affected oil accumulation, and linkage disequilibrium signatures suggested that the locus has been subject to artificial selection by breeding in oilseed B. napus forms. Comparative analysis of haplotype diversity groups (haplogroups) between three different ecotypes of the allopolyploid B. napus and its A-subgenome diploid progenitor, Brassica rapa, indicated that introgression of the bnnye1 deletion from Asian B. rapa into winter-type B. napus may have simultaneously improved its adaptation to cooler environments experienced by autumn-sown rapeseed.
DOI:10.1016/j.scienta.2012.09.004URL [本文引用: 1]
DOI:10.1016/j.pbi.2015.01.006URLPMID:25637954 [本文引用: 1]
The plant metabolome is the readout of plant physiological status and is regarded as the bridge between the genome and the phenome of plants. Unraveling the natural variation and the underlying genetic basis of plant metabolism has received increasing interest from plant biologists. Enabled by the recent advances in high-throughput profiling and genotyping technologies, metabolite-based genome-wide association study (mGWAS) has emerged as a powerful alternative forward genetics strategy to dissect the genetic and biochemical bases of metabolism in model and crop plants. In this review, recent progress and applications of mGWAS in understanding the genetic control of plant metabolism and in interactive functional genomics and metabolomics are presented. Further directions and perspectives of mGWAS in plants are also discussed.
DOI:10.1007/s00122-018-3079-5URLPMID:29497767 [本文引用: 1]
KEY MESSAGE: Thirty significant associations between 22 SNPs and five plant architecture component traits in Chinese upland cotton were identified via GWAS. Four peak SNP loci located on chromosome D03 were simultaneously associated with more plant architecture component traits. A candidate gene, Gh_D03G0922, might be responsible for plant height in upland cotton. A compact plant architecture is increasingly required for mechanized harvesting processes in China. Therefore, cotton plant architecture is an important trait, and its components, such as plant height, fruit branch length and fruit branch angle, affect the suitability of a cultivar for mechanized harvesting. To determine the genetic basis of cotton plant architecture, a genome-wide association study (GWAS) was performed using a panel composed of 355 accessions and 93,250 single nucleotide polymorphisms (SNPs) identified using the specific-locus amplified fragment sequencing method. Thirty significant associations between 22 SNPs and five plant architecture component traits were identified via GWAS. Most importantly, four peak SNP loci located on chromosome D03 were simultaneously associated with more plant architecture component traits, and these SNPs were harbored in one linkage disequilibrium block. Furthermore, 21 candidate genes for plant architecture were predicted in a 0.95-Mb region including the four peak SNPs. One of these genes (Gh_D03G0922) was near the significant SNP D03_31584163 (8.40 kb), and its Arabidopsis homologs contain MADS-box domains that might be involved in plant growth and development. qRT-PCR showed that the expression of Gh_D03G0922 was upregulated in the apical buds and young leaves of the short and compact cotton varieties, and virus-induced gene silencing (VIGS) proved that the silenced plants exhibited increased PH. These results indicate that Gh_D03G0922 is likely the candidate gene for PH in cotton. The genetic variations and candidate genes identified in this study lay a foundation for cultivating moderately short and compact varieties in future Chinese cotton-breeding programs.
DOI:10.1111/pbi.12734URLPMID:28371164 [本文引用: 1]
DOI:10.1111/nph.14154URLPMID:27596807 [本文引用: 1]
Genome-wide association studies (GWAS) have been used extensively to dissect the genetic regulation of complex traits in plants. These studies have focused largely on the analysis of common genetic variants despite the abundance of rare polymorphisms in several species, and their potential role in trait variation. Here, we conducted the first GWAS in Populus deltoides, a genetically diverse keystone forest species in North America and an important short rotation woody crop for the bioenergy industry. We searched for associations between eight growth and wood composition traits, and common and low-frequency single-nucleotide polymorphisms detected by targeted resequencing of 18 153 genes in a population of 391 unrelated individuals. To increase power to detect associations with low-frequency variants, multiple-marker association tests were used in combination with single-marker association tests. Significant associations were discovered for all phenotypes and are indicative that low-frequency polymorphisms contribute to phenotypic variance of several bioenergy traits. Our results suggest that both common and low-frequency variants need to be considered for a comprehensive understanding of the genetic regulation of complex traits, particularly in species that carry large numbers of rare polymorphisms. These polymorphisms may be critical for the development of specialized plant feedstocks for bioenergy.
DOI:10.1186/s12864-017-4160-1URLPMID:29115920 [本文引用: 1]
DOI:10.1038/s41467-019-09134-9URLPMID:30858362 [本文引用: 2]
Brassica napus (2n = 4x = 38, AACC) is an important allopolyploid crop derived from interspecific crosses between Brassica rapa (2n = 2x = 20, AA) and Brassica oleracea (2n = 2x = 18, CC). However, no truly wild B. napus populations are known; its origin and improvement processes remain unclear. Here, we resequence 588 B. napus accessions. We uncover that the A subgenome may evolve from the ancestor of European turnip and the C subgenome may evolve from the common ancestor of kohlrabi, cauliflower, broccoli, and Chinese kale. Additionally, winter oilseed may be the original form of B. napus. Subgenome-specific selection of defense-response genes has contributed to environmental adaptation after formation of the species, whereas asymmetrical subgenomic selection has led to ecotype change. By integrating genome-wide association studies, selection signals, and transcriptome analyses, we identify genes associated with improved stress tolerance, oil content, seed quality, and ecotype improvement. They are candidates for further functional characterization and genetic improvement of B. napus.
DOI:10.1126/science.1253435URLPMID:25146293 [本文引用: 1]
Oilseed rape (Brassica napus L.) was formed ~7500 years ago by hybridization between B. rapa and B. oleracea, followed by chromosome doubling, a process known as allopolyploidy. Together with more ancient polyploidizations, this conferred an aggregate 72x genome multiplication since the origin of angiosperms and high gene content. We examined the B. napus genome and the consequences of its recent duplication. The constituent An and Cn subgenomes are engaged in subtle structural, functional, and epigenetic cross-talk, with abundant homeologous exchanges. Incipient gene loss and expression divergence have begun. Selection in B. napus oilseed types has accelerated the loss of glucosinolate genes, while preserving expansion of oil biosynthesis genes. These processes provide insights into allopolyploid evolution and its relationship with crop domestication and improvement.
DOI:10.1038/srep14407URLPMID:26394547 [本文引用: 2]
Silique length (SL) and seed weight (SW) are important yield-associated traits in rapeseed (Brassica napus). Although many quantitative trait loci (QTL) for SL and SW have been identified in B. napus, comparative analysis for those QTL is seldom performed. In the present study, 20 and 21 QTL for SL and SW were identified in doubled haploid (DH) and DH-derived reconstructed F2 populations in rapeseed, explaining 55.1-74.3% and 24.4-62.9% of the phenotypic variation across three years, respectively. Of which, 17 QTL with partially or completely overlapped confidence interval on chromosome A09, were homologous with two overlapped QTL on chromosome C08 by aligning QTL confidence intervals with the reference genomes of Brassica crops. By high density selective genotyping of DH lines with extreme phenotypes, using a Brassica single-nucleotide polymorphism (SNP) array, the QTL on chromosome A09 was narrowed, and aligned into 1.14-Mb region from 30.84 to 31.98 Mb on chromosome R09 of B. rapa and 1.05-Mb region from 27.21 to 28.26 Mb on chromosome A09 of B. napus. The alignment of QTL with Brassica reference genomes revealed homologous QTL on A09 and C08 for SL. The narrowed QTL region provides clues for gene cloning and breeding cultivars by marker-assisted selection.
DOI:10.1186/s12870-016-0759-7URLPMID:27000872 [本文引用: 1]
BACKGROUND: Yield of rapeseed is determined by three components: silique number, seed number per silique and thousand seed weight. Seed number per silique and thousand seed weight are influenced by silique length, seed density, silique breadth, silique thickness and silique volume. Some QTLs for silique traits have been reported in B. napus, however, no studies have focused on the six agronomic traits (seed number per silique, silique length, silique breadth, silique thickness, seed density and silique volume) simultaneously, and the genetic determinism of such complex traits have not been fully elucidated. RESULTS: In this study, the six silique traits were evaluated using 348 lines of a doubled haploid population, the KN population. The results showed that 2, 4, 1, 1 and 2 QTLs explaining > 10 % of phenotypic variation were obtained for silique length, silique breadth, silique thickness, seed number per silique and silique volume, respectively. Notably, three major effect QTLs (cqSB-C6-1, cqSB-C6-2 and cqSV-C6-3) were identified in at least three environments, and 17 unique QTLs controlling at least two traits were obtained. A high-density consensus map containing 1225 markers was constructed for QTL comparison by combining the KN map with other five published maps. The comparative results revealed that 14, 13 and 11 QTLs for silique breadth, silique thickness and silique volume might be the potential new QTLs because few QTLs for these traits were reported in B. napus. In addition, potential new QTLs for silique length (11), seed number per silique (6) and seed density (5) were also identified. Twenty-five candidate genes underlying 27 QTLs for silique related traits were obtained. CONCLUSIONS: This study constructed QTL analysis in B. napus, and obtained 60 consensus QTLs for six silique related traits. The potential new QTLs will enhance our understanding of the genetic control of silique traits, and the stable QTLs provided the targets for improving seed yield in future. These findings provided comprehensive insights into the genetic network affecting silique traits at QTL level in B. napus.
DOI:10.1186/1471-2164-9-390URLPMID:18713468 [本文引用: 1]
BACKGROUND: DNA Microarrays are regarded as a valuable tool for basic and applied research in microbiology. However, for many industrially important microorganisms the lack of commercially available microarrays still hampers physiological research. Exemplarily, our understanding of protein folding and secretion in the yeast Pichia pastoris is presently widely dependent on conclusions drawn from analogies to Saccharomyces cerevisiae. To close this gap for a yeast species employed for its high capacity to produce heterologous proteins, we developed full genome DNA microarrays for P. pastoris and analyzed the unfolded protein response (UPR) in this yeast species, as compared to S. cerevisiae. RESULTS: By combining the partially annotated gene list of P. pastoris with de novo gene finding a list of putative open reading frames was generated for which an oligonucleotide probe set was designed using the probe design tool TherMODO (a thermodynamic model-based oligoset design optimizer). To evaluate the performance of the novel array design, microarrays carrying the oligo set were hybridized with samples from treatments with dithiothreitol (DTT) or a strain overexpressing the UPR transcription factor HAC1, both compared with a wild type strain in normal medium as untreated control. DTT treatment was compared with literature data for S. cerevisiae, and revealed similarities, but also important differences between the two yeast species. Overexpression of HAC1, the most direct control for UPR genes, resulted in significant new understanding of this important regulatory pathway in P. pastoris, and generally in yeasts. CONCLUSION: The differences observed between P. pastoris and S. cerevisiae underline the importance of DNA microarrays for industrial production strains. P. pastoris reacts to DTT treatment mainly by the regulation of genes related to chemical stimulus, electron transport and respiration, while the overexpression of HAC1 induced many genes involved in translation, ribosome biogenesis, and organelle biosynthesis, indicating that the regulatory events triggered by DTT treatment only partially overlap with the reactions to overexpression of HAC1. The high reproducibility of the results achieved with two different oligo sets is a good indication for their robustness, and underlines the importance of less stringent selection of regulated features, in order to avoid a large number of false negative results.
DOI:10.3389/fpls.2018.00390URLPMID:29643859 [本文引用: 1]
Plant height (PH), branch initiation height (BIH), and stem diameter (SD) are three stem-related traits that play crucial roles in plant architecture and lodging resistance. Herein, we show one doubled haploid (DH) population obtained from a cross between Y689 (one Capsella bursa-pastoris derived Brassica napus intertribal introgression) and Westar (B. napus cultivar) that these traits were significantly positively correlated with one another and with flowering time (FT). Based on a high-density SNP map, a total of 102 additive quantitative trait loci (QTL) were identified across six environments. Seventy-two consensus QTL and 49 unique QTL were identified using a two-round strategy of QTL meta-analysis. Notably, a total of 19 major QTL, including 11 novel ones, were detected for these traits, which comprised two QTL clusters on chromosomes A02 and A07. Conditional QTL mapping was performed to preliminarily evaluate the genetic basis (pleiotropy or tight linkage) of the co-localized QTL. In addition, QTL by environment interactions (QEI) mapping was performed to verify the additive QTL and estimate the QEI effect. In the genomic regions of all major QTL, orthologs of the genes involved in phytohormone biosynthesis, phytohormone signaling, flower development, and cell differentiation in Arabidopsis were proposed as candidate genes. Of these, BnaA02g02560, an ortholog of Arabidopsis GASA4, was suggested as a candidate gene for PH, SD, and FT; and BnaA02g08490, an ortholog of Arabidopsis GNL, was associated with PH, BIH and FT. These results provide useful information for further genetic studies on stem-related traits and plant growth adaptation.
DOI:10.1038/srep36452URLPMID:27811979 [本文引用: 1]
Harvest index (HI), the ratio of seed mass to total biomass of the aboveground plant parts, is an important trait for harvestable yield of crops. Unfortunately, HI of Brassica napus is lower than that of other economically important crops. To identify candidate genes associated with high HI, a genome-wide association study of HI and four HI-related traits was conducted with 520 B. napus accessions cultivated in both Yunnan and Chongqing. We detected 294 single nucleotide polymorphisms significantly associated with the abovementioned traits, including 79 SNPs that affected two or more traits. Differentially expressed genes between extremely high- and low-HI accessions were identified in 8 tissues at two cultivated regions. Combination of linkage disequilibrium and transcriptome analyses revealed 33 functional candidate genes located within the confidence intervals of significant SNPs associated with more than one trait, such as SHOOT GRAVITROPISM 5 (Bna.SGR5), ATP-CITRATE LYASE A-3 (Bna.ACLA-3) and CAROTENOID CLEAVAGE DIOXYGENASE 1 (Bna.CCD1), their orthologs in the Arabidopsis thaliana have been shown to play key roles in photosynthesis, inflorescence, and silique development. Our results provide insight into the molecular mechanisms underlying establishment of high-HI B. napus and lay a foundation for characterization of candidate genes aimed at developing high-HI B. napus varieties.
DOI:10.1021/acs.jafc.7b01226URLPMID:28650150 [本文引用: 1]
Seed coat color is an extremely important breeding characteristic of Brassica napus. To elucidate the factors affecting the genetic architecture of seed coat color, a genome-wide association study (GWAS) of seed coat color was conducted with a diversity panel comprising 520 B. napus cultivars and inbred lines. In total, 22 single-nucleotide polymorphisms (SNPs) distributed on 7 chromosomes were found to be associated with seed coat color. The most significant SNPs were found in 2014 near Bn-scaff_15763_1-p233999, only 43.42 kb away from BnaC06g17050D, which is orthologous to Arabidopsis thaliana TRANSPARENT TESTA 12 (TT12), an important gene involved in the transportation of proanthocyanidin precursors into the vacuole. Two of eight repeatedly detected SNPs can be identified and digested by restriction enzymes. Candidate gene mining revealed that the relevant regions of significant SNP loci on the A09 and C08 chromosomes are highly homologous. Moreover, a comparison of the GWAS results to those of previous quantitative trait locus (QTL) studies showed that 11 SNPs were located in the confidence intervals of the QTLs identified in previous studies based on linkage analyses or association mapping. Our results provide insights into the genetic basis of seed coat color in B. napus, and the beneficial allele, SNP information, and candidate genes should be useful for selecting yellow seeds in B. napus breeding.
DOI:10.1186/s13068-019-1557-xURLPMID:31528204 [本文引用: 1]
Background: Increasing seed oil content is one of the most important targets for rapeseed (Brassica napus) breeding. However, genetic mechanisms of mature seed oil content in Brassica napus (B. napus) remain little known. To identify oil content-related genes, a genome-wide association study (GWAS) was performed using 588 accessions. Results: High-throughput genome resequencing resulted in 385,692 high-quality single nucleotide polymorphism (SNPs) with a minor allele frequency (MAF) > 0.05. We identified 17 loci that were significantly associated with seed oil content, among which 12 SNPs were distributed on the A3 (11 loci) and A1 (one loci) chromosomes, and five novel significant SNPs on the C5 (one loci) and C7 (four loci) chromosomes, respectively. Subsequently, we characterized differentially expressed genes (DEGs) between the seeds and silique pericarps on main florescences and primary branches of extremely high- and low-oil content accessions (HO and LO). A total of 64 lipid metabolism-related DEGs were identified, 14 of which are involved in triacylglycerols (TAGs) biosynthesis and assembly. Additionally, we analyzed differences in transcription levels of key genes involved in de novo fatty acid biosynthesis in the plastid, TAGs assembly and lipid droplet packaging in the endoplasmic reticulum (ER) between high- and low-oil content B. napus accessions. Conclusions: The combination of GWAS and transcriptome analyses revealed seven candidate genes located within the confidence intervals of significant SNPs. Current findings provide valuable information for facilitating marker-based breeding for higher seed oil content in B. napus.
DOI:10.1007/s11274-016-2181-6URLPMID:27909993 [本文引用: 1]
Chlorophyll is a commercially important natural green pigment responsible for the absorption of light energy and its conversion into chemical energy via photosynthesis in plants and algae. This bioactive compound is widely used in the food, cosmetic, and pharmaceutical industries. Chlorophyll has been consumed for health benefits as a nutraceutical agent with antioxidant, anti-inflammatory, antimutagenic, and antimicrobial properties. Microalgae are photosynthesizing microorganisms which can be extracted for several high-value bioproducts in the biotechnology industry. These microorganisms are highly efficient at adapting to physicochemical variations in the local environment. This allows optimization of culture conditions for inducing microalgal growth and biomass production as well as for changing their biochemical composition. The modulation of microalgal culture under controlled conditions has been proposed to maximize chlorophyll accumulation. Strategies reported in the literature to promote the chlorophyll content in microalgae include variation in light intensity, culture agitation, and changes in temperature and nutrient availability. These factors affect chlorophyll concentration in a species-specific manner; therefore, optimization of culture conditions has become an essential requirement. This paper provides an overview of the current knowledge on the effects of key environmental factors on microalgal chlorophyll accumulation, focusing on small-scale laboratory experiments.
DOI:10.1074/jbc.M406763200URLPMID:15322131 [本文引用: 1]
Identification of membrane proteomes remains challenging. Here, we present a simple, fast, and scalable off-line procedure based on three-phase partitioning with butanol to fractionate membrane proteomes in combination with both in-gel and in-solution digestions and mass spectrometry. This should help to further accelerate the field of membrane proteomics. Using this new strategy, we analyzed the salt-stripped thylakoid membrane of chloroplasts of Arabidopsis thaliana. 242 proteins were identified, at least 40% of which are integral membrane proteins. The functions of 86 proteins are unknown; these include proteins with TPR, PPR, rhodanese, and DnaJ domains. These proteins were combined with all known thylakoid proteins and chloroplast (associated) envelope proteins, collected from primary literature, resulting in 714 non-redundant proteins. They were assigned to functional categories using a classification developed for MapMan (Thimm, O., Blasing, O., Gibon, Y., Nagel, A., Meyer, S., Kruger, P., Selbig, J., Muller, L. A., Rhee, S. Y., and Stitt, M. (2004) Plant J. 37, 914-939), updated with information from primary literature. The analysis elucidated the likely location of many membrane proteins, including 190 proteins of unknown function, holding the key to better understanding the two membrane systems. The three-phase partitioning procedure added a new level of dynamic resolution to the known thylakoid proteome. An automated strategy was developed to track possible ambiguous identifications to more than one gene model or family member. Mass spectrometry search results, ambiguities, and functional classifications can be searched via the Plastid Proteome Database.
DOI:10.1042/BJ20101538URLPMID:21083539 [本文引用: 1]
The outer antenna of higher-plant PSI (Photosystem I) is composed of four complexes [Lhc (light-harvesting complex) a1-Lhca4] belonging to the light-harvesting protein family. Difficulties in their purification have so far prevented the determination of their properties and most of the knowledge about Lhcas has been obtained from the study of the in vitro reconstituted antennas. In the present study we were able to purify the native complexes, showing that Lhca2/3 and Lhca1/4 form two functional heterodimers. Both dimers show red-fluorescence emission with maxima around 730 nm, as in the intact PSI complex. This indicates that the dimers are in their native state and that LHCI-680, which was previously assumed to be part of the PSI antenna, does not represent the native state of the system. The data show that the light-harvesting properties of the two dimers are functionally identical, concerning absorption, long-wavelength emission and fluorescence quantum yield, whereas they differ in their high-light response. Implications of the present study for the understanding of the energy transfer process in PSI are discussed. Finally, the comparison of the properties of the native dimers with those of the reconstituted complexes demonstrates that all of the major properties of the Lhcas are reproduced in the in vitro systems.
DOI:10.1093/pcp/pcx009URLPMID:28184910 [本文引用: 1]
The light-harvesting complex I (LHCI) proteins in Arabidopsis thaliana are encoded by six genes. Major LHCI proteins (Lhca1-Lhca4) harvest light energy and transfer the resulting excitation energy to the PSI core by forming a PSI supercomplex. In contrast, the minor LHCI proteins Lhca5 and Lhca6 contribute to supercomplex formation between the PSI supercomplex and the chloroplast NADH dehydrogenase-like (NDH) complex, although Lhca5 is also solely associated with the PSI supercomplex. Lhca6 was branched from Lhca2 during the evolution of land plants. In this study, we focused on the molecular evolution involved in the transition from a major LHCI, Lhca2, to the linker protein Lhca6. To elucidate the domains of Lhca6 responsible for linker function, we systematically swapped domains between the two LHCI proteins. To overcome problems due to the low stability of chimeric proteins, we employed sensitive methods to evaluate supercomplex formation: we monitored NDH activity by using Chl fluorescence analysis and detected NDH-PSI supercomplex formation by using protein blot analysis in the form of two-dimensional blue-native (BN)/SDS-PAGE. The stromal loop of Lhca6 was shown to be necessary and sufficient for linker function. Chimeric Lhca6, in which the stromal loop was substituted by that of Lhca2, was not functional as a linker and was detected at the position of the PSI supercomplex in the BN-polyacrylamide gel. The stromal loop of Lhca6 is likely to be necessary for the interaction with chloroplast NDH, rather than for the association with the PSI supercomplex.
DOI:10.1111/j.1365-313X.2008.03401.xURLPMID:18182030 [本文引用: 1]
We surveyed differential gene expression patterns during early photomorphogenesis in both wild-type and mutant Arabidopsis defective in HY5, an influential positive regulator of the responses of gene expression to a light stimulus, to identify light-responsive genes whose expression was HY5 dependent. These gene-expression data identified light-regulated zinc finger protein 1 (LZF1), a gene encoding a previously uncharacterized C2C2-CO B-box transcriptional regulator. HY5 has positive trans-activating activity toward LZF1 and binding affinity to LZF1 promoter in vivo. HY5 is needed but not sufficient for the induction of LZF1 expression. Anthocyanin content is significantly diminished in lzf1 under far red, which is the most efficient light for the induction of LZF1. The expression of PAP1/MYB75 is elevated in plants overexpressing LZF1, which leads to the hyperaccumulation of anthocyanin in transgenic Arabidopsis. The transition from etioplast to chloroplast and the accumulation of chlorophyll were notably compromised in the lzf1 mutant. We provide molecular evidence that LZF1 influences chloroplast biogenesis and function via regulating genes encoding chloroplast proteins. In the absence of HY5, mutation of LZF1 leads to further reduced light sensitivity for light-regulated inhibition of hypocotyl elongation and anthocyanin and chlorophyll accumulation. Our data indicate that LZF1 is a positive regulator functioning in Arabidopsis de-etiolation.
DOI:10.1104/pp.111.175042URL [本文引用: 1]
Light regulates multiple aspects of growth and development in plants. Transcriptomic changes govern the expression of signaling molecules with the perception of light. Also, the 26S proteasome regulates the accumulation of positive and negative regulators for optimal growth of Arabidopsis (Arabidopsis thaliana) in the dark, light, or light/dark cycles. BBX22, whose induction is both light regulated and HY5 dependent, is a positive regulator of deetiolation in Arabidopsis. We found that during skotomorphogenesis, the expression of BBX22 needs to be tightly regulated at both transcriptional and posttranslational levels. During photomorphogenesis, the expression of BBX22 transiently accumulates to execute its roles as a positive regulator. BBX22 protein accumulates to a higher level under short-day conditions and functions to inhibit hypocotyl elongation. The proteasome-dependent degradation of BBX22 protein is tightly controlled even in plants overexpressing BBX22. An analysis of BBX22 degradation kinetics shows that the protein has a short half-life under both dark and light conditions. COP1 mediates the degradation of BBX22 in the dark. Although dispensable in the dark, HY5 contributes to the degradation of BBX22 in the light. The constitutive photomorphogenic development of the cop1 mutant is enhanced in cop1BBX22ox plants, which show a short hypocotyl, high anthocyanin accumulation, and expression of light-responsive genes. Exaggerated light responsiveness is also observed in cop1BBX22ox seedlings grown under short-day conditions. Therefore, the proper accumulation of BBX22 is crucial for plants to maintain optimal growth when grown in the dark as well as to respond to seasonal changes in daylength.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}