 ,*河南农业大学农学院, 河南郑州 450046
,*河南农业大学农学院, 河南郑州 450046Cloning and Function Analysis of ZmNAOD Gene in Maize
MA Chen-Yu, ZHAN Wei-Min, LI Wen-Liang, ZHANG Meng-Di, XI Zhang-Ying,*Agronomy College, Henan Agricultural University, Zhengzhou 450046, Henan, China通讯作者:
第一联系人:
收稿日期:2018-03-13接受日期:2018-06-12网络出版日期:2018-07-02
| 基金资助: |
Received:2018-03-13Accepted:2018-06-12Online:2018-07-02
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (1523KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
马晨雨, 詹为民, 李文亮, 张梦迪, 席章营. 玉米ZmNAOD基因的克隆与功能分析[J]. 作物学报, 2018, 44(10): 1433-1441. doi:10.3724/SP.J.1006.2018.01433
MA Chen-Yu, ZHAN Wei-Min, LI Wen-Liang, ZHANG Meng-Di, XI Zhang-Ying.
玉米(Zea mays L.)是全世界总产量最高的作物, 具有粮食、饲料、工业原料等多种用途, 在农业生产中占据重要地位。据联合国粮农组织(http://www. fao.org/)统计, 在全球范围内, 1998年以后, 玉米的总产量超过稻谷和小麦, 成为世界第一大粮食作物; 当前我国的玉米单位面积产量和发达国家相比还有较大的差距, 在玉米产量及相关性状功能基因的研究方面还相对落后, 用于分子设计育种的基因资源还比较少。因此, 挖掘玉米产量及相关性状的功能基因, 研究其遗传控制机制, 分析其基因功能和遗传调控网络对于进一步提高玉米单产具有重要意义。
籽粒大小是影响玉米产量的主要因素, 也是当代育种重点选育的性状之一[1]。相对而言, 大粒的种子能够提供较多的养分来应对发芽过程中的各种逆境, 从而确保苗齐苗壮, 为高产稳产奠定基础[2]。光周期的敏感性是影响玉米生育期和地理分布的主要性状[3], 研究光周期调控途径的内在机制是调控生育期和打破热带玉米种质利用障碍的关键, 对我国玉米种质资源的扩增和创新具有重要作用。
乙酰鸟氨酸脱酰酶(N-Acetylornithine deacetylase, NAOD)是鸟氨酸合成途径中的关键酶, 参与鸟氨酸合成的线性途径(linear pathway)[4,5,6], 催化N-乙酰鸟氨酸与水生成醋酸盐和鸟氨酸的反应, 在植物中参与果实发育、逆境应答、光周期调节等过程。鸟氨酸是重要的储氮化合物, 它是调节多种生长发育过程的多胺类化合物(polyamines, PAs)的前体物质之一。在植物中, PAs影响细胞分裂、花期、果实、根的形成以及个体衰老等过程[7,8,9,10,11]。
Molesini等[12]克隆得到了拟南芥的AtNAOD基因, 通过对AtNAOD基因缺失的atnaod突变体表型的调查发现, 其花期比野生型拟南芥提前, 不育荚果数目增多; 进一步研究发现, 在atnaod突变体中, 由于鸟氨酸合成途径的受阻, 花中的鸟氨酸含量比野生型低45%, 乙酰鸟氨酸的含量约比野生型高16%, 从而引起了精胺、亚精胺等PAs含量的变化, 并推测这是造成atnaod突变体花期提前和不育荚果数目增多的原因[13]; 在木瓜中, 与NAOD相似的酰化氨基酸水解酶调控果实成熟[14]; 番茄中, 单倍体果实中的NAOD基因与乙烯诱导花蕾形成相关[15]。
本研究通过RT-PCR的方法从玉米自交系昌7-2中克隆得到了一个乙酰鸟氨酸脱酰酶基因ZmNAOD, 拟从ZmNAOD的序列、时空表达、转基因拟南芥的表型等方面来研究ZmNAOD基因的功能。
1 材料与方法
1.1 试验材料
1.1.1 植物材料及处理 2016年6 月将玉米自交系昌7-2种植于人工气候室和郑州毛庄河南农业大学科教园区(34°51′54.66″N, 113°35′12.11″E)。在温室中分别于三叶期取叶片和根, 十叶期将整株幼苗剥开取茎, 3次重复; 对田间种植的材料, 标注每株的抽雄日期, 选3株生长良好且长势一致的植株, 于抽雄后第5天取其雄穗; 选长势一致的植株自交授粉, 标记授粉日期, 于授粉后第0、第5、第10、第15、第20、第25、第30、第35、第40天, 分别取3株果穗, 剥离果穗中部的籽粒, 液氮冷冻后保存于-80℃冰箱中备用。1.1.2 主要试剂和菌株 RNA 提取试剂盒为北京天恩泽柱式植物RNAout 2.0; cDNA第1 链合成试剂盒为诺唯赞HiScript II Q RT Suppermix for qPCR; qRT-PCR试剂盒为Promega生物技术有限公司的GO Taq qPCR Master Mix, 限制性核酸内切酶购自New England Biolabs公司, Premix Taq酶、pMD18-T载体和T4连接酶均购自TaKaRa (大连)公司; 大肠杆菌感受态细胞DH5α和根癌农杆菌感受态细胞GV3101购自上海唯地生物技术有限公司, 其他生化试剂购自北京索莱宝科技有限公司。
1.2 玉米ZmNAOD基因的克隆与序列分析
基于B73的GRMZM2G181273的mRNA序列, 应用Oligo 7设计克隆引物NAOD-cDNA-F: 5°-CCG ATCCTTTCCCCGTCTGA-3°、NAOD-cDNA-R: 5°-A GGGAGTATTGGTCTATCCCAT-3°, 以昌7-2授粉后5 d籽粒的总RNA 反转录的cDNA 第1 链为模板进行RT-PCR 扩增。PCR 体系(25.0 μL)含cDNA 1 μL、LA Taq DNA 酶(0.1 U μL-1) 12.5 μL、上下游引物(10 μmol L-1)各1 μL、ddH2O 9.5 μL。PCR扩增程序为95℃预变性5 min; 95℃变性50 s, 55℃退火50 s, 72℃延伸90 s, 35个循环; 72℃再延伸10 min。PCR 产物经1%琼脂糖凝胶电泳检测及胶回收纯化后, 与pMD18-T克隆载体16℃隔夜连接(约10 h), 转化大肠杆菌DH5α 感受态细胞, 将PCR检测呈阳性的菌落送华大基因测序。利用ProtParam tool在线软件分析ZmNAOD基因编码的蛋白的分子量、等电点等理化性质; 利用Signalp 4.0 server软件[16]进行信号肽预测; 利用TMHMM Server v.2.0在线软件进行跨膜结构分析; 利用SOPMA软件进行蛋白的二级结构预测; 运用Netphos 2.0 server软件进行磷酸化位点预测; 用WoLF PSORTII Prediction进行亚细胞定位预测; 利用MEGA 7.0 (NJ, 1000 BootStrap)[17]构建系统进化树。
1.3 ZmNAOD基因启动子区域的克隆与序列分析
根据玉米参考基因组(B73 RefGen_v3)第5染色体GRMZM2G181273上游的基因组序列信息 (198 620 000~198 623 032), 应用Oligo 7设计重叠引物P-ZmNAOD-F1: 5°-AATCTGCTGTTACCTTGC G-3°、P-ZmNAOD-R1: 5°-GTTGGAACTTGCTGCTT GGA-3°, P-ZmNAOD-F2: 5°-CTCCAACCCCTTACA TGC-3°、P-ZmNAOD-R2: 5°-CGGCCCAAATGTACA CCT-3°, 以昌7-2叶片的DNA为模板进行扩增, 采用25.0 μL的PCR体系, PCR程序为95℃预变性5 min; 95℃变性50 s, 分别用55℃、57℃退火50 s, 72℃延伸1 min, 35个循环; 72℃再延伸10 min。PCR产物经电泳、胶回收、载体连接和转化大肠杆菌后测序, 将两段序列比对拼接去除冗余序列后获得 ZmNAOD基因的启动子序列, 利用PlantCARE[18]在线软件分析启动子序列。1.4 ZmNAOD基因的时空表达分析
以昌7-2不同器官(根、茎、叶、雄穗、籽粒)和授粉后不同天数的籽粒中的cDNA 为模板, 以玉米18S rRNA为内参基因, 内参基因引物[19]为18S-F: 5°-CCTGCGGCTTAATTGACTC-3°、18S-R: 5°-GTTA GCAGGCTGAGGTCTCG-3°; 在ZmNAOD基因的编码序列中跨内含子设计qRT-PCR引物Q-NAOD-F: 5°-TTGAATACCCCGGCAC-3°、Q-NAOD-R: 5°-TCC AAGACAGTCGGTT-3°。反应体系含荧光定量Mix Go Taq qPCR Master Mix 2 × 12.5 μL, 引物(10 μmol L-1)各1 μL, cDNA 模板1 μL, 加DNase free水至终体积25 μL。使用罗氏LightCycler 480 II型实时荧光定量PCR仪扩增, 反应程序为94℃预变性 180 s; 94℃ 变性20 s, 58℃退火 20 s, 72℃延伸 40 s, 40个循环; 72℃读取荧光值; 循环结束后60℃保温60 s; 由熔解曲线判定PCR 反应的特异性。每个样品进行3次生物学重复及3次PCR 重复, 参照2-ΔΔCt法[20]计算相对表达量。1.5 ZmNAOD基因过表达载体的构建与遗传转化
以含有目标基因的阳性菌液为模板, 利用引物O-NAOD-F: 5°-GAAGATCTATGGCATCTCCGG-3°, O-NAOD-R: 5°-CGGACTAGTTCAGACCTCTTCT-3°扩增带有Bgl II与Spe I酶切位点的ZmNAOD基因的CDS片段; 然后, 对胶回收片段及含有CaMV35S启动子的过表达载体质粒pCAMBIA1304 在37℃双酶切4 h, 用T4连接酶16℃连接过夜(约10 h), 构建重组质粒pCAMBIA 1304-ZmNAOD。将菌液PCR呈阳性及测序正确的重组阳性质粒转入根癌农杆菌菌株GV3101 感受态细胞中, 涂布于含有卡那霉素(50 μg mL-1)和利福平(50 μg mL-1)的YEP固体培养基上, 经28℃培养72 h, 挑取单克隆点进行PCR 检测及质粒双酶切鉴定。将验证正确的阳性重组菌株GV3101 pCAMBIA1304-ZmNAOD 菌液加入含50 μg mL-1卡那霉素和50 μg mL-1利福平的YEP 液体培养基中, 于28℃振荡培养过夜(约16 h)。离心收集菌体后, 用含有200 μmol L-1 Silwet L-77的MS液体培养基重悬菌体至OD值为0.8, 使用floral dip方法[21]侵染拟南芥, 得到T0代转基因种子。1.6 纯合转基因拟南芥植株的筛选与表型鉴定
将T0代转基因种子用6.25%的NaClO溶液浸泡15 min后, 用灭菌ddH2O清洗5次, 然后平铺于含有35 μg mL-1潮霉素B的MS培养基上进行筛选; 在T1代提取拟南芥基因组DNA, 检测载体是否插入到拟南芥基因组中; 在T2和T3代, 取其叶片提取RNA进行qRT-PCR鉴定, 直至选出纯合转基因植株。取T3代纯合转基因植株的根、茎、叶、花序、荚果, 用灭菌水清洗干净后, 用吸水纸吸干水分, 保存于-80℃冰箱备用。将在MS培养皿中正常生长14 d的转基因拟南芥和野生型拟南芥放置在黑暗环境下培养10 d, 并分别调查15株转基因株系和野生型拟南芥根的长度。选择ZmNAOD基因表达量相对较高的3个转基因株系进行表型鉴定, 每个株系种植26株, 以野生型为对照, 记录每个单株出现花蕾的日期与第1朵花开花的日期。成熟时, 收获野生型拟南芥和转基因拟南芥的种子, 自然风干后, 从每个材料中随机选取1000粒净种子测定千粒重, 重复3次; 随机选取100粒净种子, 用Image J图像识别软件[22]测量每粒种子的长和宽, 重复3次。
2 结果与分析
2.1 玉米ZmNAOD基因的克隆与序列分析
玉米ZmNAOD基因的CDS全长为1344 bp, 编码447个氨基酸(图1)。该蛋白质的分子式为C2195H3441N585O661S18, 相对分子质量为49.179 kD, 理论等电点为5.45, 其中赖氨酸和亮氨酸含量最高, 均为9.4%; 有59个带负电荷的氨基酸残基(Asp + Glu)、45个带正电荷的氨基酸残基(Arg + Lys), 总体带负电荷, 属于稳定的亲水蛋白。保守结构域预测显示, 该蛋白具有高度保守的peptidase_M20结构域中的M20_ArgE-ralated亚族结构, 该亚族可能与脱乙酰基酶活动相关; 没有信号肽序列及跨膜结构, 含有18个磷酸化位点。其蛋白质的二级结构中, 含有36.42%的α-螺旋, 34.90%的无规则卷曲, 19.91%的延伸链和8.95%的β-转角; 亚细胞定位预测显示, 该蛋白有可能位于细胞质中。将该蛋白的氨基酸序列在UniProt数据库中进行BLAST并构建系统进化树(图2)表明, ZmNAOD编码的蛋白与高粱中的SORBI_3004G275600相似性最高, 为97.00%; 该蛋白与拟南芥中的AtNAOD (At4g17830)蛋白的相似度为60.54%, 在拟南芥中AtNAOD蛋白的表达与开花时间及籽粒发育有关, 其他蛋白功能尚无研究。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1ZmNAOD的CDS序列及编码的氨基酸序列
Fig. 1DNA coding sequence and amino acid alignments of ZmNAOD
图2
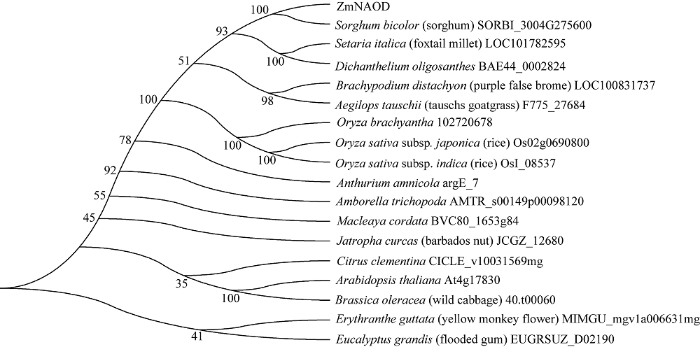 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2植物中NAOD蛋白的系统进化树
Fig. 2Phylogenetic analysis of NAOD proteins in plants
2.2 玉米ZmNAOD基因启动子区域的克隆与分析
以昌7-2基因组DNA为模板进行启动子区域的TA克隆, 将PCR产物测序后分别得到一条867 bp和一条1658 bp的片段, 将两段序列比对拼接去除冗余序列后, 最终得到一条2401 bp的序列, 通过序列比对确认为ZmNAOD基因的启动子区域。PlantCARE分析显示, 在该启动子区域含有典型的TATA-box与CAAT-box, 还有胚乳表达元件Skn-1_motif、GCN4_motif, 昼夜节律调控元件circadian, 光响应元件G-box、Sp1、ATCT-motif、LAMP-element、Box II、CATT-motif、GAG-motif、GA-motif、I-box、TGGCA-motif, 与激素相关的顺式作用元件ABRE、AuxRR-core、CGTCA-motif、TGACG-motif、TCA-element, 与逆境响应相关的HSE、LTR、MBS、TC-rich repeats, 及其他诱导型和组织特异性启动子元件等(表1)。
Table 1
表1
表1ZmNAOD启动子区域调控元件预测
Table 1
| 功能 Function | 元件名称(核心序列) Element (core sequence) |
|---|---|
| 胚乳表达元件 cis-acting regulatory element required for endosperm expression | Skn-1_motif (GTCAT), GCN4_motif (TGTGTCA) |
| 昼夜节律控制元件 cis-acting regulatory element involved in circadian control | Circadian (CAANNNNATC) |
| 光响应相关元件 cis-acting regulatory element involved in light responsiveness | G-box (CACGTC), Sp1 (CC(G/A)CCC) |
| 与光响应相关保守 DNA模块 Part of a conserved DNA module involved in light responsiveness | ATCT-motif (AATCTGATCG) |
| 部分光响应相关元件 Part of a light responsive element | LAMP-element (CTTTATCA), Box II (GTGAGGTAATAT), CATT-motif (GCATTC), GAG-motif (AGAGATG), GA-motif (ATAGATAA), I-box (aAGATAAGA), TGGCA-motif (GATGGAAGTGGCA) |
| 脱落酸响应元件 cis-acting element involved in the abscisic acid responsiveness | ABRE (AGTACGTGGC) |
| 生长素响应相关元件 cis-acting regulatory element involved in auxin responsiveness | AuxRR-core (GGTCCAT) |
| 茉莉酸甲酯响应元件 cis-acting regulatory element involved in the MeJA-responsiveness | CGTCA-motif (CGTCA), TGACG-motif (TGACG) |
| 水杨酸响应元件 cis-acting element involved in salicylic acid responsiveness | TCA-element (TCAGAAGAGG) |
| 热响应相关元件 cis-acting element involved in heat stress responsiveness | HSE (AAAAAATTTC) |
| 低温响应元件 cis-acting element involved in low-temperature responsiveness | LTR (CCGAAA) |
| MYB结合位点 MYB binding site involved in drought-inducibility | MBS (TAACTG) |
| 逆境响应元件 cis-acting element involved in defense and stress responsiveness | TC-rich repeats (ATTTTCTTCA) |
新窗口打开|下载CSV
2.3 ZmNAOD基因的时空表达分析
ZmNAOD基因的表达没有器官特异性, 在根、茎、叶、雄穗和籽粒中均有表达, 其中在雄穗中的表达量最高, 其次是籽粒, 然后是叶、茎和根(图3)。图3
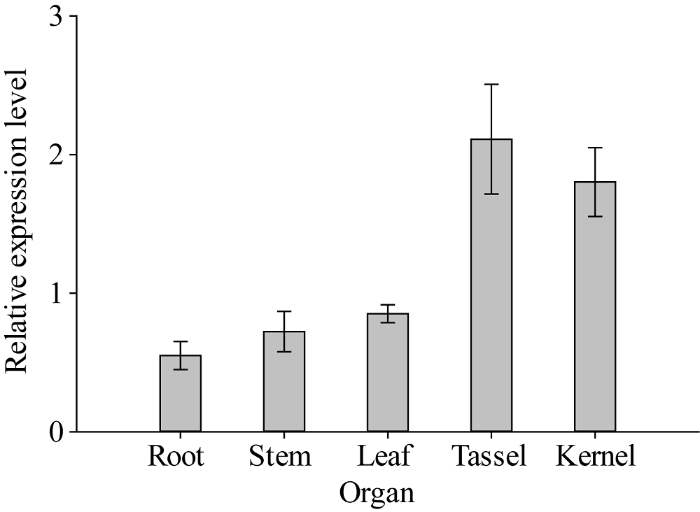 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3ZmNAOD基因在昌7-2不同器官中的表达
Fig. 3Relative expression of ZmNAOD gene in different organs in Chang 7-2
在授粉后0~15 d内, 籽粒中ZmNAOD基因的表达量呈快速上升趋势, 在授粉后15 d达到最高值, 之后迅速下降, 20 d后表达量很低(图4)。
图4
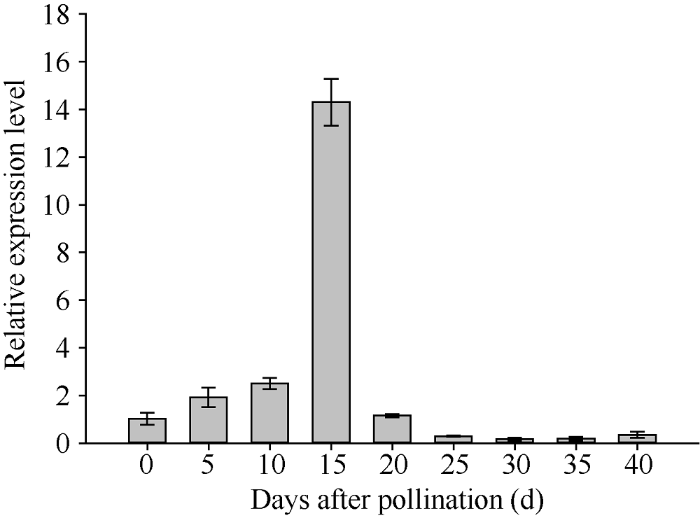 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4ZmNAOD基因在授粉后不同天数的籽粒中的表达
Fig. 4Relative expression of ZmNAOD gene in kernel at different days after pollination
2.4 ZmNAOD基因过表达载体的构建及转基因拟南芥的获得
将pCAMBIA1304载体和目的基因双酶切产物连接, 经转化大肠杆菌、菌液检测和测序后, 从阳性转化子提取质粒, 用Bgl II和Spe I进行双酶切电泳检测, 电泳结果显示存在一条约10 000 bp的载体片段条带和一条约1500 bp的目的基因条带。用滴蘸法将ZmNAOD基因转化拟南芥, 通过抗性筛选, 获得有潮霉素抗性的拟南芥, 在T1代提取拟南芥基因组DNA进行潮霉素基因的PCR鉴定, 收获具有抗性基因的种子; 从T2代取30株转ZmNAOD基因拟南芥叶片提取RNA, 反转录为cDNA, 以ZmNAOD基因在野生型拟南芥叶片中的表达量为基准进行qRT-PCR鉴定, 选择表达量相对较高的5个单株收获种子在T3代种植为株系; 在T3代提取5个株系的cDNA进行qRT-PCR鉴定表明, 获得了能够稳定转录的转基因植株(图5), 选择ZmNAOD基因表达量相对较高的T3-17、T3-23、T3-26株系收获种子进行表型鉴定。图5
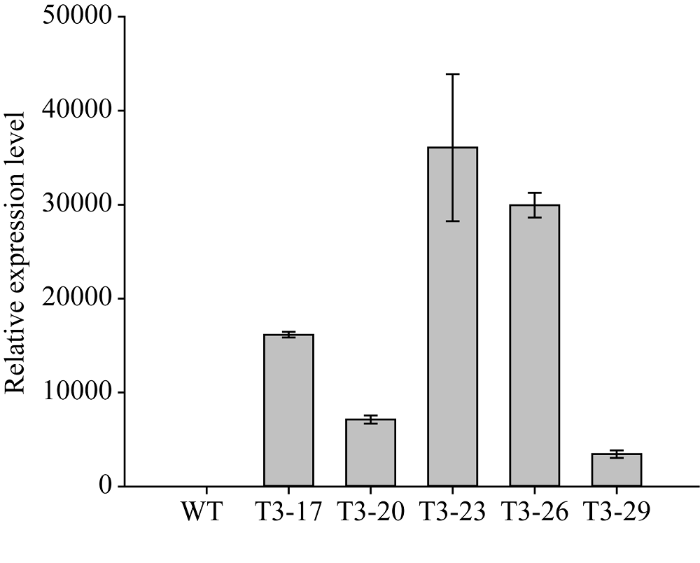 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5ZmNAOD基因在拟南芥野生型和转基因株系中的表达
Fig. 5Relative expression of ZmNAOD gene in wild type (WT) and transgenic lines
2.5 转基因拟南芥的表型鉴定
2.5.1 ZmNAOD基因在转基因拟南芥不同器官中的表达 选择ZmNAOD表达量相对较高的T3-23株系, 以ZmNAOD基因在转基因拟南芥叶片中的表达量为基准计算其在不同器官中的相对表达量。结果显示, ZmNAOD基因在根中的表达量最高, 随后是果荚、叶、花序, 在茎中的表达量最低(图6)。图6
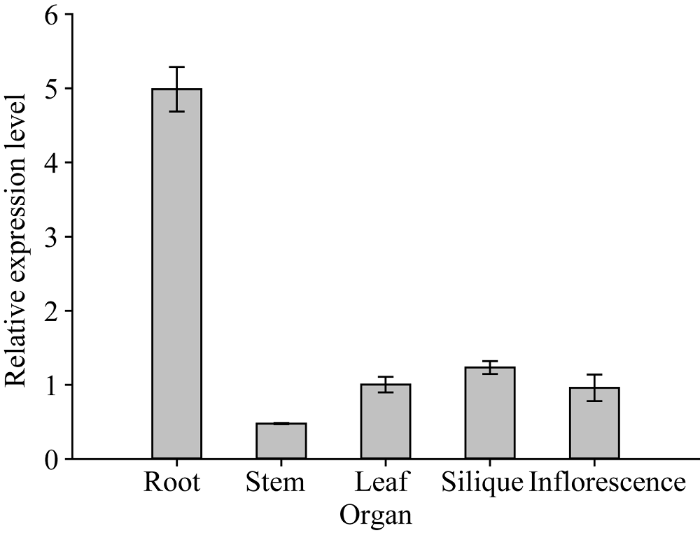 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6ZmNAOD基因在转基因拟南芥不同器官中的表达
Fig. 6Relative expression of ZmNAOD gene in different organs of transgenic plant
2.5.2 暗处理后拟南芥的根长鉴定 将野生型拟南芥种子和T3代转基因种子点播于MS培养基中, 在22℃、16 h光照/8 h黑暗的条件下培养14 d, 转移至黑暗条件下培养10 d后, 转基因株系的根长为(5.71±0.16) cm, 野生型拟南芥的根长为(4.37±0.31) cm, 前者显著大于后者。
2.5.3 转基因拟南芥的花期鉴定 野生型拟南芥及3个转基因株系(T3-17、T3-23、T3-26)用于生育期调查的有效株数依次为26、24、24、15。野生型拟南芥出现第1朵花序的天数为(37.60±0.47) d, 第1朵花开花的天数为(48.23±0.55) d; 3个转基因拟南芥株系出现第1朵花序的天数分别为(36.08±0.42) d、(34.80±1.08) d、(36.00±0.53) d, 第1朵花开花的天数分别为(46.96±0.64) d、(43.00±0.72) d、(46.00±0.65) d (表2)。转基因拟南芥株系出现第1朵花序的天数及第1朵花开花的天数均显著早于野生型拟南芥(图7)。
Table 2
表2
表2野生型拟南芥和转基因株系的生育期
Table 2
| 野生型 Wild type | T3-17 | T3-23 | T3-26 | |
|---|---|---|---|---|
| 第1朵可见花序的天数 Days of the first buds visible (d) | 37.60±0.47 | 36.08±0.42* | 34.80±1.08** | 36.00±0.53* |
| 第1朵花开花的天数 Days of the first flower visible (d) | 48.23±0.55 | 46.96±0.64* | 43.00±0.72** | 46.00±0.65** |
新窗口打开|下载CSV
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7种植45 d后转基因拟南芥和野生型拟南芥的表型对比
1, 2: 转基因株系T3-23; 3, 4: 野生型植株。
Fig. 7Flowering phenotype observed in transgenic lines and wild type lines at 45 days after sowing
1, 2: transgenic lines T3-23; 3, 4: wild type lines.
2.5.4 转基因拟南芥籽粒性状的鉴定 野生型拟南芥籽粒的长度为(0.7857±0.0380) mm, 宽度为(0.6682±0.0259) mm, 千粒重为(13.353±0.122) mg; 转基因拟南芥T3-17株系种子的长度为(0.8936± 0.0449) mm, 宽度为(0.6798±0.0128) mm, 千粒重为(14.233±0.031) mg; 转基因拟南芥T3-23株系种子的长度为(0.9004±0.0527) mm, 宽度为(0.6656± 0.0315) mm, 千粒重为(14.249±0.137) mg; 转基因拟南芥T3-26株系种子的长度为(0.9029±0.0564) mm, 宽度为(0.6646±0.0220) mm, 千粒重为(14.222±0.152) mg (表3)。转基因拟南芥的粒长和千粒重均显著大于野生型, 粒宽差异不显著。
Table 3
表3
表3转基因拟南芥籽粒的表型分析
Table 3
| 野生型 Wild type | 转基因T3-17 Transgenic T3-17 | 转基因T3-23 Transgenic T3-23 | 转基因T3-26 Transgenic T3-26 | |
|---|---|---|---|---|
| 粒长 Seed length (mm) | 0.7857±0.0380 | 0.8936±0.0449** | 0.9004±0.0527** | 0.9029±0.0564** |
| 粒宽 Seed width (mm) | 0.6682±0.0259 | 0.6798±0.0128 | 0.6656±0.0315 | 0.6646±0.0220 |
| 千粒重 Thousand-seed weight (mg) | 13.353±0.122 | 14.233±0.031** | 14.249±0.137** | 14.222±0.152** |
新窗口打开|下载CSV
3 讨论
乙酰鸟氨酸脱酰酶是鸟氨酸合成途径中的一种关键酶, 参与鸟氨酸合成中的线性途径, 催化N-乙酰鸟氨酸与水生成醋酸盐和鸟氨酸的反应。在植物中有很多推测的NAOD蛋白的编码序列, 其中的一些蛋白质与生长发育调节相关。Molesini等[8]克隆得到拟南芥的AtNAOD基因, 该基因的缺失使不育荚果数增加, 使花期提前。番茄单倍体果实中NAOD基因与花蕾形成有关[15]。本研究显示, ZmNAOD基因在玉米雄穗和籽粒中的表达量均较高, 在授粉后不同天数的籽粒中的表达呈现0~15 d迅速上升之后快速下降的趋势, ZmNAOD基因的启动子区域含有多种与胚乳表达相关的元件, 过表达转基因拟南芥籽粒的千粒重显著大于野生型。这些结果表明, ZmNAOD基因可能参与玉米籽粒发育的调控。雄穗和雌穗花序均是由花序分生组织(inflorescence meristem, IM)分化而来的[23], 有很多基因同时对雄穗和雌穗性状起着调控作用, 例如fasciated ea2 (fea2)基因对玉米雄花小穗的数量产生影响, 同时还影响雌穗的行数[24,25]; unbranched2 (ub2)和unbranched3 (ub3)基因的双缺失突变体的雄穗分枝数和穗行数都会增加[26]。本研究中, ZmNAOD基因在雄穗和籽粒中的表达量均较高, 可能是该基因同时参与雄穗和雌穗发育的调控。在ZmNAOD基因启动子区域发现的GCN4-motif和E-box顺式作用元件是许多籽粒特异表达基因的启动子部分常有的元件[27]。张冬梅等[28]的研究结果表明, 玉米自交系昌7-2籽粒的平均灌浆速率在授粉后前15 d迅速增快, 随后开始逐渐降低, 这与本研究中ZmNAOD基因在授粉后不同天数的籽粒中的表达趋势基本一致。可能是因为ZmNAOD基因表达量的增加, 催化了N-乙酰鸟氨酸与水生成醋酸盐和鸟氨酸的反应, 使得鸟氨酸快速积累, 进而引起植物体内PAs含量的变化, 导致干物质积累速率改变[10,13], 灌浆速率增加。过表达转基因拟南芥籽粒的千粒重显著大于野生型, 可能与NAOD参与植物果实发育的机制[29]有关。
本研究中, ZmNAOD基因的启动子区域含有大量的光响应元件, ZmNAOD基因过表达的转基因拟南芥的花期与野生型拟南芥相比显著提前, ZmNAOD基因在转基因拟南芥根中的表达量最高, 经过10 d的黑暗处理后的转基因拟南芥根长显著大于野生型拟南芥。Qiao等[30]克隆得到一个玉米肌动蛋白解聚因子基因ZmADF3, 并构建过表达载体1302-ZmADF3将其转入拟南芥植株, 暗培养8 d后发现转基因株系的下胚轴长度比野生型增加55.8%, 在ZmADF3的启动子区域也发现了许多与光照有关的调控元件, 推测ZmADF3基因的功能与光响应相关。本研究发现黑暗条件能够促进转基因拟南芥根的伸长, 说明光照可能对ZmNAOD基因的表达起着调控作用。ZmNAOD基因的启动子区域的G-box、GA motif、GAG motif、I-box等元件常在光调控基因的启动子区域被发现, 它们对光调控的转录活动至关重要[31,32,33]。这些结果表明, ZmNAOD基因的表达可能与玉米光周期的调节相关。
4 结论
克隆了玉米乙酰鸟氨酸脱酰酶基因ZmNAOD, 该基因启动子区域含有与胚乳发育、光响应等有关的顺式作用元件。ZmNAOD基因在昌7-2的不同器官中均有表达, 在授粉后不同天数的籽粒中的表达量先上升后下降, 与昌7-2的灌浆速率的变化基本一致; ZmNAOD基因在拟南芥中的过表达使花期提前, 使转基因株系种子长度和千粒重显著增加, 推测ZmNAOD基因的表达可能与玉米的籽粒发育和光周期有关。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/ng.169URLPMID:18604208 [本文引用: 1]

The domestication of crops involves a complex process of selection in plant evolution and is associated with changes in the DNA regulating agronomically important traits. Here we report the cloning of a newly identified QTL, qSW5 (QTL for seed width on chromosome 5), involved in the determination of grain width in rice. Through fine mapping, complementation testing and association analysis, we found that a deletion in qSW5 resulted in a significant increase in sink size owing to an increase in cell number in the outer glume of the rice flower; this trait might have been selected by ancient humans to increase yield of rice grains. In addition, we mapped two other defective functional nucleotide polymorphisms of rice domestication-related genes with genome-wide RFLP polymorphisms of various rice landraces. These analyses show that the qSW5 deletion had an important historical role in artificial selection, propagation of cultivation and natural crossings in rice domestication, and shed light on how the rice genome was domesticated.
DOI:10.1126/science.1104863URLPMID:15681384 [本文引用: 1]
Abstract Improved phylogenies and the accumulation of broad comparative data sets have opened the way for phylogenetic analyses to trace trait evolution in major groups of organisms. We arrayed seed mass data for 12,987 species on the seed plant phylogeny and show the history of seed size from the emergence of the angiosperms through to the present day. The largest single contributor to the present-day spread of seed mass was the divergence between angiosperms and gymnosperms, whereas the widest divergence was between Celastraceae and Parnassiaceae. Wide divergences in seed size were more often associated with divergences in growth form than with divergences in dispersal syndrome or latitude. Cross-species studies and evolutionary theory are consistent with this evidence that growth form and seed size evolve in a coordinated manner.
DOI:10.1038/nrg3291URLPMID:22898651 [本文引用: 1]
Plants respond to the changing seasons to initiate developmental programmes precisely at particular times of year. Flowering is the best characterized of these seasonal responses, and in temperate climates it often occurs in spring. Genetic approaches in Arabidopsis thaliana have shown how the underlying responses to changes in day length (photoperiod) or winter temperature (vernalization) are conferred and how these converge to create a robust seasonal response. Recent advances in plant genome analysis have demonstrated the diversity in these regulatory systems in many plant species, including several crops and perennials, such as poplar trees. Here, we report progress in defining the diverse genetic mechanisms that enable plants to recognize winter, spring and autumn to initiate flower development.
DOI:10.1016/j.plaphy.2005.06.007URLPMID:20202022020202020202020 [本文引用: 1]
Arabidopsis genes encoding enzymes for each of the eight steps in l-arginine (Arg) synthesis were identified, based upon sequence homologies with orthologs from other organisms. Except for N-acetylglutamate synthase (NAGS; EC 2.3.1.1), which is encoded by two genes, all remaining enzymes are encoded by single genes. Targeting predictions for these enzymes, based upon their deduced sequences, and subcellular fractionation studies, suggest that most enzymes of Arg synthesis reside within the plastid. Synthesis of the l-ornthine (Orn) intermediate in this pathway from l-glutamate occurs as a series of acetylated intermediates, as in most other organisms. An N-acetylornithine:glutamate acetyltransferase (NAOGAcT; EC 2.3.1.35) facilitates recycling of the acetyl moiety during Orn formation (cyclic pathway). A putative N-acetylornithine deacetylase (NAOD; EC 3.5.1.16), which participates in the inear pathway for Orn synthesis in some organisms, was also identified. Previous biochemical studies have indicated that allosteric regulation of the first and, especially, the second steps in Orn synthesis (NAGS; N-acetylglutamate kinase (NAGK), EC 2.7.2.8) by the Arg end-product are the major sites of metabolic control of the pathway in organisms using the cyclic pathway. Gene expression profiling for pathway enzymes further suggests that NAGS, NAGK, NAOGAcT and NAOD are coordinately regulated in response to changes in Arg demand during plant growth and development. Synthesis of Arg from Orn is further coordinated with pyrimidine nucleotide synthesis, at the level of allocation of the common carbamoyl-P intermediate.
DOI:10.1016/0031-9422(88)80404-7URL [本文引用: 1]
The biosynthesis of ornithine, and the biosynthesis and metabolism of arginine, are complex processes that have been shown to be strictly compartmented
DOI:10.1042/BJ20030002URLPMID:12633501 [本文引用: 1]
Abstract N -Acetylglutamate (NAG) fulfils distinct biological roles in lower and higher organisms. In prokaryotes, lower eukaryotes and plants it is the first intermediate in the biosynthesis of arginine, whereas in ureotelic (excreting nitrogen mostly in the form of urea) vertebrates, it is an essential allosteric cofactor for carbamyl phosphate synthetase I (CPSI), the first enzyme of the urea cycle. The pathway that leads from glutamate to arginine in lower organisms employs eight steps, starting with the acetylation of glutamate to form NAG. In these species, NAG can be produced by two enzymic reactions: one catalysed by NAG synthase (NAGS) and the other by ornithine acetyltransferase (OAT). In ureotelic species, NAG is produced exclusively by NAGS. In lower organisms, NAGS is feedback-inhibited by L-arginine, whereas mammalian NAGS activity is significantly enhanced by this amino acid. The NAGS genes of bacteria, fungi and mammals are more diverse than other arginine-biosynthesis and urea-cycle genes. The evolutionary relationship between the distinctly different roles of NAG and its metabolism in lower and higher organisms remains to be determined. In humans, inherited NAGS deficiency is an autosomal recessive disorder causing hyperammonaemia and a phenotype similar to CPSI deficiency. Several mutations have been recently identified in the NAGS genes of families affected with this disorder.
[本文引用: 1]
DOI:10.1146/annurev.pp.40.060189.001315URL [本文引用: 2]
do polyamines have roles in plant development?
DOI:10.1104/pp.104.041699URL [本文引用: 1]
The cellular polyamines putrescine, spermidine, and spermine are ubiquitous in nature and have been implicated in a wide range of growth and developmental processes. There is little information, however, on mutant plants or animals defective in the synthesis of polyamines. The Arabidopsis genome has two genes encoding spermidine synthase, SPDS1 and SPDS2. In this paper, we describe T-DNA insertion mutants of both of these genes. While each mutant allele shows normal growth, spds1-1 spds2-1 double-mutant seeds are abnormally shrunken and they have embryos that are arrested morphologically at the heart-torpedo transition stage. These seeds contain significantly reduced levels of spermidine and high levels of its precursor, putrescine. The embryo lethal phenotype of spds1-1 spds2-1 is complemented by the wild-type SPDS1 gene. In addition, we observed a nearly identical seed phenotype among an F2seed population from the cross between the spds2-1 allele and SPDS1 RNA interference transgenic lines. These data provide the first genetic evidence indicating a critical role of the spermidine synthase in plant embryo development.
DOI:10.1093/treephys/26.6.791URLPMID:16510395 [本文引用: 2]
To investigate the role of polyamines in pre- and post-harvest fruit development of 'Akatsuki' peach (Prunus persica (L.) Batsch.) we measured polyamine concentrations, activities of polyamine biosynthetic enzymes and expression of genes encoding these enzymes. Concentrations of the free polyamines, putrescine (Put), spermidine (Spd) and spermine (Spm) in pre-harvest fruit peaked 16 days after full bloom (DAF) and then progressively decreased until harvest with the exception of Put, which showed a second peak at 94 DAF, just before the onset of ethylene production. In post-harvest fruit, minor changes in concentrations of Spd and Spm were observed, whereas Put concentration peaked on the harvest day, followed by an abrupt decrease and a subsequent 2-fold increase, which was opposite to the fluctuating pattern of ethylene production. Activities of arginine decarboxylase (ADC) and ornithine decarboxylase (ODC) peaked during the first stage of fruit development and then decreased until 80 DAF, after which the activities were below detection limits, suggesting that Put is synthesized during the early stage of fruit development. Activity of S-adenosylmethionine decarboxylase (SAMDC) decreased progressively until the end of S2. Expression levels of five putative polyamine biosynthetic genes, ADC, ODC, SAMDC, spermidine synthase (SPDS) and spermine synthase (SPMS), in pre-harvest and post-harvest fruit did not coincide precisely with the observed changes in enzymatic activities and polyamine concentrations. The possible role of polyamines during peach fruit development and the relationship between polyamines and ethylene biosynthesis are discussed.
DOI:10.1007/s00425-014-2055-9URLPMID:24659098 [本文引用: 1]
Compelling evidence indicates that free polyamines (PAs) (mainly putrescine, spermidine, spermine, and its isomer thermospermine), some PA conjugates to hydroxycinnamic acids, and the products of PA...
[本文引用: 1]
[本文引用: 2]
DOI:10.1186/1471-2229-12-242URLPMID:3562526 [本文引用: 1]
Background Papaya ( Carica papaya L.) is a commercially important crop that produces climacteric fruits with a soft and sweet pulp that contain a wide range of health promoting phytochemicals. Despite its importance, little is known about transcriptional modifications during papaya fruit ripening and their control. In this study we report the analysis of ripe papaya transcriptome by using a cross-species (XSpecies) microarray technique based on the phylogenetic proximity between papaya and Arabidopsis thaliana . Results Papaya transcriptome analyses resulted in the identification of 414 ripening-related genes with some having their expression validated by qPCR. The transcription profile was compared with that from ripening tomato and grape. There were many similarities between papaya and tomato especially with respect to the expression of genes encoding proteins involved in primary metabolism, regulation of transcription, biotic and abiotic stress and cell wall metabolism. XSpecies microarray data indicated that transcription factors (TFs) of the MADS -box, NAC and AP2 / ERF gene families were involved in the control of papaya ripening and revealed that cell wall-related gene expression in papaya had similarities to the expression profiles seen in Arabidopsis during hypocotyl development. Conclusion The cross-species array experiment identified a ripening-related set of genes in papaya allowing the comparison of transcription control between papaya and other fruit bearing taxa during the ripening process.
DOI:10.1186/1756-0500-2-143URLPMID:2718906 [本文引用: 2]
Fruit normally develops from the ovary after pollination and fertilization. However, the ovary can also generate seedless fruit without fertilization by parthenocarpy. Parthenocarpic fruit development has been obtained in tomato (Solanum lycopersicum) by genetic modification using auxin-synthesising gene(s) (DefH9-iaaM;DefH9-RI-iaaM) expressed specifically in the placenta and ovules. We have performed a cDNA Amplified Fragment Length Polymorphism (cDNA-AFLP) analysis on pre-anthesis tomato flower buds (0.5 cm long) collected fromDefH9-iaaMandDefH9-RI-iaaMparthenocarpic and wild-type plants, with the aim to identify genes involved in very early phases of tomato fruit development. We detected 212 transcripts differentially expressed in auxin-ipersynthesising pre-anthesis flower buds, 65 of them (31%) have unknown function. Several differentially expressed genes show homology to genes involved in protein trafficking and protein degradation via proteasome. These processes are crucial for auxin cellular transport and signaling, respectively. The data presented might contribute to elucidate the molecular basis of the fruiting process and to develop new methods to confer parthenocarpy to species of agronomic interest. In a recently published work, we have demonstrated that one of the genes identified in this screening, corresponding to #109 cDNA clone, regulates auxin-dependent fruit initiation and its suppression causes parthenocarpic fruit development in tomato.
DOI:10.1038/nmeth.1701URLPMID:21959131 [本文引用: 1]
Nat Methods. 2011 Sep 29;8(10):785-6. doi: 10.1038/nmeth.1701. Letter
DOI:10.1093/molbev/msw054URLPMID:27004904 [本文引用: 1]
Abstract. We present the latest version of the Molecular Evolutionary Genetics Analysis (M ega ) software, which contains many sophisticated methods and tools
DOI:10.1093/nar/30.1.325URL [本文引用: 1]
[本文引用: 1]
DOI:10.1006/meth.2001.1266URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00122-005-0133-xURLPMID:16395569 [本文引用: 1]
Maize ( Zea mays L.) ear inflorescence architecture is directly relevant to grain yield components, and tassel architecture is relevant to hybrid seed production. The objectives of this study were to (1) determine heritabilities and correlations of a comprehensive set of tassel and ear inflorescence architecture traits in a set of (Illinois Low Protein B73) B73 S 1 families, (2) identify chromosomal positions of QTL affecting tassel and ear architecture, and (3) identify possible candidate genes associated with these QTL. For tassel traits, the number of detected QTL ranged from one to five, and explained between 6.5 and 35.9% of phenotypic variation. For ear traits, the number of detected QTL ranged from one to nine and phenotypic variation explained by those QTL varied between 7.9 and 53.0%. We detected QTL for tassel architecture traits that required calculation of ratios from measured traits. Some of these calculated traits QTL were detected in regions that did not show QTL for the measured traits, suggesting that calculation of ratios may reveal developmentally relevant patterns of tassel architecture. We detected a QTL on chromosome 7 for tassel branch number near the gene ramosa1 ( ra1 ), which is known to control tassel branch number, making ra1 a candidate gene for tassel branch number. We detected QTL for several traits on chromosomes 6, 8, and 9, where no inflorescence architecture genes have been mapped, thus providing initial information towards new gene discovery for control of inflorescence architecture.
[本文引用: 1]
DOI:10.1038/ng.2534URLPMID:23377180 [本文引用: 1]
Domestication of cereal crops, such as maize, wheat and rice, had a profound influence on agriculture and the establishment of human civilizations. One major improvement was an increase in seed number per inflorescence, which enhanced yield and simplified harvesting and storage(1,2). The ancestor of maize, teosinte, makes 2 rows of kernels, and modern varieties make similar to 8-20 rows(3). Kernel rows are initiated by the inflorescence shoot meristem, and shoot meristem size is controlled by a feedback loop involving the CLAVATA signaling proteins and the WUSCHEL transcription factor(4,5). We present a hypothesis that variation in inflorescence meristem size affects kernel row number (KRN), with the potential to increase yield. We also show that variation in the CLAVATA receptor-like protein FASCIATED EAR2 leads to increased inflorescence meristem size and KRN. These findings indicate that modulation of fundamental stem cell proliferation control pathways has the potential to enhance crop yields.
[本文引用: 1]
DOI:10.2307/3869791URLPMID:8281041 [本文引用: 1]
The sulfhydryl (SH) modifying reagent N-ethylmaleimide (NEM) was applied to the internodal cells of Chara corallina to study the role of SH residues in the activity of the plasma membrane H+ pump. NEM (1 mu M) caused a marked depolarizing shift of the resting potential by 64+/-10 mV (n=7) together with depression of the conductance peak at around -200 mV, indicating a marked depression of the H+ pump activity. This effect of NEM was partly reversible, the membrane repolarized and the conductance peak was restored after extracellular washing. The H+ pump inhibitor, dicyclohexylcarbodiimide (DCCD), caused no additive membrane depolarization and/or depression of the H+ pump conductance, in the presence of NEM. This suggests that NEM blocks the H+ pump and that SH residues play a pivotal role in maintaining the H+ pump activity in Chara corallina.
[本文引用: 1]
[本文引用: 1]
In: ?stergaard L, ed.
URL [本文引用: 1]
Fruit development and seed dispersal are major topics within plant and crop sciences research with important developments in research being reported regularly. Drawing together reviews by some of the world's leading experts in these areas, the Editor of this volume, Lars Ostergaard has provided a volume which is an essential purchase for all those working in plant and crop sciences worldwide.
[本文引用: 1]
[本文引用: 1]
DOI:10.1146/annurev.pp.46.060195.002305URL [本文引用: 1]
DOI:10.1006/scel.1994.1040URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}