 ,1,2,*
,1,2,*Proteomic analysis of drought stress response on drought resistance for Vicia faba L. variety ‘Qinghai 13’ in Qinghai Plateau of China
LI Ping1,2, HOU Wan-Wei1,2, LIU Yu-Jiao,1,2,*通讯作者:
第一联系人:
收稿日期:2018-05-29接受日期:2018-10-8网络出版日期:2018-11-03
| 基金资助: |
Received:2018-05-29Accepted:2018-10-8Online:2018-11-03
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (2437KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李萍, 侯万伟, 刘玉皎. 青海高原耐旱蚕豆品种青海13号响应干旱胁迫蛋白质组学分析[J]. 作物学报, 2019, 45(2): 267-275. doi:10.3724/SP.J.1006.2019.84075
LI Ping, HOU Wan-Wei, LIU Yu-Jiao.
干旱是胁迫因素中对植物的生长发育影响最大的因子之一, 而对农作物造成的损更是严重[1], 在众多非生物胁迫中占首位。有****在关于植物的抗性研究中指出在水分胁迫下植物能够产生一系列包括形态、生理生化及分子生物学等多方面的变化而表现出抗旱能力, 这些适应性调节反应是植物自身的保护性选择[2,3]。蛋白质组学是研究生物体在特定时间或空间内所表达的全部蛋白质的特征, 是揭示植物与胁迫之间相互作用分子机制的一种新手段。随着蛋白质组学的发展和植物蛋白质组学研究的日趋深入, 以通过研究植物在非生物逆境胁迫过程中抗逆相关蛋白质的表达而进一步了解逆境胁迫的伤害机制及植物的逆境适应机制的逆境蛋白质组学研究成为热点[4,5,6]。双向凝胶电泳是蛋白质组学研究的一个重要支撑技术, 结合质谱技术, 仍是目前最常用和可靠的蛋白质分离鉴定技术平台, 涉及豆科作物逆境胁迫后蛋白质组的变化, 屡有对获得的差异蛋白进行功能研究的报道。Mohammadi等[7]对干旱胁迫和PEG渗透胁迫处理的大豆幼苗研究发现, 胁迫处理下叶片中代谢相关蛋白表达上调, 能量和蛋白质合成相关蛋白下调表达, 叶片、根、下胚轴这3个器官中蛋氨酸合成酶的mRNA与蛋白水平均下调, 蛋氨酸合成酶是一种干旱反应蛋白, 其下调表达抑制了大豆幼苗的正常生长。蛋白质组学在菜豆干旱胁迫[8]、冷胁迫[9]和紫花苜蓿[10]盐胁迫、箭舌豌豆[11]化学胁迫等方面得以广泛应用, 为深入开展豆科作物抗逆蛋白质组学研究提供了参考和借鉴。本课题组前期对水分胁迫下蚕豆叶片蛋白质组变化初探发现, 胁迫后的叶片差异蛋白中下调表达的占大多数且主要与胁迫防御、代谢和能量、细胞骨架以及氧化平衡有关, 上调表达的相对较少, 主要参与蛋白折叠与聚集以及光合系统[12]。
蚕豆(Vicia faba L.)属蝶形花科野豌豆属, 是世界上重要的豆科作物, 也是青海高原重要的粮食、蔬菜、副食、饲料和养地兼有作物, 在该省农业经济生产和生态保护中占有重要的地位。本研究采用人工控水模拟干旱胁迫, 研究叶片蛋白质水平的响应, 发掘与蚕豆抵御干旱胁迫相关蛋白, 为后续的抗旱基因克隆和抗逆分子育种工作提供有力技术支持和理论基础。
1 材料与方法
1.1 试验材料及处理
青海13号是农业生产中表现强抗旱性的蚕豆品种, 由青海省农林科学院提供。挑选色泽正常无病斑的种子, 经10%次氯酸溶液消毒及蒸馏水漂洗, 置玻璃培养皿中25℃恒温光照培养催芽, 24 h后挑选发芽一致的蚕豆种子种植在抗旱棚中, 播种4周后待蚕豆幼苗长至二叶一心时进行不同梯度人工控水模拟干旱胁迫处理。第1天, 对第10排蚕豆(DS9)停止供水, 由其蒸腾作用开始进行自然干旱; 第2天, 对第9排蚕豆(DS8)停止供水开始自然干旱; 以此类推, 直到第2排蚕豆(DS1)开始干旱胁迫, 以正常供水为对照(CK)。1.2 IEF/SDS-PAGE双向电泳技术及质谱分析
参照改良后的TCA/丙酮法[13], 略有改进, 提取叶片总蛋白。第一向IEF采用17 cm, pH 3~10 (NL)固相胶条, 上样量900 μg; 第二向采用12%的聚丙烯酰胺分离胶进行SDS-PAGE电泳。电泳结束后以考马斯亮蓝染色经UMAX Power look 2100 XL型光密度扫描仪扫描, 以300 dpi分辨率采集图像。采用PD Quest 8.0.1软件比较处理组和对照组的叶片总蛋白2-DE图谱, 通过差异蛋白点检测, 凝胶图谱标准化处理, 蛋白质点匹配和生物统计, 确定差异表达的蛋白质点, 实验重复3次数据进行t-test检验, 倍数变化>2或<0.5为差异蛋白。挖取差异表达蛋白质点送上海鹿明生物有限公司进行MALDI- TOF/TOF串联质谱分析, 采用ABI5800串联飞行时间质谱仪进行质谱点靶鉴定, 获得的PMF的质谱扫描范围为800~3500 Da, 对选择强度最大的10个峰进行二级质谱, 将一级和二级质谱数据整合并使用GPS 3.6 (Applied Biosystems)和Mascot2.3(Matrix Science)对质谱数据进行分析和蛋白鉴定。进行NCBInr-Other green plants数据库检索, 消化多肽的酶为胰蛋白酶, 允许最大 切位点为1, 固定修饰为Carbamidomethyl (C), 可变修饰为Acetyl (Protein N-term)、Deamidated (NQ)、Dioxidation (W)和Oxidation (M), MS tolerance为0.0001 Da, MS/MS tolerance为0.3 Da, Protein score C.I.%大于95%为鉴定成功。2 结果与分析
2.1 干旱胁迫最适时期确定
土壤含水量(SWC)是表征干旱胁迫程度的一个重要的正相关指标。通过图1可以看出, SWC呈显著下降趋势, 从42.67% (SWCCK)下降到6.7% (SWCDS9),与对照相比各处理间差异极显著, 说明本实验采用的不同干旱胁迫梯度设置成功, 从DS1到DS9干旱胁迫程度逐渐加强。其中, DS3的SWC下降迅速, 出现一个“折点”, 胁迫继续增加, DS4和DS5与DS6至DS9胁迫间差异不显著。由此可见, 可在后续蚕豆抗旱性研究中直接选择DS3作为干旱胁迫的最适处理期。植物叶片相对含水量(RWC)是衡量植物持水能力强弱的重要指标, 在一定程度上体现植物抗旱能力。由图1可以看出, 随着胁迫时间的延长, 蚕豆叶片RWC呈下降趋势, 但与正常供水对照相比, 直到干旱胁迫DS4时其RWC含量才与CK差异显著。值得注意的是, 在干旱胁迫处理下, SWC在DS3处理期极显著减小(P<0.01)的情况下蚕豆叶片的RWC与对照相比在0.05水平上无显著差异, 这也是实验材料青海13号具有抗旱性的一个生理表现, 选择此材料非常具有代表性。图1
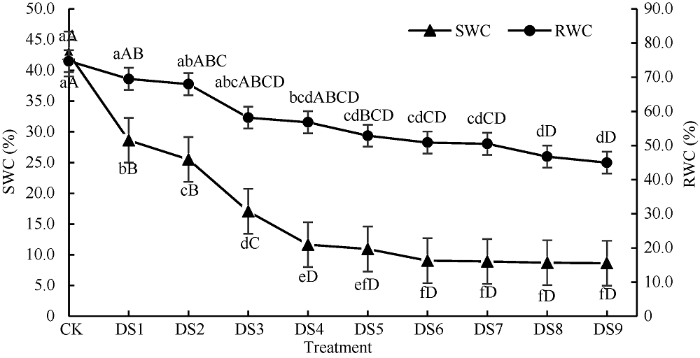 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1不同梯度干旱胁迫下土壤含水量(SWC)和蚕豆叶片的相对含水量(RWC)变化
大写字母表示不同胁迫处理间0.01水平上显著性差异, 小写字母表示0.05水平上差异显著。DS1~DS9为不同梯度干旱胁迫处理, 从DS1到DS9干旱胁迫程度逐渐加强。
Fig. 1Change of SWC and RWC under different drought stress levels
The letter denotes differences between stress degree: the same letters indicate significant difference at the 0.05 probability level; Capital letters indicate significant difference at the 0.01 probability level. DS1-DS9 are drought stress treatments and the drought stress degrees gradually strengthened from DS1 to DS9.
2.2 双向电泳图谱构建和质谱分析
DS3胁迫处理是适宜的胁迫条件, 故取此处理时期的蚕豆叶片进行下一步分析。利用本课题组前期建立的改良后的TCA/丙酮法分别提取对照组和处理组叶片总蛋白进行双向电泳, 经考马斯亮蓝染色后获得最终电泳图谱(图2)。图2
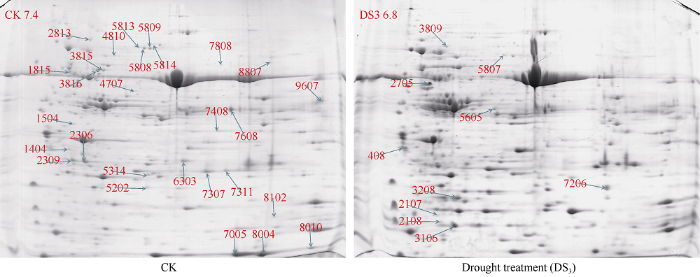 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2干旱胁迫下蚕豆叶片蛋白质组双向电泳图谱
Fig. 22-DE profile of proteins from leaves of broad bean under drought stress
应用PD Quest8.0.1软件对凝胶图谱进行分析, 结合人工筛选, t-检测, 共有38个蛋白点的表达发生显著变化(Fold>2.0或<0.5, P<0.05)。处理组相对于正常对照组有10 个差异蛋白点的相对表达量上调,占总差异蛋白点的26.3%, 其中特异表达的差异蛋白1个(3809)。而28个差异蛋白点的相对表达量下调, 占总差异蛋白点的73.7%, 其中消失的蛋白点7个。挖取蚕豆叶片2-DE凝胶对差异蛋白点进行MALDI-TOF-TOF质谱分析, 通过MASCOT数据库对获得的数据信息进行搜索比对, Mowse Score大于65分的蛋白即为鉴定的蛋白。通过分析成功鉴定了38个差异蛋白点中的27个, 鉴定率为71.1%, 这些成功鉴定的蛋白点的具体信息列于表1。鉴定结果表明, 有多个蛋白点被鉴定为一个相同蛋白, 如叶绿素a/b结合蛋白AB80 (gi|115788: 蛋白点2036和2309), ATP合酶CF1 (gi|528749836: 蛋白点3815和3816), 质体转酮醇酶(gi|357445031: 蛋白点5808和5809), 碳酸酐酶叶绿体X1 (gi|502090577: 蛋白点6303、7307和7311), 核酮糖-二磷酸羧化小链3C (gi|132097: 蛋白点7005和8004), 最后27个成功鉴定的蛋白代表了21个非冗余的蛋白质。
Table 1
表1
表1干旱胁迫下蚕豆叶片中差异表达蛋白的MALDI-TOF/TOF鉴定结果
Table 1
| 蛋白点 Spot No. | 登录号 Accession No. | 分子量/等电点 Theoretical Mr (kD)/pI | 序列覆盖率 Sequence coverage (%) | 评分 Score | 蛋白名称 Protein name | 物种来源 Source of species |
|---|---|---|---|---|---|---|
| A, up-regulated protein spots | ||||||
| 2705 | gi|15667623 | 15.923/5.78 | 18 | 230 | Drought inducible 22 kD protein | 甘蔗Saccharum officinarum |
| 3106 | gi|380005612 | 20.731/5.79 | 31 | 717 | Superoxide dismutase | 蚕豆Vicia faba |
| 3208 | gi|83776798 | 16.256/5.82 | 19 | 332 | 17.5 kDa class I HSP, partial | 花生Arachis hypogaea |
| 3809 | gi|308810206 | 85.783/8.69 | 1 | 54 | Shikimate dehydrogenase substrate binding,N-terminal, partial (ISS) | 绿藻类Ostreococcus tauri |
| 5605 | gi|357508933 | 39.616/5.91 | 9 | 158 | O-acetylserine (thiol) lyase | 蒺藜状苜蓿Medicago truncatula |
| 5807 | gi|357481949 | 65.668/5.7 | 5 | 237 | Stress-inducible protein, putative | 蒺藜状苜蓿Medicago truncatula |
| 7206 | gi|75220301 | 23.908/6.16 | 13 | 212 | Full=Kunitz-type trypsin inhibitor-like 2 protein; | 豌豆Pisum sativum |
| 408 | gi|571556750 | 21.914/4.36 | 35 | 493 | Nascent polypeptide-associated complex subunit alpha-like protein 1 | 大豆Glycine max |
| B, down-regulated protein spots | ||||||
| 1815 | gi|3913031 | 56.446/5.34 | 13 | 387 | Full=1,4-alpha-D-glucan maltohydrolase | 紫花苜蓿Medicago sativa |
| 2306 | gi|115788 | 28.692/5.47 | 25 | 411 | Full=Chlorophyll a-b binding protein AB80, chloroplastic; | 豌豆Pisum sativum |
| 2309 | gi|115788 | 28.692/5.47 | 25 | 415 | Full=Chlorophyll a-b binding protein AB80, chloroplastic; | 豌豆Pisum sativum |
| 2813 | gi|1045394920 | 86.972/5.46 | 13 | 643 | Hypothetical protein TSUD_183880 | 三叶草Trifolium subterraneum |
| 3815 | gi|528749836 | 55.812/5.22 | 20 | 659 | ATP synthase CF1 alpha subunit (plastid) | 蚕豆Vicia faba |
| 3816 | gi|528749836 | 55.812/5.22 | 20 | 760 | ATP synthase CF1 alpha subunit (plastid) | 蚕豆Vicia faba |
| 4707 | gi|75308025 | 43.565/5.5 | 17 | 323 | Full=S-adenosylmethionine synthase 2; Short=AdoMet synthase 2; | 茱萸Elaeagnus umbellata |
| 5202 | gi|217071344 | 31.249/6.59 | 8 | 309 | Unknown | 蒺藜状苜蓿Medicago truncatula |
| 5314 | gi|729390274 | 29.386/8.65 | 16 | 426 | Chlorophyll a-b binding protein 8, chloroplastic-like | 醉蝶花Tarenaya hassleriana |
| 5808 | gi|357445031 | 80.087/6 | 11 | 480 | Plastid transketolase | 蒺藜状苜蓿Medicago truncatula |
| 5809 | gi|357445031 | 80.087/5.78 | 11 | 532 | Plastid transketolase | 蒺藜状苜蓿Medicago truncatula |
| 6303 | gi|502090577 | 37.493/7.04 | 25 | 570 | Carbonic anhydrase, chloroplastic isoform X1 | 鹰嘴豆Cicer arietinum |
| 7005 | gi|132097 | 20.402/9.24 | 39 | 354 | Full=Ribulose bisphosphate carboxylase small chain 3C, chloroplastic; Short=RuBisCO small subunit 3C; AltName: Full=PSS15; Flags: Precursor | 豌豆Pisum sativum |
| 7307 | gi|502090577 | 37.493/7.04 | 31 | 723 | Carbonic anhydrase, chloroplastic isoform X1 | 鹰嘴豆Cicer arietinum |
| 7311 | gi|502090577 | 37.493/7.04 | 31 | 735 | Carbonic anhydrase, chloroplastic isoform X1 | 鹰嘴豆Cicer arietinum |
| 7608 | gi|20729 | 43.696/8.93 | 24 | 847 | Unnamed protein product | 豌豆Pisum sativum |
| 8004 | gi|132097 | 20.402/9.24 | 31 | 448 | Full=Ribulose bisphosphate carboxylase small chain 3C, chloroplastic; Short=RuBisCO small subunit 3C; AltName: Full=PSS15; Flags: Precursor | 豌豆Pisum sativum |
| 8010 | gi|502082899 | 19.055/9.93 | 20 | 219 | Photosystem II repair protein PSB27-H1, chloroplastic | 鹰嘴豆Cicer arietinum |
| 1404 | gi|502133626 | 37.498/7.63 | 29 | 541 | Haloacid dehalogenase-like hydrolase domain-containing protein At3g48420 | 鹰嘴豆Cicer arietinum |
新窗口打开|下载CSV
2.3 差异蛋白的功能及生物信息学分析
根据鉴定出来的21个差异蛋白所参与的代谢途径和生化功能将其分为7类(图3)。(1)参与信号转导的2个蛋白, 占所有蛋白的10%, 分别为22 kD干旱诱导蛋白(drought inducible 22 kD protein, spot 2705)和应激诱导蛋白(stress-inducible protein, spot 5807)。(2)参与自由基清除的1个蛋白, 超氧化物歧化酶(superoxide dismutase, spot 3106)。(3)参与防卫反应的1个蛋白, 17.5 kD一级热激蛋白(17.5 kD classⅠHSPza, partial, spot 3208)。(4)参与代谢的8个蛋白占到所有蛋白的38%, 属于最大一类, 分别为莽草酸酯脱氢酶(Shikimate dehydrogenase substrate binding, N-terminal, partial (ISS), spot 3809)、乙酰丝氨酸裂解酶(0-acetylserin(thiol)lyase, spot 5605)、胰蛋白酶抑制剂(Kunitz-type trypsin inhibitor-like 2 protein, spot 7206)、1,4-前体D-麦芽糖水解酶(1,4-alpha- D-glucan maltohydrolase, spot 1815)、ATP合酶CF1 (ATP synthase CF1 alpha subunit, spot 3815)、S-腺苷甲硫氨酸合酶(S-adenosylmethionine synthase 2, spot 4707)、质体转酮醇酶(plastid transketolase, spot 5808)和脱卤素酶水解酶(haloacid dehalogenase-like hydrolase domain-containing protein At3g48420, spot 1404)。(5)参与蛋白加工的1个蛋白, nascent肽-关联复合体亚基蛋白I (nascent polypeptide-associated complex subunit alpha-like protein 1, spot 408)。(6)参与光合的5个蛋白, 分别是叶绿体a/b结合蛋白AB80 (chlorophyll a/b binding protein AB80, spot 2306)、叶绿体a/b结合蛋白8 (chlorophyll a/b binding protein 8, chloroplastic-like, spot 5314)、RuBisCO小亚基3C (Ribulose bisphosphate carboxylase small chain 3C, spot 7005)、碳酸酐酶、叶绿体同型X1 (carbonic anhydrase, chloroplastic isoform X1, spot 7307)和光合系统II修补蛋白PSB27-H1 (photosystem II repair protein PSB27-H1, spot 8010)。(7)未知功能蛋白3个, 假定蛋白TSUD-183880 (hypothetical protein TSUD_183880, spot 2813)、未知蛋白(gi|217071344, spot 5202)和未命名的蛋白(unnamed protein product, spot 7608)。图3
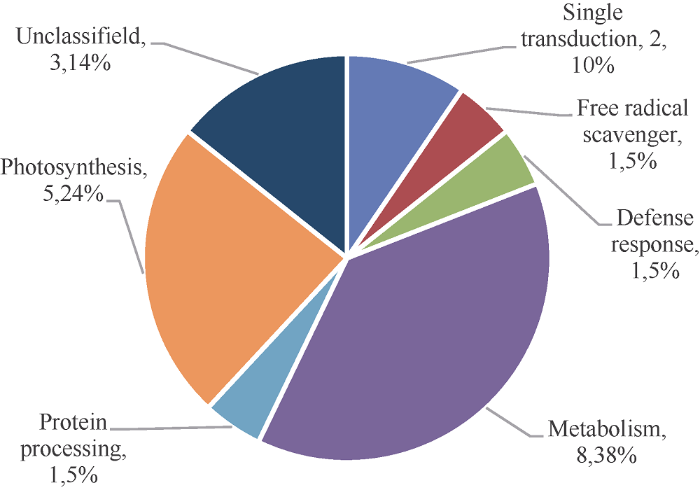 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3蚕豆叶片中鉴定的21个差异蛋白的功能分类
Fig. 3Functional classification of the 21 differential protein spots identified from broad bean
基于主流的数据库David 6.7和QuickGO (http://www.ebi.ac.uk/QuickGO/)将21个差异表达蛋白点进行质谱搜库检索得到ID, 同源映射到模式植物拟南芥上进行GO (gene ontology)富集差异表达蛋白生物信息学分析(图4)。从图中可以看出, 这21个差异蛋白在生物过程富集分析中参与了20个生物学过程, 主要参与光合系统、有机化合物代谢、有机物生物合成、响应非生物刺激等生物过程; 在细胞定位富集中可以定位到类囊体、细胞质、细胞器部分、胞内细胞器、核糖核蛋白复合物、外部包装结构、蛋白酶体核心复合物7个细胞中, 其中在细胞质、细胞器部分及胞内细胞器富集蛋白百分比均超过50%; 在分子功能富集分析中, 21个差异蛋白在未折叠蛋白、前蛋白、核苷磷酸、核苷酸、核苷、核糖核苷酸、同类蛋白、几丁质、热休克蛋白等的结合及转移酶活性、阴离子配位和异构酶活性中发挥功能, 其中参与核苷磷酸结合和核苷酸结合的蛋白富集百分比均为38.2%, 参与核苷结合和核糖核苷酸结合的蛋白富集百分比均为31.1%, 而参与阴离子配位的蛋白富集百分比为34.6%。
图4
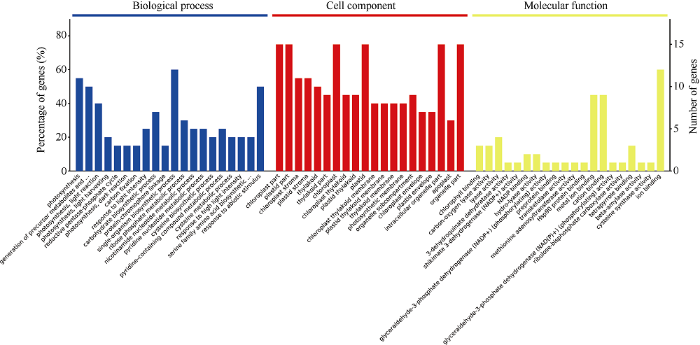 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4差异蛋白GO功能分类
Fig. 4GO Functional category distribution of difference expressed proteins
3 讨论
植物在遭受干旱等逆境胁迫后基因表达发生改变,表现出原来一些蛋白的合成受到抑制, 也会新合成一些蛋白[14,15], 它们是植物抗旱一系列生理生化反应的基础, 因此分析胁迫前后蛋白组的变化对从蛋白质水平了解植物的抗逆机制是很有帮助的[16,17,18]。比起蛋白的增加或新蛋白的出现, 干旱更容易引起蛋白含量的减少或丢失[19], 本研究中双向电泳分析结果也得出了相同的结论, 在分离到的38个差异蛋白点中, 下调表达蛋白数量高于上调表达蛋白, 其中消失的蛋白有7个(spot 1504、spot 5202、spot 5314、spot 5814、spot 7311、spot 7808、spot 8102),说明干旱胁迫抑制了蚕豆植物体某些蛋白的合成来提高对干旱环境的适应性, 亦或提高一些蛋白的表达量(8个上调表达蛋白), 同时又诱导产生一些新的蛋白如莽草酸之脱氢酶(Shikimate dehydrogenase substrate binding, N-terminal, partial (ISS), spot 3809), 从而提高蚕豆植株对干旱的耐受能力。利用MALDI-TOF/TOF串联质谱分析技术成功鉴定了21个非冗余的干旱响应相关差异蛋白, 涉及信号转导、防卫反应、光合作用、能量代谢、自由基清除、蛋白加工等过程, 还有部分获得的蛋白为未知功能蛋白。本研究中参与信号转导的Drought inducible 22 kD protein (spot 2705)和stress-inducible protein (spot 5807)等蛋白表达丰度均上调, 推测蛋白酶体的增加有利于清除在干旱胁迫条件下产生的非正常蛋白。Desclos等[20]发现油菜干旱诱导的22 kD蛋白同时具有水溶性叶绿素结合蛋白活性和胰蛋白酶抑制剂活性, 它能通过保持蛋白的完整性和正常的光合作用保护油菜幼嫩组织不受逆境的影响。这说明Drought inducible 22 kD protein和Stress-inducible protein表达量的上升对青海13号蚕豆的抗旱性发挥了重要的作用。在植物应对非生物胁迫中活性氧(ROS)伤害植物的膜系统和体内大分子, 是新陈代谢的有毒副产物, 而超氧化物歧化酶(SOD)是可解除胁迫条件下植物体内产生的多余活性氧分子的一类重要的活性氧清除酶, 此类表达量的增加使植物减少或免除胁迫对植物的伤害, 提高其抗逆性[21,22]。我们发现, 干旱胁迫下超氧化物歧化酶(superoxide dismutase, spot 3106)被诱导表达, 这与豌豆[23]、水稻[24]等作物中的研究结果一致, 是青海13号蚕豆品种对干旱胁迫的一种应激性生理响应, 推测这也是青海13号具有强抗旱性的原因所在。热激蛋白(heat shock protein, HSP)是干旱胁迫物质形成的必需因子, 表达量的升高是植物适应胁迫的一种机制,有利于维持胁迫调节下蛋白的稳定性和维持植物生存与生长, 可以提高植株的抗逆能力, 在胁迫中的作用己经被广泛地研究[25,26]。研究者Sato和Yokoya[27]发现超表达sHSP17.7基因可提高干旱处理的水稻植株抗旱性, 在水稻叶片蛋白质组分析中也发现干旱胁迫调节多个热激蛋白上调表达[28,29]。本研究中发现干旱胁迫使17.5 kD一级热激蛋白(spot 3208)表达上调, 这说明青海13号蚕豆在干旱胁迫下通过诱导热激蛋白的增加来对逆境适应起保护作用这也是该品种具有抗旱性的原因所在。氧乙酰丝氨酸裂解酶(0-acetylserin(thiol)lyase)是半胱氨酸合成最后一步的关键酶, 在半胱氨酸的合成调节中占据核心地位, 植物体内的一些含硫化合物(如谷胱甘肽)可通过一些生化反应途径淬灭这些游离基团, 从而提高植物体的抗逆性, 我们在干旱胁迫处理下发现了差异上调表达蛋白中包括此蛋白(spot 5605)。干旱诱导蛋白是指植物在受到干旱胁迫时新合成或合成增多的一类蛋白, 干旱诱导蛋白的形成是植物抵御干旱胁迫的主动保护机制[30]。本研究发现在分离鉴定的众多差异蛋白中, 莽草酸酯脱氢酶(shikimate dehydrogenase substrate binding, N-terminal, partial (ISS), spot 3809)是新产生的一类干旱诱导蛋白。莽草酸途径是存在于植物中的一条重要的代谢途径, 与植物应对环境刺激相关联, 而莽草酸酯脱氢酶是一类促进莽草酸途径的关键性酶之一[31,32], 我们推测此干旱诱导蛋白对青海13号蚕豆的抗旱性有贡献。植物抵抗环境胁迫的一个重要机制是调节基因的表达, 严顺平[24]在水稻抗逆性研究中推测新生肽结合蛋白复合物的α亚基(α-NAC)可能参与了水稻抗逆的基因表达的调节。也有证据表明新生肽结合复合物的α亚基可以作为转录的激活子[33], 在我们的研究中α-NAC (spot 408)在干旱胁迫下表达量上调, 这可能影响了整个NAC的功能, 维持胁迫下基因转录、蛋白质翻译和定位的正常进行, 保持了蚕豆正常的生理功能, 表现出了抗旱性。
光合作用是绿色植物产生糖类等各种有机物的起始反应,是蚕豆产量构成的唯一来源, 对于干旱、盐渍等很多非生物胁迫来说都是一个非常敏感的生物学过程[34,35], 所以在本研究中发现干旱胁迫处理引起许多参与光合作用的蛋白表达发生变化并不奇怪。我们通过蛋白质组学的分析发现,很多参与光合作用的蛋白在干旱胁迫下被降解或者差异表达, 这也表明了光合作用对干旱胁迫信号也是非常敏感的。Chlorophyll a/b binding protein AB80 (spot2306)、chlorophyll a/b binding protein 8 (spot 5314)和photosystem II repair protein PSB27-H1 (spot 8010)都是与光合系统密切相关的蛋白, 蛋白表达的下降也验证了在干旱胁迫下, 植物叶片的光合作用受到抑制。ATP合酶CFI亚基(spot 3815)是与能量代谢相关的酶, 这些ATP合酶亚基在干旱胁迫下被降解将不可避免地影响光合磷酸化产生ATP进一步影响光合作用的卡尔文循环。在干旱胁迫条件下, 植物通过改变体内的初级代谢如碳的能量代谢使其达到新的平衡状态[36], 本研究发现参与糖代谢途径的磷酸戊糖途径的关键酶, 转酮醇酶(transketolase, TK, spot 5808)在干旱胁迫下表达量下调, 说明干旱胁迫下磷酸戊糖途径受到抑制。S-腺苷甲硫氨酸合酶2 (SAMS2)是植物合成木质素的关键酶, 其下降表达表明木质素以及其他相关的次生代谢产物的合成下降, 以利于减少不必要的能量和物质的消耗。本研究中干旱胁迫下SAMS2 (spot 4707)蛋白表达下调, 前人在研究水稻响应盐胁迫和干旱胁迫的蛋白质组变化时也发现SAMS2的蛋白水平被下调[37], 推测此蛋白也与青海13号蚕豆抗旱性有关联。
4 结论
在干旱胁迫下, 青海 13 号蚕豆叶片与信号转导、自由基清除、防卫反应、能量代谢及蛋白加工等相关的蛋白主要表现为上调表达中发现了一个新的干旱胁迫响应蛋白莽草酸酯脱氢酶(shikimate dehydrogenase substrate binding, N-terminal, partial (ISS), spot 3809), 推测是青海13号蚕豆具有抗旱性机制所在。这些为我们深入了解蚕豆对干旱胁迫的响应提供了新的线索, 也是利用遗传等手段进一步研究这些基因功能的开始。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1023/B:COLL.0000023121.03238.e8URLPMID:17921517 [本文引用: 1]

Abstract Dehydration or water-deficit is one of the most important environmental stress factors that greatly influences plant growth and development and limits crop productivity. Plants respond and adapt to such stress by altering their cellular metabolism and activating various defense machineries. Mechanisms that operate signal perception, transduction, and downstream regulatory events provide valuable information about the underlying pathways involved in environmental stress responses. The nuclear proteins constitute a highly organized, complex network that plays diverse roles during cellular development and other physiological processes. To gain a better understanding of dehydration response in plants, we have developed a comparative nuclear proteome in a food legume, chickpea (Cicer arietinum L.). Three-week-old chickpea seedlings were subjected to progressive dehydration by withdrawing water and the changes in the nuclear proteome were examined using two-dimensional gel electrophoresis. Approximately 205 protein spots were found to be differentially regulated under dehydration. Mass spectrometry analysis allowed the identification of 147 differentially expressed proteins, presumably involved in a variety of functions including gene transcription and replication, molecular chaperones, cell signaling, and chromatin remodeling. The dehydration responsive nuclear proteome of chickpea revealed a coordinated response, which involves both the regulatory as well as the functional proteins. This study, for the first time, provides an insight into the complex metabolic network operating in the nucleus during dehydration.
DOI:10.1104/pp.100.3.1486URLPMID:16653148 [本文引用: 1]
Under progressive drought stress, Brassica napus displays differential leaf modifications. The oldest leaves, developed before the onset of water deficit, wilt gradually, whereas the youngest leaves harden. Hardening was distinguished by leaf turgor and bluish wax bloom when the shoot water potential was below -3 MPa and the leaf water saturation deficit was about 60%. This adaptive change was accompanied by modifications in two-dimensional protein profiles. Ten percent of the polypeptides had altered abundance or were unique to drought-stressed plants. Two-dimensional analysis of in vitro translation products did not reveal a general decrease in mRNA population. A 22-kD double polypeptide was increased by progressive or rapid water stress and salinity and disappeared upon rehydration. These polypeptides have a common N-terminal sequence, which does not reveal homology with any known water-stress protein but which contains the signature motif of soybean Kunitz trypsin inhibitors. Immunoprecipitation allowed these polypeptides to be identified on two-dimensional gels of in vitro translation products. They appeared to be synthesized as a 24-kD precursor, and their transcript was present in the control well-watered leaves, where the polypeptides were never detected, indicating a possible translational regulation. A putative function of this protein, named BnD22, in the retardation of drought-induced leaf senescence is discussed.
DOI:10.1146/annurev.arplant.47.1.377URL [本文引用: 1]
DOI:10.1023/A:1024711428760URL [本文引用: 1]
Host range and pathogenicity of a range of Aphanomyces spp. isolates obtained from pea roots but also from a range of other field-grown leguminous crops in southern Sweden was investigated. The Aphanomyces euteiches isolates originating from pea and the few obtained isolates originating from alfalfa, green bean and yellow sweet-clover were highly pathogenic only to pea. The A. euteiches isolated from common vetch differed from these isolates by being weakly pathogenic to pea and other legumes, but highly pathogenic to common vetch. Vetch isolates also formed a well-defined separate cluster based on principal component analysis of pathogenicity pattern on tested crops. Oospores of A. euteiches were observed in root tissue of pea as well as common vetch, alfalfa, green bean, broad bean, red clover and yellow sweet-clover in the greenhouse pathogenicity tests. An Aphanomyces sp. that morphologically differed from A. euteiches , was frequently isolated from several leguminous plants, but was non-pathogenic to all tested crops in the pathogenicity tests. In isozyme analysis the banding pattern of these isolates resembled the pattern of A. cladogamus. Another, different and so far unidentified Aphanomyces sp. from roots of green bean and broad bean, was also non-pathogenic to the tested legume species. Based on the isolates tested, the results obtained suggest that the population of Aphanomyces spp. infecting legume roots in Sweden consists of a pea-specific and a vetch-specific group of A. euteiches . Two other groups comprised (i) Aphanomyces sp. isolates that resembled A. cladogamus , and (ii) isolates, which resembled neither A. euteiches nor A. cladogamus . In addition, the host range of Swedish A. euteiches isolates was not as broad as reported for A. euteiches isolates from other countries.
DOI:10.1016/j.jprot.2012.03.055URLPMID:22516432 [本文引用: 1]
78 Translational proteomics is an emerging sub-discipline of the proteomics field. 78 Translational plant proteomics has already existed for a long time though not by name. 78 We have defined translational plant proteomics. 78 Translational plant proteomics is driven by advances in plant proteomics. 78 We envisage global engagement of the plant proteomics scientific community.
DOI:10.3724/SP.J.1006.2017.01337URL [本文引用: 1]
采用桶栽方式,对抗旱性强的F172和抗旱性弱的YL6甘蔗品种在苗期进行重度干旱胁迫处理后,应用蛋白质双向电泳技术进行差异蛋白质分析,分别找出差异显著的28和20个差异蛋白点,其中部分呈现上调表达,部分呈现下调表达,还有部分新增的蛋白点,因品种抗性不同而表现各异,F172叶片中的差异蛋白主要表现为上调表达,而YL6中大多表现为下调表达。在重度干旱胁迫下,抗旱性不同的甘蔗品种蛋白质丰度变化有显著差异。采用MALDI-TOF-TOF/MS鉴定所获得的差异蛋白,从YL6、F172中分别鉴定出18、14个蛋白的氨基酸序列,对所鉴定的蛋白质根据功能分为8类。YL6中参与自由基清除的2个,参与光合作用的6个,参与细胞生长和分裂的1个,参与基础代谢的6个,参与防卫反应的2个,未知功能蛋白1个。F172中参与自由基清除的1个,参与光合作用的2个,参与细胞生长和分裂的2个,参与基础代谢的4个,参与信号转导的2个,参与蛋白加工的1个,未知功能蛋白2个,其中22 k D干旱诱导蛋白的丰度明显提高,而在YL6中则检测不到此蛋白。这说明在干旱胁迫下抗旱性不同的甘蔗品种在蛋白质组成上有很大差异,推测这是不同甘蔗品种间抗旱性差异的重要分子基础。
DOI:10.3724/SP.J.1006.2017.01337URL [本文引用: 1]
采用桶栽方式,对抗旱性强的F172和抗旱性弱的YL6甘蔗品种在苗期进行重度干旱胁迫处理后,应用蛋白质双向电泳技术进行差异蛋白质分析,分别找出差异显著的28和20个差异蛋白点,其中部分呈现上调表达,部分呈现下调表达,还有部分新增的蛋白点,因品种抗性不同而表现各异,F172叶片中的差异蛋白主要表现为上调表达,而YL6中大多表现为下调表达。在重度干旱胁迫下,抗旱性不同的甘蔗品种蛋白质丰度变化有显著差异。采用MALDI-TOF-TOF/MS鉴定所获得的差异蛋白,从YL6、F172中分别鉴定出18、14个蛋白的氨基酸序列,对所鉴定的蛋白质根据功能分为8类。YL6中参与自由基清除的2个,参与光合作用的6个,参与细胞生长和分裂的1个,参与基础代谢的6个,参与防卫反应的2个,未知功能蛋白1个。F172中参与自由基清除的1个,参与光合作用的2个,参与细胞生长和分裂的2个,参与基础代谢的4个,参与信号转导的2个,参与蛋白加工的1个,未知功能蛋白2个,其中22 k D干旱诱导蛋白的丰度明显提高,而在YL6中则检测不到此蛋白。这说明在干旱胁迫下抗旱性不同的甘蔗品种在蛋白质组成上有很大差异,推测这是不同甘蔗品种间抗旱性差异的重要分子基础。
DOI:10.1016/j.jprot.2011.12.041URLPMID:22245419 [本文引用: 1]
Changes in protein levels in drought-stressed soybean seedlings were analyzed using a proteomics approach. Three-day-old soybean seedlings were subjected to drought stress or treated with 10% polyethylene glycol (PEG) as osmotic stress. After treatment, the proteins were extracted from the leaf, hypocotyl, and root and separated using two-dimensional polyacrylamide gel electrophoresis. The root was the most drought-responsive organ, with the levels of 32, 13, and 12 proteins changing in response to drought stress, PEG treatment, and both, respectively. In the leaves of PEG-treated and drought-stressed seedlings, metabolism-related proteins increased and energy production- and protein synthesis-related proteins decreased. For 3 proteins present in all organs in drought-stressed plants, mRNA was differentially regulated: heat shock protein 70 and actin isoform B were upregulated, and methionine synthase was downregulated. mRNA expression patterns reflected those of protein levels, suggesting transcriptional regulation of these proteins. Western blot analysis confirmed the increase in ascorbate peroxidase in drought-stressed plants. The downregulation of mRNA and decreased protein levels of methionine synthase in the leaves, hypocotyl, and roots of drought-stressed plants, but not in other treatments, indicated that methionine synthase is a drought response protein. These results also suggest that the decreased methionine synthase in response to drought stress can impair the soybean seedling growth.Highlights? Metabolism-related proteins increased in the roots than in other organs of drought-stressed plants. ? HSP 70 and actin isoform B, and methionine synthase were regulated by transcriptional level under drought. ? Western blot analysis confirmed the increase in ascorbate peroxidase in drought-stressed plants. ? Decreased methionine synthase in response to drought stress can impair soybean seedling growth.
DOI:10.1016/j.jprot.2012.09.021URLPMID:23026550 [本文引用: 1]
78 2D-DIGE approach is used to reveal changes in protein levels of common bean under drought. 78 Leaves of two cultivars with contrasting response to drought are analyzed. 78 Proteins with different biological functions are identified. 78 A protein–protein interaction network is proposed for both cultivars.
DOI:10.1016/j.jplph.2013.10.020URLPMID:24594390 [本文引用: 1]
Plants respond to different environmental cues in a complex way, entailing changes at the cellular and physiological levels. An important step to understand the molecular foundation of stress response in plants is the analysis of stress-responsive proteins. In this work we attempted to investigate and compare changes in the abundance of proteins in the roots of bean (Phaseolus vulgaris L.) germinating under long continuous chilling conditions (10 C, 16 days), exposed to short rapid chilling during germination (10 C, 24h), as well as subjected to recovery from stress (25 C, 24h). The results we obtained indicate that germination under continuous chilling causes alterations in the accumulation of the proteins involved in stress response, energy production, translation, vesicle transport, secondary metabolism and protein degradation. The subsequent recovery influences the accumulation of the proteins implicated in calcium-dependent signal transduction pathways, secondary metabolism and those promoting cell division and expansion. Subjecting the germinating bean seeds to short rapid chilling stress resulted in a transient changes in the relative content of the proteins taking part in energy production, DNA repair, RNA processing and translation. Short stress triggers also the mechanisms of protection against oxidative stress and promotes expression of anti-stress proteins. Subjecting bean seeds to the subsequent recovery influences the abundance of the proteins involved in energy metabolism, protection against stress and production of phytohormones. The exposure to long and short chilling did not result in the alterations of any proteins common to both treatments. The same situation was observed with respect to the recovery after stresses. Bean response to chilling is therefore strongly correlated with the manner and length of exposure to low temperature, which causes divergent proteomic alterations in the roots.
DOI:10.14088/j.cnki.issn0439-8114.2015.21.057URL [本文引用: 1]
6 d龄紫花苜蓿(Mesicago sativa L.)幼苗在0、200 mmol/L NaCl处理9 d后,采用双向电泳分别分离了其根部蛋白组. 最终每块胶中有超过900个蛋白质点被分离,采用MALDI-TOF-TOF/MS质谱技术结合MASCOT软件在线检索,成功鉴定21个表达量发生1.5倍以上变化的蛋白质点,为19种不同的蛋白质. 生物信息学分析表明,这些差异表达蛋白质主要参与胁迫防御、碳水化合物和能量代谢、次生代谢、转录和翻译调控、信号传导和离子转运等调控途径.
DOI:10.14088/j.cnki.issn0439-8114.2015.21.057URL [本文引用: 1]
6 d龄紫花苜蓿(Mesicago sativa L.)幼苗在0、200 mmol/L NaCl处理9 d后,采用双向电泳分别分离了其根部蛋白组. 最终每块胶中有超过900个蛋白质点被分离,采用MALDI-TOF-TOF/MS质谱技术结合MASCOT软件在线检索,成功鉴定21个表达量发生1.5倍以上变化的蛋白质点,为19种不同的蛋白质. 生物信息学分析表明,这些差异表达蛋白质主要参与胁迫防御、碳水化合物和能量代谢、次生代谢、转录和翻译调控、信号传导和离子转运等调控途径.
URL [本文引用: 1]
为探讨箭舌豌豆根响应镉胁迫的分子机理,以两个镉耐性差异的箭舌豌豆品种(镉耐性品种‘L3’,镉敏感品种‘ZM’)为材料,通过双向电泳和基质辅助激光解析/电离时间飞行质谱技术以及NCBI数据库搜索,分析25μmol·L~(-1) CdCl_2处理5 d后两个品种根蛋白质的差异表达。结果表明,镉胁迫下‘L3’和‘ZM’根共有56个蛋白点的表达量发生显著变化,其中5个蛋白点在两个品种根中共同上调,1个蛋白点共同下调,4个蛋白点为镉处理后两个品种中共同新出现。28个差异表达蛋白点得到鉴定,这些蛋白质的功能可分为七类:(1)抗氧化与解毒相关蛋白;(2)呼吸与能量代谢相关蛋白;(3)胁迫响应相关蛋白;(4)信号转导相关蛋白;(5)细胞壁合成相关蛋白;(6)细胞增殖相关蛋白;(7)未知功能蛋白。镉胁迫下耐性品种‘L3’较敏感品种‘ZM’鉴定到更多抗氧化解毒、胁迫响应和细胞增殖蛋白的上调表达,这可能是耐性品种‘L3’较敏感品种‘ZM’具有更强镉耐性的重要原因;生长素、水杨酸和乙烯诱导的信号转导途径可能参与箭舌豌豆对镉胁迫的响应。
URL [本文引用: 1]
为探讨箭舌豌豆根响应镉胁迫的分子机理,以两个镉耐性差异的箭舌豌豆品种(镉耐性品种‘L3’,镉敏感品种‘ZM’)为材料,通过双向电泳和基质辅助激光解析/电离时间飞行质谱技术以及NCBI数据库搜索,分析25μmol·L~(-1) CdCl_2处理5 d后两个品种根蛋白质的差异表达。结果表明,镉胁迫下‘L3’和‘ZM’根共有56个蛋白点的表达量发生显著变化,其中5个蛋白点在两个品种根中共同上调,1个蛋白点共同下调,4个蛋白点为镉处理后两个品种中共同新出现。28个差异表达蛋白点得到鉴定,这些蛋白质的功能可分为七类:(1)抗氧化与解毒相关蛋白;(2)呼吸与能量代谢相关蛋白;(3)胁迫响应相关蛋白;(4)信号转导相关蛋白;(5)细胞壁合成相关蛋白;(6)细胞增殖相关蛋白;(7)未知功能蛋白。镉胁迫下耐性品种‘L3’较敏感品种‘ZM’鉴定到更多抗氧化解毒、胁迫响应和细胞增殖蛋白的上调表达,这可能是耐性品种‘L3’较敏感品种‘ZM’具有更强镉耐性的重要原因;生长素、水杨酸和乙烯诱导的信号转导途径可能参与箭舌豌豆对镉胁迫的响应。
URL [本文引用: 1]
对1种抗旱性蚕豆品种“尕大豆’,进行幼苗期抗旱性研究。以正常供水为对照,测定了不同胁迫时间(7d,14d)、不同胁迫程度(轻度LS,重度WS)下幼苗形态及生理生化指标,并对胁迫条件7d、WS处理下的蚕豆叶片进行差异蛋白组学分析。结果表明,水分胁迫降低了“尕大豆”幼苗的地上部分和地下部分的生长,14项形态及生理指标在不同胁迫程度(LS,WS)、不同胁迫历时(7d,14d)下的表现各有差异。其中,在长历时胁迫下,植株地下部分(根长,侧根数)和CAT含量出现增长趋势,且达到极显著水平(p〈0.01)。这正是“尕大豆”表现抗旱性的原因。对7d、WS胁迫处理下的叶片进行双向电泳差异蛋白组学分析,经t检验发现了50个差异表达蛋白点(表达量〉2或〈0.5)。采用MALDI.TOF/TOF分析,30个蛋白点成功鉴定(25个下调,5个上调)。对所鉴定的蛋白质按其所参与的代谢途径和生化功能分为5类,其中调节蛋白占到所有蛋白的46.7%,属于最大一类,代谢和能量占到23.3%,功能未知仅占3.3%,为最小一类。从功能分类上,可以得出下降表达的蛋白主要和胁迫防御、代谢和能量、细胞骨架以及氧化平衡有关,而上升表达的蛋白主要参与了蛋白折叠与聚集以及光合系统。
URL [本文引用: 1]
对1种抗旱性蚕豆品种“尕大豆’,进行幼苗期抗旱性研究。以正常供水为对照,测定了不同胁迫时间(7d,14d)、不同胁迫程度(轻度LS,重度WS)下幼苗形态及生理生化指标,并对胁迫条件7d、WS处理下的蚕豆叶片进行差异蛋白组学分析。结果表明,水分胁迫降低了“尕大豆”幼苗的地上部分和地下部分的生长,14项形态及生理指标在不同胁迫程度(LS,WS)、不同胁迫历时(7d,14d)下的表现各有差异。其中,在长历时胁迫下,植株地下部分(根长,侧根数)和CAT含量出现增长趋势,且达到极显著水平(p〈0.01)。这正是“尕大豆”表现抗旱性的原因。对7d、WS胁迫处理下的叶片进行双向电泳差异蛋白组学分析,经t检验发现了50个差异表达蛋白点(表达量〉2或〈0.5)。采用MALDI.TOF/TOF分析,30个蛋白点成功鉴定(25个下调,5个上调)。对所鉴定的蛋白质按其所参与的代谢途径和生化功能分为5类,其中调节蛋白占到所有蛋白的46.7%,属于最大一类,代谢和能量占到23.3%,功能未知仅占3.3%,为最小一类。从功能分类上,可以得出下降表达的蛋白主要和胁迫防御、代谢和能量、细胞骨架以及氧化平衡有关,而上升表达的蛋白主要参与了蛋白折叠与聚集以及光合系统。
DOI:10.7666/d.y1333475URL [本文引用: 1]
蛋白质组学研究在功能基因组时代发挥着越来越重要的作用。双向电泳(2-DE)技术是其关键技术之一,具有重复性好、极敏感和分辨率高等特点。马铃薯(Solanum tuberosum L.)是世界上第四大粮食作物,对其块茎的发生发育过程进行研究,了解其块茎发育的机理,将为马铃薯的栽培、育种和生产提供理论依据。马铃薯块茎的发育是一个复杂而又有规律的过程,块茎蛋白质的数量、种类、丰度等在不同发育阶段存在着一定的差异,而这些差异蛋白质又参与基因的表达和块茎某些重要性状的调控。因此,对马铃薯块茎蛋白质组进行研究有助于我们对块茎生长和发育机理的理解,也对马铃薯块茎产量及品质等性状的提高具有一定的现实意义。 深入研究块茎蛋白质在发育过程中的差异表达,首先需要建立起马铃薯块茎蛋白质的2-DE体系,而样品的制备则是2-DE的关键环节,直接影响2-DE的分辨率和重复性。实验中,我们采用三氯乙酸/丙酮沉淀法,改良三氯乙酸/丙酮沉淀法,酚提取法和两步沉淀四种方法提取马铃薯块茎蛋白质,进行2-DE分离,对四种方法提取样品蛋白质的获得量、纯度及2-DE蛋白质图谱分辨率进行了比较。结果表明:两步沉淀法提取块茎蛋白质的获得量和纯度较其它三种提取方法高,2-DE蛋白质图谱较清晰、分辨率高。两步沉淀法提取马铃薯块茎蛋白质及其2-DE电泳技术的建立,为研究马铃薯块茎蛋白质组学提供了技术支撑。 用两步沉淀法提取马铃薯块茎发育过程中四个阶段的蛋白质,即匍匐茎的诱导和发生,匍匐茎的伸长,匍匐茎纵向生长的停止和块茎的发生及膨大阶段,并进行双向电泳分析。结果表明:马铃薯块茎蛋白质主要集中在分子量在15.5 kD-87.2 kD范围,等电点范围在3.8-7.2区域内。(107.8 kD,pI 4.9)、(37.7 kD,pI 7.3)、(42.8 kD,pI 6.2)、(21.3 kD,pI 8.9)、(37.7 kD,pI 6.0)、(33.8 kD,pI 7.2)、(23.7 kD,pI 5.1)和(77.3 kD,pI 6.4)八个蛋白质点只在第一发育阶段特异表达,在随后的发育过程中不再出现;在匍匐茎的伸长阶段,新出现了十三个蛋白质点,其中(25.2 kD,pI 8.6)、(26.3 kD,pI 7.7)和(30.4 kD,pI 6.1)三个点在随后的发育过程又消失,另外三个蛋白质点(51.3 kD,pI 5.8)、(51.6 kD,pI 6.7)和(50.9 kD,pI 4.2)的丰度在该阶段明显下调;在匍匐茎纵向生长的停止阶段,出现十个新蛋白质点,其中五个蛋白质点(45.1 kD,pI 6.8)、(45.3 kD,pI 4.8)、(44.3 kD,pI 6.1)、(44.7 kD,pI 5.2)和(42.9 kD,pI 7.3)只在该阶段特异表达,在丰度明显上调的十四个蛋白质点中,(22.7 kD,pI 8.8)在该发育阶段的丰度表现为最高,在块茎的发生及膨大阶段该蛋白质点的丰度又下调,另有十七个蛋白质点的丰度明显下调,其中变化最明显的为(61.3 kD,pI 6.8)和(58.5 kD,pI 6.4)两个点;在第四发育阶段即块茎开始膨大时有二十二个新蛋白质点出现,且都分布在较低分子量(53 kD-23 kD)范围内,丰度明显上调和下调的蛋白质点分别有十九和九个;Patatin蛋白在整个发育过程中其丰度明显增加。在块茎的整个发育过程中,这些蛋白质可能是这些发育阶段特异表达的蛋白质,它们的出现和消失可能与相应的发育阶段密切相关。
DOI:10.7666/d.y1333475URL [本文引用: 1]
蛋白质组学研究在功能基因组时代发挥着越来越重要的作用。双向电泳(2-DE)技术是其关键技术之一,具有重复性好、极敏感和分辨率高等特点。马铃薯(Solanum tuberosum L.)是世界上第四大粮食作物,对其块茎的发生发育过程进行研究,了解其块茎发育的机理,将为马铃薯的栽培、育种和生产提供理论依据。马铃薯块茎的发育是一个复杂而又有规律的过程,块茎蛋白质的数量、种类、丰度等在不同发育阶段存在着一定的差异,而这些差异蛋白质又参与基因的表达和块茎某些重要性状的调控。因此,对马铃薯块茎蛋白质组进行研究有助于我们对块茎生长和发育机理的理解,也对马铃薯块茎产量及品质等性状的提高具有一定的现实意义。 深入研究块茎蛋白质在发育过程中的差异表达,首先需要建立起马铃薯块茎蛋白质的2-DE体系,而样品的制备则是2-DE的关键环节,直接影响2-DE的分辨率和重复性。实验中,我们采用三氯乙酸/丙酮沉淀法,改良三氯乙酸/丙酮沉淀法,酚提取法和两步沉淀四种方法提取马铃薯块茎蛋白质,进行2-DE分离,对四种方法提取样品蛋白质的获得量、纯度及2-DE蛋白质图谱分辨率进行了比较。结果表明:两步沉淀法提取块茎蛋白质的获得量和纯度较其它三种提取方法高,2-DE蛋白质图谱较清晰、分辨率高。两步沉淀法提取马铃薯块茎蛋白质及其2-DE电泳技术的建立,为研究马铃薯块茎蛋白质组学提供了技术支撑。 用两步沉淀法提取马铃薯块茎发育过程中四个阶段的蛋白质,即匍匐茎的诱导和发生,匍匐茎的伸长,匍匐茎纵向生长的停止和块茎的发生及膨大阶段,并进行双向电泳分析。结果表明:马铃薯块茎蛋白质主要集中在分子量在15.5 kD-87.2 kD范围,等电点范围在3.8-7.2区域内。(107.8 kD,pI 4.9)、(37.7 kD,pI 7.3)、(42.8 kD,pI 6.2)、(21.3 kD,pI 8.9)、(37.7 kD,pI 6.0)、(33.8 kD,pI 7.2)、(23.7 kD,pI 5.1)和(77.3 kD,pI 6.4)八个蛋白质点只在第一发育阶段特异表达,在随后的发育过程中不再出现;在匍匐茎的伸长阶段,新出现了十三个蛋白质点,其中(25.2 kD,pI 8.6)、(26.3 kD,pI 7.7)和(30.4 kD,pI 6.1)三个点在随后的发育过程又消失,另外三个蛋白质点(51.3 kD,pI 5.8)、(51.6 kD,pI 6.7)和(50.9 kD,pI 4.2)的丰度在该阶段明显下调;在匍匐茎纵向生长的停止阶段,出现十个新蛋白质点,其中五个蛋白质点(45.1 kD,pI 6.8)、(45.3 kD,pI 4.8)、(44.3 kD,pI 6.1)、(44.7 kD,pI 5.2)和(42.9 kD,pI 7.3)只在该阶段特异表达,在丰度明显上调的十四个蛋白质点中,(22.7 kD,pI 8.8)在该发育阶段的丰度表现为最高,在块茎的发生及膨大阶段该蛋白质点的丰度又下调,另有十七个蛋白质点的丰度明显下调,其中变化最明显的为(61.3 kD,pI 6.8)和(58.5 kD,pI 6.4)两个点;在第四发育阶段即块茎开始膨大时有二十二个新蛋白质点出现,且都分布在较低分子量(53 kD-23 kD)范围内,丰度明显上调和下调的蛋白质点分别有十九和九个;Patatin蛋白在整个发育过程中其丰度明显增加。在块茎的整个发育过程中,这些蛋白质可能是这些发育阶段特异表达的蛋白质,它们的出现和消失可能与相应的发育阶段密切相关。
DOI:10.1007/s11738-009-0331-2URL [本文引用: 1]
The leaf protein pattern from drought-tolerant and drought-sensitive wheat varieties subjected to severe soil drought but with the possibility for recover from stress was studied by two-dimensional polyacrylamide gel electrophoresis (2D-PAGE). The spots representing Rubisco large subunit (RLS) were identified using polyclonal antibodies against Rubisco and immunoblotting. Some qualitative and quantitative differences in the 2D-PAGE protein map of wheat varieties were revealed under drought conditions. Three days recovery of wheat plants were not enough for restoring RLS quantity to the level of controls after 7 days drought, especially in the drought-sensitive variety Miziya. There are contradictory data in the literature concerning increased or diminished RLS level in drought stressed plants. A comparison of RLS after SDS-PAGE and 2D-PAGE was made. The revealed protein pattern depended on the presence or absence of protease inhibitors in the extraction buffer, on the procedure of extraction, and on the degree of stress.
DOI:10.1002/1615-9861(200209)2:9<1131::AID-PROT1131>3.0.CO;2-1URL [本文引用: 1]
DOI:10.1016/j.jprot.2011.12.026URLPMID:22230808 [本文引用: 1]
The trace proteome of a Ginger drink, stated to be produced with a ginger root extract, has been investigated via capture with combinatorial peptide ligand libraries (ProteoMiner). Although in traces, we could confirm the presence of five grape proteins and one apple protein, but not even the faintest trace of any ginger root proteins. The first two findings are correct, as the producer stated that this beverage had been reinforced with 12% grape juice and 6% apple juice, but the absence of even traces of ginger proteins does not permit the classification of this beverage as a ginger extract on a proteomics scale. However, organoleptic tasting has confirmed the presence of a ginger extract, due to its piquant and tongue-biting taste. Nevertheless, any ginger root extract must be considered as a minor component as compared to the presence of grape and apple juice. At the light of these findings, it is hoped that the competent authorities will in the future make compulsory the proper labelling also of beverages so that all amounts of compounds utilized will be clearly stated in the label, including the presumptive main component.Graphical abstractGinger Ale or Ginger Rogers? Even though we might be going against our findings reported inside, we confess that we much prefer the latter! Chapeau to her, in fact op Hat to the fabulous duo Ginger and Fred.
DOI:10.3389/fpls.2014.00627URLPMID:4235293 [本文引用: 1]
Flooding stress has a negative impact on soybean cultivation because it severely impairs growth and development. To understand the flooding responsive mechanism in early stage soybeans, a glycoproteomic technique was used. Two-day-old soybeans were treated with flooding for 2 days and roots were collected. Globally, the accumulation level of glycoproteins, as revealed by cross-reaction with concanavalin A decreased by 2 days of flooding stress. Glycoproteins were enriched from total protein extracts using concanavalin A lectin resin and analyzed using a gel-free proteomic technique. One-hundred eleven and 69 glycoproteins were identified without and with 2 days of flooding stress, respectively. Functional categorization of these identified glycoproteins indicated that the accumulation level of proteins related to protein degradation, cell wall, and glycolysis increased, while stress-related proteins decreased under flooding stress. Also the accumulation level of glycoproteins localized in the secretory pathway (e.g., peroxidases and plycosyl hydrolases) decreased under flooding stress. Out of 23 common glycoproteins between control and flooding conditions, peroxidases and glycosyl hydrolases were decreased by 2 days of flooding stress. mRNA expression levels of proteins in the endoplasmic reticulum and N-glycosylation related proteins were downregulated by flooding stress. These results suggest that flooding might negatively affect the process of N-glycosylation of proteins related to stress and protein degradation; however glycoproteins involved in glycolysis are activated.
DOI:10.1016/j.euprot.2014.05.005URL [本文引用: 1]
Salinity stress is one of the major abiotic stresses that limit agricultural yield. To understand salt-responsive protein networks in soybean seedling, the extracted proteins from seedling roots of two different genotypes (Lee 68 and Jackson) were analyzed under salt stress by two-dimensional polyacrylamide gel electrophoresis. Sixty-eight differentially expressed proteins were detected and identified. The identified proteins were involved in 13 metabolic pathways and cellular processes. Proteins correlated to brassinosteroid and gilbberellin signalings were significantly increased only in the genotype Lee 68 under salt stress; abscisic acid content was positively correlated with this genotype; proteins that can be correlated to Ca2+signaling were more strongly enhanced by salt stress in the seedling roots of genotype Lee 68 than in those of genotype Jackson; moreover, genotype Lee 68 had stronger capability of reactive oxygen species scavenging and cell K+/Na+homeostasis maintaining in seedling roots than genotype Jackson under salt stress. Since the genotype Lee 68 has been described in literature as being tolerant and Jackson as sensitive, we hypothesize that these major differences in the genotype Lee 68 might contribute to salt tolerance. Combined with our previous comparative proteomics analysis on seedling leaves, the similarities and differences between the salt-responsive protein networks found in the seedling leaves and roots of both the genotypes were discussed. Such a result will be helpful in breeding of salt-tolerant soybean cultivars.
DOI:10.1016/S0958-1669(99)80004-4URLPMID:10047502 [本文引用: 1]
23 years after O'Farrel developed two-dimensional gel electrophoresis we still debate if the technique can be improved, or if there are other alternative separation technologies that can challenge its central position in proteomic projects. These questions are relevant as the pharmaceutical industry expects proteomic studies to provide novel protein targets for drug discovery and diagnostics. In our opinion, there are various aspects of the technology that can be improved, including resolution, sample preparation and detection, but so far there is no alternative technique(s) available, or any under development, that can replace it.
DOI:10.1104/pp.108.116905URLPMID:18552235 [本文引用: 1]
Despite its water-soluble chlorophyll-binding protein (WSCP) function, the putative trypsin inhibitor (TI) activity of the Brassica napus drought 22 kD (BnD22) protein and its physiological function in young leaves during leaf nitrogen (N) remobilization promoted by stressful conditions remains an enigma. Therefore, our objectives were to determine (1) if BnD22 is related to the 19-kD TI previously detected in B. napus young leaves, and (2) if the levels of BnD22 transcripts, BnD22 protein, and TI activity in young leaves are associated with plant responses to stress conditions (N starvation and methyl jasmonate [MeJA] treatments) that are able to modulate leaf senescence. Compared to control, N starvation delayed initiation of senescence and induced 19-kD TI activity in the young leaves. After 3 d with MeJA, the 19-kD TI activity was 7-fold higher than the control. Using two-dimensional electrophoresis gel, TI activity, and electrospray ionization liquid chromatography tandem mass spectrometry analysis, it was demonstrated that two 19-kD proteins with isoelectric points 5.0 and 5.1 harboring TI activity correspond to BnD22 perfectly. BnD22 gene expression, TI activities, and BnD22 protein presented similar patterns. Using polyclonal anti-WSCP antibodies of Brassica oleracea, six polypeptides separated by two-dimensional electrophoresis were detected in young leaves treated with MeJA. Electrospray ionization liquid chromatography tandem mass spectrometry analysis of six polypeptides confirms their homologies with WSCP. Results suggest that BnD22 possesses dual functions (WSCP and TI) that lead to the protection of younger tissues from adverse conditions by maintaining metabolism (protein integrity and photosynthesis). By sustaining sink growth of stressed plants, BnD22 may contribute to a better utilization of recycling N from sources, a physiological trait that improves N-use efficiency.
DOI:10.1071/PP00158URL [本文引用: 1]
The antioxidant defence system of soybean (Glycine maxL.) nodules and roots was studied in plants subjected to three different concentrations (50, 100 and 200 M ) of CdCl 2 . Cadmium (Cd)-induced oxidative stress was determined by lipid peroxidation, which remained unaltered in nodules and roots treated with 50 M Cd(II). No changes were observed in nodules treated with 100 M Cd(II), while a 20% increase was found in roots and 200 M Cd(II) produced an increase of about 55% in both tissues. The soluble antioxidant defence, reduced glutath one(GSH) and the corresponding reduced/oxidised glutathione (GSH/GSSG) ratio showed different behaviour in both tissues, decreasing in roots with 100 and 200 M Cd(II). No changes were observed in the GSH/GSSG ratio in nodules under the three Cd treatments. However, ascorbate content and ascorbate/dehydroascorbate (As/DAs) ratio were diminished in nodules and roots subjected to the three Cd concentrations. Regarding the antioxidant enzymes, it was found that, except for catalase, a general decrease in nodule enzyme activities was produced only under the 200 M Cd(II) treatment. Nevertheless, root enzymatic antioxidant defences showed significant increments in L -ascorbate peroxidase (APOX), dehydroascorbate reductase (DHAR) and glutathione reductase (GR) activities under lower Cd treatment, while the enzyme activities decreased with the two higher concentrations of CdCl 2 . These results suggest that soybean roots were more affected than nodules by Cd treatments, although the higher Cd concentration produced oxidative stress and deleterious effects in antioxidant defence system in both tissues.
DOI:10.1016/j.plaphy.2004.01.005URLPMID:15051047 [本文引用: 1]
To investigate whether re-aeration after a short-term hypoxic pre-treatment (for 2, 12 or 24 h) induces oxidative stress, the temporal sequence of physiological reactions, including the level of free radicals, hydrogen peroxide production, and changes in antioxidative enzymes, was characterized in roots of hydroponically grown lupine (Lupinus luteus L., cv. Juno) seedlings. By using electron paramagnetic resonance (EPR), we found that the exposure of hypoxically grown roots (hypoxic pre-treatment for 12 and 24 h) to air caused an increase in the level of free radicals. The amount of hydrogen peroxide also tended to increase when hypoxically pre-treated roots were re-aerated, which attests to a higher production of reactive oxygen species. Re-aeration caused a higher activity of superoxide dismutase (SOD, EC 1.15.1.1) and catalase (CAT, EC 1.11.1.6), whereas the activity of peroxidase (POX, EC 1.11.1.7) was only slightly influenced. The roots were less tolerant to longer hypoxic pre-treatments, with a significant decrease in viability, associated with death of root tips immediately after hypoxic stress. Roots exposed to hypoxia for 2 h showed less pronounced responses and their viability was not affected by hypoxic stress and re-aeration. These results indicate that re-aeration following short-term hypoxia imposes a mild oxidative stress. This led us to conclude that re-oxygenation stress per se was not the key factor for cell death in root tips.
DOI:10.1046/j.1365-3040.2000.00602.xURL [本文引用: 1]
ABSTRACT Using two cultivars of Pisum sativum L. with different sensitivity to NaCl, the effect of long-term (15 d) NaCl (70 m M ) treatments on the activity and expression of the foliar ascorbate lutathione cycle enzymes, superoxide dismutase isozymes and their mRNAs was evaluated and related to their ascorbate and glutathione contents. High-speed supernatant (soluble) fractions, enriched for cytosolic components of the antioxidant system, were used. In this fraction from the NaCl-tolerant variety (cv Granada), the activities of ascorbate peroxidase (APX), glutathione reductase (GR), monodehydroascorbate reductase (MDHAR), Mn-superoxide dismutase (Mn-SOD) and dehydroascorbate reductase (DHAR) increased, while CuZn-SOD activity remained constant. In the NaCl-sensitive plants (cv Challis), salinity did not produce significant changes in APX, MDHAR and GR activities. Only DHAR activity was induced in cv Challis, whereas soluble CuZn-SOD activity decreased by about 35%. Total ascorbate and glutathione contents decreased in both cultivars, but the decline was greater in NaCl-sensitive plants. This difference between the two cultivars was more pronounced when the transcript levels of some these enzymes were examined. Transcript levels for mitochondrial Mn-SOD, chloroplastic CuZn-SOD and phospholipid hydroperoxide glutathione peroxidase (PHGPX), cytosolic GR and APX were strongly induced in the NaCl-tolerant variety but not in the NaCl-sensitive variety. These data strongly suggest that induction of antioxidant defences is at least one component of the tolerance mechanism of peas to long-term salt-stress.
URL [本文引用: 2]
蛋白质组学研究在功能基因组时代发挥着越来越重要的作用。双向电泳是其关键技术之一。水稻(Oryza sativa L.)不仅是世界上最重要的粮食作物,也是单子叶植物的模式植物。我们改进和优化水稻蛋白的提取方法及电泳条件,得到了高分辨率和高重复性的双向电泳图谱。在根、叶、悬浮细胞的双向电泳图谱上分别检测到至少1150、1000、1050个蛋白点。重复样品图谱间的相关系数在0.87和0.94之间。 盐胁迫是影响全世界作物产量的主要非生物胁迫之一,全世界约有20%的土地或50%的灌溉地受盐害的影响。三周大小的日本晴水稻幼苗(cv. Nipponbare)在150 mM的NaCl处理24、48、72 h后,根中有34个和20个蛋白点分别被上调和下调。质谱分析共鉴定出12个蛋白点,代表了10种不同的蛋白,其中3个点是同一个蛋白(烯醇化酶)。4个蛋白是已知的盐胁迫响应蛋白,另6个蛋白是新的盐胁迫响应蛋白,包括UDP葡萄糖焦磷酸化酶、根的谷氨酰胺合成酶、推测的新生肽结合蛋白复合物的α亚基、细胞色素C氧化酶6b-1亚基、推测的剪接因子类似蛋白、推测的肌动蛋白结合蛋白。这些蛋白参与了碳水化合物代谢、氮代谢、能量代谢、氧自由基的清除、mRNA和蛋白质的加工、细胞骨架的稳定。 低温胁迫是另一种主要的非生物胁迫,影响很多重要粮食作物的产量和地理分布。三周大小的水稻幼苗在6℃处理6h或24h后,生长受到抑制,叶片卷曲,相对电导率升高,净光合速率下降。用双向电泳共检测到1000多个叶片可溶性蛋白,其中31个被下调,66个被上调。质谱分析共鉴定出85个差异表达蛋白,包括已知的和新的低温胁迫响应蛋白。多个蛋白在低温胁迫下被降解,尤其是光合机构蛋白,有19个蛋白点均被鉴定为Rubisco大亚基的降解片段。这些蛋白参与信号转导,RNA加工,蛋白质翻译、蛋白质加工,氧化还原平衡,光合作用,光呼吸,碳、氮、硫及能量代谢。用定量PCR分析了44个基因的表达情况,结果表明mRNA与蛋白质水平的变化并不一致。另外,我们用Pro-QDiamond染料共检测到41个下调的和26个上调的磷酸化蛋白。
URL [本文引用: 2]
蛋白质组学研究在功能基因组时代发挥着越来越重要的作用。双向电泳是其关键技术之一。水稻(Oryza sativa L.)不仅是世界上最重要的粮食作物,也是单子叶植物的模式植物。我们改进和优化水稻蛋白的提取方法及电泳条件,得到了高分辨率和高重复性的双向电泳图谱。在根、叶、悬浮细胞的双向电泳图谱上分别检测到至少1150、1000、1050个蛋白点。重复样品图谱间的相关系数在0.87和0.94之间。 盐胁迫是影响全世界作物产量的主要非生物胁迫之一,全世界约有20%的土地或50%的灌溉地受盐害的影响。三周大小的日本晴水稻幼苗(cv. Nipponbare)在150 mM的NaCl处理24、48、72 h后,根中有34个和20个蛋白点分别被上调和下调。质谱分析共鉴定出12个蛋白点,代表了10种不同的蛋白,其中3个点是同一个蛋白(烯醇化酶)。4个蛋白是已知的盐胁迫响应蛋白,另6个蛋白是新的盐胁迫响应蛋白,包括UDP葡萄糖焦磷酸化酶、根的谷氨酰胺合成酶、推测的新生肽结合蛋白复合物的α亚基、细胞色素C氧化酶6b-1亚基、推测的剪接因子类似蛋白、推测的肌动蛋白结合蛋白。这些蛋白参与了碳水化合物代谢、氮代谢、能量代谢、氧自由基的清除、mRNA和蛋白质的加工、细胞骨架的稳定。 低温胁迫是另一种主要的非生物胁迫,影响很多重要粮食作物的产量和地理分布。三周大小的水稻幼苗在6℃处理6h或24h后,生长受到抑制,叶片卷曲,相对电导率升高,净光合速率下降。用双向电泳共检测到1000多个叶片可溶性蛋白,其中31个被下调,66个被上调。质谱分析共鉴定出85个差异表达蛋白,包括已知的和新的低温胁迫响应蛋白。多个蛋白在低温胁迫下被降解,尤其是光合机构蛋白,有19个蛋白点均被鉴定为Rubisco大亚基的降解片段。这些蛋白参与信号转导,RNA加工,蛋白质翻译、蛋白质加工,氧化还原平衡,光合作用,光呼吸,碳、氮、硫及能量代谢。用定量PCR分析了44个基因的表达情况,结果表明mRNA与蛋白质水平的变化并不一致。另外,我们用Pro-QDiamond染料共检测到41个下调的和26个上调的磷酸化蛋白。
DOI:10.1002/pmic.200401101URLPMID:15712235 [本文引用: 1]
Abstract Drought is one of the major factors limiting the yield of sugar beet ( Beta vulgaris L.). The identification of candidate genes for marker-assisted selection (MAS) could greatly improve the efficiency of breeding for increased drought tolerance. Drought-induced changes in the proteome could highlight important genes. Two genotypes of sugar beet (7112 and 7219-P.69) differing in genetic background were cultivated in the field. A line-source sprinkler irrigation system was used to apply irrigated and water deficit treatments beginning at the four-leaf stage. At 157 days after sowing, leaf samples were collected from well-watered and drought-stressed plants for protein extraction and to measure shoot biomass and leaf relative water content. Changes induced in leaf proteins were studied by two-dimensional gel electrophoresis and quantitatively analyzed using image analysis software. Out of more than 500 protein spots reproducibly detected and analyzed, 79 spots showed significant changes under drought. Some proteins showed genotype-specific patterns of up- or downregulation in response to drought. Twenty protein spots were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS), leading to identification of Rubisco and 11 other proteins involved in redox regulation, oxidative stress, signal transduction, and chaperone activities. Some of these proteins could contribute a physiological advantage under drought, making them potential targets for MAS.
DOI:10.1016/j.tplants.2004.03.006URLPMID:15130550 [本文引用: 1]
Abiotic stresses usually cause protein dysfunction. Maintaining proteins in their functional conformations and preventing the aggregation of non-native proteins are particularly important for cell survival under stress. Heat-shock proteins (Hsps)/chaperones are responsible for protein folding, assembly, translocation and degradation in many normal cellular processes, stabilize proteins and membranes, and can assist in protein refolding under stress conditions. They can play a crucial role in protecting plants against stress by re-establishing normal protein conformation and thus cellular homeostasis. Here, we summarize the significance of Hsps and chaperones in abiotic stress responses in plants, and discuss the co-operation among their different classes and their interactions with other stress-induced components.
DOI:10.1007/s00299-007-0470-0URLPMID:17968552 [本文引用: 1]
Exposure of rice ( Oryza sativa L.) seedlings to a high temperature (42°C) for 24 h resulted in a significant increase in tolerance to drought stress. To try to determine the mechanisms of acquisition of tolerance to drought stress by heat shock, the rice small heat-shock protein gene, sHSP17.7 , the product of which was shown to act as molecular chaperones in vitro and in vivo in our previous study, was overexpressed in the rice cultivar “Hoshinoyume”. Western and Northern blot analyses showed higher expression levels of sHSP17.7 protein in three transgenic lines than in one transgenic line. Drought tolerance was assessed in these transgenic lines and wild-type plants by withholding water for 602days for evaluation of the ability of plants to continue growth after water-stress treatments. Although no significant difference was found in water potential of seedlings between transgenic lines and wild-type plants at the end of drought treatments, only transgenic seedlings with higher expression levels of sHSP17.7 protein could regrow after rewatering. Similar results were observed in survival rates after treatments with 30% polyethylene glycol (PEG) 3640 for 302days. These results suggest that overproduction of sHSP17.7 could increase drought tolerance in transgenic rice seedlings.
DOI:10.7666/d.y1598163URL [本文引用: 1]
干旱是影响很多重要作物产量和地理分布的主要非生物胁迫之一。水稻(Oryzasativa L.)不仅是世界上最重要的粮食作物,也是单子叶植物的模式种。利用组学方法研究水稻对干旱胁迫的应答机制,进而研究抗旱机理,对于全面揭示植物干旱适应的遗传基础,发掘干旱应答相关基因、蛋白和代谢物具有重要的意义。我们通过关联蛋白质组、转录组和代谢组的数据,运用生物信息学手段探讨了水稻在干旱胁迫下的整体反应、分析水稻抗旱的分子机制。 四周大小的早稻品种IRAT109幼苗停止浇水3天之后,水稻叶片出现卷曲,相对电导率和脯氨酸含量升高,叶水势和土壤含水量下降。 用双向电泳对水稻叶片全蛋白进行检测,发现1200多个可溶性蛋白,经T检验发现71个蛋白点丰度发生1.5倍以上显著变化(48个上调,23个下调)。将这些差异蛋白点进行MALDI-TOF-TOF-MS分析,有60个蛋白点得到有效的鉴定。大部分蛋白质是以前报道的与干旱等逆境胁迫相关,有4个是首次发现的与干旱应答有关的蛋白质,分别为腺苷高半胱氨酸水解酶(adenosylhomocysteinase)、醇酮酸还原异构酶(ketol-acid reductoisomerase)、蔗糖-UDP葡糖基转移酶2(sucroce-UDPglucosyltransferase 2)、甲酸脱氢酶(formate dehydrogenase)。对所鉴定的蛋白质通过Expasy、KEGG和NCBI数据库分析以及相关的文献查询对其功能进行了分类,发现这些蛋白质的功能涉及调节因子(regulatory factor)、过氧化物平衡(redoxhomeostasis)、代谢和能量(metabolism and energy)和细胞骨架(cytoskeleton)等六个方面,其中与调节因子类相关的蛋白最多,占总蛋白数的40%。 通过cDNA芯片对水稻叶片mRNA的表达情况进行调查,总共发现4427个变化倍数在2倍以上的差异表达基因,其中有1812个上调基因、2615个下调基因。随机挑选40个差异基因设计引物进行荧光定量PCR(RT-PCR)验证,发现两者的相关系数为0.95。通过Gene Ontology(GO)分类系统对这些差异基因进行分类,发现这些基因主要分布在7个类别:细胞(cell)、细胞组分(cell part)、细胞器(organelle)、结合功能(binding)、催化功能(catalyitc)、亚细胞过程(cellular process)和代谢过程(metabolic process),整体上与差异蛋白的GO功能分类结果相吻合。相关性分析发现所有差异蛋白与其对应mRNA的表达水平的相关性系数只有0.08,同时各功能类蛋白的相关系数却不尽相同:调节因子类为-0.35、过氧化物类为0.03、能量与代谢类为0.02、抗旱相关类为0.82。 利用气相色谱(GC)和气相色谱-质谱(GC-MS)联用的方法,对水稻叶片的全代谢物进行调查。共发现129个代谢物,主成分分析(PCA)发现胁迫组和对照组可以明显分开。T检验发现有38个代谢物含量发生1.5倍以上显著变化,主要包括氨基酸、糖类和有机酸等。采用生物信息学方法对56个差异蛋白所参与的代谢通路(pathway)进行基于离散分布的显著性分析,总共发现13个代谢通路在干旱胁迫下受这些差异蛋白的调控,其中最显著代谢通路为光合作用中的碳固定(carbonfixation)过程。通过KEGG数据库对碳固定进行映射(mapping)分析,发现38个差异代谢物中的天冬氨酸(旱胁迫之后上调2.7倍)正好是最显著代谢途径碳固定中的一个组分。
DOI:10.7666/d.y1598163URL [本文引用: 1]
干旱是影响很多重要作物产量和地理分布的主要非生物胁迫之一。水稻(Oryzasativa L.)不仅是世界上最重要的粮食作物,也是单子叶植物的模式种。利用组学方法研究水稻对干旱胁迫的应答机制,进而研究抗旱机理,对于全面揭示植物干旱适应的遗传基础,发掘干旱应答相关基因、蛋白和代谢物具有重要的意义。我们通过关联蛋白质组、转录组和代谢组的数据,运用生物信息学手段探讨了水稻在干旱胁迫下的整体反应、分析水稻抗旱的分子机制。 四周大小的早稻品种IRAT109幼苗停止浇水3天之后,水稻叶片出现卷曲,相对电导率和脯氨酸含量升高,叶水势和土壤含水量下降。 用双向电泳对水稻叶片全蛋白进行检测,发现1200多个可溶性蛋白,经T检验发现71个蛋白点丰度发生1.5倍以上显著变化(48个上调,23个下调)。将这些差异蛋白点进行MALDI-TOF-TOF-MS分析,有60个蛋白点得到有效的鉴定。大部分蛋白质是以前报道的与干旱等逆境胁迫相关,有4个是首次发现的与干旱应答有关的蛋白质,分别为腺苷高半胱氨酸水解酶(adenosylhomocysteinase)、醇酮酸还原异构酶(ketol-acid reductoisomerase)、蔗糖-UDP葡糖基转移酶2(sucroce-UDPglucosyltransferase 2)、甲酸脱氢酶(formate dehydrogenase)。对所鉴定的蛋白质通过Expasy、KEGG和NCBI数据库分析以及相关的文献查询对其功能进行了分类,发现这些蛋白质的功能涉及调节因子(regulatory factor)、过氧化物平衡(redoxhomeostasis)、代谢和能量(metabolism and energy)和细胞骨架(cytoskeleton)等六个方面,其中与调节因子类相关的蛋白最多,占总蛋白数的40%。 通过cDNA芯片对水稻叶片mRNA的表达情况进行调查,总共发现4427个变化倍数在2倍以上的差异表达基因,其中有1812个上调基因、2615个下调基因。随机挑选40个差异基因设计引物进行荧光定量PCR(RT-PCR)验证,发现两者的相关系数为0.95。通过Gene Ontology(GO)分类系统对这些差异基因进行分类,发现这些基因主要分布在7个类别:细胞(cell)、细胞组分(cell part)、细胞器(organelle)、结合功能(binding)、催化功能(catalyitc)、亚细胞过程(cellular process)和代谢过程(metabolic process),整体上与差异蛋白的GO功能分类结果相吻合。相关性分析发现所有差异蛋白与其对应mRNA的表达水平的相关性系数只有0.08,同时各功能类蛋白的相关系数却不尽相同:调节因子类为-0.35、过氧化物类为0.03、能量与代谢类为0.02、抗旱相关类为0.82。 利用气相色谱(GC)和气相色谱-质谱(GC-MS)联用的方法,对水稻叶片的全代谢物进行调查。共发现129个代谢物,主成分分析(PCA)发现胁迫组和对照组可以明显分开。T检验发现有38个代谢物含量发生1.5倍以上显著变化,主要包括氨基酸、糖类和有机酸等。采用生物信息学方法对56个差异蛋白所参与的代谢通路(pathway)进行基于离散分布的显著性分析,总共发现13个代谢通路在干旱胁迫下受这些差异蛋白的调控,其中最显著代谢通路为光合作用中的碳固定(carbonfixation)过程。通过KEGG数据库对碳固定进行映射(mapping)分析,发现38个差异代谢物中的天冬氨酸(旱胁迫之后上调2.7倍)正好是最显著代谢途径碳固定中的一个组分。
DOI:10.7666/d.y2004679URL [本文引用: 1]
干旱是影响作物生长和产量的重要非生物胁迫之一,对干旱敏感型作物的影响尤其明显。水稻(Oryza sativa L.)是世界上种植最广泛的粮食作物之一,同时也是一种重要的模式作物。以蛋白质组学为代表的组学研究对于系统地了解水稻抗旱的分子机制有着十分重要的作用。 旱稻品种IRAT109具有极强的抗旱能力,被广泛地用于抗旱基因的发掘以及水稻抗旱性的遗传改良。本研究以该材料为核心,以蛋白质双向电泳和质谱鉴定为主要技术手段,结合形态学和生理学调查,分析了叶片在全根渗透胁迫和分根渗透胁迫下的响应。主要研究结果如下: 1.全根渗透胁迫引起了叶片ABA和脯氨酸含量的上升,并伴随有叶片卷曲和质膜损伤等胁迫症状,而分根渗透胁迫只诱导了ABA以及脯氨酸含量的上升,但是并没有引起明显的叶片损伤,表明分根渗透胁迫诱导了植物渗透胁迫信号的产生,并引发了包括渗透调节在内的一系列抗逆反应。 2.采用双向电泳分别将全根渗透胁迫,分根渗透胁迫与对照组之间的叶片蛋白表达谱进行了比较,总共发现了67个差异表达蛋白点,其中36个仅在全根渗透胁迫表现差异,15个仅在分根渗透胁迫表现差异。此外,有11个蛋白点在两种处理下都表现上升表达,有3个蛋白点都表现下降表达,还有2个蛋白点在不同的处理中表现完全相反的表达模式。采用串联质谱成功鉴定了67个差异蛋白中的58个。按照其功能,这些蛋白被归于胁迫防御、光合作用等12类。 3.分根渗透胁迫诱导的差异表达蛋白主要参与光合作用、碳水化合物代谢等。具体包括促使了核酮糖1,5-二磷酸羧化/加氧酶(Rubisco)大亚基的降解,增强了参与糖酵解和三羧酸循环关键酶-顺乌头酸酶和3-磷酸甘油醛脱氢酶以及ATP合成酶的表达。此外,分根渗透胁迫还特异诱导了邻苯氨甲酸盐合成酶和镍结合蛋白2A的上升表达。其中,邻苯氨甲酸盐合成酶的上升表达可能和ABA预处理克服胁迫对植物的生长抑制有关;而对于镍结合蛋白2A,虽然前人也有将其基因列为水稻抗旱候选基因,但该蛋白在植物抵御干旱以及其它胁迫中的作用还知之甚少。 4.全根渗透胁迫诱导了大量分子伴侣及具有分子伴侣功能的热激蛋的上升表达,有利于维持胁迫环境下其它蛋白的稳定性和维持植物生存与生长。此外,全根渗透胁迫还诱导了单脱氢抗坏血酸还原酶、过氧化物酶以及谷胱甘肽-S-转移酶等氧化平衡类蛋白的上升表达,有助于清除胁迫诱导产生的活性氧离子,降低渗透胁迫对植物的危害。另外,S-腺苷甲硫氨酸合成酶和咖啡酸3-O-甲基转移酶等参与次生代谢的蛋白在全根渗透胁迫表达下降,表明木质素以及其它相关次生代谢产物的合成下降。这有助于植物减少不必要的能量和物质消耗。 为了更系统地了解旱稻品种IRAT109对干旱胁迫的响应,本研究采用蛋白质组与转录组和代谢组分析相结合的策略,探究旱稻品种IRAT109幼苗的叶片在干旱胁迫下的综合反应。主要研究结果如下: 1.双向电泳分析发现干旱胁迫引起了71个蛋白点的表达发生明显差异。采用串联质谱成功鉴定了其中的60个,包括38个上升表达的蛋白点以及22个下降表达的蛋白点。这些蛋白按照其功能被归于蛋白折叠和聚集、碳水化合物代谢等11个类别。从蛋白功能分类以及所属蛋白的调节方式上分析,我们可以看出下降表达的蛋白主要与翻译相关,而上升表达的蛋白主要参与了蛋白折叠与聚集以及氧化平衡。 2.转录组分析发现了4756个差异表达基因,包括2528个上升表达基因和2228个下降表达基因。这些差异表达基因有的属于信号转导和转录以及蛋白翻译等调节性基因,有的属于氧化平衡、碳水化合物代谢以及氨基酸代谢等功能性基因。此外,还有一些类别仅仅在差异表达基因中才有发现,比如参与核苷酸代谢和脂类代谢的基因,甚至还有很多非编码蛋白基因受干旱胁迫诱导差异表达。 3.使用KEGG数据库对差异表达蛋白和基因参与的pathway进行分析,总共发现了47个被显著改变的pathway,其中有5个pathway在蛋白质和mRNA水平上都有发现。在我们发现的这些pathway中,很多都属于基础代谢(碳固定、尿素循环、脂肪酸代谢、氨基酸合成)。此外,也有几个属于次生代谢(苯丙烷合成、柠檬精油和松萜降解等)。 4.采用GC-MS技术进行的代谢谱分析发现了37个明显变化的代谢产物,分属于6大类,包括有机酸(38%)、糖类(19%)、氨基酸(16%)、脂肪酸(14%)、糖醇(5%)以及其它类别(8%)。 5.根据差异表达蛋白和基因的pathway分析结果并结合所发现的差异表达代谢产物,我们对植物在干旱胁迫下的代谢调节反应有了一个更全面的认识:干旱胁迫下,光合作用受到抑制,导致能量和碳水化合物输出减少。然而,植物需要大量的能量和物质用于其它一些生物学过程,比如活性氧清除和渗透物质合成等以抵御胁迫。额外的能量与物质的需求和较低的供应成为了一对矛盾,此时,需要动用植物的储备物质来解决这一矛盾。碳水化合物和脂肪酸是植物叶片中最重要的两种储备物质。干旱胁迫加速了糖酵解以及脂肪酸降解,促进了三羧酸循环以及各种氨基酸的合成,形成了从碳水化合物和脂肪酸向氨基酸方向的能量和物质的转移和利用。该过程一方面有助于累积包括脯氨酸在内的各种氨基酸,增进植物细胞的渗透调节能力,同时也为植物体合成新的蛋白做好了准备。另外,储备物质通过糖酵解以及三羧酸循环等过程,产生大量的ATP,也为植物应对干旱胁迫提供了能量。
DOI:10.7666/d.y2004679URL [本文引用: 1]
干旱是影响作物生长和产量的重要非生物胁迫之一,对干旱敏感型作物的影响尤其明显。水稻(Oryza sativa L.)是世界上种植最广泛的粮食作物之一,同时也是一种重要的模式作物。以蛋白质组学为代表的组学研究对于系统地了解水稻抗旱的分子机制有着十分重要的作用。 旱稻品种IRAT109具有极强的抗旱能力,被广泛地用于抗旱基因的发掘以及水稻抗旱性的遗传改良。本研究以该材料为核心,以蛋白质双向电泳和质谱鉴定为主要技术手段,结合形态学和生理学调查,分析了叶片在全根渗透胁迫和分根渗透胁迫下的响应。主要研究结果如下: 1.全根渗透胁迫引起了叶片ABA和脯氨酸含量的上升,并伴随有叶片卷曲和质膜损伤等胁迫症状,而分根渗透胁迫只诱导了ABA以及脯氨酸含量的上升,但是并没有引起明显的叶片损伤,表明分根渗透胁迫诱导了植物渗透胁迫信号的产生,并引发了包括渗透调节在内的一系列抗逆反应。 2.采用双向电泳分别将全根渗透胁迫,分根渗透胁迫与对照组之间的叶片蛋白表达谱进行了比较,总共发现了67个差异表达蛋白点,其中36个仅在全根渗透胁迫表现差异,15个仅在分根渗透胁迫表现差异。此外,有11个蛋白点在两种处理下都表现上升表达,有3个蛋白点都表现下降表达,还有2个蛋白点在不同的处理中表现完全相反的表达模式。采用串联质谱成功鉴定了67个差异蛋白中的58个。按照其功能,这些蛋白被归于胁迫防御、光合作用等12类。 3.分根渗透胁迫诱导的差异表达蛋白主要参与光合作用、碳水化合物代谢等。具体包括促使了核酮糖1,5-二磷酸羧化/加氧酶(Rubisco)大亚基的降解,增强了参与糖酵解和三羧酸循环关键酶-顺乌头酸酶和3-磷酸甘油醛脱氢酶以及ATP合成酶的表达。此外,分根渗透胁迫还特异诱导了邻苯氨甲酸盐合成酶和镍结合蛋白2A的上升表达。其中,邻苯氨甲酸盐合成酶的上升表达可能和ABA预处理克服胁迫对植物的生长抑制有关;而对于镍结合蛋白2A,虽然前人也有将其基因列为水稻抗旱候选基因,但该蛋白在植物抵御干旱以及其它胁迫中的作用还知之甚少。 4.全根渗透胁迫诱导了大量分子伴侣及具有分子伴侣功能的热激蛋的上升表达,有利于维持胁迫环境下其它蛋白的稳定性和维持植物生存与生长。此外,全根渗透胁迫还诱导了单脱氢抗坏血酸还原酶、过氧化物酶以及谷胱甘肽-S-转移酶等氧化平衡类蛋白的上升表达,有助于清除胁迫诱导产生的活性氧离子,降低渗透胁迫对植物的危害。另外,S-腺苷甲硫氨酸合成酶和咖啡酸3-O-甲基转移酶等参与次生代谢的蛋白在全根渗透胁迫表达下降,表明木质素以及其它相关次生代谢产物的合成下降。这有助于植物减少不必要的能量和物质消耗。 为了更系统地了解旱稻品种IRAT109对干旱胁迫的响应,本研究采用蛋白质组与转录组和代谢组分析相结合的策略,探究旱稻品种IRAT109幼苗的叶片在干旱胁迫下的综合反应。主要研究结果如下: 1.双向电泳分析发现干旱胁迫引起了71个蛋白点的表达发生明显差异。采用串联质谱成功鉴定了其中的60个,包括38个上升表达的蛋白点以及22个下降表达的蛋白点。这些蛋白按照其功能被归于蛋白折叠和聚集、碳水化合物代谢等11个类别。从蛋白功能分类以及所属蛋白的调节方式上分析,我们可以看出下降表达的蛋白主要与翻译相关,而上升表达的蛋白主要参与了蛋白折叠与聚集以及氧化平衡。 2.转录组分析发现了4756个差异表达基因,包括2528个上升表达基因和2228个下降表达基因。这些差异表达基因有的属于信号转导和转录以及蛋白翻译等调节性基因,有的属于氧化平衡、碳水化合物代谢以及氨基酸代谢等功能性基因。此外,还有一些类别仅仅在差异表达基因中才有发现,比如参与核苷酸代谢和脂类代谢的基因,甚至还有很多非编码蛋白基因受干旱胁迫诱导差异表达。 3.使用KEGG数据库对差异表达蛋白和基因参与的pathway进行分析,总共发现了47个被显著改变的pathway,其中有5个pathway在蛋白质和mRNA水平上都有发现。在我们发现的这些pathway中,很多都属于基础代谢(碳固定、尿素循环、脂肪酸代谢、氨基酸合成)。此外,也有几个属于次生代谢(苯丙烷合成、柠檬精油和松萜降解等)。 4.采用GC-MS技术进行的代谢谱分析发现了37个明显变化的代谢产物,分属于6大类,包括有机酸(38%)、糖类(19%)、氨基酸(16%)、脂肪酸(14%)、糖醇(5%)以及其它类别(8%)。 5.根据差异表达蛋白和基因的pathway分析结果并结合所发现的差异表达代谢产物,我们对植物在干旱胁迫下的代谢调节反应有了一个更全面的认识:干旱胁迫下,光合作用受到抑制,导致能量和碳水化合物输出减少。然而,植物需要大量的能量和物质用于其它一些生物学过程,比如活性氧清除和渗透物质合成等以抵御胁迫。额外的能量与物质的需求和较低的供应成为了一对矛盾,此时,需要动用植物的储备物质来解决这一矛盾。碳水化合物和脂肪酸是植物叶片中最重要的两种储备物质。干旱胁迫加速了糖酵解以及脂肪酸降解,促进了三羧酸循环以及各种氨基酸的合成,形成了从碳水化合物和脂肪酸向氨基酸方向的能量和物质的转移和利用。该过程一方面有助于累积包括脯氨酸在内的各种氨基酸,增进植物细胞的渗透调节能力,同时也为植物体合成新的蛋白做好了准备。另外,储备物质通过糖酵解以及三羧酸循环等过程,产生大量的ATP,也为植物应对干旱胁迫提供了能量。
DOI:10.3969/j.issn.1001-7461.2004.03.018URL [本文引用: 1]
从渗透调节物质、清除活性氧、保护生物大分子及其膜结构的蛋白质、转录因子及其他酶类的基因工程等方面 ,综述了目前用于植物抗旱基因工程的研究进展及存在的一些问题。
DOI:10.3969/j.issn.1001-7461.2004.03.018URL [本文引用: 1]
从渗透调节物质、清除活性氧、保护生物大分子及其膜结构的蛋白质、转录因子及其他酶类的基因工程等方面 ,综述了目前用于植物抗旱基因工程的研究进展及存在的一些问题。
DOI:10.1016/S1360-1385(97)84622-5URL [本文引用: 1]
Abstract The shikimate pathway, a collection of seven enzymatic reactions whose end product is chorismate, has been studied for many years in a variety of microorganisms and plants. In microbial systems, the end product of the pathway is used primarily for the synthesis of aromatic amino acids. In plants, chorismate is the precursor not only for the synthesis of aromatic amino acids, but also for many secondary metabolites with diverse physiological roles. Because of the dual involvement of the shikimate pathway in primary and secondary metabolism, there are long-standing questions regarding its subcellular location, and its potential coordinate regulation with other pathways that use chorismate or its products in response to environmental or developmental demands.
DOI:10.1016/S0168-9452(01)00410-1URL [本文引用: 1]
Pepper ( Capsicum annuum L.) plants growing in a nutrient solution with excess copper, showed an increase in shikimate dehydrogenase (SKDH, EC 1.1.1.25) and peroxidase (EC 1.11.1.7) activities in the hypocotyl. In the roots, peroxidase was also induced, but SKDH activity per organ was depleted rather than enhanced. Copper stress caused stunting in the plants, reflected by a decrease in the fresh weight of all the organs. In the hypocotyl, the induction of both enzymatic activities was associated with the accumulation of soluble phenolics and lignin. The two SKDH isozymes present in the control hypocotyls (SKDH-3 and SKDH-4) increased in a similar proportion after copper stress. In the case of peroxidases, two new isozymes (PRX-A2 and PRX-A4) were detected in copper-stressed hypocotyls, and the other two isoperoxidases, PRX-B and PRX-A3, were enhanced c. 10 and three times, respectively, with respect to the control. The application of the chelator EDTA was able to counteract all the stress effects of the metal cited above. The role of these enzymes in phenolic metabolism under heavy metal stress is discussed.
DOI:10.1007/PL00012490URL [本文引用: 1]
DOI:10.1093/aob/mcn125URL [本文引用: 1]
DOI:10.1016/j.jplph.2004.01.013URL [本文引用: 1]
DOI:10.1146/annurev.arplant.50.1.571URLPMID:15012220 [本文引用: 1]
74 Abstract68Many plants increase in freezing tolerance upon exposure to low nonfreezing temperatures, a phenomenon known as cold acclimation. In this review, recent advances in determining the nature and function of genes with roles in freezing tolerance and the mechanisms involved in low temperature gene regulation and signal transduction are described. One of the important conclusions to emerge from these studies is that cold acclimation includes the expression of certain cold-induced genes that function to stabilize membranes against freeze-induced injury. In addition, a family of Arabidopsis transcription factors, the CBF/DREB1 proteins, have been identified that control the expression of a regulon of cold-induced genes that increase plant freezing tolerance. These results along with many of the others summarized here further our understanding of the basic mechanisms that plants have evolved to survive freezing temperatures. In addition, the findings have potential practical applications as freezing te...
DOI:10.1016/j.bbapap.2010.01.004URLPMID:20079886 [本文引用: 1]
Enhanced salt tolerance of rice seedlings by abscisic acid (ABA) pretreatment was observed from phenotypic and physiological analyses. Total proteins from rice roots treated with ABA plus subsequent salt stress were analyzed by using proteomics method. Results showed that, 40 protein spots were uniquely upregulated in the seedlings under the condition of ABA pretreatment plus subsequent salt stress, whereas only 16 under the condition of salt treatment. About 78% (31 spots) of the 40 protein spots were only upregulated in the presence of the subsequent salt stress, indicating that plants might have an economical strategy to prevent energy loss under a false alarm. The results also showed that more enzymes involved in energy metabolism, defense, primary metabolism, etc. were upregulated uniquely in ABA-pretreated rice seedlings, suggesting more abundant energy supply, more active anabolism (nitrogen, nucleotide acid, carbohydrate, etc), and more comprehensive defense systems in ABA-pretreated seedlings than in salt stressed ones.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}