 ,1,**, 王芳,1,**, 方俐,1,**, 杨涛1, 张红岩1, 黄宇宁1, 王栋1,3, 季一山1, 徐东旭2, 李冠1, 郭瑞军1, 宗绪晓,1,*
,1,**, 王芳,1,**, 方俐,1,**, 杨涛1, 张红岩1, 黄宇宁1, 王栋1,3, 季一山1, 徐东旭2, 李冠1, 郭瑞军1, 宗绪晓,1,*An integrated high-density SSR genetic linkage map from two F2 population in Chinese pea
LIU Rong,1,**, WANG Fang,1,**, FANG Li,1,**, YANG Tao1, ZHANG Hong-Yan1, HUANG Yu-Ning1, WANG Dong1,3, JI Yi-Shan1, XU Dong-Xu2, LI Guan1, GUO Rui-Jun1, ZONG Xu-Xiao,1,*通讯作者:
收稿日期:2020-02-8接受日期:2020-04-15网络出版日期:2020-05-09
| 基金资助: |
Received:2020-02-8Accepted:2020-04-15Online:2020-05-09
| Fund supported: |
作者简介 About authors
刘荣, E-mail:
王芳, E-mail:
方俐, E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1354KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘荣, 王芳, 方俐, 杨涛, 张红岩, 黄宇宁, 王栋, 季一山, 徐东旭, 李冠, 郭瑞军, 宗绪晓. 利用2个F2群体整合中国豌豆高密度SSR遗传连锁图谱[J]. 作物学报, 2020, 46(10): 1496-1506. doi:10.3724/SP.J.1006.2020.04028
LIU Rong, WANG Fang, FANG Li, YANG Tao, ZHANG Hong-Yan, HUANG Yu-Ning, WANG Dong, JI Yi-Shan, XU Dong-Xu, LI Guan, GUO Rui-Jun, ZONG Xu-Xiao.
豌豆(Pisum sativum L.)属于豆科(Leguminosae/ Fabaceae), 野豌豆族(Vicieae), 豌豆属(Pisum), 染色体数为2n = 2x = 14, 基因组大小约为4.45 Gb[1,2]。豌豆富含蛋白质和多种营养元素, 是经济上最重要的食用豆类作物之一, 在世界范围内广泛种植, 既可以作为谷物和蔬菜供人类食用, 又可作为牲畜的饲料[3,4]。根据FAO的统计, 2018年, 豌豆(包括青豌豆和干豌豆)在全球食用豆类作物中的总产量仅次于普通菜豆; 同时, 中国青豌豆的总产量居世界首位, 而干豌豆的总产量仅次于加拿大和俄罗斯[5]。此外, 豌豆因其固氮能力而被认为是一种环境友好型作物, 在可持续农业系统中起着至关重要的作用[6]。
高密度遗传连锁图谱是功能基因定位、比较基因组学以及分子辅助育种等研究的重要工具[7]。以前, 人们一直致力于利用包括RFLP、RAPD、SSR和SNP在内的多种分子标记基于不同类型的群体来构建豌豆的遗传连锁图谱[8,9,10,11,12,13,14,15]。最近, 有****针对豌豆开发了基于高密度SNP的遗传连锁图谱, 并为鉴定重要农艺性状的遗传基础提供了强大的工具[16,17,18,19]。此外, 新近公布的豌豆参考基因组也为理解豌豆关键农艺性状的分子基础并促进其育种改良奠定了重要基础[20]。
SSR标记因其具有信息量丰富、共显性遗传、多等位基因、基因组覆盖广等特性, 同时在相近物种之间具有可重复性和可移植性[21,22], 在遗传多样性评估和物种亲缘关系鉴定[23,24]、遗传连锁图谱构建[25,26]、标记辅助选择[27,28]、DNA指纹图谱鉴定[29,30]等方面具有显著优势。相比基因组SSR, 位于基因区的EST-SSR因其具有更高的可转移性、较低的开发成本以及与基因的密切关系, 而越来越受到人们的重视[11,21]。然而, 尽管针对豌豆的遗传连锁作图研究已有很长的历史, 并且在豌豆中已经构建了几十种具有不同标记的遗传连锁图谱[31], 但公众可获得的可用于豌豆遗传研究的SSR上图标记较少, 同时基于遗传独特的中国豌豆种质[1,32-33]的遗传连锁图谱仍然有限。值得注意的是, 过去基于中国豌豆种质构建的遗传连锁图谱包括157个SSR标记, 分布在11个连锁群中, 全长1518 cM, 标记数量较少, 需要进一步加密并完善至7个连锁群[15]。
与单个遗传连锁图谱相比, 整合遗传连锁图谱由于整合了多个群体的信息而具有多种优势[34,35], 例如具有更高的标记密度, 更完整的基因组覆盖范围, 可对不同群体进行标记共线性比较等, 在许多作物包括豌豆中均有应用[16,20,36-37]。因此, 本研究的目的如下: 1)筛选豌豆中可移植转换的SSR标记, 用于豌豆的遗传研究和分子作图。2)对我们以往基于G0003973×G0005527 F2群体, 构建的遗传连锁图谱进行加密。3)基于W6-22600×W6-15174 F2群体, 构建新的遗传连锁图谱。4)结合上述2个基于中国种质的遗传连锁图谱信息, 构建一张豌豆整合SSR遗传连锁图谱。
1 材料与方法
1.1 作图群体
本研究利用基于中国豌豆种质为亲本的2个F2群体进行遗传连锁作图。群体1 (PSP1)与本实验室之前的研究相同[15], 来自母本G0003973 (耐寒)和父本G0005527 (不耐寒)之间的杂交, 由190个F2个体组成。群体2 (PSP2)则是以母本W6-22600 (多小叶)与父本W6-15174 (无小叶)进行杂交, 由480个F2个体组成。1.2 SSR标记筛选
利用本实验室自主开发[38]和文献获取[39,40,41]的12,491个SSR标记(包括11,145个基因组SSR和1346个EST-SSR), 对PSP1的亲本及随机选择的4个F2个体进行全基因组扫描, 筛选出多态性SSR标记用于遗传连锁作图。此外, 从以往研究中已发表的豌豆遗传连锁图谱中, 选择具有已知连锁群位置的125个SSR标记[10,42-44], 利用PSP1和PSP2的亲本和4个随机选择的F2个体来筛选锚定标记。1.3 DNA提取和PCR扩增
在2个F2群体种植当年, 收集每个F2个体植株的嫩叶, 经液氮速冻后, 使用改良的CTAB方法[45]提取基因组DNA。用NanoDrop 2000检测DNA浓度并稀释到工作液浓度50 ng μL-1后, 于-20℃保存备用。PCR扩增反应体系为10 μL, 包含1.5 μL基因组DNA (50 ng μL-1)、5 μL 2×Taq PCR Master Mix (Genstar, 中国北京)、0.5 μL正向引物 (2 μmol L-1)、0.5 μL反向引物 (2 μmol L-1)和2.5 μL ddH2O。PCR产物通过8%非变性聚丙烯酰胺凝胶电泳(PAGE)分离, 并通过0.1%硝酸银染色。根据片段大小记录等位基因状态, SSR标记状态编码如下: 与父本相同的带型记为“AA”, 与母本相同的带型记为“BB”; 具有双亲带型的记为“AB”; 缺失或无效的带型记为“-”。只有那些能够扩增出清晰条带并可以显示亲本多态性的SSR标记才被选择用于后续的基因分型。
1.4 遗传连锁图谱构建
分别对PSP1和PSP2的所有F2个体进行基因分型, 并去除缺失数据超过20%的标记或个体。使用χ2分析来检测标记偏分离状况, 并使用Bonferroni校正对P = 0.05的显著性水平进行校正, 在进一步的遗传作图中排除显著偏分离的标记。利用Kosambi作图函数对2个群体构建遗传连锁图谱, LOD>2。在以往公布的豌豆遗传连锁图谱的基础上, 通过筛选得到的锚定标记对每个连锁群进行分组[10,42-44]。然后, 利用共有标记将这2个群体的信息整合到一张遗传连锁图谱上。以上所有分析均利用QTL IciMapping V4.0软件完成[46]。遗传连锁图谱和物理图谱利用MapChart V2.3软件进行可视化展示[47]。然后, 本研究以新近发表的豌豆基因组为参考(Caméor genome build 1a) [20], 利用KnowPulse网站(https:// knowpulse.usask.ca/blast/nucleotide/nucleotide)的BL ASTn工具对50个共有标记的扩增片段序列进行比对, 参数选取默认参数, E-value设为1e-3。2 结果与分析
2.1 多态性标记筛选
利用本实验室自主开发[38]和文献获取[39,40,41]的12,491个SSR标记(包括11,145个基因组SSR和1346个EST-SSR), 对PSP1的亲本及随机选择的4个F2个体进行全基因组扫描, 初步筛选出扩增条带清晰且在父母本间呈多态性差异的954个多态性SSR标记, 用于PSP1群体190个F2个体的基因分型, 最终得到729个在190份F2群体单株中有清晰条带的多态性标记, 用于后续遗传连锁图谱的构建。然后利用这729个多态性标记对PSP2的亲本及随机选择的4个F2个体进行多态性检测, 最终在480个F2个体中成功筛选了103个多态性标记用于PSP2的遗传连锁图谱构建。此外, 本研究还利用125个已知遗传连锁群位置信息的可公开获得的SSR标记[10,39-41]在2个群体中筛选锚定标记, 分别在PSP1和PSP2中鉴定出11个和17个锚定标记, 其中有3个标记为2个群体共有的锚定标记, 共计25个锚定标记, 这些标记可在后续遗传连锁图谱构建中用于分配连锁群(表1)。
Table 1
表1
表1筛选得到的25个锚定标记在以往发表豌豆遗传连锁图谱的分布
Table 1
| 连锁群 Linkage group | 锚定标记的数目 Number of anchor markers | 锚定标记的名称 Name of anchor markers |
|---|---|---|
| I | 5 | AA67, AD147, AF016458, D21, PsAS2 |
| II | 3 | AA332, AD83, D23 |
| III | 4 | AA355, AD270, PSAJ3318, PSBLOX13.2 |
| IV | 4 | AA430942, AA122, AA285, AA315 |
| V | 1 | PSGAPA1 |
| VI | 5 | AA335, AB71, AD160, AD60, PSGSR1 |
| VII | 3 | AB65, AF004843, PSAB60 |
新窗口打开|下载CSV
2.2 遗传连锁图谱构建
获得所有样本的基因型数据后, 首先对所有标记进行数据完整性和偏分离检测。对于PSP1作图群体, 740个标记(包含11个锚定标记)中, 有43个标记缺失信息大于20%, 占总标记数的5.81%, 而有80个标记检测到偏分离现象, 占总标记数的10.81%。因此, 排除这123个标记后, 共有617个标记用于后续的遗传连锁图谱分析。针对PSP1构建的遗传连锁图谱, 将603个标记分配到7条连锁群上。根据11个锚定标记, PSP1图谱的7条连锁群分别对应于以往发表的遗传连锁图谱的6条连锁群[10,39-41], 有2条连锁群均对应于LGI, 而缺少对应于LGV的连锁群, 可能是由于缺乏LGV的锚定标记。此外, 该图谱全长5330.6 cM, 相邻标记之间的平均距离为8.9 cM。每条连锁群的长度从494.9 cM (LGII)到904.7 cM (LGIV)不等, 平均为761.5 cM; 每条连锁群的标记数目从62 (LGII)到113 (LGI-2)不等, 平均为86个标记(图1和表2)。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1豌豆PSP1群体遗传连锁图谱
粗体代表锚定标记, 标记名称以“e”开头的为EST-SSR标记。
Fig. 1Genetic linkage map of PSP1 population in pea
The bold labels on the marker name represent anchor markers, and marker names started with “e” represent EST-SSR markers.
Table 2
表2
表2利用豌豆PSP1和PSP2群体构建的2个遗传连锁图谱的标记分布
Table 2
| 连锁群 Linkage group | 上图标记数目 Number of mapped markers | 图谱长度 Map length (cM) | 平均标记密度 Average marker density | |||
|---|---|---|---|---|---|---|
| PSP1 | PSP2 | PSP1 | PSP2 | PSP1 | PSP2 | |
| I | 74 | 11 | 751.0 | 137.4 | 10.1 | 12.5 |
| II | 62 | 13 | 494.9 | 160.6 | 8.0 | 12.4 |
| III | 90 | 20 | 855.3 | 158.4 | 9.5 | 7.9 |
| IV | 97 | 14 | 904.7 | 119.0 | 9.3 | 8.5 |
| I-2/V | 113 | 19 | 822.5 | 173.3 | 7.3 | 9.1 |
| VI | 70 | 21 | 658.6 | 175.0 | 9.4 | 8.3 |
| VII | 97 | 20 | 843.6 | 203.4 | 8.7 | 10.2 |
| 平均Mean | 86 | 17 | 761.5 | 161.0 | 8.9 | 9.8 |
| 总计Total | 603 | 118 | 5330.6 | 1127.1 | 62.3 | 68.9 |
新窗口打开|下载CSV
对于PSP2作图群体来说, 120个标记(包含17个锚定标记)中, 仅有1个标记由于缺失大于20%而被排除在后续分析之外。剩余的119个标记被用于进一步的连锁作图分析, 结果发现上图的118个标记分布在7条连锁群上。根据17个锚定标记, 这7条连锁群刚好完全对应于以往发表的遗传连锁图谱的7个连锁群[10,39-41]。此外, 该图谱的累积长度为1127.1 cM, 相邻标记之间的平均遗传距离为9.8 cM (图2和表2)。每条连锁群的长度从119.0 cM (LG IV)到203.4 cM (LGVII)不等, 平均为161.0 cM; 每条连锁群的标记数目从11 (LGI)到21 (LGVI)不等, 平均为17个标记(图2和表2)。
图2
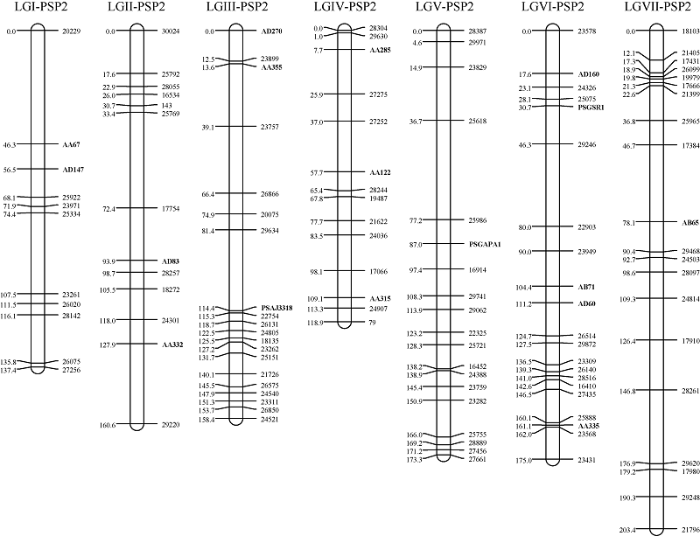 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2豌豆PSP2群体遗传连锁图谱
粗体代表锚定标记。
Fig. 2Genetic linkage map of PSP2 population in pea
The bold labels on the marker name represent anchor markers.
2.3 整合遗传连锁图谱构建
基于以上2个遗传连锁图谱, 通过两两比较共发现了53个共有标记, 每个连锁群上的共有标记数为3 (LGI)到14 (LGVII)不等(表3)。利用上述2个遗传连锁图上的53个共有标记, 我们构建了一张包含668个SSR标记的整合遗传连锁图谱(标记信息详见附表1), 分布在7条连锁群上, 累积长度为6592.6 cM, 相邻标记之间的平均距离为10.0 cM。每条连锁群的长度从682.7 cM (LGII)到1077.2 cM (LGIII)不等, 平均为941.8 cM。在这7条连锁群中, 分布在LGV的标记数量最多, 有125个标记, 同时标记密度也最低, 为8.1 cM; 另有3条连锁群包含的标记数也都超过了100, 分别是LGIV (104)、LGVII (103)和LGIII (102); 而标记数量最少的连锁群为LGII, 仅有68个标记, 累积长度也是最小的, 仅有682.7 cM (图3和表3)。Table 3
表3
表3豌豆整合遗传连锁图谱上的标记分布
Table 3
| 连锁群 Linkage group | 共有标记数目 Number of bridge markers | 上图标记数目 Number of mapped markers | 图谱长度 Map length (cM) | 平均标记密度 Average marker density |
|---|---|---|---|---|
| I | 3 | 82 | 976.8 | 11.9 |
| II | 7 | 68 | 682.7 | 10.0 |
| III | 8 | 102 | 1077.2 | 10.6 |
| IV | 7 | 104 | 1058.9 | 10.2 |
| V | 7 | 125 | 1018.2 | 8.1 |
| VI | 7 | 84 | 801.8 | 9.5 |
| VII | 14 | 103 | 977.0 | 9.5 |
| 平均Mean | 7.6 | 95 | 941.8 | 10.0 |
| 总计Total | 53 | 668 | 6592.6 | 69.8 |
新窗口打开|下载CSV
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3豌豆PSP1和PSP2整合遗传连锁图谱
粗体代表锚定标记, 下画线代表共有标记, 粗体加下画线代表共有锚定标记。标记名称以“e”开头的为EST-SSR标记。
Fig. 3Genetic linkage map integrated with PSP1 and PSP2 information in pea
The bold, underlined and bold underlined labels on the marker name represent anchor markers, common markers and common anchor markers, respectively. Marker names started with “e” represent EST-SSR markers.
Supplementary table 1
附表1
附表1豌豆整合遗传连锁图谱上图标记信息
Supplementary table 1
| 序号 No. | 标记名称 Marker name | 标记类型 Marker type | 正向引物序列 Forward primer sequence (5°-3°) | 反向引物序列 Reverse primer sequence (5°-3°) | 退火温度Tm (℃) | 片段大小Band size (bp) | 连锁群Linkage group | ||
|---|---|---|---|---|---|---|---|---|---|
| 整合图谱Integrated map | PSP1 G0003973× G0005527 | PSP2 W6-22600×W6-15174) | |||||||
| 1 | 27256 | 基因组SSR | CATTCCATTCCATACATCCATTT | TGAAGTTGAAGCAGCCATTG | 60 | 153 | I | I | |
| 2 | 26075 | 基因组SSR | CTATGGCACCATCTCTTGGAC | AACACAATGTATGTGGTGCAAAT | 59 | 186 | I | I | |
| 3 | 28142 | 基因组SSR | TGCAAGCATATTGCCTTTTC | TCAGTGGTTGCTAGCTGTTGA | 59 | 176 | I | I | |
| 4 | 26020 | 基因组SSR | CTCTCAAATTTGGGGTCCTC | TCATTGCGTCTCAACCTCAG | 59 | 129 | I | I | |
| 5 | 25334 | 基因组SSR | TGAAGATGTGACAAACAACAGAAA | TCCTTCTCTGTTCCCACCAC | 60 | 140 | I | I | I |
| 6 | 23971 | 基因组SSR | GCGTGTTGATGCTGAAAGAA | TCAAAATTGGGGTGTGACAA | 60 | 131 | I | I | |
| 7 | 25922 | 基因组SSR | CCAAGGGAAAACCCCTTCTA | GTGTGAAAGCTTATTGTATATGTATCG | 58 | 176 | I | I | |
| 8 | AD147 | 锚定标记 | AGCCCAAGTTTCTTCTGAATCC | AAATTCGCAGAGCGTTTGTTAC | 61 | 330 | I | I-2 | I |
| 9 | AA67 | 锚定标记 | CCCATGTGAAATTCTCTTGAAGA | GCATTTCACTTGATGAAATTTCG | 51 | 370 | I | I | |
| 10 | 20229 | 基因组SSR | GCGAAGACTTCCGACCATTA | CACGGTCAAGGCCTACATTT | 60 | 181 | I | I | I |
| 11 | 18339 | 基因组SSR | TGGTTGAACTGGAACGAGTG | TGAAATTGCAATGTAAGCATGA | 59 | 137 | I | I | |
| 12 | e1154 | EST-SSR | GCCATGCGACCATATTTACC | ATGGCCACAGAAACGAAAAC | I | I | |||
| 13 | e1111 | EST-SSR | TTCTTCGTCGGAGGATGAGT | TTGAGAGGAGATTGGAGAAAAA | I | I | |||
| 14 | 28832 | 基因组SSR | CTCACGCGTTGAAGATACCA | CACCGCCATTGTAGTACAGC | I | I | |||
| 15 | 27227 | 基因组SSR | GCATTCCATGAATTGCATCT | TTGCTGATTTCTTACTTGTTGTCA | 59 | 167 | I | I | |
| 16 | e734 | EST-SSR | AGGCAGTGACTGAATCATCGT | AATGGCTTTGAGGCAGAGAG | I | I | |||
| 17 | 18591 | 基因组SSR | AGGGCCGAATGCTAAGTGAT | TTTTGAACCCTGGAGGGAGT | 61 | 155 | I | I | |
| 18 | 21662 | 基因组SSR | GAACATTATCGGAATCAACAGC | CCACACAAAAATGAACAACACA | 59 | 158 | I | I | |
| 19 | 17158 | 基因组SSR | CTCCCGAGTCTTGGCTAATG | AGGCGCTCATAAACAGTTCC | 59 | 175 | I | I | |
| 20 | e1171 | EST-SSR | AGTCCCATCCCACGAAAAAT | CTCTTTCAAATCCCCCAACA | I | I | |||
| 21 | PEACPLHPPS | 基因组SSR | GTGGCTGATCCTGTCAACAA | CAACAACCAAGAGCAAAGAAAA | 52 | 120 | I | I | |
| 22 | e613 | EST-SSR | CAATAATTTCACACACACCAAGAA | TCCAAAGAATCCTAAGAAACATGA | I | I | |||
| 23 | 24312 | 基因组SSR | TGCCATATGCATTTCATGGT | AAGCCCCTTTTCATCTTCAA | 59 | 210 | I | I | |
| 24 | e1013 | EST-SSR | ACACCAACGATGACCCATTT | CCGATCCACAAACCGTTATT | I | I | |||
| 25 | 29847 | 基因组SSR | TTGTTCCTACCGTGCTTTCA | TCTTATTGGCCTGCACATCTT | I | I | |||
| 26 | 29428 | 基因组SSR | CCATCCACATCCTTCCAAGT | CACTCAACCCACGGAAAAGT | I | I | |||
| 27 | e700 | EST-SSR | GGTTTCGGTTGATCATGGTA | GGAACTTTCTCTCGGGATCA | I | I | |||
| 28 | e1189 | EST-SSR | TGGCAATTGCGATGATTAGA | TTCATCCGTTTCATGATGTTG | I | I | |||
| 29 | 25805 | 基因组SSR | CCAACTTACTTTTGCTTATCTGGT | TGGGTCCATGACAAAGACAA | 58 | 124 | I | I | |
| 30 | 25096 | 基因组SSR | CAACATTGTTATCATCAAAACTCAA | GGCGACAATCGATCTCAAGT | 58 | 192 | I | I | |
| 31 | 28733 | 基因组SSR | TGGCCTAGGTTTTTGTGTCC | GCATCTCAAAAGGGCATTATTT | 59 | 116 | I | I | |
| 32 | 23546 | 基因组SSR | TCCACCTTGTTGCCCTAATC | TGAATGCTTCTCAGATACAAAATGA | 60 | 155 | I | I | |
| 33 | 22611 | 基因组SSR | TGCAAATGTGCAATGAATGA | GGCGGACATGAGAAGGAATA | 60 | 187 | I | I | |
| 34 | 27835 | 基因组SSR | CATCACTTGGGATTTCTTGAGAG | AGGGCAATGGTAATCAGCAC | 60 | 138 | I | I | |
| 35 | 27301 | 基因组SSR | TGTCGGAAATTAAGAGGTGGA | TGGAAAAGTAAGCGGTGAACA | 60 | 119 | I | I | |
| 36 | e1226 | EST-SSR | CACACCAGGTATCAATCTGTGAA | CGTTCCGCTTTTCACTCTCT | I | I | |||
| 37 | 16881 | 基因组SSR | ATGGGCTTTAGGGGAAGAAA | AAAAGCAGCACATGGAGGAC | 60 | 133 | I | I | |
| 38 | 21678 | 基因组SSR | CCCTTCAGCAACAATCACTG | TGCCTCAGATTTGGAATGGT | 59 | 151 | I | I | |
| 39 | 20646 | 基因组SSR | TCTCACATGTTGTTATTTCTTTCTCA | TGATGTTCCCCAGATTTTCA | 59 | 120 | I | I | |
| 40 | PsAS2 | 锚定标记 | CTAATCACACGTTTAGGACCGG | CGAAATCCAAACCGAACCTAATCC | 52 | 300 | I | I | |
| 41 | e888 | EST-SSR | GGCTTCTCCATTTGTGGTTC | GCCAATGGAGGTTCTACAGC | I | I | |||
| 42 | 18011 | 基因组SSR | GACCAACGACTTGGACATCA | GGTGAGTTCCTAAGATGAATCAGA | 59 | 197 | I | I | |
| 43 | e811 | EST-SSR | TTTTGTGGGTCTCTCTTCACC | CACCACACATGCAACACTCA | I | I | |||
| 44 | 27246 | 基因组SSR | CCAGGTTAAAACGATGATTTTTG | ACTTTTCCCCTTGGTTGGAC | 60 | 180 | I | I | |
| 45 | 3023 | 基因组SSR | GGTGCAAAATTTGAGGTGCT | CACACACGACTACACACACTACG | 59 | 153 | I | I | |
| 46 | 27167 | 基因组SSR | GGCACAACTACAACCCACAA | GGTTCAGGAATGGGTTCAGA | 59 | 194 | I | I | |
| 47 | e799 | EST-SSR | TGCAGGCTTTAGAAGTTGTTCA | CTCAGCAGCCACAATTACACA | I | I | |||
| 48 | 5897 | 基因组SSR | GGCAATAACTTAAGAGTACTAAGGAAA | AGGGTGTCGTCGTGTGTGTA | 57 | 282 | I | I | |
| 49 | 27361 | 基因组SSR | CTGAAACGGTTTGCATTGTG | TCCAACCACTTCTTAACAACCT | 58 | 113 | I | I | |
| 50 | 22209 | 基因组SSR | AATCCACAACCCCGTCAATA | CAAAAGAGACCTTCTTCCTCTCA | 59 | 164 | I | I | |
| 51 | 22259 | 基因组SSR | CATCATGGCTCAATCTCAACA | TTCCCAAATTCCTTCGTTCA | 60 | 110 | I | I | |
| 52 | 22406 | 基因组SSR | GGAAAGAGTTATGGCAATGGAC | TGGTGGTGGAGCTAAGTGTG | 60 | 174 | I | I | |
| 53 | 24555 | 基因组SSR | CGCTTATGTAGCCCCTTTTG | GGCCAAAGGAGATTTGTGTC | 60 | 123 | I | I | |
| 54 | 24499 | 基因组SSR | AAAACAAACAAAACCGCAATG | TAGCCATCACCAAAGCAACA | 60 | 169 | I | I | |
| 55 | e624 | EST-SSR | CCTTAGCAAGTTTGTCTTTGAGTG | TGCAATGACATGATGGAAGAA | I | I | |||
| 56 | 29029 | 基因组SSR | TAGGAGAGCGAGGAGCAAAG | CCACCAAAAGCAAGAATGTG | I | I | |||
| 57 | e884 | EST-SSR | TTCTTTCCGCCATGAGATTC | GAGAGCAAGGGTTTGGAACA | I | I | |||
| 58 | 28438 | 基因组SSR | GGAATGACGAAGTAACCACCT | GATGCAAGTGCAACCTTTGA | 58 | 185 | I | I | |
| 59 | 17628 | 基因组SSR | GGTTTTGTTTGCCGTTGATT | CCACCCCCAAACTTCCTTAT | 60 | 153 | I | I | |
| 60 | 17605 | 基因组SSR | CGCCCTTCATCATCATCTTC | AGAGTCGGTCCCTCCAACAT | 61 | 150 | I | I | |
| 61 | 1356 | 基因组SSR | CACGTGCACATACACACTCTT | TGTGTCAGAGCATGTGTTCG | 57 | 106 | I | I | |
| 62 | 171 | 基因组SSR | CAAACACACACGCACACAAA | CGTGTGAGCGTGCATAAGT | 58 | 70 | I | I | |
| 63 | 28654 | 基因组SSR | AGCGACGTGAATATCACAATG | GTTATCGCGGCGTGTAAATC | 59 | 134 | I | I | |
| 64 | e658 | EST-SSR | TGGTTTCTCTGCCAAAACAG | TGATGAGTGGTGACGCAAAT | I | I | |||
| 65 | e562 | EST-SSR | CAAGATGCTTCTGATTCAGTGTC | AGGATTTGAGCTTGGGAGGT | I | I | |||
| 66 | 17989 | 基因组SSR | CAGAGCCGGAGTTCTGGATA | TTTGGTTGACATTAGCACATGA | 59 | 195 | I | I | |
| 67 | 26333 | 基因组SSR | AAACACACGACATGTTTCCTTTT | TCACTGCAATTCGTCGATGT | 60 | 116 | I | I | |
| 68 | D21 | 锚定标记 | TATTCTCCTCCAAAATTTCCTT | GTCAAAATTAGCCAAATTCCTC | 51 | 200 | I | I | |
| 69 | 18928 | 基因组SSR | TGAATGTGGAAAGGAGGAATG | AGGGTCACCACTTTGGAGAG | 59 | 178 | I | I | |
| 70 | 18529 | 基因组SSR | GAATGTGCGTCCAACATCCT | AGATTTTGATGCGGAAGAGC | 59 | 151 | I | I | |
| 71 | 1863 | 基因组SSR | GCACACGAATACAGTCACGC | GTGTGTTGACGTGCGAGTTT | 60 | 118 | I | I | |
| 72 | e997 | EST-SSR | GCCTGGAGTGTTGAAGAGGA | CCATCACAATTTCCCACACA | I | I | |||
| 73 | 1752 | 基因组SSR | GCACGCACACGAATACAGTC | GACGTCGTGAGTTTGCATGT | 60 | 115 | I | I | |
| 74 | AF016458 | 锚定标记 | CACTCATAACATCAACTATCTTTC | CGAATCTTGGCCATGAGAGTTGC | 54 | I | I | ||
| 75 | 23261 | 基因组SSR | CTGCTTTTGGGGTTTGGTTA | GCAATGCAACTCCTCAACAA | 60 | 156 | I | I | I |
| 76 | 22699 | 基因组SSR | CAACATGCCATTCTGGCTAA | GCCGAAACCCCATGTAGAC | 60 | 157 | I | I | |
| 77 | 24895 | 基因组SSR | AAGAAAGTTGCGTTGGATGTG | GTTTTGTACCGCCCAACACT | 60 | 148 | I | I | |
| 78 | 24731 | 基因组SSR | AGAAAATGGCCCACGAATTA | TGCATTGCATTGTGTTTGTG | 59 | 204 | I | I | |
| 79 | 25106 | 基因组SSR | AAGGCCAAACAGAAAGGAGA | CAATGTCCAAGAAAGATCCAGTT | 59 | 178 | I | I | |
| 80 | 4083 | 基因组SSR | TGCAAACTCACACGTCAACA | GTGCGTGTGCGAAGTACG | 60 | 191 | I | I | |
| 81 | 1753 | 基因组SSR | GCACGCACACGAATACAGTC | TCGTAGTTTGCATTGTGCGT | 60 | 115 | I | I | |
| 82 | 25341 | 基因组SSR | AATGCTTCTTCCACGGTCAC | TTCGCTCGAGTTCGATTCTT | 60 | 184 | I | I | |
| 83 | 25454 | 基因组SSR | TTCCAAGCAAGCGTTGAAGT | TCAAGAGAGACTTTTCAAGAGGTT | 58 | 204 | II | II | |
| 84 | 29797 | 基因组SSR | TGTGATCAGGTGCTCCCATA | GCGACAAATTATGGCTATGC | II | II | |||
| 85 | 26436 | 基因组SSR | TTGCCTTGCCAACTTTTAGG | CTTGCTTCTGCGCCATAAAT | 60 | 195 | II | II | |
| 86 | e581 | EST-SSR | CCTTGATGCCACAAATGAGA | TTGCCACTTTCTCAAAAACTCA | II | II | |||
| 87 | S217 | 基因组SSR | CACTCAACTCACCGACCTCA | GACGGATGGAAATTGGTGTC | 52 | 1035 | II | II | |
| 88 | e956 | EST-SSR | CGAGCGTGAGACTGTGATGT | TCCACCGGTTCAACTTCAAT | II | II | |||
| 89 | e344 | EST-SSR | ATGCAACCGGCGCAGTAT | CCACCTTTTCCTCGCTTTTT | 61 | 159 | II | II | |
| 90 | e967 | EST-SSR | TGACACTTTCGTGTACTGTGTTTTT | TTCCAAAAGCCTCTCTTTCATC | II | II | |||
| 91 | 20922 | 基因组SSR | AAAAGGAGAACACATTTTATAATAGCA | TGCTCTTAAAGGCGACAATG | 58 | 146 | II | II | |
| 92 | 29141 | 基因组SSR | TTCTTTCTGCTAGGAGCCACT | CAAAGCCATCACCCTACACA | II | II | |||
| 93 | 24959 | 基因组SSR | ATCCTCACCGGTTTGATGAC | TGGAGAGTGATAGAGAAAAATTGTG | 59 | 115 | II | II | |
| 94 | 25059 | 基因组SSR | ATGGATTGCGGATAGCTCAA | CAGCAGTTGTTCGCAGGTAA | 60 | 189 | II | II | |
| 95 | 24301 | 基因组SSR | TTGTGTTTTCCGGAGAGGTC | TCCCTCCCAACCTTGAATTT | 60 | 142 | II | II | II |
| 96 | AA332 | 锚定标记 | TGAAAATAAAGGCATGCAAATA | TGATTAGTCAACTTGTTGTGGA | 51 | 255 | II | II | |
| 97 | 29220 | 基因组SSR | GGGGCAGATTTGTGGTATTG | TTCTTCTTCCTCACGTCTTTCTTT | II | II | |||
| 98 | AD83 | 锚定标记 | CACATGAGCGTGTGTATGGTAA | GGGATAAGAAGAGGGAGCAAAT | 61 | 270 | II | II | |
| 99 | 18272 | 基因组SSR | CCCCAACATTTCTCTAGGTAACA | TTCTTCGCAGCTCGGTAAGT | 59 | 131 | II | II | II |
| 100 | S42 | 基因组SSR | AGTTTCGGGTTCCTTGGAGT | GTTGGCGTAGAACGATCCAT | 53 | 211 | II | II | |
| 101 | e198 | EST-SSR | ACCATCACCACCAACAACAC | CTGCATCTGGAGAGGGAGAG | 59 | 188 | II | II | |
| 102 | e14 | EST-SSR | TCCGCAATGTTCTCTCGAAT | GGAGGTCTCCGCATTATCAA | 60 | 188 | II | II | |
| 103 | 21776 | 基因组SSR | AACGGATATGCATGGAGAGG | AAAACACGACCATCCTTTGTG | 60 | 172 | II | II | |
| 104 | 21939 | 基因组SSR | GGTCCTCAAGCACCACCTAA | TGGGCGTCACTACTTAACTTTT | 58 | 114 | II | II | |
| 105 | 28257 | 基因组SSR | AAGGGCTGACGGTCTAACTG | GAACTGACGGACGCTAGAGG | 59 | 161 | II | II | II |
| 106 | 16237 | 基因组SSR | GCAAACGAAGCAGGCTTATC | TTGGCTGATCCTGAAACTGA | 59 | 152 | II | II | |
| 107 | 18260 | 基因组SSR | AACCTTGAAATGGAGGTACATGA | GACCATGATCGGATGTTGTG | 60 | 129 | II | II | |
| 108 | 21641 | 基因组SSR | CGATTTACCGTCCTTCATCA | ACGGTCTCCCATGTGTTTGT | 59 | 166 | II | II | |
| 109 | 3015 | 基因组SSR | ACACATGCACACCCCCTTC | TGCGTGTGTACGTGTGTACG | 60 | 153 | II | II | |
| 110 | D23 | 锚定标记 | ATGGTTGTCCCAGGATAGATAA | GAAAACATTGGAGAGTGGAGTA | 51 | 170 | II | II | |
| 111 | e1031 | EST-SSR | CAACACAAGAACTTTGCACCT | TTGATCCACCTGCATCATTG | II | II | |||
| 112 | e1215 | EST-SSR | GAGACAGAAGACGGCGAAAG | TGCCAAGAGTCAGGAGATTG | II | II | |||
| 113 | 16433 | 基因组SSR | CACCGCAAACATAGCAAAAA | TCTCATAGCTGCGAGGTTCA | 60 | 127 | II | II | |
| 114 | e617 | EST-SSR | GCCAAACGGCTTTAAAACTTC | TCGCTGTTGGAAAGAGAAGAA | II | II | |||
| 115 | 20723 | 基因组SSR | CTCACTTCACGTGCGCTATC | GCAGGAGCAGCTTGATTTTC | 60 | 152 | II | II | |
| 116 | 16437 | 基因组SSR | TTGTTTTTGTTGTTCTTGTTGTTG | TTTTCGGGTTTTGCTTATGG | 59 | 128 | II | II | |
| 117 | 17531 | 基因组SSR | TGCAGGGGTGTGTGTTACAT | TGAACATGGTGAAATGGATTG | 59 | 140 | II | II | |
| 118 | 18237 | 基因组SSR | GGGATATGAGAAGGCGATACC | TGGTTGTAGGATGTGGGATTT | 59 | 127 | II | II | |
| 119 | 17754 | 基因组SSR | AGCAACGGGCAACCTTATAG | CCTTTTGTTTGGAAGCTCAA | 58 | 169 | II | II | II |
| 120 | e850 | EST-SSR | TTTCTTCTCCCAAACTACCTCAT | ATGCATGAACCAACCCATCT | II | II | |||
| 121 | 17713 | 基因组SSR | AAAAAGGGGAAAGCAGGAGA | TTGACTGTGAGGCTGGTTTG | 60 | 164 | II | II | |
| 122 | 21945 | 基因组SSR | TTCACGCTCATCGCTAAGAC | TTCGAATCCTCCCTTCTTGA | 59 | 113 | II | II | |
| 123 | e478 | EST-SSR | GCCACCAACCAATTCAACTT | TGGGTATTGGGAATGGAAAA | II | II | |||
| 124 | e36 | EST-SSR | GCAGGGTGGGTATATCTGTGA | GTGGTCCAATTCCTTTTTGC | 59 | 219 | II | II | |
| 125 | e26 | EST-SSR | TTTTGTCCCCGCGTTTAATA | CATTCATGCCACAAAAATGG | 60 | 155 | II | II | |
| 126 | C24 | 基因组SSR | GCTACTGGAGGAGGCTTTCA | GCCTTCTACACAACGGCTTC | 52 | 162 | II | II | |
| 127 | 28383 | 基因组SSR | TCGATTGTTATTGTGTTTCCTCTC | TGAGATCAAGTGGGGGAAAA | 60 | 150 | II | II | |
| 128 | e770 | EST-SSR | GGTTTGAAAGGACCCCTAGC | GTTACCGATGGCCATGAATC | II | II | |||
| 129 | 143 | 基因组SSR | ACATGCACACGTACACGCA | AGTTGGCGTGCAGTAGAGGT | 60 | 69 | II | II | II |
| 130 | 16534 | 基因组SSR | TTGCAAATATACCAATTCCAAAA | ATTGGAGCCTGGTGAAGACC | 58 | 139 | II | II | |
| 131 | 28055 | 基因组SSR | GCCAGCAATTTTAGCATTACG | TTAGCTCAGCCCGGTAAAAA | 60 | 163 | II | II | |
| 132 | 25792 | 基因组SSR | GACGGAAACGAAATCGAAGA | TCAAAATTCACGCACACGAT | 60 | 110 | II | II | II |
| 133 | 30024 | 基因组SSR | AGAGTGCCATCCCTTCAATG | GAACGTTTGGTTGGAGGAGA | II | II | |||
| 134 | 25769 | 基因组SSR | GCAGCAAGATGGTTGGTAGTT | GACGTCGTAGTCGCCATCTC | 59 | 144 | II | II | II |
| 135 | e929 | EST-SSR | TGAGGAGGGAGATGGAGAAA | GAAGGCAAACCTACCAACCTC | II | II | |||
| 136 | 30218 | 基因组SSR | GGACGTGTCCCACTCCTATG | GGAAGGATAAAAACGTTGCAATA | II | II | |||
| 137 | 30416 | 基因组SSR | GACACATGGAGCCACAAAAA | GAATGGAGGGGAGAGATGAA | II | II | |||
| 138 | e577 | EST-SSR | TGTCATTTCTTTTTAGTTCCTTTCAA | CCTTCGCTTGATTCTTCACC | II | II | |||
| 139 | 16570 | 基因组SSR | CAAACACCAACCACCACAGT | AAGGGGAGACGAAGTGGAGT | 59 | 143 | II | II | |
| 140 | 27099 | 基因组SSR | GGTACACCCACCGATACACC | TCTAGACGCGGAGAGGGTAA | 60 | 140 | II | II | |
| 141 | 27112 | 基因组SSR | GCAACAAGATTTTGACGTTTTT | GACGCTACCAACCGCTTTAG | 58 | 129 | II | II | |
| 142 | 28687 | 基因组SSR | CACGGAAGGCCCTACTTACA | GTGGCGAGTAGAGCGTAAGG | 60 | 189 | II | II | |
| 143 | 28704 | 基因组SSR | TTCTGCAGTCAGCTTCAACTTC | TAGTCACGGAAGCGATTCAA | 59 | 147 | II | II | |
| 144 | 22690 | 基因组SSR | GGTTCATCTGCACCCAAGTT | GGCAACTCTCTCACACACACA | 60 | 138 | II | II | |
| 145 | 24812 | 基因组SSR | GACCAAACCACCTCACAGATG | TGGCTCCTTTCTCATTTCTAACA | 60 | 140 | II | II | |
| 146 | 23154 | 基因组SSR | AAGACGAGGTGGCATGGTAG | GAGAATGCATGCTTCAATCAA | 59 | 183 | II | II | |
| 147 | 28785 | 基因组SSR | GATCCACCCAATTCCCTTTT | TGTATTGCAGCCGCTTTATG | 60 | 208 | II | II | |
| 148 | 19680 | 基因组SSR | GCCAACCCAACAATCTCAAC | CATTGGAACCAGATCGAACC | 60 | 148 | II | II | |
| 149 | 22052 | 基因组SSR | GTTACCGATGGCCATGAATC | AGCGAGTGAAGAGGGAAGTG | 60 | 147 | II | II | |
| 150 | 1078 | 基因组SSR | CACACGCACAGACACACGTA | GTGTGCGTATGCGTTACTGC | 60 | 99 | II | II | |
| 151 | 29138 | 基因组SSR | GCAGATTGAAACCAAAACGAC | TCGCAACCTGCACTTTCTTA | III | III | |||
| 152 | e763 | EST-SSR | ATGCTTTGATGGGCTCAACT | TCCCAAACATGCTAGCAAAA | III | III | |||
| 153 | 19532 | 基因组SSR | CCATGTTTGAATTCGGAGGA | GCGCGATGATTCAAGGTTTA | 61 | 165 | III | III | |
| 154 | 19656 | 基因组SSR | CCAACGTTGTTGTTTTAGTGG | CCAGAGTCGTGGAGCCCTAT | 58 | 169 | III | III | |
| 155 | 27876 | 基因组SSR | TGTTGTTGCCCATCAATCAT | AATCACACGAGGGATTGGAC | 60 | 149 | III | III | |
| 156 | 18323 | 基因组SSR | CAGACAATGGCAATTATTTGGTAA | CTGCTGTTGCTTCGATTTCA | 60 | 136 | III | III | |
| 157 | 21881 | 基因组SSR | CCATTCCCAACAATTTCCAC | GTGAGGTCCGGTTCTACAGG | 60 | 119 | III | III | |
| 158 | 30088 | 基因组SSR | TACTGGATCCGGATGAGGAC | TCGCATCAAAGCAAAAACCT | III | III | |||
| 159 | e605 | EST-SSR | AGCACTTGCTACGGCAATTT | AAACCTAGAAATAACGATGCAAAA | III | III | |||
| 160 | 24265 | 基因组SSR | GTTTGCGGCCAAACAATATC | TTTGCATGAGTGCACCTCTC | 60 | 181 | III | III | |
| 161 | 29627 | 基因组SSR | AGAAGACAACGACCGAGTGG | AACCGCATAACCGCAATTTA | III | III | |||
| 162 | 27019 | 基因组SSR | GCAGTTTCCACACTTTAAGTCCA | TGGGTGTGTTAACAGGGTGA | 60 | 155 | III | III | |
| 163 | e1208 | EST-SSR | AACCATTGCGCGTCTTTTAC | AGACCACCGCCATAACATTC | III | III | |||
| 164 | 28130 | 基因组SSR | CGAATTTGGTTGCGACGAC | TCGCGCCTCTTTAGGAATAA | 60 | 208 | III | III | |
| 165 | 27378 | 基因组SSR | GCCAATTATTCCCTCCAGGT | TTCGAAGGTTCTCCATCACC | 60 | 147 | III | III | |
| 166 | 28180 | 基因组SSR | TCGTTCATTTAACTTCGTGAGGTA | AGAGAGTTCTCCGCCAAACA | 60 | 201 | III | III | |
| 167 | 24903 | 基因组SSR | TGGTGTCAACTTTTTGATGTTCA | GACAAGTTGCTTTTGCTCCAC | 60 | 147 | III | III | |
| 168 | 26850 | 基因组SSR | TCACAGACAGTACACAAAGTTTTCTT | GCGAGGGAGAACAGAAACAG | 59 | 160 | III | III | III |
| 169 | 24521 | 基因组SSR | AGGGAACCCCCAATTGACTA | CCAGAAACTGGGGTTGTGTT | 60 | 208 | III | III | |
| 170 | 23311 | 基因组SSR | TTTCAGAATGGTGCAGGGTAT | AGGATCTCAGTATACATGCGTAAA | 57 | 168 | III | III | |
| 171 | 24540 | 基因组SSR | CTCCCTCATGAGTCGTGACC | ATCAAAGGGGGAAGGTGAAG | 60 | 143 | III | III | |
| 172 | 26575 | 基因组SSR | GAAAATAAACAGTTGGCAACAAAA | CCACTCCAAACCCTTCAGGT | 59 | 205 | III | III | |
| 173 | 21726 | 基因组SSR | GGTGATGGAGAAAAGGGTGA | TGCATGCAGTCAAATCAAAA | 59 | 157 | III | III | III |
| 174 | 23518 | 基因组SSR | CAAGGACGACGACAACAACA | GTGCCGACGTTCAAGAAAAT | 60 | 202 | III | III | |
| 175 | e632 | EST-SSR | AAACCTCTCTACAGCACCAACAC | GGGAGAGATTGTTTGAAGTATAGAA | III | III | |||
| 176 | 28406 | 基因组SSR | CCGATTGTGCAGCAAGAGTA | ACGATGCACATGCAGAATTT | 59 | 129 | III | III | |
| 177 | 27038 | 基因组SSR | CGTCTACCTCCGACGATAGC | TTCGCCAGATATATACAATAAAAAGA | 58 | 187 | III | III | |
| 178 | 25151 | 基因组SSR | GCCTTCGAGGCATCCTAAT | TGGAACCATAAGATTGGTGAA | 58 | 157 | III | III | III |
| 179 | 27270 | 基因组SSR | GGACCATTACCCTCCCATTT | TTCCTTCCGTTTTGCAGTCT | 60 | 194 | III | III | |
| 180 | 28194 | 基因组SSR | TGGGGTCTTAAAGTTGTGACTTC | CCGAAGGTGGGATGAAACTA | 60 | 148 | III | III | |
| 181 | 28645 | 基因组SSR | CTGAAATCGGAGTGGTCACA | GGTGAAGCCCTTAGCTACCA | 59 | 188 | III | III | |
| 182 | 17193 | 基因组SSR | CACAGCCATACCCAAGTTACAA | GGTTGCGAGGGATGAGAATA | 60 | 178 | III | III | |
| 183 | e814 | EST-SSR | GGTTGGTCCAATCCAACATC | AGAACGAACACACACGAAACA | III | III | |||
| 184 | e494 | EST-SSR | GACCCGTCTGGACTGGTAAA | TTGAAAGATGCGGAGTGATG | III | III | |||
| 185 | 29436 | 基因组SSR | TCTAGCAGCATTGGGGAAAC | TAATGAGGGGAAGGGGATTT | III | III | |||
| 186 | 27710 | 基因组SSR | AACCACAGAAAACTGCCAAGT | TGAGAACCAAAAGCAGGTCA | 59 | 110 | III | III | |
| 187 | 29074 | 基因组SSR | TCGTCGAATGGTTGAAGAGA | TTCGCAAGTGAAGGAAAAAGA | III | III | |||
| 188 | 29307 | 基因组SSR | GCGATTCCAGATGTCAGGTT | CGTCTCCCTACCAGCAAAAA | III | III | |||
| 189 | 24534 | 基因组SSR | GTGGTGTGAAAGGGGTTTTG | CTTGCATTGGATTCCCTTTG | 60 | 206 | III | III | |
| 190 | 22543 | 基因组SSR | TTTCACGTACGTTCCCAACTC | CCAGATCAACCACCTAACTTCA | 59 | 160 | III | III | |
| 191 | 23262 | 基因组SSR | GGTGACGGAGGTGATAGAGG | TAGCAAATGCAAACCCAACA | 60 | 183 | III | III | III |
| 192 | 24701 | 基因组SSR | TATGCTGGAGTGTGGAGTGG | TCAATCAATTCAACGGTACAGA | 58 | 111 | III | III | |
| 193 | 24784 | 基因组SSR | TTTAGACGGCCTTCGTTAGTG | CTGAGCCTAAAGGGCTGAAA | 59 | 115 | III | III | |
| 194 | 27254 | 基因组SSR | GAAGGCCTCTAACGGTGAAA | AATCAAACAGAGGCCACCAG | 59 | 122 | III | III | |
| 195 | 22754 | 基因组SSR | GGAACGACAACACGAACCTC | GACACGTTATGCGCACACTC | 60 | 161 | III | III | III |
| 196 | 26131 | 基因组SSR | GGAAACGGTGGAAGATGAAA | TTGGCAAAAGGGATGAGAAG | 60 | 175 | III | III | |
| 197 | 24805 | 基因组SSR | TGCAGCAGATCAACCAAAAC | TTTTGAACTAAGGTGGTCTCAATC | 59 | 144 | III | III | |
| 198 | 18135 | 基因组SSR | CTTCAACCAACTGCGAGTGA | TCATTTGAGTTTTGCCATGTTC | 60 | 120 | III | III | III |
| 199 | PSAJ3318 | 锚定标记 | CAGTGGTGACAGCAGGGCCAAG | CCTACATGGTGTACGTAGACAC | 58 | III | III | ||
| 200 | 29634 | 基因组SSR | CAATTAACAAACGCAGCCTTA | TTAGCCCGTGGATTTTCAAC | III | III | |||
| 201 | 26866 | 基因组SSR | CGATACATTAAGGGCGGAAC | TGACTCATTCGCATTTGGAGT | 59 | 186 | III | III | |
| 202 | 23757 | 基因组SSR | TGAAAGAGGGGAATTGAGAGA | TCAGGTTACAAGCCCGAGAT | 59 | 187 | III | III | |
| 203 | AA355 | 锚定标记 | AGAAAAATTCTAGCATGATACTG | GGAAATATAACCTCAATAACACA | 51 | 180 | III | III | III |
| 204 | 23899 | 基因组SSR | CCCCATCCTTGTGAACAAAT | ACGGTGTTTTGGTGGTGAAT | 60 | 151 | III | III | |
| 205 | AD270 | 锚定标记 | CTCATCTGATGCGTTGGATTAG | AGGTTGGATTTGTTGTTTGTTG | 51 | 290 | III | III | |
| 206 | 20075 | 基因组SSR | GCCAGTCCCTTGAGTTAGGA | TGTTTCACGTGTTCCCCATA | 59 | 122 | III | III | III |
| 207 | e696 | EST-SSR | CGCTATCACTCTCTCTTTCTCTTTT | ATCGGAGGACGAGGTCTTTT | III | III | |||
| 208 | e1021 | EST-SSR | AAAAAGCTGAAACATTCAGACC | GGTCCATTCATTCTGCAGTG | III | III | |||
| 209 | e673 | EST-SSR | GGAAGACGGAGTGGTGGTAA | GTCGTCGTGGTGCTTCTCTC | III | III | |||
| 210 | 16524 | 基因组SSR | CCAGAGGATGTGAACCAGGTA | TTCAACCAAGCTGAACCCTTA | 60 | 138 | III | III | |
| 211 | 4826 | 基因组SSR | AACATGCGTCTGTCGTGTCT | TAGTGGGTGTGCGTGTGAGT | 59 | 220 | III | III | |
| 212 | 27093 | 基因组SSR | TCGTCATTCTCCCTCCAATC | TATGATGTCCACGCGTTTTT | 59 | 128 | III | III | |
| 213 | 19688 | 基因组SSR | GAATCGGGTCGCTGAGATAG | ACCTCCACCGTACCATTCAA | 60 | 133 | III | III | |
| 214 | e998 | EST-SSR | GGCAGACTGGTCTCAACTCC | ATCATCGTTGGTGGAATCGT | III | III | |||
| 215 | 2875 | 基因组SSR | TATTAGCACCCCTCACGTCC | TTTCCCCTTCCTTCCAATCT | 60 | 148 | III | III | |
| 216 | 30442 | 基因组SSR | CCCACTCCATCAGTCTCTCC | AGAGGACCGGTGATGTGTTC | III | III | |||
| 217 | e446 | EST-SSR | AACCAGAGATGAGTGGAAAAATG | GCCAACACCAGAGTTTGAATC | III | III | |||
| 218 | 27160 | 基因组SSR | CTGCAGTTGCGTGTAGAGGA | TTGAATGATGATATAAATGCAATGAC | 59 | 135 | III | III | |
| 219 | e472 | EST-SSR | TCCATCACCAGGCATAGGTC | GCCGGTAGTGAGAAGGATTG | III | III | |||
| 220 | 864 | 基因组SSR | CACATACATGCAATCAAGCG | GTGTGTTACGTGCGTGTGTG | 59 | 92 | III | III | |
| 221 | 24861 | 基因组SSR | TTCACAACCCCTCTCACTACA | TGGAGGGATGGTTTACAATGA | 58 | 183 | III | III | |
| 222 | 30382 | 基因组SSR | TTGGTGAGGCCTTGATTTTT | GCCAGTGGGGATTAGAGACC | III | III | |||
| 223 | 17773 | 基因组SSR | TTCCACACGAGGCTATTTTC | TGCAAAAGCGACATCTTGAC | 58 | 170 | III | III | |
| 224 | 18363 | 基因组SSR | CATGCATGGAGTTGGAAGAG | GTCCCAAAATGCAGCCAATA | 59 | 139 | III | III | |
| 225 | 29125 | 基因组SSR | TCAGAGGTGTCATCGGTCTG | TTTCAAATAAGTTTTGAACAAAGTGT | III | III | |||
| 226 | e999 | EST-SSR | GTTTAGGAGCCTTGGGGTGT | TCCAAACTCCGGCTTCTCTA | III | III | |||
| 227 | 3647 | 基因组SSR | GGGGTCTTACAACACACGCT | AGGCAGAGGTGTGAGCATCT | 60 | 176 | III | III | |
| 228 | 6144 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | GTCGCCCTCGATTCTCATAC | 60 | 297 | III | III | |
| 229 | 4535 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | TGTGTGTACGTGTTCACCCTT | 59 | 206 | III | III | |
| 230 | 3374 | 基因组SSR | GGGGTCTTACAACACACGCT | TGAGCTAATCTCTCCGGGAA | 60 | 165 | III | III | |
| 231 | 2169 | 基因组SSR | GTTGTGTGTGTGCGTGTTCA | GGGGTCTTACAACACACGCT | 60 | 126 | III | III | |
| 232 | 1935 | 基因组SSR | GGGGTCTTACAACACACGCT | TGTGTGCGTGTTCACCTTTT | 60 | 120 | III | III | |
| 233 | 2303 | 基因组SSR | GGGGTCTTACAACACACGCT | TGTTGTGTGTGTGCGTGTTC | 60 | 130 | III | III | |
| 234 | 4653 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | TGTGTGCGTGTTCTACCCTT | 59 | 211 | III | III | |
| 235 | 5347 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | AGCATCTCTCCGGTAACCCT | 60 | 248 | III | III | |
| 236 | 2008 | 基因组SSR | GGGGTCTTACAACACACGCT | TGTGTGTGCGTGTTCTACCTT | 59 | 122 | III | III | |
| 237 | 5412 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | GTGAGCAATCTCTCCGGGTA | 60 | 252 | III | III | |
| 238 | 2051 | 基因组SSR | GGGGTCTTACAACACACGCT | TGTGTGTGTCGTGTTCTACCTTT | 60 | 123 | III | III | |
| 239 | 5400 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | GTGAGCAATCTCTCCGGAAC | 60 | 251 | III | III | |
| 240 | 5348 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | AGCAATCTCTCCGGTACCCT | 60 | 248 | III | III | |
| 241 | 4670 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | TGTGTGTGTCGTGTTCTACCC | 60 | 212 | III | III | |
| 242 | 5540 | 基因组SSR | CAGAAAAGGAAGCAAGGTGC | AGGCAGAGGTTGTGAGCAAT | 60 | 260 | III | III | |
| 243 | 21506 | 基因组SSR | ATAGGGGGAGCAGGACCTAA | TTTGACTTGTGGAAAGGAAGTT | 58 | 151 | III | III | |
| 244 | 25416 | 基因组SSR | CGCCCAATTGGATTATGATT | TGCTCAATGCACACTTACTAGC | 58 | 145 | III | III | |
| 245 | e1197 | EST-SSR | TCCAACCGTTAAACACTCTTCA | GCATGAAGGGCTCTGAGTTC | III | III | |||
| 246 | PSBLOX13.2 | 锚定标记 | CTGCTATGCTATGTTTCACATC | CTTTGCTTGCAACTTAGTAACAG | 56 | III | III | ||
| 247 | 18562 | 基因组SSR | TTCTTCTGCTGCTGCTCAAA | AAAACAAAAACCACAACCAAAAA | 60 | 154 | III | III | |
| 248 | 21610 | 基因组SSR | CGATTGATGCCGTGTCTAAG | TTTCAAGTTTCTTCTAGATTTTGTCA | 58 | 114 | III | III | |
| 249 | 27991 | 基因组SSR | AATACAGCTGGACCCCACAC | GCAGGCCATTTCATTTCATT | 60 | 121 | III | III | |
| 250 | 22724 | 基因组SSR | CCCAAGAAGAAGGATGGTGA | GAGCATTCTGGTGCTGTTGA | 60 | 152 | III | III | |
| 251 | 26028 | 基因组SSR | CGGCGAGATTTATTGACGAT | CAACGTGGCAAGCAAGTAGA | 60 | 186 | III | III | |
| 252 | 4397 | 基因组SSR | TAATCTCTCCGGGAACCCTT | TGATATGATGCCATGAGGGA | 60 | 201 | III | III | |
| 253 | 20084 | 基因组SSR | CTCCCTCCCGAATGTAATCA | CGGGACGATCAACTTTGTCT | 60 | 111 | IV | IV | |
| 254 | 16758 | 基因组SSR | CCCTTCAACAAAGCCTAACG | AGGGTGCGAAGGAGGTTAGT | 60 | 115 | IV | IV | |
| 255 | 21712 | 基因组SSR | CGGCGGGTTTGATAGAGTTA | CTTCACCCTTGCAACAAACA | 60 | 166 | IV | IV | |
| 256 | 21774 | 基因组SSR | GCAAGTTCCCAATCGTCCTA | TCAAAAGCAAGGTCCCATTC | 60 | 123 | IV | IV | |
| 257 | e671 | EST-SSR | TTGCCTCATTTATCATTCTCTTATG | CAAAAGGTTATCTAGCTACGACTTGA | IV | IV | |||
| 258 | 29964 | 基因组SSR | CAATTCATGACGAAATTGACAAA | CATGGAGATGGAGAGTTCAAAA | IV | IV | |||
| 259 | e996 | EST-SSR | GCCGGTAAACGATCCATCTA | TGCAGCCACACTCCTTTACA | IV | IV | |||
| 260 | 19632 | 基因组SSR | AATGTAATTAACCCACGAAGTTG | TGCCAAAGCTCTCTCATCCT | 57 | 201 | IV | IV | |
| 261 | 21547 | 基因组SSR | TGAAAGCCTCAAAGCAACAA | CCATGGCATGTGCTAGTGTAG | 59 | 156 | IV | IV | |
| 262 | 21250 | 基因组SSR | GTGCAATTTTCACACAGTGG | ACGAAGGTTGGAGCATGATT | 58 | 144 | IV | IV | |
| 263 | 22067 | 基因组SSR | TATGCTCAGAGGGGCATAGG | TTACGACGATGAGCGACTTG | 60 | 158 | IV | IV | |
| 264 | 18533 | 基因组SSR | TCCAAAATGCGTGTCATCAT | TGACCGACACATTCATCTTCA | 60 | 151 | IV | IV | |
| 265 | 26592 | 基因组SSR | TCCGATCCTGGTAAAGTGGT | TCCAAAATGCGTGTCATCAT | 59 | 174 | IV | IV | |
| 266 | 26369 | 基因组SSR | GCTGAAACGTGGGAAACATT | TGGTTAGTGTTTGAAGGGTCTG | 59 | 206 | IV | IV | |
| 267 | 25046 | 基因组SSR | TCCTTTGTCAGTGGGAATTTTT | AGGATCATGGTTGTCGAGTTG | 60 | 173 | IV | IV | |
| 268 | 24606 | 基因组SSR | CCGAACAAGATGAACCACCT | AACAACATCGTGTGTGTTTGTCT | 59 | 208 | IV | IV | |
| 269 | 23750 | 基因组SSR | CTCGTTGTACAATCCAGATGAA | CACCGTCCACCTTCTCATTT | 58 | 209 | IV | IV | |
| 270 | 22599 | 基因组SSR | GGGTGTGAGGCAGTTGAAAT | AAATACCGAACCGAACCACTT | 60 | 141 | IV | IV | |
| 271 | 24038 | 基因组SSR | GCCCCCACTTTTTCAACA | CTCCTGACACAAGGCCCTAC | 59 | 145 | IV | IV | |
| 272 | 18438 | 基因组SSR | GATTGAGCCGTGCCAATATC | GATCCCACCCTAGAGGAAAAA | 59 | 145 | IV | IV | |
| 273 | 22352 | 基因组SSR | CCAACATCTTCCTCATCACCT | TGAGAGTCGCAGTCGGATAA | 59 | 129 | IV | IV | |
| 274 | 16512 | 基因组SSR | TAAGCCCGACGCTTCTATTC | GTGCCTCAGTTTCCGTTTGT | 59 | 136 | IV | IV | |
| 275 | AA430942 | 锚定标记 | CTGGAATTCTTGCGGTTTAAC | CGTTTTGGTTACGATCGAGCTA | 54 | IV | IV | ||
| 276 | 17219 | 基因组SSR | TCATGTGCATGTGATGAAGAAA | GGTGTACCCATGTGCCATTT | 60 | 181 | IV | IV | |
| 277 | 28178 | 基因组SSR | TCAACCCATACTCTTGGAATCA | CCGGAGATTCCACAAATAACA | 59 | 139 | IV | IV | |
| 278 | 28085 | 基因组SSR | TGCTTGCAACGTTTCTTTCTT | ACCCCTCCCTGAAAGGAGTA | 60 | 142 | IV | IV | |
| 279 | e538 | EST-SSR | CTTCCCTTTCTTCCCTTTCA | GTTCGGAAGGATCGATTTGA | IV | IV | |||
| 280 | 28374 | 基因组SSR | TCCACGGTCTTGCTATGTGT | CTGGTTGCACATCAGGGTAG | 59 | 187 | IV | IV | |
| 281 | e578 | EST-SSR | AGCAGCTCATATTCTCTGTCCA | GCAGAAGCAGGATCTAGGGTAG | IV | IV | |||
| 282 | e656 | EST-SSR | AGCAGCTCATATTCTCTGTCCA | AGCAGAAGCAGAAGCAGGAT | IV | IV | |||
| 283 | e650 | EST-SSR | GCAGCTCATATTCTCTGTCCA | AGCAGAAGCAGAAGCAGGAT | IV | IV | |||
| 284 | 29955 | 基因组SSR | TCAAGTGCATTGGGAGAGACT | AAAAACCGACCCATAATCAATTT | IV | IV | |||
| 285 | e1073 | EST-SSR | CTTGTTTCGCTCGGTACCTC | CTATTGCAGGCAGTCCTGGT | IV | IV | |||
| 286 | 24844 | 基因组SSR | TTTCGTTTTCCCATTCTGGT | CCCCCTTACACACGAATCTG | 59 | 121 | IV | IV | |
| 287 | 26018 | 基因组SSR | TTGAGCTGCTCGCTGTAAGA | CACCAAACTGTTTCTTCACACA | 59 | 146 | IV | IV | |
| 288 | 26179 | 基因组SSR | GGAAGGTGAAATTCCGTTGA | AGCAAGTTGGTTCGGAAAAA | 60 | 166 | IV | IV | |
| 289 | 24485 | 基因组SSR | GTGTCAAAGCTCGCCCTAAT | ATTGGTACACTTGCCGAGAA | 58 | 127 | IV | IV | |
| 290 | e1130 | EST-SSR | GCTGCTACCGTGGATTGTCT | TTGAAGAGGTGTGGTGTGGA | IV | IV | |||
| 291 | e1027 | EST-SSR | GTCTCGGTCCGAACCATTTA | TCTTCTGATCAACAAAAGTAACAACA | IV | IV | |||
| 292 | S5 | 基因组SSR | TGTGGGGCTTGTTACACTGA | AGCTACCATAACAGACAAAACC | 54 | 205 | IV | IV | |
| 293 | 22276 | 基因组SSR | ATGCGGCATTTTGCTTTATC | TTGGTCTGCAAATCGAAACA | 60 | 127 | IV | IV | |
| 294 | 29630 | 基因组SSR | CCTACCTTAAGTCGCCCATGT | TCCAAACATAGGCTTCGTCTC | IV | IV | IV | ||
| 295 | AA285 | 锚定标记 | TCGCCTAATCTAGATGAGAATA | CTTAACATTTTAGGTCTTGGAG | 51 | 248 | IV | IV | |
| 296 | 28304 | 基因组SSR | TTTTCAGCTGATCGGATATCTACA | GGCAGCATCTTGAAAATCGT | 60 | 132 | IV | IV | IV |
| 297 | e535 | EST-SSR | AAAAACCAAGCACACCCATAA | GCAAGACACAGCACAAAAACA | IV | IV | |||
| 298 | 27384 | 基因组SSR | TTGTGCCAACAAAAATCACAA | CAGAATGCCGTTCACTTTTCT | 59 | 115 | IV | IV | |
| 299 | e561 | EST-SSR | TCCGATCTTGCTTCTGAATCT | TTCATCAACCCAGACGCATA | IV | IV | |||
| 300 | B83 | 基因组SSR | CCCTCTCCCATCTCATCTCA | AAAGAAAGTAGAGATCCAGCACTGA | 54 | 205 | IV | IV | |
| 301 | 24635 | 基因组SSR | CCCTCTCCCATCTCATCTCA | AGAGATCCCAGCACTGATTG | 58 | 152 | IV | IV | |
| 302 | 23788 | 基因组SSR | TGGAGAAATTATGAGAATGTTCAATG | GCACGTCGACACACACAAAC | 61 | 184 | IV | IV | |
| 303 | 24392 | 基因组SSR | GCGGAAACAGGAGAGAGAGA | GCACGTGCTCCATCATAGAC | 59 | 121 | IV | IV | |
| 304 | e828 | EST-SSR | AACCCCATTTTCAATATTTTTCA | GGATTGTTCTCCGCATCTTC | IV | IV | |||
| 305 | 18358 | 基因组SSR | CCTGAACCGATTTTGGTGAT | ATTCCGCCCTCTTTCACTTC | 60 | 138 | IV | IV | |
| 306 | 27275 | 基因组SSR | TGCTCACATTAACCAAAAGCAC | TGGATGGGTATGTCCCATTT | 60 | 147 | IV | IV | IV |
| 307 | 27252 | 基因组SSR | TAATGCCGACTGTGTGCTGT | GCAATTCAGCAAAAAGGAGAA | 59 | 111 | IV | IV | |
| 308 | AA122 | 锚定标记 | GGGTCTGCATAAGTAGAAGCCA | AAGGTGTTTCCCCTAGACATCA | 61 | 190 | IV | IV | |
| 309 | 28244 | 基因组SSR | TGGGAGAGGGGATAACTGAA | CATGTTGTTTGGTGCGTTTC | 59 | 182 | IV | IV | |
| 310 | 19487 | 基因组SSR | CCACCTGCTCAATTCCAAAT | GGCGAAGCGAATCTAACATC | 60 | 178 | IV | IV | IV |
| 311 | 22506 | 基因组SSR | CGAAACATGCACAACCATTT | TGAACGTTCTGACCCAGATG | 59 | 208 | IV | IV | |
| 312 | 22693 | 基因组SSR | CGACAACAACAACCACATCA | CTCCATCGAACGAAAGGAAC | 59 | 148 | IV | IV | |
| 313 | 24465 | 基因组SSR | TCAAGCAGAAGAGTCGACCA | TAGCTATGTTCCCGCCAAAT | 60 | 161 | IV | IV | |
| 314 | 25164 | 基因组SSR | CCAAATACAAGCATTAATAGGGAGA | TGGTCGACTCTTCTGCTTGA | 60 | 110 | IV | IV | |
| 315 | 25419 | 基因组SSR | TGCAAGTCCTGATGCAAGTC | GCGATTCAGGATTGGCTTAC | 60 | 140 | IV | IV | |
| 316 | 27605 | 基因组SSR | AAATGAACGGAAACAGAAAGAA | ACATAGCACACGCAGCAAAC | 58 | 155 | IV | IV | |
| 317 | 29578 | 基因组SSR | CAAGTCATGAACGTCTCAAAAGA | TGGACGCGTTTTAAAGTTCC | IV | IV | |||
| 318 | 27854 | 基因组SSR | TCCTTCATCAAAACGCAACA | ATTGACGTTCAAGCGGGTAG | 60 | 138 | IV | IV | |
| 319 | e155 | EST-SSR | TTTCTCGTTGCACTCATCCA | TCGGTTGTCGTTTCTTGTTG | 60 | 174 | IV | IV | |
| 320 | 21866 | 基因组SSR | GCAGCCTTCAAATCCTCTTC | AAAACGCGCTTACGCTTCTA | 59 | 139 | IV | IV | |
| 321 | 29760 | 基因组SSR | TGTGCCTCAGAGATGTTCAAA | AGAGGTGGTGCGGTGACTAT | IV | IV | |||
| 322 | 18049 | 基因组SSR | ACCCCTCTTTGCTAGGGTGA | ACCACACATCTCGCACACAT | 60 | 202 | IV | IV | |
| 323 | e342 | EST-SSR | CACAACAACCCCTCCAAAAC | TTTGGATTTTCGCTTGGGTA | 60 | 189 | IV | IV | |
| 324 | e546 | EST-SSR | TGACAGTGAGTGAGTGGCTTCT | TTGCGGGTGAAAAGAAAAAG | IV | IV | |||
| 325 | 21805 | 基因组SSR | TGGGAATGTGAAGTGGTGAA | TGTGGTGTGGTTGGTTTCTG | 60 | 128 | IV | IV | |
| 326 | 21622 | 基因组SSR | ACAGCATGAAATGCGTGAAA | TCGTCATCCCAACTTCATCA | 60 | 139 | IV | IV | IV |
| 327 | AA315 | 锚定标记 | AGTGGGAAGTAAAAGGTGTAG | TTTCACTAGATGATATTTCGTT | 51 | IV | IV | ||
| 328 | 79 | 基因组SSR | GCTCAGTCAGCCCGTCATA | GTGCGTGTGTGCGTGTGT | 60 | 66 | IV | IV | |
| 329 | 17066 | 基因组SSR | CTCTCCCCCACACCTGATAA | GAGGACCCAGTAGGGATCGT | 60 | 159 | IV | IV | |
| 330 | 24036 | 基因组SSR | GAAGGACCAAATCAATTCTCTAAA | ACCGACGTCAACGACTGATA | 58 | 196 | IV | IV | IV |
| 331 | 24423 | 基因组SSR | CATCCCACTCTAACCGCACT | GCATAATCGGCTCTCTCTCC | 59 | 184 | IV | IV | |
| 332 | 25717 | 基因组SSR | CGTGCATGCATGTGTATGTT | TCACCGATCAACACCAATTT | 59 | 192 | IV | IV | |
| 333 | 28434 | 基因组SSR | GTTTTCAATCGATCCGTCCA | TTCCACCGTCTTCTTCAACA | 59 | 164 | IV | IV | |
| 334 | e1213 | EST-SSR | TTGGTTTCCGGTTAAAATGA | CAATCCCATTCACACCACAA | IV | IV | |||
| 335 | e782 | EST-SSR | CATTGAGTTTGAGGATGAGGA | CCCATAACCATATCTCACAGTTCA | IV | IV | |||
| 336 | e336 | EST-SSR | CCCCAAACCATATCCCTACA | TTCCATTCCCAAACTCACTTG | 60 | 170 | IV | IV | |
| 337 | 21227 | 基因组SSR | CGGATTCAACAAGCAGAACA | CGAGAATGGAGGAAGAAGTTG | 60 | 153 | IV | IV | |
| 338 | S236 | 基因组SSR | AAATGGCCGTTTTATGATCG | CGGAGCTGAACCTTCTGGTA | 53 | 604 | IV | IV | |
| 339 | e182 | EST-SSR | TGGTAACCCTAGCAATCATCA | CTCTTTGGCAACAACATCTCA | 58 | 241 | IV | IV | |
| 340 | 28173 | 基因组SSR | TGCATTGCTAATAACATTAGAACCAT | TTCCTTTTAAGCAAGGTGAGGT | 59 | 197 | IV | IV | |
| 341 | 29016 | 基因组SSR | TTTCAAAGGCAAGGCAAAAC | CACCTCGCAAAATTGGACTT | IV | IV | |||
| 342 | e273 | EST-SSR | CAACAACTTCTACAGCAGCAA | GCAGTAGCATCTGGCTGTGA | 57 | 152 | IV | IV | |
| 343 | e1121 | EST-SSR | AACAACGGCAACAACAACAA | GTGGCCTTAGTCCCAAGAAA | IV | IV | |||
| 344 | e908 | EST-SSR | TGCAGTGATGAAGTGGTTGA | CACTGCTCCATATCCCACAA | IV | IV | |||
| 345 | 27057 | 基因组SSR | TGACCCTAGCAATTAGGATTTGA | ACCATGCCTCCAAAAACTTG | 60 | 160 | IV | IV | |
| 346 | 24907 | 基因组SSR | AAGCAATCCTAATCCATGTGTG | CATCCTTTCCGCCTTTGTTA | 59 | 134 | IV | IV | IV |
| 347 | 22155 | 基因组SSR | ACCCGAGTCAGTCGCTTATG | AACACGGCTTCAATTTACGA | 58 | 135 | IV | IV | |
| 348 | e878 | EST-SSR | CGCATTTTCACTCCACACAC | CGTTCGGAACATCCAAGGT | IV | IV | |||
| 349 | o79 | 基因组SSR | TTGTCTTCACCACCTTAC | GATCATCAGCCAATAGTT | 52 | 300 | IV | IV | |
| 350 | 27068 | 基因组SSR | TTTCGGGCGTCAAATAATTC | GCCACACCTCCAAATGAGTT | 60 | 152 | IV | IV | |
| 351 | 20339 | 基因组SSR | CCTTCCGTGACCAAGAGAAA | GGTGGATGAGATGGATGAATG | 60 | 146 | IV | IV | |
| 352 | e307 | EST-SSR | CTTGTCAGCTTGGCATTCAA | GCGAGTTTCCATTCATCACA | 60 | 177 | IV | IV | |
| 353 | e360 | EST-SSR | CTTGCCCAAATTCAAGCTGT | CTCTATTACACAAATGCCAGTG | 55 | 239 | IV | IV | |
| 354 | B110 | 基因组SSR | CCTCTTCAACGGTACGAGGA | TTGCAGAGAGACGAGAGAGAAA | 56 | 208 | IV | IV | |
| 355 | 27300 | 基因组SSR | CGGCAGTATTTGCAACAAGA | CCTCAAGGCCAGATGATTTT | 59 | 159 | IV | IV | |
| 356 | e1029 | EST-SSR | TCATTGCATGCCATTCTTTC | CGAAATAGAGAAAAGATAGAACCAG | IV | IV | |||
| 357 | 29971 | 基因组SSR | CGAAATTGAATGCAGGAATG | CAACCTCCAAACTCCAAACAA | V | V | |||
| 358 | 28387 | 基因组SSR | GGCTCCATATCATGTTTCTATGC | AAAAGGAGGGAACATGGAAGA | 60 | 201 | V | I-2 | V |
| 359 | 22059 | 基因组SSR | ATCTTCCGCAACAACACACA | ACGTGAAACGGCACAGTATT | 58 | 203 | V | I-2 | |
| 360 | 27937 | 基因组SSR | ACAAGGCATGGTATGGTGGA | TGAACACAAATTGCAGCCTAA | 59 | 124 | V | I-2 | |
| 361 | S220 | 基因组SSR | AGCTCTTTCTTCCACCACCA | CAGGTTCCAGCTGAGAGGAG | 55 | 1001 | V | I-2 | |
| 362 | e59 | EST-SSR | GAGGGTTTCCCGACTTCATT | TAAAGGTTTTCGCCACCATC | 60 | 154 | V | I-2 | |
| 363 | e723 | EST-SSR | GGGGGTGTCTTACGTTGATG | CCCCAAAACCAGCTGAACTA | V | I-2 | |||
| 364 | 29094 | 基因组SSR | CCCCAAAACCAGCTGAACTA | GGGGGTGTCTTACGTTGATG | V | I-2 | |||
| 365 | 18391 | 基因组SSR | CCATCCTCCACGTGTCTCTT | TCGCATATCCAAATGCAAAC | 60 | 142 | V | I-2 | |
| 366 | e121 | EST-SSR | AGCTCCATTTTGGAGTTTGT | CCTGAACCTGATTATAGCCAAGA | 56 | 211 | V | I-2 | |
| 367 | e619 | EST-SSR | AAGTCTCTCATACCTAACCAACCAC | GCAGCCAAATTTGAGGAAGA | V | I-2 | |||
| 368 | 20702 | 基因组SSR | GTTCTCCATCGCCTTCTTTC | TGTGTTATGCCGAGCTTTTG | 59 | 151 | V | I-2 | |
| 369 | e611 | EST-SSR | CCACAACCCCCTCTCTCTC | CTGCGAATTCGGAAAGAAAC | V | I-2 | |||
| 370 | e1060 | EST-SSR | AGAAGTTTTGTTGGTGCAAAGA | TGCTCATTTCTTTACCTTTCTTGA | V | I-2 | |||
| 371 | e1233 | EST-SSR | CGTTCCTTGTCTCTCCTCAAA | ATTCCCAACATGCACCATTT | V | I-2 | |||
| 372 | 22184 | 基因组SSR | GGGCGAAAACAACTTCCATA | CCTGGATGCTCCCAAAATAA | 60 | 149 | V | I-2 | |
| 373 | 26117 | 基因组SSR | CATCGGGCGAGATAACAAAT | TTCCAAGCCTCACTTTCTCC | 59 | 202 | V | I-2 | |
| 374 | 26857 | 基因组SSR | TGCTACAAGTCTAAATACAACACTCTT | CGGGAAGAGAATGATGAGGA | 57 | 133 | V | I-2 | |
| 375 | 24882 | 基因组SSR | TTTCTGGAACCTCGCAAAAC | TTGCCTCAATTTGGAGACCT | 60 | 173 | V | I-2 | |
| 376 | 23829 | 基因组SSR | CGCTCGGCCATGTAACTTAT | GGAAATGGGACTGAAACTGG | 59 | 199 | V | I-2 | V |
| 377 | 24112 | 基因组SSR | TTGATCATCCTCTCGCTTTT | TGTTGTCGTCATCAAAACACAG | 57 | 187 | V | I-2 | |
| 378 | 23525 | 基因组SSR | TGTGCTTTTCTCTTGGCTTCT | CCAGAGGAACCACAAGGTGT | 59 | 125 | V | I-2 | |
| 379 | 24512 | 基因组SSR | AAGCGTACGTGGCAAGAAAT | TCCCTGGGAGAGATGAAAGA | 60 | 171 | V | I-2 | |
| 380 | 24236 | 基因组SSR | CAAACCTTCTTTATTTCCATTTCA | ACTTCTGGTCCACGCAAAAC | 59 | 152 | V | I-2 | |
| 381 | e738 | EST-SSR | CCAATGGACTAGGTGGTGGA | TGATGGATGGGGTGATCATA | V | I-2 | |||
| 382 | 25394 | 基因组SSR | AATGGGTTTTGCTACGTGGT | GGGTGAGTGGAGAAAGCACT | 59 | 142 | V | I-2 | |
| 383 | 25076 | 基因组SSR | GCTTGCAAGTGTGCGTGTAT | CCAGCCAAATGCACAATAAA | 60 | 181 | V | I-2 | |
| 384 | 25618 | 基因组SSR | TTCCATCGTGAACCTTCCTC | CACACGACTTGCAATGTTCC | 60 | 204 | V | I-2 | V |
| 385 | e122 | EST-SSR | TCCACCGACATCTCTTCTCA | AGGTGGTGGTTGTTGTTGGT | 59 | 180 | V | I-2 | |
| 386 | 19657 | 基因组SSR | TCCAAACCCTAGTTAGAGAAAGAA | AGCACCATCATCGTTCATCA | 58 | 188 | V | I-2 | |
| 387 | 28929 | 基因组SSR | GGACTTTTGCGGGTATGAAA | TGTCTCTTTAGATTCGTTCCAAAA | V | I-2 | |||
| 388 | e5 | EST-SSR | ATTAGGGCCGGATAATTTGG | TCCTCAGCAGCTGTCTCAAA | 60 | 162 | V | I-2 | |
| 389 | 25089 | 基因组SSR | ATTCTTGTTGGCGAAACACC | TTGCATTACCCAAAGCTCCT | 60 | 185 | V | I-2 | |
| 390 | 24228 | 基因组SSR | GCAAATTTTCGTTAAATGGATGT | GACAACCTGGAGACGCATTC | 59 | 134 | V | I-2 | |
| 391 | 26521 | 基因组SSR | TGTCTAAGGGTGACAAAAGATCA | TGAACCCGCTCTTCCTTACT | 59 | 159 | V | I-2 | |
| 392 | 25851 | 基因组SSR | AGGCAACACGAGGACGAATA | CGACGGAATTGAAAAACAAAA | 60 | 152 | V | I-2 | |
| 393 | 25986 | 基因组SSR | CAATAGGCCGCGTAAGAAAA | TTGCCATCGATTTGATTTGA | 60 | 158 | V | I-2 | V |
| 394 | 22848 | 基因组SSR | GTGGTGGAAGAGCGTTTGAT | CATGGTGCGTTAACCCAGTT | 60 | 190 | V | I-2 | |
| 395 | 22829 | 基因组SSR | TAGAAGGTTGCCTTGGGTGT | GCCCACCAAAGAAATCAAAA | 60 | 114 | V | I-2 | |
| 396 | 24850 | 基因组SSR | TGGCACACATCTTCAATACAAA | GCACAACCGTTTTTGGTTCT | 59 | 137 | V | I-2 | |
| 397 | 26012 | 基因组SSR | TGGCCCTCAACCTTGTATGT | GCAACACAGAACAAAGCACAA | 60 | 138 | V | I-2 | |
| 398 | 29460 | 基因组SSR | TTCCTGACGCGGACATTAAC | GGAAATTCGGCAAGGACTTA | V | I-2 | |||
| 399 | e599 | EST-SSR | GCAATTTTCTCACTCCATCTCC | AAAGGAAAGCAACTCGGTGA | V | I-2 | |||
| 400 | 29839 | 基因组SSR | GAACCTCGTTTTTGCATCCT | AATGATAGGGGTTGCCACAT | V | I-2 | |||
| 401 | 21936 | 基因组SSR | TGTTGTTTGTTTGGTTGAGGA | CGTTGGCAAACATCATTATCA | 59 | 187 | V | I-2 | |
| 402 | 22325 | 基因组SSR | TGCGAGGGATGAGTTTCTTT | TGTGTGGCCAAATCGAAGTA | 60 | 187 | V | I-2 | V |
| 403 | 25721 | 基因组SSR | AAAAGGAAGCAACTCGGTGA | GCAATTTTCTCACTCCATCTCC | 60 | 168 | V | V | |
| 404 | 16452 | 基因组SSR | CGATGGTTGCTGTTGTGAGA | ACCCCAAACAAACACCAATG | 61 | 129 | V | V | |
| 405 | 24388 | 基因组SSR | GGTCTGGGTCTTTGGCCTAT | AGCATTGCAACGAAGGTTTC | 60 | 126 | V | V | |
| 406 | 23759 | 基因组SSR | GGGGTGACAGTGTAGGGTTTT | TAGGCACACGCTTTCATGTT | 60 | 157 | V | I-2 | V |
| 407 | 23282 | 基因组SSR | TGGTGATGCATGATCATTTAGA | TACAACCCCACCCTGATTGT | 59 | 152 | V | V | |
| 408 | 25755 | 基因组SSR | TTTTCCAACTAAGGTTGTTTCTTTC | CAAAAGGAGGAGGCTGAAGA | 59 | 150 | V | V | |
| 409 | 28889 | 基因组SSR | ATTTGTGGTGCAAGCCTTCT | AAAATTGTACATGGACTCCTTTCTC | V | V | |||
| 410 | 27456 | 基因组SSR | GCACATCCCATTTTTCCAGT | TTCATTACTTTGATAGTGTTCACAAA | 57 | 120 | V | V | |
| 411 | 27661 | 基因组SSR | ATTCATCTCTTTTCCTAAACAAAAAT | CACACCTCCACGTTCATCTC | 57 | 152 | V | V | |
| 412 | 29062 | 基因组SSR | TCACCATCGTGAGCAAGTTC | GGATGTTACGCCCACAATG | V | V | |||
| 413 | 29741 | 基因组SSR | GCAAAAAGCATTGTCCATTTC | GCCTAATCTACAAACGGCTGAG | V | V | |||
| 414 | PSGAPA1 | 锚定标记 | GACATTGTTGCCAATAACTGG | GGTTCTGTTCTCAATACAAG | 51 | V | V | ||
| 415 | 16914 | 基因组SSR | AACCTCGAGCAACAACAGGT | TTAGGTTGGCGTTTTTGGTC | 60 | 149 | V | I-2 | V |
| 416 | e691 | EST-SSR | GATTTAATGCGCGGTTGATG | TGAGTGAAATCATGGGTGGA | V | I-2 | |||
| 417 | 28482 | 基因组SSR | CCGACACACTCCTCAACAAA | TCATCAGGATGAGGACACTCC | 60 | 155 | V | I-2 | |
| 418 | 29694 | 基因组SSR | GAGTGCCTGATCCAAGAGGA | CTCTAAAGGGTGGCAACGAC | V | I-2 | |||
| 419 | 28277 | 基因组SSR | AACACAAGCGCGTTAGTTGA | GACCAGAGTCGAAGCGAAAC | 60 | 183 | V | I-2 | |
| 420 | 29430 | 基因组SSR | TGCGATTTTTCAGTGAGGTG | AACGCAGGTGATGAGCCTAT | V | I-2 | |||
| 421 | 27008 | 基因组SSR | CGAGCAACAGACTGCAAAAA | GCCAACTTTCAATGTTTGACATA | 59 | 132 | V | I-2 | |
| 422 | 16549 | 基因组SSR | CAATGAGATGCTGGCGATAA | GTTCGGTGTTGTGGGTTTTT | 60 | 140 | V | I-2 | |
| 423 | e506 | EST-SSR | CCCCTTTATCCCCCTATTTC | CCTCAACACCAATGAACCAC | V | I-2 | |||
| 424 | e938 | EST-SSR | CTCCTCCTCTGATCCCTTCA | AAATTTCGATCAGGGGTTCC | V | I-2 | |||
| 425 | e625 | EST-SSR | GCTCCAATGGCTTCCTAACA | AACAAGGGGCAATCACAATC | V | I-2 | |||
| 426 | e800 | EST-SSR | AATCGCCAAAGGGTTTGTTT | CGCTTTGGTTCTAGCAGGAT | V | I-2 | |||
| 427 | e1095 | EST-SSR | TATCCATTGCCAGCAGCATA | AATCGCCAAAGGGTTTGTTT | V | I-2 | |||
| 428 | 157 | 基因组SSR | CACATCGACAGAGACATACGA | GTGTGTGTGATGTGTGTGGTG | 57 | 70 | V | I-2 | |
| 429 | 21491 | 基因组SSR | ACACGGGATCGAGCTTTAGA | TCCTTTCCTCTAACTTCTTCCTTCT | 59 | 175 | V | I-2 | |
| 430 | 3644 | 基因组SSR | CACACGCAGAATCACACGTA | CGTGTGTGTGCATGTAATCG | 59 | 176 | V | I-2 | |
| 431 | 3790 | 基因组SSR | GCGTGTGTACATGTGTGTGC | GCGACTTGCACAAGCAGA | 60 | 182 | V | I-2 | |
| 432 | 4696 | 基因组SSR | TGCATGTGCATGTAATCGTG | TCACACGCACGTACAAATCA | 60 | 213 | V | I-2 | |
| 433 | 5849 | 基因组SSR | ACATTCACACACACACGCAA | AAGCTGTGTGCACGTGAGTT | 60 | 278 | V | I-2 | |
| 434 | 3919 | 基因组SSR | GCACACGCAAACTCACAAGT | TGCGTGTGTGCATTTGTTTA | 60 | 186 | V | I-2 | |
| 435 | 919 | 基因组SSR | GCACACGCAAACTCACACTT | GCGCATGTGCATTCGTGT | 60 | 94 | V | I-2 | |
| 436 | 4190 | 基因组SSR | ACACGCATGCACGATTACAT | TGTCCGTGTACGAGCTTTTG | 60 | 194 | V | I-2 | |
| 437 | 4013 | 基因组SSR | ACACGCATGCACGATTACAT | GTACGAGCTTTTGTCACGCA | 60 | 189 | V | I-2 | |
| 438 | 3772 | 基因组SSR | CATGCACGATTACATGCACA | CGTAGCTTTGTCACGCATGT | 60 | 181 | V | I-2 | |
| 439 | 4156 | 基因组SSR | ACACGCATGCACGATTACAT | GTGTCGTGTACGAGCTTTGC | 60 | 193 | V | I-2 | |
| 440 | 3835 | 基因组SSR | ACACGCATGCACGATTACAT | GCTTTGTCACGCGATGTGTA | 60 | 183 | V | I-2 | |
| 441 | 4427 | 基因组SSR | ACACGCATGCACGATTACAT | CGTACGTGTCCGTGTACGAG | 60 | 202 | V | I-2 | |
| 442 | 3955 | 基因组SSR | ACACGCATGCACGATTACAT | ACGTAGCTTTTGTCACGCAT | 58 | 187 | V | I-2 | |
| 443 | 4014 | 基因组SSR | ACACGCATGCACGATTACAT | GTGTACGAGCTTTGTCACGC | 60 | 189 | V | I-2 | |
| 444 | 3567 | 基因组SSR | TGCATATGTGTGTGTCTGCG | ATACACATGCGTGCAAAAGC | 60 | 173 | V | I-2 | |
| 445 | 4274 | 基因组SSR | CACACAGAAGCACACGCC | GCATGTGTGTGTGCGATGTA | 60 | 197 | V | I-2 | |
| 446 | 4043 | 基因组SSR | ACACGCATGCACGATTACAT | CGTGTACGTAGCTTTGCACG | 60 | 190 | V | I-2 | |
| 447 | 3695 | 基因组SSR | ACACGCATGCACGATTACAT | TTGTCACGCATGTGTATGTGTT | 60 | 178 | V | I-2 | |
| 448 | 3771 | 基因组SSR | ACACGCATGCACGATTACAT | GCTTTGTCACGCATGTGTATG | 60 | 181 | V | I-2 | |
| 449 | 1683 | 基因组SSR | GTGCGCATGGTGCATATAAA | CATATACACACACGCACGCA | 60 | 113 | V | I-2 | |
| 450 | 3807 | 基因组SSR | ACACGCATGCACGATTACAT | GCTTTGCACGCATGTGTAGT | 60 | 182 | V | I-2 | |
| 451 | 4114 | 基因组SSR | ATACACGCATGGCACGATTA | GTACGAGCTTTTGTCACGCA | 60 | 192 | V | I-2 | |
| 452 | 3600 | 基因组SSR | CACATGCACACGCACACTTA | TGTCCGTGTACGAGCTTTTG | 60 | 174 | V | I-2 | |
| 453 | 4080 | 基因组SSR | ACACGCATGCACGATTACAT | TGTACGAGCTTTTGTCACGC | 60 | 191 | V | I-2 | |
| 454 | 3770 | 基因组SSR | ACACGCATGCACGATTACAT | AGCTTTGCACGCATGTGTAT | 59 | 181 | V | I-2 | |
| 455 | 3808 | 基因组SSR | ACACGCATGCACGATTACAT | GCTTTTGCACGCATGTGTAT | 60 | 182 | V | I-2 | |
| 456 | 3921 | 基因组SSR | ACACGCATGCACGATTACAT | TACGAGCTTTTGTCACGCAC | 60 | 186 | V | I-2 | |
| 457 | 3377 | 基因组SSR | CACATGCACACGCACACTTA | CGTAGCTTTGTCACGCATGT | 60 | 165 | V | I-2 | |
| 458 | 3597 | 基因组SSR | CGGGTTCACGTATGTGTGTT | ACGCGTATATTCACACGCAC | 59 | 174 | V | I-2 | |
| 459 | 3923 | 基因组SSR | TACACGCATGCACGATTACA | CGTAGCTTTGTCACGCATGT | 60 | 186 | V | I-2 | |
| 460 | 4825 | 基因组SSR | GCATGTGCATTTAATCGTGC | TGCACACGTACACACAAATCA | 60 | 220 | V | I-2 | |
| 461 | 3298 | 基因组SSR | TATGCGTGTGTGTGCTTGTG | CACACATACACGTGTGAACCC | 60 | 162 | V | I-2 | |
| 462 | 3181 | 基因组SSR | CACATACACATGCGTGCAAA | CGTGTGGTCATGTACGTGTG | 59 | 158 | V | I-2 | |
| 463 | 24331 | 基因组SSR | AATGGCGCACTTCACTTTCT | CCGTTAACGCCTAGCTCAAG | 60 | 137 | V | I-2 | |
| 464 | 25387 | 基因组SSR | GGCTCATGCATCTACCACCT | ATCCCGACGTTCACATTTTC | 60 | 170 | V | I-2 | |
| 465 | 24186 | 基因组SSR | GGTGGATCCTCCTTTTGTCA | TCCCAATCACCACTTCTTCA | 59 | 167 | V | I-2 | |
| 466 | 19075 | 基因组SSR | CACGAGTACAACATGGAGTGAAG | CAAGCTCAACCTCCTCATACC | 59 | 187 | V | I-2 | |
| 467 | e531 | EST-SSR | CACCTCCACCCTTTCACCT | CTGGAGGTGGGAGATTGTCT | V | I-2 | |||
| 468 | 28857 | 基因组SSR | CCGAAATGTTCCGAAGAGAG | TTTCAATTCAATGCCGAAAT | V | I-2 | |||
| 469 | 27046 | 基因组SSR | AAAGAAGGGGATGCGAGAAG | GCTCAAGTCAGTCGGACCAC | 61 | 145 | V | I-2 | |
| 470 | 30087 | 基因组SSR | CGCATACACTGAGGTAACACC | AATACACCGGAAGAGGACCA | V | I-2 | |||
| 471 | o65 | 基因组SSR | ACCGCAACAACAGGATAAT | TGAGGTGAAATCGGAAGAC | 53 | 143 | V | I-2 | |
| 472 | e1035 | EST-SSR | TTTTGCACCCCCTTATGTCT | CCACAAAACTCGGGTGAAAT | V | I-2 | |||
| 473 | 19252 | 基因组SSR | CAATATTGATCGGAATTTGTTTC | TGCGGTTTGATTGAGTTTGA | 58 | 199 | V | I-2 | |
| 474 | 29379 | 基因组SSR | AGGCACGTTGGTGCTAGACT | CCTCAATGATCCCAAAGCAC | V | I-2 | |||
| 475 | e178 | EST-SSR | CGAAGAAGATCAAAATCACCAC | AGCTTCAGGGGTTTCTTTCC | 59 | 195 | V | I-2 | |
| 476 | e135 | EST-SSR | GTTCGTTCGTTCGTTCCTTG | TCTGTCTGGAAATGAAATGG | 56 | 219 | V | I-2 | |
| 477 | AD147 | 锚定标记 | AGCCCAAGTTTCTTCTGAATCC | AAATTCGCAGAGCGTTTGTTAC | 61 | 330 | V | I-2 | I |
| 478 | e921 | EST-SSR | AAGGGGTGATCAAGCATCAA | TTGAGGGAACATGAAGAAATCA | V | I-2 | |||
| 479 | e915 | EST-SSR | GTGGACTCGGATTGGGACTA | GCATCGACGACGAAGAAGAC | V | I-2 | |||
| 480 | 111 | 基因组SSR | CGCACAGCAACACACACAT | GCAGTTAGTGCGTGCGTG | 60 | 68 | V | I-2 | |
| 481 | 22913 | 基因组SSR | TCCAACAAACTCAGCCACAG | CAATGGTGGTGGTGCTCATA | 60 | 175 | V | I-2 | |
| 482 | 23383 | 基因组SSR | TGGAGAAATTGGTGGTGACA | TGCAACCATGTTCTTGTTCC | 60 | 141 | VI | VI | |
| 483 | 23611 | 基因组SSR | TGCAAATGTGCAATGAATGA | GGCGGACATGAGAAGGAATA | 60 | 187 | VI | VI | |
| 484 | 25953 | 基因组SSR | GGCCACAACCGTGATGAG | GGATCCAAGACCGAGACAAC | 60 | 117 | VI | VI | |
| 485 | 24326 | 基因组SSR | AAAACGAGAGGCTCGAAACA | ACTAAAACCTCGCGCATCAC | 60 | 185 | VI | VI | VI |
| 486 | 17422 | 基因组SSR | ACCACAAATGCTTCCGCTTA | GTTGTTGTTGCTGCTGCTGT | 60 | 200 | VI | VI | |
| 487 | 22197 | 基因组SSR | GTTGTTGTTGCTGCTGCTGT | ACCACAAATGCTTCCGCTTA | 60 | 200 | VI | VI | |
| 488 | 29622 | 基因组SSR | AACTTCTGCAGTGGCATGTG | CAAAACAACCTATAAGGATGGAAAA | VI | VI | |||
| 489 | 29331 | 基因组SSR | GGGTGGACCGAATATTTCAA | CGTCACCTCTACCGAAGCTC | VI | VI | |||
| 490 | e124 | EST-SSR | GCTTCTGAACCAAGCACACA | AACAATCCCATGTATCAGCAAC | 59 | 235 | VI | VI | |
| 491 | e1097 | EST-SSR | CCCTTCTCATGGGGAATGAT | TAGTCCATGGAAGCGGAAAA | VI | VI | |||
| 492 | e1105 | EST-SSR | TAGTCCATGGAAGCGGAAAA | CCCTTCTCATGGGGAATGAT | VI | VI | |||
| 493 | 27153 | 基因组SSR | GGGAGCGATGCACATAGTATT | GCCCTACAACGAGTGACACA | 59 | 134 | VI | VI | |
| 494 | 26929 | 基因组SSR | CACATTCACGACGAGGACAG | GCACACTGTAAGCACTTTTCTCA | 60 | 142 | VI | VI | |
| 495 | e853 | EST-SSR | CTTCCCGGGTAAGAACAACA | GCTATGGTTCAGGCGTTTCT | VI | VI | |||
| 496 | 23637 | 基因组SSR | AAGAGGCTCGTGACCCAATA | TGCATTGCATCCTTCAAGAG | 60 | 192 | VI | VI | |
| 497 | AD160 | 锚定标记 | ACCAGTCAAATGGTTAGAAAGT | GAATGGAAAAGAGAATCAAGTT | 51 | 190 | VI | VI | VI |
| 498 | 23578 | 基因组SSR | AAGGAAGGTGGTGTGGAATG | CAATATTACTCAGCCATTAATTAACCT | 58 | 205 | VI | VI | |
| 499 | PSGSR1 | 锚定标记 | TGAAACCACCATTCTCTGGA | AAGACCCCACTTGAAAATTACTTC | 58 | VI | VI | ||
| 500 | 25075 | 基因组SSR | CATCACTCACTCGCCAAAAA | AACCATCTTTGCCAGGTACG | 60 | 192 | VI | VI | VI |
| 501 | e709 | EST-SSR | TGTGCAACCGAGATTGGTAA | CGCCAAAAATACTGATTCACTTC | VI | VI | |||
| 502 | 27176 | 基因组SSR | TGCCTTCAGGTTTTCAAGGT | TGATGAAAGCAATTTTCATGACTT | 60 | 147 | VI | VI | |
| 503 | 29246 | 基因组SSR | CCCTTGCTTGGGTAAGAAATC | GTGCCGGGTATGTATCTGGT | VI | VI | VI | ||
| 504 | 4816 | 基因组SSR | CGTCATCATTGTTCGTCATTCT | GGTCGTAGGGTGTGTCGTCT | 60 | 219 | VI | VI | |
| 505 | 2089 | 基因组SSR | ACACACGTCACACACACGTC | TGATGTGTATGCGTGATGGA | 59 | 124 | VI | VI | |
| 506 | 28621 | 基因组SSR | CGTTTTCACATTCGCTAACC | TGGAGAAAGGTTTCCTGATGA | 58 | 171 | VI | VI | |
| 507 | 27428 | 基因组SSR | GCACGCCTGACTTCTTCTTT | AAATGGATTGCGACGTGATT | 60 | 174 | VI | VI | |
| 508 | 24560 | 基因组SSR | GATAAAGGCAGCGACAGAGG | AATGAAGTGCAAGCCCAAAT | 60 | 202 | VI | VI | |
| 509 | 23737 | 基因组SSR | CGTGCAACCATAGCAAGAGA | AAACCGCTCAAGCTCAGGTA | 60 | 182 | VI | VI | |
| 510 | 26025 | 基因组SSR | ATGCACTCAAAGGCCATCAT | CACTTGCAGAGCGAGAGAAA | 59 | 113 | VI | VI | |
| 511 | 24906 | 基因组SSR | TCGAGTCAATCGCTCAGAAC | TGCCCAGATGTCATAAGGTG | 59 | 134 | VI | VI | |
| 512 | 16445 | 基因组SSR | TCAAACCGCTGAAAAACAAA | GCGGTGGGAGGGAGATAC | 59 | 128 | VI | VI | |
| 513 | 16397 | 基因组SSR | AGGGCCAGGTTTATTTCCAC | TTTCCCAATGGCAAGTTAGC | 60 | 123 | VI | VI | |
| 514 | e715 | EST-SSR | CGTTGAAACAGCGATTCTGA | TTTCTTCAATACCTCAATGGTTC | VI | VI | |||
| 515 | 28153 | 基因组SSR | TGGGTTGTCGTGTTGTTGTT | AACACTCCCAACTCCATTTTT | 58 | 183 | VI | VI | |
| 516 | 27491 | 基因组SSR | TCCTAACCAACCAATAACACGAT | TTGAGGATTTCGGTGACCTC | 60 | 180 | VI | VI | |
| 517 | C58 | 基因组SSR | TCACGTGCTTGTCGTTCTTC | TAAGAAACCGCCATGGAT TT | VI | VI | |||
| 518 | B117 | 基因组SSR | ACATCAGGGAAGAACGCATC | GAGGGTGAAGACCAGCTTTG | VI | VI | |||
| 519 | 30379 | 基因组SSR | TGTTGGCAGGAAACTCTTCA | AGCCACAAATTTCGTTGTGTT | VI | VI | |||
| 520 | 24575 | 基因组SSR | TTGTGAGCACATTGGAGGAG | GGATTGTGTTGGTTAGAGAAAGAGA | 60 | 161 | VI | VI | |
| 521 | 23468 | 基因组SSR | CGGCAGCATCTACACAAGAG | ACGTTGAAGACTCCGTCACC | 60 | 171 | VI | VI | |
| 522 | 29440 | 基因组SSR | TGTTCCCCTTTAATTTTCATCCT | AAGAAGCCGTCACGAAATGT | VI | VI | |||
| 523 | 30004 | 基因组SSR | TCTTTGCGGATATGCATTTTA | CGGGTGAGGACTGAAAACTC | VI | VI | |||
| 524 | 2614 | 基因组SSR | ATGTGTGTGCGTGTGTGTTG | GATTGTTATGTGCTGCGTGG | 60 | 140 | VI | VI | |
| 525 | 3494 | 基因组SSR | GCACCGCTCTGACACTCATA | TGAGAGTGGAGTGGCTGAAG | 59 | 170 | VI | VI | |
| 526 | 3244 | 基因组SSR | CTTCCCCTCGCAATTTATGA | ATGTGTGTGCGTGTGTGTTG | 60 | 160 | VI | VI | |
| 527 | 4583 | 基因组SSR | ACACCATTGCACCATTCTGA | TGCGTGTGTTGTGAGTGAGA | 60 | 208 | VI | VI | |
| 528 | 4581 | 基因组SSR | ACACCATTGCACCATTCTGA | GTGCGTGTGTGTTGTGAGTG | 60 | 208 | VI | VI | |
| 529 | 4629 | 基因组SSR | ACACCATTGCACCATTCTGA | GTGCGTGTGTGTGTGAGTGA | 60 | 210 | VI | VI | |
| 530 | 4811 | 基因组SSR | ATGTGTGTGCGTGTGTGTTG | ACACCATTGCACCATTCTGA | 60 | 219 | VI | VI | |
| 531 | e93 | EST-SSR | ATGGCCTTTGCAATTACAGG | GCTGATGTTGGCCAAGGTAT | 60 | 203 | VI | VI | |
| 532 | e771 | EST-SSR | TCCGGCAAGATATTGGAAAA | CTGCAGAGGCTGTCACTCAA | VI | VI | |||
| 533 | e1109 | EST-SSR | TCCGGCAAGATATTGGAAAA | GCTTGGATCGCAGGAAAATA | VI | VI | |||
| 534 | 20896 | 基因组SSR | TGATGACCCTGCAAATTCAA | TGCACCACTGTCAGGTGATT | 60 | 131 | VI | VI | |
| 535 | e596 | EST-SSR | TCCCTCATTCTCCCTTTTCA | GACGGCGCTGATGATAGACT | VI | VI | |||
| 536 | e1098 | EST-SSR | AGAGGACGTGTTGCTGTGTG | CACAGAATTGGCAGAAACAGAG | VI | VI | |||
| 537 | 27272 | 基因组SSR | TGTAGCGGCACACTTTGAGA | GATCTCTGCCACCCATCTTC | 60 | 190 | VI | VI | |
| 538 | 28790 | 基因组SSR | GCTGTGGGGGTTTAATCAGA | CCGCAATCCTTCAAGAACTC | 60 | 133 | VI | VI | |
| 539 | 29200 | 基因组SSR | ATGCTGATGAAATGCGAATG | CATCTGTACCCGGACCTTTG | VI | VI | |||
| 540 | 27844 | 基因组SSR | GCTTCAAGCTACCAAGTGGA | CCTCACGGGCTCTACCATAC | 58 | 118 | VI | VI | |
| 541 | 29877 | 基因组SSR | GTCGTGGGGAAAAGGTATCA | GGTACGACAACCCTACCTTTG | VI | VI | |||
| 542 | e975 | EST-SSR | AGCAGCTCCTACTCCTTCTCC | GCGCAAATCCTATTCCAAAG | VI | VI | |||
| 543 | e940 | EST-SSR | GCGCAAATCCTATTCCAAAG | ATTTCAGCAGCTCCTACTCCT | VI | VI | |||
| 544 | 19691 | 基因组SSR | TTGTAAGACCGACTCGTCCA | CGGTCTGAGGTTGTTGTGAA | 59 | 146 | VI | VI | |
| 545 | 16588 | 基因组SSR | CGGTCTGAGGTTGTTGTGAA | TTGTAAGACCGACTCGTCCA | 59 | 146 | VI | VI | |
| 546 | 25488 | 基因组SSR | TGAAGAATGAGCTTCAATTTTTGT | GGGTGCAATCATGAGTGTTG | 59 | 145 | VI | VI | |
| 547 | 26625 | 基因组SSR | GCTCCATCACGGTGAGTTTT | TCCCACTTTCACGATGTTCA | 60 | 191 | VI | VI | |
| 548 | 25280 | 基因组SSR | CTCTCTGCCCACTGCTCTG | TCTCACGTTGGGATGCTAAA | 59 | 122 | VI | VI | |
| 549 | 25711 | 基因组SSR | AAGGTTTTGAAATAAATGAAGTTTG | TGAAAGCCCACTTGATCTTC | 57 | 188 | VI | VI | |
| 550 | 25888 | 基因组SSR | AAGGGGGAGAGAGGTGGTTA | TCGCCTTTTCTTTCTTCTTCA | 59 | 164 | VI | VI | VI |
| 551 | AA335 | 锚定标记 | ACGCACACGCTTAGATAGAAAT | ATCCACCATAAGTTTTGGCATA | 61 | 220 | VI | VI | |
| 552 | 23568 | 基因组SSR | TCCCTCATTCTCCCTTTTCA | GACGGCGCTGATGATAGACT | 60 | 127 | VI | VI | VI |
| 553 | 23081 | 基因组SSR | ACCCTTGCTTTGCCACATAA | TGTTGCTCTTTTGTTGAGTTGA | 59 | 149 | VI | VI | |
| 554 | 23431 | 基因组SSR | GCAACAACAGCAACCTCTGA | TGAAGGTGGAACTTGGTTTTG | 60 | 145 | VI | VI | VI |
| 555 | 27435 | 基因组SSR | TTGATGCTCTTCTTCCATTCAA | CATACAAAACACACAAAAAGGATTG | 60 | 171 | VI | VI | |
| 556 | 16410 | 基因组SSR | AAGGTCATGCTTCTTCATCTCT | GGGTGAGGTGTTATGGCACT | 57 | 125 | VI | VI | |
| 557 | 28516 | 基因组SSR | CCAAAATTCATGCATGGTACG | TCCAGTGGCTCATAGAGGAGA | 60 | 169 | VI | VI | |
| 558 | 26140 | 基因组SSR | TTGTGTGCAAACCACCTAGC | GATTGCATCACACGGTCAAG | 60 | 200 | VI | VI | |
| 559 | 23309 | 基因组SSR | GAAGATGGCAACGTGTCAAA | AACTCATCCTCCACCACCAC | 60 | 128 | VI | VI | |
| 560 | 29872 | 基因组SSR | TCCACTTCCACCCACAAAAT | GCAATGGAGGTTTTGCTTTT | VI | VI | |||
| 561 | 26514 | 基因组SSR | TGGGCACAAGTCTGTGAGAA | TTGGGTTGAGGTGTTTAGGTG | 60 | 153 | VI | VI | |
| 562 | AD60 | 锚定标记 | CTGAAGCACTTTTGACAACTAC | ATCATATAGCGACGAATACACC | 51 | 216 | VI | VI | |
| 563 | AB71 | 锚定标记 | CCAACCATTTGTGAGTTCCCTT | TTCGTCGAACCACGAGAATAGA | 61 | 145 | VI | VI | |
| 564 | 23949 | 基因组SSR | TCGGTCAATTTTCACGTAGC | GCAGAGAATGAAAGAAATATAAAGAAA | 58 | 142 | VI | VI | |
| 565 | 22903 | 基因组SSR | TGCTTGCCAGAATAAAAGTCC | CCTCTAGGGTTTCGGGTCTC | 59 | 181 | VI | VI | |
| 566 | 27055 | 基因组SSR | TCTGCAAACTCCCAACACTG | GGTGGTTTGGTTGCAATCTT | 60 | 206 | VII | VII | |
| 567 | e282 | EST-SSR | GGCAAGCATAAAAGGGACAC | TTCATCCAAGAACCCTCGAC | 60 | 185 | VII | VII | |
| 568 | e144 | EST-SSR | TCCATTTCCCGAGTTTATCG | AACACGAATATGCAACAAGC | 56 | 151 | VII | VII | |
| 569 | S244 | 基因组SSR | TTTAGCACAGAACAGCGTAGT | TAACGCCCTTGAGAATTTCG | 53 | 606 | VII | VII | |
| 570 | 21399 | 基因组SSR | TGATTCTAGTTCATTTCACAAACACA | CGTCCTGCACCTAGCTTCTT | 60 | 153 | VII | VII | VII |
| 571 | 26099 | 基因组SSR | GCTCTTCGTAACGCTCACAA | TGACGACGGAGACTGAGTTG | 59 | 201 | VII | VII | |
| 572 | 19979 | 基因组SSR | CACCAGAAAATTTGTTATCAAAAAGA | GCACCCTGGGAAATTACAAA | 60 | 161 | VII | VII | VII |
| 573 | 24547 | 基因组SSR | CGGCAGAATTAGGGTTTTGA | TCAATTCCGAACCACCTTTC | 60 | 198 | VII | VII | |
| 574 | e968 | EST-SSR | ACCGCTTGAACTCCAAACAA | GAAGGTAACAACGCCGAGAA | VII | VII | |||
| 575 | 18013 | 基因组SSR | TCAATTCCGAACCACCTTTC | CGGCAGAATTAGGGTTTTGA | 60 | 198 | VII | VII | |
| 576 | S144 | 基因组SSR | TTTTCTCACCGCGCTTATTT | AACAACCACCGAAGACGAAG | 54 | 235 | VII | VII | |
| 577 | 20828 | 基因组SSR | CATGGATCCCAACAGAAACA | TGGTTTTTACCCGAGACTGG | 59 | 144 | VII | VII | |
| 578 | e876 | EST-SSR | GCAGATTGACTGCTCATGATGT | AGCTGATTATTGGGCACCTG | VII | VII | |||
| 579 | 21405 | 基因组SSR | TCCACCATCTAATCCCCTCTT | AGCTGATTATTGGGCACCTG | 60 | 147 | VII | VII | VII |
| 580 | 18103 | 基因组SSR | CATGTGCATGTGCAAGTACG | CTTTAAAATGCCCGGACAAA | 60 | 209 | VII | VII | |
| 581 | 17431 | 基因组SSR | TTCACAATTCACCACCAATCA | CCAACGTCAGGTACGATTCA | 60 | 201 | VII | VII | VII |
| 582 | 17666 | 基因组SSR | GTGCATTGGCTCGTACTCAA | TCCACAATATAGCCCAGACCA | 60 | 158 | VII | VII | VII |
| 583 | S85 | 基因组SSR | TTCCAACCATGGAAGCTTTT | TTCTTCGTCGGGTACAGTGA | 54 | 188 | VII | VII | |
| 584 | e255 | EST-SSR | TGGAGAAGGGCAAAATATCG | TCTCAACACCACATCAAGGAA | 59 | 249 | VII | VII | |
| 585 | 25965 | 基因组SSR | TGATTCGTAGACCCCACACA | AAGGTTAATGTCTTCTTTTTGAAGTT | 57 | 150 | VII | VII | VII |
| 586 | 17384 | 基因组SSR | AGTAGCGGTGTGTGGTTGTG | GGGAAGAAAAAGGTTGGAAGA | 60 | 197 | VII | VII | VII |
| 587 | 30253 | 基因组SSR | CTACGTTTGGCCCTTGTGTT | GGCCCTAAATCTAAAATGAACAA | VII | VII | |||
| 588 | B179 | 基因组SSR | ACCGACGCTTCAATGGTATT | TTCATCTCCGACCCTACACC | 55 | 267 | VII | VII | |
| 589 | e499 | EST-SSR | GACTCCACGCACAGAACTGA | GGAGAGGGGAGTGAATGTGA | VII | VII | |||
| 590 | e544 | EST-SSR | CACTACACAAGAAGCAAAGAAAAA | ATTCCTTTCCGGTCCATTTC | VII | VII | |||
| 591 | e1002 | EST-SSR | CCACGCGTGACAAGTAAAGA | ATTCCTTTCCGGTCCATTTC | VII | VII | |||
| 592 | 21420 | 基因组SSR | GGTGGTCGACGTATCGAAGT | TCAATGTTGTTGCGCTTACAT | 59 | 142 | VII | VII | |
| 593 | 18255 | 基因组SSR | TGTCAAATCCAATAAAAACACACA | TTTGTGCACACCGTCAATTT | 59 | 129 | VII | VII | |
| 594 | e643 | EST-SSR | CACCACCACTCTCACACCAT | TGCATTGCGAGAGTAAGACAA | VII | VII | |||
| 595 | e825 | EST-SSR | AGTTTCCGCCATCAACATTT | CCTACCTGCATTCACAACCAT | VII | VII | |||
| 596 | 17225 | 基因组SSR | GTTGCAAGCTGCTACCATCA | AGACGGATCCAACAATCTCC | 59 | 182 | VII | VII | |
| 597 | 17593 | 基因组SSR | CATCCTCCTCCTCCATACCA | TCATCATCAATGCAAAGGACA | 60 | 148 | VII | VII | |
| 598 | 24407 | 基因组SSR | GGGATCAAAGCAACCCTTTT | CATGGCAAGGAAGACCGTAG | 60 | 208 | VII | VII | |
| 599 | 24810 | 基因组SSR | TGAGCGAGGTAGGAAGAACC | CCTCTACAGTGGCCCTCTCA | 59 | 132 | VII | VII | |
| 600 | 25354 | 基因组SSR | ACCCTTGGGGCTTACAATTC | GACGTGGCTGGACATAACAA | 60 | 184 | VII | VII | |
| 601 | 25799 | 基因组SSR | CTGCAGAAGGCCCTGTTCTA | GATTCTTCATTCTCAACACACATTG | 60 | 137 | VII | VII | |
| 602 | 25430 | 基因组SSR | GCACTTGACGATGCATTTGA | GGGAAAGGGAACGATTTCAT | 60 | 210 | VII | VII | |
| 603 | 23294 | 基因组SSR | GGAGGAGGATGACGATGAAA | AGGGCTACCGGACTGAAACT | 60 | 172 | VII | VII | |
| 604 | 24142 | 基因组SSR | GCAGCCATGGTTGATTGATT | TCAAGAACATTACTTTTTCCCTCT | 58 | 194 | VII | VII | |
| 605 | 27051 | 基因组SSR | TGGTGGTGGAGAGTGATTGA | TCTGGTGGTACTTCCTCCAAAT | 60 | 208 | VII | VII | |
| 606 | e1114 | EST-SSR | GTGCGGCTTCATTTTCAACT | TTCTCAACTGGTTGGTTCCATA | VII | VII | |||
| 607 | e1209 | EST-SSR | AGTGCGGCTTCATTTTCAAC | TCAATTTGATCCATGCAGTAGG | VII | VII | |||
| 608 | AF004843 | 锚定标记 | CCATTTCTGGTTATGAAACCG | CTGTTCCTCATTTTCAGTGGG | 54 | VII | VII | ||
| 609 | 23606 | 基因组SSR | GGGTTTGCCTCTTTTTCTCTC | ATCGTCAAAACTGCCCAAAC | 59 | 113 | VII | VII | |
| 610 | 21345 | 基因组SSR | TGCAATGCATGTTGATACGTC | GCAAAAACTCAAACTCAAACTCAA | 60 | 152 | VII | VII | |
| 611 | 27583 | 基因组SSR | TGCACAGAGGATGGTTCTCA | TGGATTGAGCCTCTTGTCCT | 60 | 110 | VII | VII | |
| 612 | 25610 | 基因组SSR | TTTGGTCGTTGCCAAACTAA | AGGACATACCGAGCCAGATG | 59 | 133 | VII | VII | |
| 613 | 26656 | 基因组SSR | AGAAGAGCGTCGGGAAGAGT | CCATGACGGAAAACAACCTT | 60 | 188 | VII | VII | |
| 614 | 25728 | 基因组SSR | TGGACAAATCTCGTGCAAAG | CCAACTTCCCATCTCCAACA | 60 | 164 | VII | VII | |
| 615 | 24471 | 基因组SSR | CAACACAACCTCCTCCAGGT | TAAGCCATTCCCACCTTCTG | 60 | 121 | VII | VII | |
| 616 | 24734 | 基因组SSR | CGCATGAGAGGATAATGATGAA | CCATAGCTTTCCCGAATCAG | 60 | 183 | VII | VII | |
| 617 | 24588 | 基因组SSR | AAATAGATGAGAAAGAGAGAGATTACG | CGCACTTCCATTCACATGAT | 57 | 137 | VII | VII | |
| 618 | 24602 | 基因组SSR | TGAGTGGGCGTGTGATTTAG | TTGCACTGTCGCATTTGAGT | 60 | 118 | VII | VII | |
| 619 | 24503 | 基因组SSR | AAAGGAGATCACCTATGAGAGAGAA | AATTGATTTGGGATCTTGGATA | 57 | 154 | VII | VII | VII |
| 620 | 28097 | 基因组SSR | CAAAATCGGCAGGATTTACAA | TTTGCCTATGAACCTAAAACCAT | 59 | 124 | VII | VII | |
| 621 | 24814 | 基因组SSR | GTGCAACCAGAGCATGTTTC | CATCGACTGTGGAGACATTGA | 59 | 152 | VII | VII | |
| 622 | AB65 | 锚定标记 | CTCGTATCCAAAGATTCGTAGA | AGGGTTAATCGGAGTTTTATGA | 51 | VII | VII | ||
| 623 | 29468 | 基因组SSR | AGTACTCTCGCCGCATCAGT | CACCCCACTTGAGCATATCA | VII | VII | VII | ||
| 624 | 28967 | 基因组SSR | CTCGTCCTCATCGAAAAGCTA | GTGAACAACGCAAGGGTTTC | VII | VII | |||
| 625 | 29500 | 基因组SSR | TGGGAATTGACGAAGAGTGTT | ATGGAGAGGGTTGCTGACAT | VII | VII | |||
| 626 | e46 | EST-SSR | AGGAGGGAGTGGTGGAGATT | CATCACGTGCTTGTGCTTCT | 60 | 227 | VII | VII | |
| 627 | e1173 | EST-SSR | TGATTCCGAATGGGAAACTT | AATCCGCAAACACATCAACA | VII | VII | |||
| 628 | e1276 | EST-SSR | TGAAACAATAGTGCTTTGTTGAAACT | TTTTCTCGTCTGCGTGTGAC | VII | VII | |||
| 629 | 22060 | 基因组SSR | CCGCCTTAGGAAGCCTAACT | CGGCTTGATAATTTGGTGCT | 60 | 181 | VII | VII | |
| 630 | e628 | EST-SSR | GCCACCCTGTTTCTGCTAAC | CTTGTGGATCGGTTCGTGAT | VII | VII | |||
| 631 | e524 | EST-SSR | TCACACCATAGAGAATAACAACAACA | TCTGAAGCCATCTCCATTCTC | VII | VII | |||
| 632 | 28608 | 基因组SSR | TCATGATTTCAAATTTCTTTCACAA | CGCCGTTGGGTAATTGTAAC | 60 | 136 | VII | VII | |
| 633 | 22101 | 基因组SSR | ACCAATCCAGACGCAAATTC | GGGGACAGTGACGAGAACAT | 60 | 156 | VII | VII | |
| 634 | 19460 | 基因组SSR | CAATCCAAACGCAAATCTAACA | AATTGCAAGCCCTACACACA | 59 | 187 | VII | VII | |
| 635 | 17910 | 基因组SSR | CATGCCTGCTTCCTTCTGTT | TTGCAATTTCAAGCCTTCAC | 59 | 187 | VII | VII | VII |
| 636 | 27398 | 基因组SSR | GAACCACATTGGGGATTCTG | AACCTGCAAAAGCCATAAGC | 59 | 180 | VII | VII | |
| 637 | 30165 | 基因组SSR | CCTTTTTACCCCTCCCTCAG | TCAAATGCAAAGGGAAACAA | VII | VII | |||
| 638 | 27755 | 基因组SSR | TGCATTTGAGCTAGTGACATTCT | AAAAACCAACCCAACCACTT | 58 | 163 | VII | VII | |
| 639 | e1193 | EST-SSR | GTTGGCGAGGAATGATTGTT | TCACACACTCTGCCATTTCA | VII | VII | |||
| 640 | PSAB60 | 基因组SSR | AATTAATGCCAATCCTAAGGTATT | GGTTGCACTATTTTCGTTCTC | 59 | VII | VII | ||
| 641 | 28054 | 基因组SSR | AGCAAAGTCACCAGCTGTTTT | TCCTGTTCTAAAACAAAAACAAGAGA | 59 | 113 | VII | VII | |
| 642 | 30124 | 基因组SSR | GACTATGTGTGTGCATGAATTTGA | GGCCATCTCGTTTCAGTACC | VII | VII | |||
| 643 | 30304 | 基因组SSR | CACGCACAGACAGATCATCA | GAAGTTGGGGATGGGAAGAT | VII | VII | |||
| 644 | 28108 | 基因组SSR | CGACAATGTTGCCAGCTATC | TTTTAGGATTTTATCGACGTTTTTC | 59 | 119 | VII | VII | |
| 645 | e291 | EST-SSR | CAAGCGTCGAAGATGAACAA | GCTGGCTGCAAAAGTTTACC | 60 | 205 | VII | VII | |
| 646 | e292 | EST-SSR | GCACATGAAAAATGCCAAAG | CTGTTGCTGTTGGTGGTGAG | 59 | 220 | VII | VII | |
| 647 | PS11824 | 基因组SSR | ACCACCACCACCGAGAAGAT | TTTGTGGCAATGGAGAAACA | 52 | 210 | VII | VII | |
| 648 | e527 | EST-SSR | TTGAAGCAGTGGCAGAGTTG | TCTCAATGAAACATAAGAATGACCTT | VII | VII | |||
| 649 | e1202 | EST-SSR | TGCATGGTTATGATGCTTGA | TCACACACCCCTTCAATTATTC | VII | VII | |||
| 650 | 23521 | 基因组SSR | CGCCAATTCCTTTTCCCTAC | AGAACTCACAGGCGATGGTC | 60 | 112 | VII | VII | |
| 651 | 24652 | 基因组SSR | GAGAAAGCGGCTGCTTAGAA | GCTGTCACCGAGAATGATGA | 60 | 190 | VII | VII | |
| 652 | 25241 | 基因组SSR | TGCAAGCAAGCAAAATTGAA | TGCATGCCCTTTATTTCTTTG | 60 | 142 | VII | VII | |
| 653 | 24944 | 基因组SSR | TCAACCGGAATCTGGAAAAC | ATGGCGCAATCCTAGTGAAC | 60 | 193 | VII | VII | |
| 654 | 22896 | 基因组SSR | ATGTACGCCATGCAGTCAAA | ACAAGATGGGCGTCGAATAC | 60 | 151 | VII | VII | |
| 655 | 24398 | 基因组SSR | TTCGATGCATGAATGACAAA | ATCGGCGGAGACTAAGATCA | 59 | 142 | VII | VII | |
| 656 | 20817 | 基因组SSR | GGCGTAGAGGGCTAAACCTT | TTCCCGAGTCCTAACTTTCTTG | 60 | 135 | VII | VII | |
| 657 | 19526 | 基因组SSR | ACTCCTGGACACCCTGAGAA | CTGACCAAGGGGACCTGTAA | 60 | 198 | VII | VII | |
| 658 | 29620 | 基因组SSR | CCACTGAAGGCTCCTGAACT | AGCGATCACCGATAGTGTCC | VII | VII | VII | ||
| 659 | e1220 | EST-SSR | ATGGTGGTGGAGGTGTGATT | CATCGCCAAATGGATCTTCT | VII | VII | |||
| 660 | 17980 | 基因组SSR | CAATTCACAACGTTCCACTCA | TTTTCGTGAAATTGAAATGACC | 59 | 194 | VII | VII | VII |
| 661 | 29248 | 基因组SSR | CAACAATGTGCATGGAAAAA | CTCGCATTGCGTAACGATAA | VII | VII | VII | ||
| 662 | 28595 | 基因组SSR | CCGGTTCATCGATAAATGTGA | TCTCAAACCCACCAACAACA | 60 | 199 | VII | VII | |
| 663 | 2629 | 基因组SSR | CAAAACACATACGCACACACA | CCCGTCATGATGTCATGTAAA | 59 | 141 | VII | VII | |
| 664 | e123 | EST-SSR | GAGCACAACTTTGCAAGCAG | ACACGTCATTTCAAACCACT | 55 | 198 | VII | VII | |
| 665 | e1249 | EST-SSR | CCCGTTTCAAATTAGAACGATAA | CACGGTTCGGCATTTATTTC | VII | VII | |||
| 666 | 23536 | 基因组SSR | GACGTTGATTGGCCTGTTTT | TGACCATGACATGCCTGTTT | 60 | 180 | VII | VII | |
| 667 | 21796 | 基因组SSR | TCTTCGCTGGGAAGTTGAGT | GGAAGCGATGTCGTTTCATT | 60 | 210 | VII | VII | VII |
| 668 | 28261 | 基因组SSR | CTCTCCCCATGGAGAACAAC | AACAGCTGAAATTGGCGTAGA | 60 | 131 | VII | VII | |
新窗口打开|下载CSV
将我们的整合图谱与以往的遗传图谱相比较, 结果发现, 本研究中的7条连锁群能够与以往遗传连锁图谱中7条连锁群相对应[10,42-44]。通过比较整合遗传图谱和2个单独的遗传图谱之间共有标记的顺序和位置, 结果发现, LGIV和LGVII这2个连锁群在不同图谱中的标记顺序存在较高的共线性, 而其他连锁群则由于作图群体差异而观察到了一些倒位和标记重排的现象。此外, 在整合图谱的LGV上, 同时包含2个锚定标记, 一个是在以往图谱中定位于LGV的PSGAPA1, 另一个是在以往图谱中定位于LGI的锚定标记AD147, 因而导致LGV的定义存疑(附图1)。
附图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图1整合图谱和2个单独图谱及其与豌豆参考基因组物理图谱之间共有标记比较(A–G)
图谱左侧数字为标记位置信息,而右侧为标记名称。遗传图谱包括PSP1、PSP2和整合图谱的图距单位为cM,参考基因组(Reference genome)的物理图谱图距单位为MB。图谱标记名称红色代表锚定标记,标记名称粗体加下划线代表共有标记。标记名称以“e”开头的为EST-SSR标记。
Supplementary Fig. 1Comparison of the common markers among the integrated map and the two individual maps as well as the physical map of the reference genome (A–G)
Numbers on the left of the map and labels on the right of the map represent the position and name of markers, respectively. The map distance unit of genetic maps including PSP1, PSP2 and integrated map is cM, while the map distance unit of physical map of the reference genome is MB. The red and underline labels represent anchor markers and common markers, respectively. Marker names that start with “e” represent EST-SSR markers.
本研究以新近发表的豌豆基因组为参考(Caméor genome build 1a)[20], 对50个共有标记的扩增片段序列进行了BLAST比对分析, 从而对本文得到的遗传图谱和豌豆的物理图谱相比较(附表2)。发现有45个标记被成功比对到豌豆的7条染色体上, 上述有争议的1条连锁群(LGI-2/LGV)被证明为LGV。其中30个标记具有唯一比对位置, 15个标记比对到2个以上的位置; 剩下的5个标记比对到了未挂载到染色体的组装支架上。此外, 还比较了3个遗传图谱和物理图谱的标记顺序(附图1), 发现chr2LG1与LGI-PSP2高度一致, chr6LG2与LGII-PSP1高度一致, chr5LG3与LGIII-Integrated map基本一致, chr4LG4与LGIV- PSP1、LGIV-PSP2和LGIV-Integrated map基本一致, chr3LG5与LGV-PSP2和LGV-Integrated map基本一致, chr1LG6与LGVI-PSP2和LGVI-Integrated map基本一致, chr7LG7与LGIV-PSP1、LGIV-PSP2和LGIV-Integrated map一致率较高。
Supplementary table 2
附表2
附表2豌豆遗传连锁图谱上共有标记BLAST比对结果
Supplementary table 2
| 标记名称Marker name | 连锁群Linkage group | 染色体/连锁群 Chromosome/LG | 比对条数 Number of blast results | 最小E-value The minimal of E-value | 最高比对率 The maximal identifies (%) | 起始位置 Start position | 终止为止 End position | 比对长度Mapped length | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 整合图谱 Integrated map | PSP1 G0003973 × G0005527 | PSP2 W6-22600×W6-15174) | ||||||||
| 20229 | I | I | I | chr2LG1 | 20 | 6.7E-94 | 100 | 21,564,671 | 21,564,851 | 180 |
| 25334 | I | I | I | chr2LG1 | 4 | 2.7E-70 | 100 | 329,941,758 | 329,941,897 | 139 |
| 23261 | I | I | I | chr2LG1 | 1 | 1.7E-79 | 100 | 410,201,631 | 410,201,786 | 155 |
| 24301 | II | II | II | chr6LG2 | 1 | 3.9E-58 | 96 | 44,747,008 | 44,747,143 | 135 |
| 28257 | II | II | II | chr6LG2 | 1 | 3.5E-55 | 93 | 89,359,789 | 89,359,941 | 152 |
| 17754 | II | II | II | chr6LG2 | 1 | 3.0E-85 | 99 | 284,992,981 | 284,993,149 | 168 |
| 25769 | II | II | II | chr6LG2 | 17 | 3.9E-58 | 95 | 379,342,213 | 379,342,360 | 147 |
| 25792 | II | II | II | chr6LG2 | 3 | 6.5E-121 | 93 | 430,274,319 | 430,274,608 | 289 |
| 26850 | III | III | III | chr5LG3 | 1 | 4.5E-80 | 99 | 95,945,216 | 95,945,375 | 159 |
| 21726 | III | III | III | chr5LG3 | 2 | 4.4E-80 | 100 | 105,392,839 | 105,392,995 | 156 |
| 25151 | III | III | III | chr5LG3 | 31 | 4.4E-80 | 100 | 128,426,677 | 128,426,833 | 156 |
| 18135 | III | III | III | chr5LG3 | 1 | 8.3E-59 | 100 | 157,545,697 | 157,545,816 | 119 |
| 22754 | III | III | III | chr5LG3 | 3 | 2.2E-82 | 100 | 178,517,012 | 178,517,172 | 160 |
| 20075 | III | III | III | chr5LG3 | 1 | 6.6E-45 | 95 | 255,763,443 | 255,763,559 | 116 |
| 29630 | IV | IV | IV | chr4LG4 | 1 | 2.3E-97 | 100 | 77,755,735 | 77,755,921 | 186 |
| 28304 | IV | IV | IV | chr4LG4 | 4 | 5.8E-64 | 99 | 77,763,197 | 77,763,328 | 131 |
| 27275 | IV | IV | IV | chr4LG4 | 5 | 1.1E-58 | 94 | 144,374,211 | 144,374,362 | 151 |
| 24036 | IV | IV | IV | chr4LG4 | 1 | 1.5E-102 | 100 | 298,729,278 | 298,729,473 | 195 |
| 19487 | IV | IV | IV | chr4LG4 | 2 | 3.6E-92 | 100 | 329,433,230 | 329,433,407 | 177 |
| 21622 | IV | IV | IV | chr4LG4 | 1 | 1.0E-69 | 100 | 411,138,053 | 411,138,191 | 138 |
| 24907 | IV | IV | IV | chr4LG4 | 1 | 7.5E-67 | 100 | 441,578,306 | 441,578,439 | 133 |
| 23829 | V | V | V | chr3LG5 | 4 | 2.8E-104 | 100 | 40,897,218 | 40,897,416 | 198 |
| 25618 | V | V | V | chr3LG5 | 10 | 8.7E-101 | 99 | 91,875,171 | 91,875,372 | 201 |
| 25986 | V | V | V | chr3LG5 | 1 | 9.0E-56 | 91 | 181,332,864 | 181,333,028 | 164 |
| 16914 | V | V | V | chr3LG5 | 1 | 2.2E-45 | 89 | 191,073,596 | 191,073,747 | 151 |
| 22325 | V | V | V | chr3LG5 | 1 | 5.4E-91 | 98 | 221,771,656 | 221,771,845 | 189 |
| 23759 | V | V | V | chr3LG5 | 1 | 4.4E-80 | 100 | 276,140,552 | 276,140,708 | 156 |
| 24326 | VI | VI | VI | chr1LG6 | 1 | 3.3E-96 | 100 | 30,662,379 | 30,662,563 | 184 |
| 25075 | VI | VI | VI | chr1LG6 | 1 | 1.0E-92 | 98 | 46,237,147 | 46,237,342 | 195 |
| 29246 | VI | VI | VI | chr1LG6 | 2 | 5.0E-72 | 100 | 63,898,496 | 63,898,638 | 142 |
| 23568 | VI | VI | VI | chr1LG6 | 1 | 7.9E-63 | 100 | 339,601,745 | 339,601,871 | 126 |
| 25888 | VI | VI | VI | chr1LG6 | 1 | 4.1E-84 | 100 | 342,366,936 | 342,367,099 | 163 |
| 23431 | VI | VI | VI | chr1LG6 | 1 | 3.5E-73 | 100 | 359,154,849 | 359,154,993 | 144 |
| 21405 | VII | VII | VII | chr7LG7 | 1 | 4.0E-47 | 90 | 23,151,033 | 23,151,166 | 133 |
| 17431 | VII | VII | VII | chr7LG7 | 2 | 7.5E-94 | 98 | 28,469,955 | 28,470,153 | 198 |
| 19979 | VII | VII | VII | chr7LG7 | 1 | 8.3E-71 | 97 | 33,247,383 | 33,247,539 | 156 |
| 17666 | VII | VII | VII | chr7LG7 | 1 | 8.1E-71 | 97 | 33,247,466 | 33,247,619 | 153 |
| 25965 | VII | VII | VII | chr7LG7 | 1 | 7.5E-49 | 89 | 79,270,351 | 79,270,512 | 161 |
| 17384 | VII | VII | VII | chr7LG7 | 1 | 4.0E-103 | 100 | 86,051,350 | 86,051,546 | 196 |
| 29468 | VII | VII | VII | chr7LG7 | 1 | 3.9E-58 | 95 | 196,186,432 | 196,186,576 | 144 |
| 24503 | VII | VII | VII | chr7LG7 | 4 | 2.6E-52 | 91 | 201,461,481 | 201,461,642 | 161 |
| 17910 | VII | VII | VII | chr7LG7 | 1 | 2.0E-68 | 97 | 329,043,834 | 329,043,990 | 156 |
| 17980 | VII | VII | VII | chr7LG7 | 1 | 1.9E-94 | 98 | 474,504,396 | 474,504,591 | 195 |
| 29248 | VII | VII | VII | chr7LG7 | 1 | 7.2E-75 | 99 | 481,640,880 | 481,641,037 | 157 |
| 21796 | VII | VII | VII | chr7LG7 | 1 | 2.7E-97 | 97 | 488,671,955 | 488,672,158 | 203 |
| 18272 | II | II | II | scaffold00024 | 1 | 9.2E-59 | 98 | 43,943 | 44,076 | 133 |
| 23262 | III | III | III | scaffold03071 | 1 | 4.7E-95 | 100 | 151,557 | 151,739 | 182 |
| 28387 | V | V | V | scaffold03509 | 2 | 2.0E-105 | 100 | 40,828 | 41,028 | 200 |
| 21399 | VII | VII | VII | scaffold00916 | 1 | 8.8E-67 | 96 | 9,846 | 10,002 | 156 |
| 29620 | VII | VII | VII | scaffold01087 | 12 | 1.1E-54 | 96 | 47,702 | 47,832 | 130 |
新窗口打开|下载CSV
3 讨论
3.1 SSR标记筛选
由于具有多态性高、多等位基因、共显性、可重复、可转移和基因组覆盖度高等特性, SSR标记包括基因组SSR和基于表达序列标签的EST-SSR已被广泛应用于种质资源鉴定评价、遗传变异检测、遗传进化分析、遗传连锁图谱构建、功能基因定位以及标记辅助育种等诸多领域[21,48-50]。通过对基因组SSR和EST-SSR的应用范围进行比较发现, 基因组SSR因为多态性较高更适合于区分遗传关系比较近的基因型, 并且在指纹图谱构建和种质资源鉴定研究中更具优势; 而从基因或表达序列标签数据生成的EST-SSR标记则有利于获得更多基因的相关信息, 在重要农艺性状的标记辅助选择中具有更大优势, 同时在近缘物种之间具有更高的可转移性[21,51]。以往的研究已经成功开发了一些SSR标记[10-12,38,44], 并用于遗传多样性评估[1,33,52-55], 遗传连锁图谱构建[10,15], 以及标记性状关联分析[32,56]等。然而, 豌豆作为具有重要经济价值和显著生态优势的食用豆类作物之一, 目前具有明确定位信息同时可公开获取的基因组SSR和EST-SSR标记数量还相对有限, 这极大地阻碍了豌豆的分子遗传研究和标记辅助育种。在本研究中, 我们利用大规模筛选从本实验室开发的12,491个SSR标记中得到了729个多态性SSR标记, 同时从125个已发表的具有明确遗传图谱定位信息的标记中筛选了25个锚定标记, 分别用于2个基于中国豌豆种质F2群体的遗传连锁图谱构建和整合。在最终得到的整合遗传图谱中, 上图标记达到668个, 包括509个基因组SSR、134个EST-SSR和25个锚定标记, 分布在7条连锁群上, 对应于豌豆的7条染色体, 相关标记信息详见附表1。这些已定位的SSR标记将为豌豆种质资源鉴定、遗传关系分析、分子遗传作图以及标记辅助选择提供宝贵资源。3.2 遗传连锁图谱构建
高密度遗传连锁图谱是进行遗传研究和分子育种的有力工具。针对豌豆的遗传连锁作图已经有很长的研究历史, 前人利用不同的分子标记和不同的作图群体已经构建了许多豌豆遗传连锁图谱[31]。随着二代测序技术的发展, 基因组SSR、EST-SSR和SNP标记的高通量开发为豌豆的分子作图奠定了重要基础[14-18,57]。最近, 有****还利用SRAP、SSR和SNP这3种标记基于F2群体构建了一张豌豆的遗传连锁图谱, 包含128个遗传标记, 分布在9个连锁群上, 然后他们利用9个SSR标记作为锚定标记, 将其中的6个连锁群与以往发表的遗传连锁图谱中的连锁群对应起来[58]。尽管在豌豆中已经构建了超过50张的遗传连锁图谱, 然而目前国际上还没有基于SSR标记构建的标记数目达500个以上的图谱, 因此豌豆SSR遗传连锁图谱无论在标记数目和密度上均有待完善[10,15]。此外, 前人的研究已经证明中国豌豆种质资源具有独特的遗传背景[1,32-33], 然而基于中国种质的豌豆遗传连锁图谱很少。过去的一项研究基于中国种质构建的豌豆遗传连锁图谱总长1518 cM, 仅包含157个SSR标记, 标记间平均距离为9.7 cM, 而且分布在11条连锁群上, 与豌豆的单倍体染色体数并不一致(2n = 2x = 14)[15]。在本研究中, 我们首先基于与以往研究[15]相同的作图群体(PSP1), 筛选了大量多态性SSR标记, 对以往的图谱进行加密。加密后的图谱累计长度扩展到5330.6 cM, 标记数目增加到603个, 标记间平均距离缩小为8.9 cM, 所有标记分布在7个连锁群上(图1和表2), 其中6条与以往发表的豌豆遗传图谱一一对应。此外, 我们还利用一个新的大样本作图群体(PSP2)构建了一张新的豌豆遗传连锁图谱, 并通过17个锚定标记将总共118个标记定位在与以前发表的豌豆遗传图谱完全一致的7条连锁群上(图2和表2)[10]。本研究基于中国豌豆种质构建的2个作图群体代表了与国外豌豆群体具有显著差异的基因组背景, 得到了2个SSR遗传连锁图谱, 与以往的研究相比[15], PSP1图谱在标记数目和密度上具有明显提高, 而PSP2在连锁群装配方面则具有明显改善, 这些图谱将为中国豌豆的标记辅助育种提供有力工具。3.3 整合遗传图谱
整合图谱凭借较高的标记数目和密度以及更完整的基因组覆盖范围等优势[34,35], 在许多作物包括豌豆中均有应用。过去几十年, 在豌豆遗传连锁作图悠久的研究历史中, 前人已经通过整合来自多个作图群体的信息而获得了很多复合遗传连锁图谱[10,16,31,36-37,42,59-61]。2015年, 法国科学家利用13.2K的基因芯片对12个豌豆RIL群体进行基因分型, 同时整合了以往图谱中的2277个标记, 构建了一个包含15,079个标记和7条连锁群的高密度一致性连锁图谱, 该图谱全长794.9 cM, 平均标记间距离0.24 cM, 为评估豌豆基因组结构、基因含量和进化历史以及候选基因鉴定提供了有力工具[16]。然而上述一致性图谱包含的EST-SSR和基因组SSR标记仅有187个, 为了在豌豆遗传连锁图谱上积累最大数量的SSR标记, 本研究通过53个共有标记将2个F2作图群体的数据合并构建了一个整合遗传连锁图谱, 覆盖范围为6592.6 cM, 包括668个SSR标记(509 基因组SSR、134 EST-SSR和25个锚定标记), 分布在7条连锁群上(图3和表3)。值得注意的是, 遗传连锁图谱的长度随标记数目的增多而延伸, 这种现象在包括豌豆在内的多个物种中均有发现, 有人推测可能与重组事件和偏分离有关[36-37,62-64]。此外, 有****认为延伸的图谱长度对图谱上标记的映射顺序几乎没有影响[61]。过去的研究发现, 由于多等位标记、数据缺失、偏分离和染色体重排等原因, 在比较不同杂交群体获得的遗传连锁图谱时, 会出现标记定位和顺序的不一致[10,36-37,65]。在本研究中, 通过对2个单独的遗传连锁图谱之间53个共有标记的比较发现, 基于2个群体构建的遗传图谱均可组装到7条连锁群上, 与以往研究中的7个连锁群具有较好的对应关系, 然而PSP1图谱的LGI-2 (包含LGI锚定标记AD147)对应于PSP2图谱的LGV (包含LGV锚定标记PSGAPA1), 导致整合图谱中LGV的不确定性(附图1)。但是由于PSP1图谱中7个连锁群有6个与以往发表的图谱一一对应, 仅剩1个连锁群(LGI-2)因为缺乏额外的锚定标记无法与以往发表的LGV相对应; 同时PSP1图谱中的LGI-2与PSP2图谱中的LGV之间存在7个共有标记, 基于上述两点原因, 我们推测LGI-2应该对应于以往发表图谱中的LGV。然后通过对50个共有标记的扩增片段序列的BLAST比对分析, 对3个遗传图谱和物理图谱进行共线性比较, 结果支持有争议的一条连锁群确实对应于以往遗传图谱中的LGV (附表2), 而该连锁群上的锚定标记AD147可能是异位所致。另一方面, 单独图谱和整合图谱共线性比较发现, 不同图谱在LGIV和LGVII这2个连锁群上的标记顺序具有高度一致性, 而在其他连锁群上则观察到标记的颠倒和错位(附图1), 推测这种不一致性可能是由不同作图群体中发生的染色体重排引起的[10,36-37]。不同遗传图谱与物理图谱的共线性比较得到了类似的结果(附图1和附表2), 物理图谱的chr4LG4和chr7LG7与3个遗传图谱在LGIV和LGVII这2个连锁群上的标记顺序具有较高的一致性, 说明这2条连锁群或染色体在不同群体的亲本间并未发生明显结构变异; 物理图谱中的chr2LG1、chr3LG5和chr1LG6与PSP2遗传图谱的LGI、LGV和LGVI基本一致, 而与PSP1遗传图谱的标记顺序全部或部分相反, 说明PSP1群体的亲本在这3条连锁群或染色体上可能发生了倒位; 物理图谱中的chr6LG2与PSP1遗传图谱LGII的标记顺序基本一致, 而与PSP2遗传图谱的标记顺序全部相反, 说明PSP2群体的亲本在这条连锁群或染色体上可能发生了倒位; 物理图谱中的chr5LG3与LGIII-Composite map的标记顺序一致, 而与PSP1和PSP2遗传图谱的标记顺序相反, 可能是参考基因组的豌豆基因型在该条染色体上发生了倒位或者错误组装。4 结论
利用2个基于中国豌豆种质的F2群体, 通过QTL IciMapping V4.0软件, 整合得到一张包含7条连锁群、668个SSR标记、总遗传距离为6592.6 cM、平均标记密度为10 cM的豌豆遗传连锁图谱。本研究将为豌豆的分子遗传研究和标记辅助育种提供有力工具。附表和附图 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuow xb.aspx。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1007/s00122-008-0887-zURL [本文引用: 4]

Twenty-one informative microsatellite loci were used to assess and compare the genetic diversity among Pisum genotypes sourced from within and outside China. The Chinese germplasm comprised 1243 P. sativum genotypes from 28 provinces and this was compared to 774 P. sativum genotypes that represented a globally diverse germplasm collection, as well as 103 genotypes from related Pisum species. The Chinese P. sativum germplasm was found to contain genotypes genetically distinct from the global gene pool sourced outside China. The Chinese spring type genotypes were separate from the global gene pool and from the other main Chinese gene pool of winter types. The distinct Chinese spring gene pool comprised genotypes from Inner Mongolia and Sha’anxi provinces, with those from Sha’anxi showing the greatest diversity. The other main gene pool within China included both spring types from other northern provinces and winter types from central and southern China, plus some accessions from Inner Mongolia and Sha’anxi. A core collection of Chinese landraces chosen to represent molecular diversity was compared both to the wider Chinese collection and to a geographically diverse core collection of Chinese landraces. The average gene diversity and allelic richness per locus of both the micro-satellite based core and the wider collection were similar, and greater than the geographically diverse core. The genetic diversity of P. sativum within China appears to be quite different to that detected in the global gene pool, including the presence of several rare alleles, and may be a useful source of allelic variation for both major gene and quantitative traits.]]>
DOI:10.3390/agronomy2020074URL [本文引用: 1]
DOI:10.1046/j.1365-2621.1999.00236.xURL [本文引用: 1]
DOI:10.4141/P00-114URL [本文引用: 1]
URL [本文引用: 1]
DOI:10.1016/j.agsy.2013.08.009URL [本文引用: 1]
[本文引用: 1]
DOI:10.1007/BF00222388URLPMID:24185876 [本文引用: 1]
An F2 population of pea (Pisum sativum L.) consisting of 174 plants was analysed by restriction fragment length polymorphism (RFLP) and random amplified polymorphic DNA (RAPD) techniques. Ascochyta pisi race C resistance, plant height, flowering earliness and number of nodes were measured in order to map the genes responsible for their variation. We have constructed a partial linkage map including 3 morphological character genes, 4 disease resistance genes, 56 RFLP loci, 4 microsatellite loci and 2 RAPD loci. Molecular markers linked to each resistance gene were found: Fusarium wilt (6 cM from Fw), powdery mildew (11 cM from er) and pea common Mosaic virus (15 cM from mo). QTLs (quantitative traits loci) for Ascochyta pisi race C resistance were mapped, with most of the variation explained by only three chromosomal regions. The QTL with the largest effect, on chromosome 4, was also mapped using a qualitative, Mendelian approach. Another QTL displayed a transgressive segregation, i.e. the parental line that was susceptible to Ascochyta blight had a resistance allele at this QTL. Analysis of correlations between developmental traits in terms of QTL effects and positions suggested a common genetic control of the number of nodes and earliness, and a loose relationship between these traits and height.
DOI:10.1007/s001220050971URL [本文引用: 1]
DOI:10.1007/s00122-005-0014-3URL [本文引用: 14]
This paper aims at providing reliable and cost effective genotyping conditions, level of polymorphism in a range of genotypes and map position of newly developed microsatellite markers in order to promote broad application of these markers as a common set for genetic studies in pea. Optimal PCR conditions were determined for 340 microsatellite markers based on amplification in eight genotypes. Levels of polymorphism were determined for 309 of these markers. Compared to data obtained for other species, levels of polymorphism detected in a panel of eight genotypes were high with a mean number of 3.8 alleles per polymorphic locus and an average PIC value of 0.62, indicating that pea represents a rather polymorphic autogamous species. One of our main objectives was to locate a maximum number of microsatellite markers on the pea genetic map. Data obtained from three different crosses were used to build a composite genetic map of 1,430cM (Haldane) comprising 239 microsatellite markers. These include 216 anonymous SSRs developed from enriched genomic libraries and 13 SSRs located in genes. The markers are quite evenly distributed throughout the seven linkage groups of the map, with 85% of intervals between the adjacent SSR markers being smaller than 10cM. There was a good conservation of marker order and linkage group assignment across the three populations. In conclusion, we hope this report will promote wide application of these markers and will allow information obtained by different laboratories worldwide in diverse fields of pea genetics, such as QTL mapping studies and genetic resource surveys, to be easily aligned.]]>
DOI:10.1111/pbr.2011.131.issue-1URL [本文引用: 2]
DOI:10.1111/pbr.2011.131.issue-1URL [本文引用: 2]
DOI:10.1186/1471-2229-13-161URLPMID:24134188 [本文引用: 1]
BACKGROUND: Field pea (Pisum sativum L.) is a self-pollinating, diploid, cool-season food legume. Crop production is constrained by multiple biotic and abiotic stress factors, including salinity, that cause reduced growth and yield. Recent advances in genomics have permitted the development of low-cost high-throughput genotyping systems, allowing the construction of saturated genetic linkage maps for identification of quantitative trait loci (QTLs) associated with traits of interest. Genetic markers in close linkage with the relevant genomic regions may then be implemented in varietal improvement programs. RESULTS: In this study, single nucleotide polymorphism (SNP) markers associated with expressed sequence tags (ESTs) were developed and used to generate comprehensive linkage maps for field pea. From a set of 36,188 variant nucleotide positions detected through in silico analysis, 768 were selected for genotyping of a recombinant inbred line (RIL) population. A total of 705 SNPs (91.7%) successfully detected segregating polymorphisms. In addition to SNPs, genomic and EST-derived simple sequence repeats (SSRs) were assigned to the genetic map in order to obtain an evenly distributed genome-wide coverage. Sequences associated with the mapped molecular markers were used for comparative genomic analysis with other legume species. Higher levels of conserved synteny were observed with the genomes of Medicago truncatula Gaertn. and chickpea (Cicer arietinum L.) than with soybean (Glycine max [L.] Merr.), Lotus japonicus L. and pigeon pea (Cajanus cajan [L.] Millsp.). Parents and RIL progeny were screened at the seedling growth stage for responses to salinity stress, imposed by addition of NaCl in the watering solution at a concentration of 18 dS m-1. Salinity-induced symptoms showed normal distribution, and the severity of the symptoms increased over time. QTLs for salinity tolerance were identified on linkage groups Ps III and VII, with flanking SNP markers suitable for selection of resistant cultivars. Comparison of sequences underpinning these SNP markers to the M. truncatula genome defined genomic regions containing candidate genes associated with saline stress tolerance. CONCLUSION: The SNP assays and associated genetic linkage maps developed in this study permitted identification of salinity tolerance QTLs and candidate genes. This constitutes an important set of tools for marker-assisted selection (MAS) programs aimed at performance enhancement of field pea cultivars.
DOI:10.1007/s00122-014-2375-yURL [本文引用: 2]
Pea (Pisum sativum L.) is one of the world's oldest domesticated crops and has been a model system in plant biology and genetics since the work of Gregor Mendel. Pea is the second most widely grown pulse crop in the world following common bean. The importance of pea as a food crop is growing due to its combination of moderate protein concentration, slowly digestible starch, high dietary fiber concentration, and its richness in micronutrients; however, pea has lagged behind other major crops in harnessing recent advances in molecular biology, genomics and bioinformatics, partly due to its large genome size with a large proportion of repetitive sequence, and to the relatively limited investment in research in this crop globally. The objective of this research was the development of a genome-wide transcriptome-based pea single-nucleotide polymorphism (SNP) marker platform using next-generation sequencing technology. A total of 1,536 polymorphic SNP loci selected from over 20,000 non-redundant SNPs identified using deep transcriptome sequencing of eight diverse Pisum accessions were used for genotyping in five RIL populations using an Illumina GoldenGate assay. The first high-density pea SNP map defining all seven linkage groups was generated by integrating with previously published anchor markers. Syntenic relationships of this map with the model legume Medicago truncatula and lentil (Lens culinaris Medik.) maps were established. The genic SNP map establishes a foundation for future molecular breeding efforts by enabling both the identification and tracking of introgression of genomic regions harbouring QTLs related to agronomic and seed quality traits.]]>
DOI:10.1016/j.cj.2014.03.004URL [本文引用: 8]
DOI:10.1111/tpj.13070URLPMID:26590015 [本文引用: 4]
Single nucleotide polymorphism (SNP) arrays represent important genotyping tools for innovative strategies in both basic research and applied breeding. Pea is an important food, feed and sustainable crop with a large (about 4.45 Gbp) but not yet available genome sequence. In the present study, 12 pea recombinant inbred line populations were genotyped using the newly developed GenoPea 13.2K SNP Array. Individual and consensus genetic maps were built providing insights into the structure and organization of the pea genome. Largely collinear genetic maps of 3918-8503 SNPs were obtained from all mapping populations, and only two of these exhibited putative chromosomal rearrangement signatures. Similar distortion patterns in different populations were noted. A total of 12 802 transcript-derived SNP markers placed on a 15 079-marker high-density, high-resolution consensus map allowed the identification of ohnologue-rich regions within the pea genome and the localization of local duplicates. Dense syntenic networks with sequenced legume genomes were further established, paving the way for the identification of the molecular bases of important agronomic traits segregating in the mapping populations. The information gained on the structure and organization of the genome from this research will undoubtedly contribute to the understanding of the evolution of the pea genome and to its assembly. The GenoPea 13.2K SNP Array and individual and consensus genetic maps are valuable genomic tools for plant scientists to strengthen pea as a model for genetics and physiology and enhance breeding.
DOI:10.1186/s12864-016-2447-2URLPMID:26892170 [本文引用: 1]
BACKGROUND: Progress in genetics and breeding in pea still suffers from the limited availability of molecular resources. SNP markers that can be identified through affordable sequencing processes, without the need for prior genome reduction or a reference genome to assemble sequencing data would allow the discovery and genetic mapping of thousands of molecular markers. Such an approach could significantly speed up genetic studies and marker assisted breeding for non-model species. RESULTS: A total of 419,024 SNPs were discovered using HiSeq whole genome sequencing of four pea lines, followed by direct identification of SNP markers without assembly using the discoSnp tool. Subsequent filtering led to the identification of 131,850 highly designable SNPs, polymorphic between at least two of the four pea lines. A subset of 64,754 SNPs was called and genotyped by short read sequencing on a subpopulation of 48 RILs from the cross 'Baccara' x 'PI180693'. This data was used to construct a WGGBS-derived pea genetic map comprising 64,263 markers. This map is collinear with previous pea consensus maps and therefore with the Medicago truncatula genome. Sequencing of four additional pea lines showed that 33 % to 64 % of the mapped SNPs, depending on the pairs of lines considered, are polymorphic and can therefore be useful in other crosses. The subsequent genotyping of a subset of 1000 SNPs, chosen for their mapping positions using a KASP assay, showed that almost all generated SNPs are highly designable and that most (95 %) deliver highly qualitative genotyping results. Using rather low sequencing coverages in SNP discovery and in SNP inferring did not hinder the identification of hundreds of thousands of high quality SNPs. CONCLUSIONS: The development and optimization of appropriate tools in SNP discovery and genetic mapping have allowed us to make available a massive new genomic resource in pea. It will be useful for both fine mapping within chosen QTL confidence intervals and marker assisted breeding for important traits in pea improvement.
DOI:10.1186/s12870-016-0956-4URLPMID:28193168 [本文引用: 2]
BACKGROUND: Marker-assisted breeding is now routinely used in major crops to facilitate more efficient cultivar improvement. This has been significantly enabled by the use of next-generation sequencing technology to identify loci and markers associated with traits of interest. While rich in a range of nutritional components, such as protein, mineral nutrients, carbohydrates and several vitamins, pea (Pisum sativum L.), one of the oldest domesticated crops in the world, remains behind many other crops in the availability of genomic and genetic resources. To further improve mineral nutrient levels in pea seeds requires the development of genome-wide tools. The objectives of this research were to develop these tools by: identifying genome-wide single nucleotide polymorphisms (SNPs) using genotyping by sequencing (GBS); constructing a high-density linkage map and comparative maps with other legumes, and identifying quantitative trait loci (QTL) for levels of boron, calcium, iron, potassium, magnesium, manganese, molybdenum, phosphorous, sulfur, and zinc in the seed, as well as for seed weight. RESULTS: In this study, 1609 high quality SNPs were found to be polymorphic between 'Kiflica' and 'Aragorn', two parents of an F6-derived recombinant inbred line (RIL) population. Mapping 1683 markers including 75 previously published markers and 1608 SNPs developed from the present study generated a linkage map of size 1310.1 cM. Comparative mapping with other legumes demonstrated that the highest level of synteny was observed between pea and the genome of Medicago truncatula. QTL analysis of the RIL population across two locations revealed at least one QTL for each of the mineral nutrient traits. In total, 46 seed mineral concentration QTLs, 37 seed mineral content QTLs, and 6 seed weight QTLs were discovered. The QTLs explained from 2.4% to 43.3% of the phenotypic variance. CONCLUSION: The genome-wide SNPs and the genetic linkage map developed in this study permitted QTL identification for pea seed mineral nutrients that will serve as important resources to enable marker-assisted selection (MAS) for nutritional quality traits in pea breeding programs.
DOI:10.3389/fpls.2018.00167URLPMID:29497430 [本文引用: 1]
Pisum fulvum, a wild relative of pea is an important source of allelic diversity to improve the genetic resistance of cultivated species against fungal diseases of economic importance like the pea rust caused by Uromyces pisi. To unravel the genetic control underlying resistance to this fungal disease, a recombinant inbred line (RIL) population was generated from a cross between two P. fulvum accessions, IFPI3260 and IFPI3251, and genotyped using Diversity Arrays Technology. A total of 9,569 high-quality DArT-Seq and 8,514 SNPs markers were generated. Finally, a total of 12,058 markers were assembled into seven linkage groups, equivalent to the number of haploid chromosomes of P. fulvum and P. sativum. The newly constructed integrated genetic linkage map of P. fulvum covered an accumulated distance of 1,877.45 cM, an average density of 1.19 markers cM(-1) and an average distance between adjacent markers of 1.85 cM. The composite interval mapping revealed three QTLs distributed over two linkage groups that were associated with the percentage of rust disease severity (DS%). QTLs UpDSII and UpDSIV were located in the LGs II and IV respectively and were consistently identified both in adult plants over 3 years at the field (Cordoba, Spain) and in seedling plants under controlled conditions. Whenever they were detected, their contribution to the total phenotypic variance varied between 19.8 and 29.2. A third QTL (UpDSIV.2) was also located in the LGIVand was environmentally specific as was only detected for DS % in seedlings under controlled conditions. It accounted more than 14% of the phenotypic variation studied. Taking together the data obtained in the study, it could be concluded that the expression of resistance to fungal diseases in P. fulvum originates from the resistant parent IFPI3260.
DOI:10.1038/s41588-019-0480-1URLPMID:31477930 [本文引用: 4]
We report the first annotated chromosome-level reference genome assembly for pea, Gregor Mendel's original genetic model. Phylogenetics and paleogenomics show genomic rearrangements across legumes and suggest a major role for repetitive elements in pea genome evolution. Compared to other sequenced Leguminosae genomes, the pea genome shows intense gene dynamics, most likely associated with genome size expansion when the Fabeae diverged from its sister tribes. During Pisum evolution, translocation and transposition differentially occurred across lineages. This reference sequence will accelerate our understanding of the molecular basis of agronomically important traits and support crop improvement.
DOI:10.1007/s10681-010-0286-9URL [本文引用: 4]
In recent years, molecular markers have been utilized for a variety of applications including examination of genetic relationships between individuals, mapping of useful genes, construction of linkage maps, marker assisted selections and backcrosses, population genetics and phylogenetic studies. Among the available molecular markers, microsatellites or simple sequence repeats (SSRs) which are tandem repeats of one to six nucleotide long DNA motifs, have gained considerable importance in plant genetics and breeding owing to many desirable genetic attributes including hypervariability, multiallelic nature, codominant inheritance, reproducibility, relative abundance, extensive genome coverage including organellar genomes, chromosome specific location and amenability to automation and high throughput genotyping. High degree of allelic variation revealed by microsatellite markers results from variation in number of repeat-motifs at a locus caused by replication slippage and/or unequal crossing-over during meiosis. In spite of limited understanding of the functions of the SSR motifs within the plant genes, SSRs are being widely utilized in plant genome analysis. Microsatellites can be developed directly from genomic DNA libraries or from libraries enriched for specific microsatellites. Alternatively, microsatellites can also be found by searching public databases such as GenBank and EMBL or through cross-species transferability. At present, EST databases are an important source of candidate genes, as these can generate markers directly associated with a trait of interest and may be transferable in close relative genera. A large number of SSR based techniques have been developed and a quantum of literature has accumulated regarding the applicability of SSRs in plant genetics and genomics. In this review we discuss the recent developments (last 4-5 years) made in plant genetics using SSR markers.
DOI:10.1590/1678-4685-GMB-2016-0027URLPMID:27561112 [本文引用: 1]
Microsatellites or Single Sequence Repeats (SSRs) are extensively employed in plant genetics studies, using both low and high throughput genotyping approaches. Motivated by the importance of these sequences over the last decades this review aims to address some theoretical aspects of SSRs, including definition, characterization and biological function. The methodologies for the development of SSR loci, genotyping and their applications as molecular markers are also reviewed. Finally, two data surveys are presented. The first was conducted using the main database of Web of Science, prospecting for articles published over the period from 2010 to 2015, resulting in approximately 930 records. The second survey was focused on papers that aimed at SSR marker development, published in the American Journal of Botany's Primer Notes and Protocols in Plant Sciences (over 2013 up to 2015), resulting in a total of 87 publications. This scenario confirms the current relevance of SSRs and indicates their continuous utilization in plant science.
DOI:10.1038/s41598-018-36877-0URLPMID:30626881 [本文引用: 1]
=0.9 for Ceriodaphnoa dubia, Asellus aquaticus, Daphnia magna, Daphnia pulex; r(2) >=0.8 for Hyalella azteca, Chironomus spec. larvae and Culex spec. larvae) to convert size measured on the spheroid counter to traditional, microscope based, length measurements, which follow the longest orientation of the body. Finally, we demonstrate semi-automated measurement of growth curves of individual daphnids (C. dubia and D. magna) over time and find that the quality of individual growth curves varies, partly due to methodological reasons. Nevertheless, this novel method could be adopted to other species and represents a step change in experimental throughput for measuring organisms' shape, size and growth curves. It is also a significant qualitative improvement by enabling high-throughput assessment of inter-individual variation of growth.]]>
DOI:10.7717/peerj.6419URLPMID:30805247 [本文引用: 1]
Salix psammophila (desert willow) is a shrub endemic to the Kubuqi Desert and the Mu Us Desert, China, that plays an important role in maintaining local ecosystems and can be used as a biomass feedstock for biofuels and bioenergy. However, the lack of information on phenotypic traits and molecular markers for this species limits the study of genetic diversity and population structure. In this study, nine phenotypic traits were analyzed to assess the morphological diversity and variation. The mean coefficient of variation of 17 populations ranged from 18.35% (branch angle (BA)) to 38.52% (leaf area (LA)). Unweighted pair-group method with arithmetic mean analysis of nine phenotypic traits of S. psammophila showed the same results, with the 17 populations clustering into five groups. We selected 491 genets of the 17 populations to analyze genetic diversity and population structure based on simple sequence repeat (SSR) markers. Analysis of molecular variance (AMOVA) revealed that most of the genetic variance (95%) was within populations, whereas only a small portion (5%) was among populations. Moreover, using the animal model with SSR-based relatedness estimated of S. psammophila, we found relatively moderate heritability values for phenotypic traits, suggesting that most of trait variation were caused by environmental or developmental variation. Principal coordinate and phylogenetic analyses based on SSR data revealed that populations P1, P2, P9, P16, and P17 were separated from the others. The results showed that the marginal populations located in the northeastern and southwestern had lower genetic diversity, which may be related to the direction of wind. These results provide a theoretical basis for germplasm management and genetic improvement of desert willow.
DOI:10.1007/s13258-019-00813-xURLPMID:30953340 [本文引用: 1]
BACKGROUND: In this study, we used phenotypic and genetic analysis to investigate Double haploid (DH) lines derived from normal corn parents (HF1 and 11S6169). DH technology offers an array of advantages in maize genetics and breeding as follows: first, it significantly shortens the breeding cycle by development of completely homozygous lines in two or three generations; and second, it simplifies logistics, including requiring less time, labor, and financial resources for developing new DH lines compared with the conventional RIL population development process. OBJECTIVES: In our study, we constructed a maize genetic linkage map using SSR markers and a DH population derived from a cross of normal corn (HF1) and normal corn (11S6169). METHODS: The DH population used in this study was developed by the following methods: we crossed normal corn (HF1) and normal corn (11S6169), which are parent lines of a normal corn cultivar, in 2014; and the next year, the F1 hybrids were crossed with a tropicalized haploid inducer line (TAIL), which is homozygous for the dominant marker gene R1-nj (Nanda and Chase in Crop Sci 6:213-215, 1966), and we harvested seeds of the haploid lines. RESULTS: A total of 200 SSR markers were assigned to 10 linkage groups that spanned 1145.4 cM with an average genetic distance between markers of 5.7 cM. 68 SSR markers showed Mendelian segregation ratios in the DH population at a 5% significance threshold. A total of 15 quantitative trait loci (QTLs) for plant height (PH), ear height (EH), ear height ratio (ER), leaf length (LL), ear length (EL), set ear length (SEL), set ear ratio (SER), ear width (EW), 100 kernel weight (100 KW), and cob color (CC) were found in the 121 lines in the DH population. CONCLUSION: The results of this study may help to improve the detection and characterization of agronomic traits and provide great opportunities for maize breeders and researchers using a DH population in maize breeding programs.
DOI:10.1111/pbr.2019.138.issue-2URL [本文引用: 1]
DOI:10.1111/pbr.2018.137.issue-6URL [本文引用: 1]
DOI:10.1007/s11032-018-0919-6URL [本文引用: 1]
DOI:10.1007/s12010-017-2643-9URLPMID:29082475 [本文引用: 1]
Euryale ferox is native to Southeast Asia and China, and it is one of the important aquatic food crops propagated mostly in eastern part of India. The aim of the present study was to characterize and evaluate the genetic diversity of ex situ collections of E. ferox germplasm from different geographical states of India using microsatellite (simple sequence repeats (SSRs)) markers. Ten SSR markers were analyzed to assess DNA fingerprinting and genetic diversity of 16 cultivated germplasm of E. ferox. Total 37 polymorphic alleles were recorded with an average of 3.7 allele frequency per primer. The polymorphic information content value varied from 0.204 to 0.735 with mean of 0.448. A high range of heterozygosity (Ho 0.228; He 0.512) was detected in the present study. The neighbor-joining (N-J) tree and the principle coordinate analysis showed that the germplasm divided in to three main clusters. The results of the present investigation comply that SSR markers are effective for computing genetic assessment of genetic diversity and similarity with classifying cultivated varieties of E. ferox. Evaluation of genetic diversity among Indian E. ferox germplasm could provide useful information for genetic improvement.
DOI:10.7717/peerj.4266URLPMID:29511604 [本文引用: 1]
Durian (Durio zibethinus) is one of the most popular tropical fruits in Asia. To date, 126 durian types have been registered with the Department of Agriculture in Malaysia based on phenotypic characteristics. Classification based on morphology is convenient, easy, and fast but it suffers from phenotypic plasticity as a direct result of environmental factors and age. To overcome the limitation of morphological classification, there is a need to carry out genetic characterization of the various durian types. Such data is important for the evaluation and management of durian genetic resources in producing countries. In this study, simple sequence repeat (SSR) markers were used to study the genetic variation in 27 durian types from the germplasm collection of Universiti Putra Malaysia. Based on DNA sequences deposited in Genbank, seven pairs of primers were successfully designed to amplify SSR regions in the durian DNA samples. High levels of variation among the 27 durian types were observed (expected heterozygosity, HE = 0.35). The DNA fingerprinting power of SSR markers revealed by the combined probability of identity (PI) of all loci was 2.3x10(-3). Unique DNA fingerprints were generated for 21 out of 27 durian types using five polymorphic SSR markers (the other two SSR markers were monomorphic). We further tested the utility of these markers by evaluating the clonal status of shared durian types from different germplasm collection sites, and found that some were not clones. The findings in this preliminary study not only shows the feasibility of using SSR markers for DNA fingerprinting of durian types, but also challenges the current classification of durian types, e.g., on whether the different types should be called
DOI:10.3389/fpls.2015.01037URLPMID:26640470 [本文引用: 3]
Pea (Pisum sativum L.) is an annual cool-season legume and one of the oldest domesticated crops. Dry pea seeds contain 22-25% protein, complex starch and fiber constituents, and a rich array of vitamins, minerals, and phytochemicals which make them a valuable source for human consumption and livestock feed. Dry pea ranks third to common bean and chickpea as the most widely grown pulse in the world with more than 11 million tons produced in 2013. Pea breeding has achieved great success since the time of Mendel's experiments in the mid-1800s. However, several traits still require significant improvement for better yield stability in a larger growing area. Key breeding objectives in pea include improving biotic and abiotic stress resistance and enhancing yield components and seed quality. Taking advantage of the diversity present in the pea genepool, many mapping populations have been constructed in the last decades and efforts have been deployed to identify loci involved in the control of target traits and further introgress them into elite breeding materials. Pea now benefits from next-generation sequencing and high-throughput genotyping technologies that are paving the way for genome-wide association studies and genomic selection approaches. This review covers the significant development and deployment of genomic tools for pea breeding in recent years. Future prospects are discussed especially in light of current progress toward deciphering the pea genome.
DOI:10.1038/s41598-017-06222-yURLPMID:28724947 [本文引用: 3]
Frost stress is one of the major abiotic stresses causing seedling death and yield reduction in winter pea. To improve the frost tolerance of pea, field evaluation of frost tolerance was conducted on 672 diverse pea accessions at three locations in Northern China in three growing seasons from 2013 to 2016 and marker-trait association analysis of frost tolerance were performed with 267 informative SSR markers in this study. Sixteen accessions were identified as the most winter-hardy for their ability to survive in all nine field experiments with a mean survival rate of 0.57, ranging from 0.41 to 0.75. Population structure analysis revealed a structured population of two sub-populations plus some admixtures in the 672 accessions. Association analysis detected seven markers that repeatedly had associations with frost tolerance in at least two different environments with two different statistical models. One of the markers is the functional marker EST1109 on LG VI which was predicted to co-localize with a gene involved in the metabolism of glycoproteins in response to chilling stress and may provide a novel mechanism of frost tolerance in pea. These winter-hardy germplasms and frost tolerance associated markers will play a vital role in marker-assisted breeding for winter-hardy pea cultivar.
DOI:10.2135/cropsci2015.07.0415URL [本文引用: 3]
DOI:10.1371/journal.pone.0028495URLPMID:22163026 [本文引用: 2]
BACKGROUND: Rye (Secale cereale L.) is an economically important crop, exhibiting unique features such as outstanding resistance to biotic and abiotic stresses and high nutrient use efficiency. This species presents a challenge to geneticists and breeders due to its large genome containing a high proportion of repetitive sequences, self incompatibility, severe inbreeding depression and tissue culture recalcitrance. The genomic resources currently available for rye are underdeveloped in comparison with other crops of similar economic importance. The aim of this study was to create a highly saturated, multilocus linkage map of rye via consensus mapping, based on Diversity Arrays Technology (DArT) markers. METHODOLOGY/PRINCIPAL FINDINGS: Recombinant inbred lines (RILs) from 5 populations (564 in total) were genotyped using DArT markers and subjected to linkage analysis using Join Map 4.0 and Multipoint Consensus 2.2 software. A consensus map was constructed using a total of 9703 segregating markers. The average chromosome map length ranged from 199.9 cM (2R) to 251.4 cM (4R) and the average map density was 1.1 cM. The integrated map comprised 4048 loci with the number of markers per chromosome ranging from 454 for 7R to 805 for 4R. In comparison with previously published studies on rye, this represents an eight-fold increase in the number of loci placed on a consensus map and a more than two-fold increase in the number of genetically mapped DArT markers. CONCLUSIONS/SIGNIFICANCE: Through the careful choice of marker type, mapping populations and the use of software packages implementing powerful algorithms for map order optimization, we produced a valuable resource for rye and triticale genomics and breeding, which provides an excellent starting point for more in-depth studies on rye genome organization.
DOI:10.1371/journal.pone.0045739URLPMID:23029214 [本文引用: 2]
A consensus genetic map of tetraploid cotton was constructed using six high-density maps and after the integration of a sequence-based marker redundancy check. Public cotton SSR libraries (17,343 markers) were curated for sequence redundancy using 90% as a similarity cutoff. As a result, 20% of the markers (3,410) could be considered as redundant with some other markers. The marker redundancy information had been a crucial part of the map integration process, in which the six most informative interspecific Gossypium hirsutumxG. barbadense genetic maps were used for assembling a high density consensus (HDC) map for tetraploid cotton. With redundant markers being removed, the HDC map could be constructed thanks to the sufficient number of collinear non-redundant markers in common between the component maps. The HDC map consists of 8,254 loci, originating from 6,669 markers, and spans 4,070 cM, with an average of 2 loci per cM. The HDC map presents a high rate of locus duplications, as 1,292 markers among the 6,669 were mapped in more than one locus. Two thirds of the duplications are bridging homoeologous A(T) and D(T) chromosomes constitutive of allopolyploid cotton genome, with an average of 64 duplications per A(T)/D(T) chromosome pair. Sequences of 4,744 mapped markers were used for a mutual blast alignment (BBMH) with the 13 major scaffolds of the recently released Gossypium raimondii genome indicating high level of homology between the diploid D genome and the tetraploid cotton genetic map, with only a few minor possible structural rearrangements. Overall, the HDC map will serve as a valuable resource for trait QTL comparative mapping, map-based cloning of important genes, and better understanding of the genome structure and evolution of tetraploid cotton.
DOI:10.1007/s11105-014-0837-7URL [本文引用: 5]
DOI:10.1007/s11032-015-0376-4URL [本文引用: 5]
DOI:10.1371/journal.pone.0139775URLPMID:26440522 [本文引用: 3]
Pea (Pisum sativum L.) is an important food legume globally, and is the plant species that J.G. Mendel used to lay the foundation of modern genetics. However, genomics resources of pea are limited comparing to other crop species. Application of marker assisted selection (MAS) in pea breeding has lagged behind many other crops. Development of a large number of novel and reliable SSR (simple sequence repeat) or microsatellite markers will help both basic and applied genomics research of this crop. The Illumina HiSeq 2500 System was used to uncover 8,899 putative SSR containing sequences, and 3,275 non-redundant primers were designed to amplify these SSRs. Among the 1,644 SSRs that were randomly selected for primer validation, 841 yielded reliable amplifications of detectable polymorphisms among 24 genotypes of cultivated pea (Pisum sativum L.) and wild relatives (P. fulvum Sm.) originated from diverse geographical locations. The dataset indicated that the allele number per locus ranged from 2 to 10, and that the polymorphism information content (PIC) ranged from 0.08 to 0.82 with an average of 0.38. These 1,644 novel SSR markers were also tested for polymorphism between genotypes G0003973 and G0005527. Finally, 33 polymorphic SSR markers were anchored on the genetic linkage map of G0003973 x G0005527 F2 population.
DOI:10.1007/s13258-011-0213-zURL [本文引用: 5]
DOI:10.1186/1471-2164-13-104URLPMID:22433453 [本文引用: 2]
BACKGROUND: Field pea (Pisum sativum L.) and faba bean (Vicia faba L.) are cool-season grain legume species that provide rich sources of food for humans and fodder for livestock. To date, both species have been relative 'genomic orphans' due to limited availability of genetic and genomic information. A significant enrichment of genomic resources is consequently required in order to understand the genetic architecture of important agronomic traits, and to support germplasm enhancement, genetic diversity, population structure and demographic studies. RESULTS: cDNA samples obtained from various tissue types of specific field pea and faba bean genotypes were sequenced using 454 Roche GS FLX Titanium technology. A total of 720,324 and 304,680 reads for field pea and faba bean, respectively, were de novo assembled to generate sets of 70,682 and 60,440 unigenes. Consensus sequences were compared against the genome of the model legume species Medicago truncatula Gaertn., as well as that of the more distantly related, but better-characterised genome of Arabidopsis thaliana L.. In comparison to M. truncatula coding sequences, 11,737 and 10,179 unique hits were obtained from field pea and faba bean. Totals of 22,057 field pea and 18,052 faba bean unigenes were subsequently annotated from GenBank. Comparison to the genome of soybean (Glycine max L.) resulted in 19,451 unique hits for field pea and 16,497 unique hits for faba bean, corresponding to c. 35% and 30% of the known gene space, respectively. Simple sequence repeat (SSR)-containing expressed sequence tags (ESTs) were identified from consensus sequences, and totals of 2,397 and 802 primer pairs were designed for field pea and faba bean. Subsets of 96 EST-SSR markers were screened for validation across modest panels of field pea and faba bean cultivars, as well as related non-domesticated species. For field pea, 86 primer pairs successfully obtained amplification products from one or more template genotypes, of which 59% revealed polymorphism between 6 genotypes. In the case of faba bean, 81 primer pairs displayed successful amplification, of which 48% detected polymorphism. CONCLUSIONS: The generation of EST datasets for field pea and faba bean has permitted effective unigene identification and functional sequence annotation. EST-SSR loci were detected at incidences of 14-17%, permitting design of comprehensive sets of primer pairs. The subsets from these primer pairs proved highly useful for polymorphism detection within Pisum and Vicia germplasm.
DOI:10.3732/ajb.1100445URL [本文引用: 5]
Methods and Results: Forty-one novel EST-SSR primers were developed and characterized for size polymorphism in 32 Pisum sativum individuals from four populations from China. In each population, the number of alleles per locus ranged from one to seven, with observed heterozygosity and expected heterozygosity ranging from 0 to 0.8889 and 0 to 0.8400, respectively. Furthermore, 53.7% of these markers could be transferred to the related species, Vicia faba.Conclusions: The developed markers have potential for application in the study of genetic diversity, germplasm appraisal, and marker-assisted breeding in pea and other legume species.]]>
[本文引用: 4]
DOI:10.1046/j.1439-0523.2001.00608.xURL [本文引用: 4]
DOI:10.1007/BF02712670URL [本文引用: 1]
DOI:10.1016/j.cj.2015.01.001URL [本文引用: 1]
DOI:10.1093/jhered/93.1.77URLPMID:12011185 [本文引用: 1]
DOI:10.1007/s00122-008-0902-4URL [本文引用: 1]
Genomic microsatellite markers are capable of revealing high degree of polymorphism. Sugarcane (Saccharum sp.), having a complex polyploid genome requires more number of such informative markers for various applications in genetics and breeding. With the objective of generating a large set of microsatellite markers designated as Sugarcane Enriched Genomic MicroSatellite (SEGMS), 6,318 clones from genomic libraries of two hybrid sugarcane cultivars enriched with 18 different microsatellite repeat-motifs were sequenced to generate 4.16Mb high-quality sequences. Microsatellites were identified in 1,261 of the 5,742 non-redundant clones that accounted for 22% enrichment of the libraries. Retro-transposon association was observed for 23.1% of the identified microsatellites. The utility of the microsatellite containing genomic sequences were demonstrated by higher primer designing potential (90%) and PCR amplification efficiency (87.4%). A total of 1,315 markers including 567 class I microsatellite markers were designed and placed in the public domain for unrestricted use. The level of polymorphism detected by these markers among sugarcane species, genera, and varieties was 88.6%, while cross-transferability rate was 93.2% within Saccharum complex and 25% to cereals. Cloning and sequencing of size variant amplicons revealed that the variation in the number of repeat-units was the main source of SEGMS fragment length polymorphism. High level of polymorphism and wide range of genetic diversity (0.16–0.82 with an average of 0.44) assayed with the SEGMS markers suggested their usefulness in various genotyping applications in sugarcane.]]>
In: Abdin M Z, Kiran U, Kamaluddin, Ali A, eds.
DOI:10.1080/13102818.2017.1400401URL [本文引用: 1]
DOI:10.1016/j.tibtech.2004.11.005URLPMID:15629858 [本文引用: 1]
Expressed sequence tag (EST) projects have generated a vast amount of publicly available sequence data from plant species; these data can be mined for simple sequence repeats (SSRs). These SSRs are useful as molecular markers because their development is inexpensive, they represent transcribed genes and a putative function can often be deduced by a homology search. Because they are derived from transcripts, they are useful for assaying the functional diversity in natural populations or germplasm collections. These markers are valuable because of their higher level of transferability to related species, and they can often be used as anchor markers for comparative mapping and evolutionary studies. They have been developed and mapped in several crop species and could prove useful for marker-assisted selection, especially when the markers reside in the genes responsible for a phenotypic trait. Applications and potential uses of EST-SSRs in plant genetics and breeding are discussed.
DOI:10.1007/s00122-008-0785-4URL [本文引用: 1]
One hundred and sixty-four accessions representing Czech and Slovak pea (Pisum sativum L.) varieties bred over the last 50years were evaluated for genetic diversity using morphological, simple sequence repeat (SSR) and retrotransposon-based insertion polymorphism (RBIP) markers. Polymorphic information content (PIC) values of 10 SSR loci and 31 RBIP markers were on average high at 0.89 and 0.73, respectively. The silhouette method after the Ward clustering produced the most probable cluster estimate, identifying nine clusters from molecular data and five to seven clusters from morphological characters. Principal component analysis of nine qualitative and eight quantitative morphological parameters explain over 90 and 93% of total variability, respectively, in the first three axes. Multidimensional scaling of molecular data revealed a continuous structure for the set. To enable integration and evaluation of all data types, a Bayesian method for clustering was applied. Three clusters identified using morphology data, with clear separation of fodder, dry seed and afila types, were resolved by DNA data into 17, 12 and five sub-clusters, respectively. A core collection of 34 samples was derived from the complete collection by BAPS Bayesian analysis. Values for average gene diversity and allelic richness for molecular marker loci and diversity indexes of phenotypic data were found to be similar between the two collections, showing that this is a useful approach for representative core selection.]]>
DOI:10.3724/SP.J.1006.2008.01330URL
利用21对豌豆多态性SSR引物, 对来自全国春、秋播区19省区市的1 221份豌豆地方品种进行遗传多样性分析, 共扩增出104条多态性带, 每对引物平均扩增出4.95个等位变异, 其中有效等位变异占62.52%。省份间SSR等位变异分布均匀, 但是省份间有效等位变异数、Shannon’s信息指数(I)差异明显, 省籍资源群间遗传多样性差异显著。遗传多样性以内蒙古资源群最高, 甘肃、四川、云南和西藏等资源群其次, 辽宁资源群最低。PCA三维空间聚类图揭示, 我国豌豆地方品种资源分化成3个基因库, 基因库I主要由春播区的内蒙古、陕西资源构成, 基因库II主要由秋播区最北端的河南资源构成, 基因库III主要由除上述省份之外的其他省区市的资源构成。UPGMA聚类分析表明, 不同省份资源群间的遗传距离变化范围为5.159~27.586, 中国豌豆地方资源据此聚类成2个组群8个亚组群, 与3个基因库的聚类结果相呼应。聚类结果显示, 我国豌豆地方品种资源群间遗传距离与其来源地生态环境相关联。
DOI:10.1016/S1671-2927(08)60208-4URL
DOI:10.3724/SP.J.1006.2008.01518URL [本文引用: 1]
Pisum sativum L.)进行遗传多样性分析与核心种质构建。共扩增出109条多态性带, 每对引物平均扩增出5.19个等位变异。SSR等位变异在各大洲间分布不均匀, 有效等位变异数、Shannon’s信息指数(I)洲际间差异明显。各大洲资源群间遗传多样性差异显著, 其中亚洲最高(I = 1.1753), 欧洲其次(I = 1.1387), 俄罗斯联邦(I = 1.0285)、美洲(I = 1.0196)、非洲(I = 0.9254)、大洋洲(I = 0.8608)依次降低。利用Popgene 1.32软件, 依豌豆栽培资源洲际间Nei78遗传距离可聚类成2个组群和4个亚组群; 基于Structure 2.2软件分析, 国外栽培豌豆资源实际由3大类群组成, 并与Popgene 1.32聚类结果呼应得较好。上述两种分析方法均表明, 国外栽培豌豆类群的遗传多样性与其地理分布相关。设计并实践了一套基于Structure分析的科学可靠、逻辑性强的核心种质构建标准化方案, 并依此构建了一套以6.57%的资源(48份)涵盖总体84.4%等位变异的国外栽培豌豆核心种质。]]>
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/1471-2164-15-126URL [本文引用: 1]
DOI:10.1007/s11032-019-0949-8URL [本文引用: 1]
DOI:10.1007/s00122-005-0205-yURL [本文引用: 1]
The identification of the molecular polymorphisms giving rise to phenotypic trait variability—both quantitative and qualitative—is a major goal of the present agronomic research. Various approaches such as positional cloning or transposon tagging, as well as the candidate gene strategy have been used to discover the genes underlying this variation in plants. The construction of functional maps, i.e. composed of genes of known function, is an important component of the candidate gene approach. In the present paper we report the development of 63 single nucleotide polymorphism markers and 15 single-stranded conformation polymorphism markers for genes encoding enzymes mainly involved in primary metabolism, and their genetic mapping on a composite map using two pea recombinant inbred line populations. The complete genetic map covers 1,458cM and comprises 363 loci, including a total of 111 gene-anchored markers: 77 gene-anchored markers described in this study, 7 microsatellites located in gene sequences, 16 flowering time genes, the Tri gene, 5 morphological markers, and 5 other genes. The mean spacing between adjacent markers is 4cM and 90% of the markers are closer than 10cM to their neighbours. We also report the genetic mapping of 21 of these genes in Medicago truncatula and add 41 new links between the pea and M. truncatula maps. We discuss the use of this new composite functional map for future candidate gene approaches in pea.]]>
DOI:10.1186/1471-2164-15-126URL
DOI:10.1007/s00122-014-2375-yURL [本文引用: 2]
Pea (Pisum sativum L.) is one of the world's oldest domesticated crops and has been a model system in plant biology and genetics since the work of Gregor Mendel. Pea is the second most widely grown pulse crop in the world following common bean. The importance of pea as a food crop is growing due to its combination of moderate protein concentration, slowly digestible starch, high dietary fiber concentration, and its richness in micronutrients; however, pea has lagged behind other major crops in harnessing recent advances in molecular biology, genomics and bioinformatics, partly due to its large genome size with a large proportion of repetitive sequence, and to the relatively limited investment in research in this crop globally. The objective of this research was the development of a genome-wide transcriptome-based pea single-nucleotide polymorphism (SNP) marker platform using next-generation sequencing technology. A total of 1,536 polymorphic SNP loci selected from over 20,000 non-redundant SNPs identified using deep transcriptome sequencing of eight diverse Pisum accessions were used for genotyping in five RIL populations using an Illumina GoldenGate assay. The first high-density pea SNP map defining all seven linkage groups was generated by integrating with previously published anchor markers. Syntenic relationships of this map with the model legume Medicago truncatula and lentil (Lens culinaris Medik.) maps were established. The genic SNP map establishes a foundation for future molecular breeding efforts by enabling both the identification and tracking of introgression of genomic regions harbouring QTLs related to agronomic and seed quality traits.]]>
DOI:10.1139/g96-061URLPMID:18469909 [本文引用: 1]
The paradigm that meiotic recombination and chiasmata have the same basis has been challenged, primarily for plants. High resolution genetic mapping frequently results in maps with lengths far exceeding those based on chiasma counts. In addition, recombination between specific homoeologous chromosomes derived from interspecific hybrids is sometimes much higher than can be explained by meiotic chiasma frequencies. However, almost the entire discrepancy disappears when proper care is taken of map inflation resulting from the shortcomings of the mapping algorithm and classification errors, the use of dissimilar material, and the difficulty of accurately counting chiasmata. Still, some exchanges, especially of short interstitial segments, cannot readily be explained by normal meiotic behaviour. Aberrant meiotic processes involving segment replacement or insertion can probably be excluded. Some cases of unusual recombination are somatic, possibly premeiotic exchange. For other cases, local relaxation of chiasma interference caused by small interruptions of homology disturbing synaptonemal complex formation is proposed as the cause. It would be accompanied by a preference for compensating exchanges (negative chromatid interference) resulting from asymmetry of the pairing chromatid pairs, so that one side of each pair preferentially participates in pairing. Over longer distances, the pairing face may switch, causing the normal random chromatid participation in double exchanges and the relatively low frequency of short interstitial exchanges. Key words : recombination frequency, map length, chiasmata, discrepancy, chromatid interference.
URLPMID:12399396
Several plant genetic maps presented in the literature are longer than expected from cytogenetic data. Here we compare F(2) and RI maps derived from a cross between the same two parental lines and show that excess heterozygosity contributes to map inflation. These maps have been constructed using a common set of dominant markers. Although not generally regarded as informative for F(2) mapping, these allowed rapid map construction, and the resulting data analysis has provided information not otherwise obvious when examining a population from only one generation. Segregation distortion, a common feature of most populations and marker systems, found in the F(2) but not the RI, has identified excess heterozygosity. A few markers with a deficiency of heterozygotes were found to map to linkage group V (chromosome 3), which is known to form rod bivalents in this cross. Although the final map length was longer for the F(2) population, the mapped order of markers was generally the same in the F(2) and RI maps. The data presented in this analysis reconcile much of the inconsistency between map length estimates from chiasma counts and genetic data.
[本文引用: 1]
URLPMID:1551583 [本文引用: 1]
We have analyzed segregation patterns of markers among the late generation progeny of several crosses of pea. From the patterns of association of these markers we have deduced linkage orders. Salient features of these linkages are discussed, as is the relationship between the data presented here and previously published genetic and cytogenetic data.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}