 ,*, 肖应辉,*湖南农业大学农学院 / 水稻油菜抗病育种湖南省重点实验室 / 南方粮油作物协同创新中心, 湖南长沙 410128
,*, 肖应辉,*湖南农业大学农学院 / 水稻油菜抗病育种湖南省重点实验室 / 南方粮油作物协同创新中心, 湖南长沙 410128Fine Mapping and Candidate Gene Analysis of Rice Blast Resistance Gene Pi47
XIAO Xiang-Yi, SHI Xue-Tao, SHENG Hao-Wen, LIU Jin-Ling,*, XIAO Ying-Hui,*Agronomy College of Hunan Agricultural University / Hunan Provincial Key Laboratory of Rice and Rapeseed Breeding for Disease Resistance / Southern Regional Collaborative Innovation Center for Grain and Oil Crops in China, Changsha 410128, Hunan, China通讯作者:
收稿日期:2017-11-21接受日期:2018-03-26网络出版日期:2018-04-27
| 基金资助: |
Received:2017-11-21Accepted:2018-03-26Online:2018-04-27
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (1350KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
肖湘谊, 史学涛, 盛浩闻, 刘金灵, 肖应辉. 水稻抗稻瘟病基因Pi47的精细定位和候选基因分析[J]. 作物学报, 2018, 44(7): 977-987. doi:10.3724/SP.J.1006.2018.00977
XIAO Xiang-Yi, SHI Xue-Tao, SHENG Hao-Wen, LIU Jin-Ling, XIAO Ying-Hui.
稻瘟病是由子囊真菌(Magnaporthe oryzae)引起的水稻最严重病害之一, 至少分布在85个国家, 每年导致水稻减产10%~30%[1]。使用化学药剂和推广抗病新品种是防治稻瘟病的主要方法。大量施用化学药剂不仅污染环境, 危及环境生物和食品安全, 还对人类健康构成潜在威胁。因此, 利用抗病基因选育抗病水稻新品种是防治稻瘟病最有效、经济和安全的措施[2]。然而, 由于稻瘟菌致病性迅速和频繁变异, 导致抗病品种种植3~5年后就丧失了抗性[3]。因此, 不断挖掘新的抗病种质和克隆新的抗病基因是水稻抗稻瘟病育种的基础和核心。
其中, 克隆稻瘟病抗性基因是解析水稻抗病分子遗传机制的重要基础。近年, 通过图位克隆技术分离了28个稻瘟病抗性基因, 分别是编码NBS- LRR蛋白的Pb1[4]、Pia[5]、Pib[6]、Pid3[7]、Pik[8]、Pik-h/Pi54[9]、Pik-m[10]、Pik-p[11]、Pish[12]、Pit[13]、Pita[14]、Piz-t[15]、Pi1[16]、Pi2[15]、Pi5[17]、Pi9[18]、Pi25[7]、Pi36[19]、Pi37[20]、Pi56[21]、Pi63[22]、PiCO39[23]、Pi50[24]、Pi65(t)[25]和Pigm[26]; 编码B-Lectin 蛋白激酶的Pid2[27]; 和编码富含脯氨酸蛋白的唯一隐性抗性基因pi21[28]。通过等位基因挖掘技术克隆了Pid3-A4、Pi54of和Pi54rh[29,30,31]。这些稻瘟病抗性基因分布于除第3染色体外的所有水稻染色体上, 其中第6染色体Pi9位点、第11染色体Pik位点和第12染色体Pita位点是稻瘟病抗性基因成簇分布的3个热点区域。Pik位点位于水稻第11染色体长臂近端粒处, 是水稻一个主效抗稻瘟病位点, 该位点具有Pik、Pikm、Pikp、Pikh、Pi1、Piks等多个抗稻瘟病等位基因。研究表明, Pik位点等位基因介导的抗性由2个紧密连锁、具有独立功能的NBS-LRR基因协同作用[10]。NBS包含3个高度保守的功能基序激酶1a、激酶2a和激酶3a, 它们与ATP或GTP结合后获得能量用于抵抗病原菌; 不同的抗病基因其LRR域也相差较大, 差别主要体现在重复序列的长度、数目和位置上; LRR域与病原小种的特异识别相关, 不同的抗病基因拥有不同的抗谱, 这跟 LRR域的不同有较大的关系[32]。最近, Zhang等[33]从全基因组范围内132个位点的332个NBS-LRR基因中鉴定到98个抗稻瘟病新基因, 揭示了水稻基因组存在丰富的NBS-LRR类抗病基因从而对不同稻瘟菌生理小种产生抗性。
湘资3150是湖南省一个地方水稻品种, 在几十年种植过程中一直保持对稻瘟病广谱、持久抗性[34]。本实验室Huang等[34]利用湘资3150与高感品种CO39杂交构建的重组自交系群体(RIL)定位了2个主效抗瘟基因Pi47和Pi48, 其中Pi47被定位于第11染色体的SSR标记RM206和RM224之间的33.5 cM染色体区域。本研究利用Pi47单基因系与感病品种CO39构建F2分离群体, 旨在对Pi47进行精细定位和候选基因预测分析, 明确Pi47与定位区间已有稻瘟病抗性基因的等位关系, 为进一步克隆分离该基因奠定基础。
1 材料与方法
1.1 植物材料与种植方法
抗性基因供体材料湘资3150 (XZ), 高感稻瘟病材料CO39, Pi47单基因系CX18、CX52和CX279, 以及单基因系与CO39的F2群体。种子均由湖南农业大学水稻基因组实验室提供。CO39背景的近等基因系IRBLk-Ka (Pik)、IRBLkh-K3 (Pikh)、IRBLkm-Ts (Pikm)、IRBLkp-K60 (Pikp), 由国际水稻研究所提供。在室温下浸种, 露白后播种于装有泥土的塑料育秧盘中(18 cm × 10 cm × 5 cm), 以CO39为感病对照。育苗土是预先经过酸化处理, 并添加草炭、N、P、K肥和杀菌剂敌克松的田间土壤, 或草炭蛙石(2∶1)混合的基质。冬春季育苗在温室内进行, 夏秋季在田间进行。于稻苗二叶期浇施(NH4)2SO4一次(2 g 盘-1), 促使秧苗嫩绿、健壮。
1.2 供试菌系
供试稻瘟菌株共17个, 包括来自中国湖南的318-2、CHL473、X2007A-3、CHL440、CHL438、X2007A-7、195-2-2、87-4、193-1-1、110-2、236-1; 来自中国广东的RB4、RB6; 来自中国福建的RB18、RB19; 来源不详的中国小种RB12; 以及来自日本的KOH。菌种均由湖南农业大学水稻基因组实验室收集保存。1.3 接种与病害调查
称取燕麦片30 g磨成粉状, 约200 g成熟番茄压榨成汁, 琼脂粉12 g, 混匀后加蒸馏水定容至1 L。灭菌并分装于培养皿中, 凝固后用薄膜封藏备用。将纯化保存的稻瘟病菌接种于培养基上, 于27℃培养箱中暗培养7 d左右。待其菌落直径达4~5 cm, 使用无菌水在超净工作台上洗掉培养基表面的气生菌丝, 晾干后置弱光下培养3~4 d即产孢。用1%的Tween 20洗下培养基表面的分生孢子, 双层纱布过滤收集, 10×10倍显微镜下调至每视野30~40个孢子为宜。
将三叶一心期的水稻幼苗移至接种箱中(50 cm × 45 cm × 35 cm), 用高压喷雾器将配制好的孢子悬浮液均匀喷施于水稻叶片上。接种后, 在温度26~28℃、相对湿度85%的黑暗条件下保湿24 h; 再在相同温度及相对湿度75%条件下光照培养5~7 d后, 参照国际水稻研究所0~9级发病评判标准[35]调查发病情况。
1.4 STS标记和CAPS标记的开发
从水稻基因组计划(Rice Genome Project, RGP)网站(1.5 DNA提取、PCR扩增及电泳检测
采用CTAB法提取DNA[34]。将DNA稀释成 20 ng μL-1, 作为PCR扩增的模板。PCR体系10 μL, 包含DNA模板1 μL (20 ng μL-1)、SSR引物1 μL (含正反向引物, 2 pmol μL-1)、10 × buffer (含Mg2+) 1 μL、dNTP (2.5 mmol L-1) 0.2 μL、Taq DNA聚合酶0.1 μL (5 U μL-1, 购自宝生物工程(大连)有限公司)、ddH2O 6.7 μL。用美国Thermo公司PCR1000 DNA热循环仪进行扩增反应, 反应程序为94℃预变性4 min, 94℃变性30 s、55℃退火30 s、72℃延伸1 min、35个循环, 72℃延伸7 min。扩增产物经8%非变性聚丙烯酰胺凝胶(PAGE)电泳分离, 银染显色, 用WD-9406型胶片观察仪读胶记录基因型数据。1.6 遗传与物理图谱的构建和候选基因预测
采用隐性群体分析法(RCA)进行遗传图谱的构建, 标记重组率r = (N1+N2/2)/N, 其中N1为各标记带有抗病亲本纯合基因型的个体数, N2为带有双亲杂合基因型的个体数, N为感病群体的总株数。各标记与基因间的遗传距离按每1%的重组率合1 cM的图距计算。最后根据各标记间的遗传距离绘制Pi47基因位点的遗传连锁图谱。利用遗传图谱构建中连锁标记信息, 在水稻日本晴参考基因组IRGSP-1.0版本数据库中进行BLAST比对, 确定各连锁标记在参考基因组上的物理位置, 进而绘制Pi47位点的物理图谱。并查找各标记所在BAC克隆, 构建Pi47位点的BAC克隆重叠群。
利用RGP在线基因预测软件RiceGAAS (
1.7 序列比对和系统进化树构建
使用Muscle (2 结果与分析
2.1 Pi47单基因系筛选与精细定位群体的构建
采用分别与Pi47连锁的标记RM206、RM224和RM5926, 以及与Pi48连锁的标记RM3102、RM1337和RM7102对CO39/XZ RIL F10群体各株系进行基因型分析, 筛选出Pi47连锁标记RM206、RM224和RM5926为抗病亲本XZ基因型、Pi48连锁标记RM3102、RM1337和RM7102为感病亲本CO39基因型的12个株系, 记为Pi47单基因系(表1)。Table 1
表1
表1CO39/湘资3150 RIL中Pi47单基因系的鉴定
Table 1
| 株系 Line | 接种菌株193-1-1的抗性表型 Phenotype inoculated with isolate 193-1-1 | Pi47连锁标记 Markers linked to Pi47 | Pi48连锁标记 Markers linked to Pi48 | |||||
|---|---|---|---|---|---|---|---|---|
| RM206 | RM224 | RM5926 | RM3102 | RM1337 | RM7102 | |||
| CX18 | R | + | + | + | - | - | - | |
| CX36 | R | + | + | + | - | - | - | |
| CX40 | R | + | + | + | - | - | - | |
| CX52 | R | + | + | + | - | - | - | |
| CX66 | R | + | + | + | - | - | - | |
| CX67 | R | + | + | + | - | - | - | |
| CX70 | R | + | + | + | - | - | - | |
| CX147 | R | + | + | + | - | - | - | |
| CX152 | R | + | + | + | - | - | - | |
| CX155 | R | + | + | + | - | - | - | |
| CX166 | R | + | + | + | - | - | - | |
| CX279 | R | + | + | + | - | - | - | |
+ : the genotype of the resistant parent; -: the genotype of the susceptible parent.
新窗口打开|下载CSV
为了进一步对Pi47基因精细定位, 将12个单基因系分别与感病亲本CO39杂交并自交构建F2群体, 选取种子量较多的CX18/CO39、CX52/CO39、CX155/CO39与CX279/CO39的F2群体进行接种鉴定。结果表明除CX155/CO39外, 其他3个群体抗感比例都接近3∶1, 符合单基因分离规律(表2)。因此, 以这3个群体接种后存活的1687株感病单株作为精细定位作图群体, 其中CX18/CO39群体308株, CX52/CO39群体247株, CX279/CO39群体1132株(表2)。
Table 2
表2
表2各F2群体抗稻瘟病表型分离分析
Table 2
| F2群体 F2 population | 抗病单株 Resistant individuals | 感病单株 Susceptible individuals | 作图群体株数 Individual number of mapping population | 卡方值 χ2 for 3:1 ratio |
|---|---|---|---|---|
| CX18/CO39 F2 | 1040 | 329 | 308 | 0.68 |
| CX52/CO39 F2 | 1116 | 340 | 247 | 2.21 |
| CX155/CO39 F2 | 1512 | 310 | 0 | 62.0 |
| CX279/CO39 F2 | 3820 | 1221 | 1132 | 1.66 |
| 总数 Total | / | / | 1687 | / |
新窗口打开|下载CSV
2.2 Pi47的精细定位
利用初定位的2个侧翼标记RM224、RM5926对CX52/CO39 F2群体中247个感病单株(表2)进行基因型分析, 用RM224鉴定到2个重组子, 重组率为0.4%; 而用RM5926鉴定到不同于RM224的11个重组子, 重组率为2.23% (表3)。表明Pi47位于RM224和RM5926之间, 与前述的初定位结果吻合(图1)。Table 3
表3
表3所用标记在3个F2群体中鉴定到的重组子数量
Table 3
| 群体 Population | 感病单株数目 No. of susceptible individuals | 重组子分布 Distribution of recombinants | |||||||
|---|---|---|---|---|---|---|---|---|---|
| RM224 | S11, S4, S32 | S10 | K33 | K107 | K134 | RM5926 | |||
| CX279/CO39F2 | 1132 | 20 | 4 | 0 | 1 | 1 | 8 | 56 | |
| CX18/CO39F2 | 308 | 5 | 2 | 0 | 0 | 0 | 1 | 8 | |
| CX52/CO39F2 | 247 | 2 | 1 | 0 | 0 | 0 | 1 | 11 | |
| 合计 Total | 1687 | 27 | 7 | 0 | 1 | 1 | 10 | 75 | |
新窗口打开|下载CSV
图1
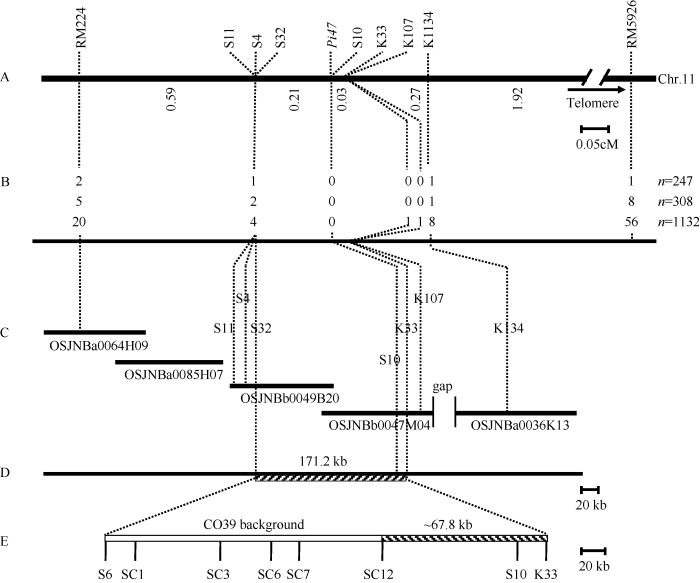 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1Pi47位点区域遗传图谱与物理图谱的构建
A: Pi47位点区域遗传图谱。中间粗实线为第11染色体Pi47位点区域示意图, 上方为定位使用标记, 下方数字为相邻两个标记间的遗传距离; B: Pi47精细定位重组各标记重组子数。从上往下依次为对应标记在CX52/CO39 (n=247)、CX18/CO39 (n=308)、CX279/CO39 (n=1132) F2群体中鉴定的重组子数目; C: Pi47位点在日本晴参考基因组中的BAC克隆重叠群构建的物理图谱。每段黑实线代
Fig. 1Genetic and physical map construction of Pi47 locus
A: the genetic map of Pi47 locus. The middle crude black line is the Pi47 locus on chromosome 11, the letter above the line is the molecular makers used in gene mapping, and the number below is the genetic distance between two adjacent markers. B: the number of recombinant of corresponding markers used in Pi47 fine mapping. The numbers from top to bottom are recombinants detected in CX52/CO39 (n=247), CX18/CO39 (n=308), CX279/CO39 (n=1132) F2 mapping population, respectively. C: the physical map of Pi47 locus in japonica rice Nipponbare reference genome. Each crude line represents one BAC clone with its clone number under the line; the crossing points are the position of markers used in fine mapping. D: final physical region of Pi47 fine mapping within 171.2 kb between S32 and S10 marks. E: genetic background analysis of Pi47 single gene lines with molecular marker.
根据文献报道[15,16], 筛选获得在抗感亲本湘资3150和CO39中表现多态的STS标记K134、K107和CAPS标记K33 (表4); 并根据定位区间参考基因组序列信息, 设计开发了4个新的CAPS标记S4、S10、S11与S32 (表4)。利用K134在CX52/CO39 F2群体247个感病单株中鉴定到来自RM5926的1个重组子, 重组率为0.2% (表3), 表明K134与RM5926位于Pi47同一侧, 而K134距离基因更近。Pi47被进一步定位在RM224和K134之间(图1)。
Table 4
表4
表4Pi47精细定位使用的标记信息
Table 4
| 标记 Marker | 引物顺序 Primer sequence (5°-3°) | 物理位置 Physical position | 扩增大小 Expect size | 酶 Enzyme | 标记类型 Marker type |
|---|---|---|---|---|---|
| RM224 | F: TCGATCGATCTTCACGAGG R: TGCTATAAAAGGCATTCGGG | 27673251 -27673372 | 122 | / | SSR |
| S11 | F: TCTTTGAACAAGAGGACCAG R: AATTACCGAATTACTTAGCG | 27824419 -27825439 | 1021 | Mva I or Hinf I | CAPS |
| S4 | F: CGAATGATGACGAGGACCCG R: ATCAGGCACGGCAGCAGAAG | 27827786 -27828843 | 1058 | Hinf I or Rsa I | CAPS |
| S32 | F: GCCCAGTAACCGTAGGAGTC R: TTTGATGAGCGGCAAAGTAG | 27835604 -27836295 | 692 | Msp I | CAPS |
| S10 | F: AGAGCAAGAAGGGCACGGTAT R: AGAATGGTCCTCCTGACATCG | 27992866 -27993970 | 1105 | Rsa I | CAPS |
| K33 | F: TTTGGTGCTCCTCTTACGGG R: TCGGAGTTCACAGAGCCAAG | 28005690 -28006842 | 1153 | Hinf I | CAPS |
| K107 | F: ACATCAATGGCTACAACT R: TGCTAACGGTGCTGGTAT | 28012753 -28012940 | 184/188 | / | STS |
| K134 | F: GATGGCGAGATGGTTGTC R: GCCTTTGAGATAGGGATTGC | 28163563 -28163742 | 180/168 | / | STS |
| RM5926 | F: TAGGTCCATCCAAATCTCGATCC R: TGGCAGAGGAGATTAGAGTAATACGG | 28957564 -28957858 | 295 | / | SSR |
| S6 | F: TCAACCGAACGCAAGAATCAC R: GGCCCTCCAGCTCATCTACCT | 27841529 -27842431 | 903 | / | STS |
| SC1 | F: CCCCACAATCCCACGAGAC R: CGGGCCGACAATCCAATAC | 27848711 -27849635 | 925 | / | STS |
| SC3 | F: TTTCCCACCGATGACCCAG R: GCTCGCCATCCTTCCCTTT | 27879005 -27879906 | 902 | / | STS |
| SC6 | F: TATTGGTAAGTGGTAACGGGTGA R: GCAGATTCGAGGAGGAAGG | 27891111 -27892550 | 1440 | / | STS |
| SC7 | F: TGGCGTGGCGGTTTGGTAG R: GCTGGTTGGGCATGGGTTG | 27909896 -27910602 | 707 | / | STS |
| SC12 | F: GGGCAACAAGCGAGCCATAA R: GTGGTGAGGCAGCGGAACAG | 27939030 -27940473 | 1444 | / | STS |
新窗口打开|下载CSV
进一步利用RM224和K134对CX18/CO39 F2与CX279/CO39 F2 群体总共1440个感病单株进行分析, 以RM224分别鉴定到来自2个群体的5个和20个重组子, 以K134分别鉴定到1个和8个重组子(表3)。因此, 以RM224和K134在3个F2感病群体总共1687个感病单株中分别鉴定到27个和10个重组子, 对应的重组率分别为0.8%和0.3%, 即与Pi47的遗传距离分别为0.8 cM和0.3 cM (图1)。
再利用K33、K107、S4、S11和S32标记对3个群体1687个感病单株进行分析, 结果用K33和K107鉴定到来自K134位点的1个重组子, 用S4、S11和S32鉴定到来自RM224位点相同的7个重组子, 而用S10未鉴定到重组(表3), 表明RM224、S4、S11和S32位于Pi47基因的同一侧, K33、K107和K134位于基因的另一侧, 与目标基因的遗传距离分别为0.21 cM和0.03 cM, 而S10则与目标基因共分离。因此, Pi47被精细定位在S32和K33之间0.24 cM的区间内(图1)。
2.3 遗传图谱与物理图谱的整合及候选基因区间的确定
根据精细定位各标记与Pi47之间的遗传距离, 构建了Pi47位点区域的标记连锁图谱(图1-A)。为了明确Pi47位点在水稻Nipponbare参考基因组中的物理图谱, 利用各标记在参考基因组中的位置信息, 获得5个BAC重叠群OSJNBa0064H09、OsJNBa0085H07、OsJNBb0049B20、OsJNBb0047M04、OsJNBa0036K13, 覆盖了Pi47基因位点RM224至K134之间约490 kb的物理图谱, 其中重叠群OsJNBb0047M04和OsJNBa0036K13之间存在1个间隙(图1-C)。Pi47基因被精细定位在S32和K33之间0.24 cM的遗传距离区间, 对应的物理距离约为171.2 kb (图1-D)。为了进一步缩小Pi47候选基因区间, 在S32和K33之间开发了6个在抗病亲本湘资3150和感病亲本CO39中表现多态的分子标记, 即S6、SC1、SC3、SC6、SC7和SC12 (表3)。基因型分析结果表明, Pi47单基因系CX279、CX18和CX52上述6个标记座位均表现为感病亲本CO39的基因型(图1-E), 表明3个单基因系中S6-SC12间的基因组片段来自感病亲本CO39, 该区间对应参考基因组物理距离约为97.5 kb (图1-E)。利用标记S10、K33分析3个单基因系基因型, 发现2个标记位点均表现出与抗病亲本湘资3150相同的基因型(图1-E)。综合上述试验结果, 推测Pi47被进一步限定在SC12和K33之间约67.8 kb的区域内(图1-E)。
2.4 候选基因的预测与克隆
根据上述精细定位的结果, 以Nipponbare参考基因组IRGSP-1.0版本为参照, 对标记SC12和K33间67.8 kb的DNA序列进行了基因预测和注释。结果表明, 该区间编码8个结构基因(表5)。其中, LOC_Os11g46180、LOC_Os11g46190和LOC_ Os11g46240 3个基因编码转座子或逆转座子蛋白; LOC_Os11g46150和LOC_Os11g46220编码功能未知的表达蛋白或假想蛋白; LOC_Os11g46200和LOC_Os11g46210则编码NBS-LRR抗病类似蛋白, 因此将后两者作为Pi47的候选功能基因(表5)。Table 5
表5
表5Pi47位点标记SC12和K33间参考基因组区域内候选基因预测
Table 5
| 基因名称 Gene name | 推测功能 Putative functions |
|---|---|
| LOC_Os11g46150 | Expressed protein |
| LOC_Os11g46180 | Transposon protein |
| LOC_Os11g46190 | Transposon protein |
| LOC_Os11g46200 | Leucine rich repeat family protein, expressed |
| LOC_Os11g46210 | NB-ARC domain containing protein, expressed |
| LOC_Os11g46220 | Hypothetical protein |
| LOC_Os11g46230 | Tetratricopeptide repeat domain containing protein |
| LOC_Os11g46240 | Retrotransposon protein |
新窗口打开|下载CSV
利用同源基因PCR克隆法, 根据LOC_ Os11g46200和LOC_Os11g46210这2个基因参考基因组序列设计同源PCR引物, 对湘资3150中同源基因进行了PCR扩增和测序。进一步利用湘资3150中同源基因序列的测序结果在NCBI DNA序列数据中比对, 发现Pi47的2个候选基因中, Pi47-1 (对应LOC_Os11g46200)与已克隆的Pik位点等位基因Pik1-KA、Pi7-1、Piks-1、Pik-2、Pikp-1、Pikm1-TS在DNA序列上的相似度在97.5%~99.8%之间, 在蛋白序列上的相似度达94.6%~99.5% (表6); Pi47-2 (对应LOC_Os11g46210)与对应等位基因Pik2-KA、Pi7-2、Piks-2、Pik-1、Pikp-2、Pikm2-TS在DNA和蛋白水平上的相似度分别为99.8%~99.9%和99.6%~99.9% (表7)。而Pi47-1、Pi47-2与Pi1的2个基因Pi1-5、Pi1-6在DNA水平上的相似度分别为99.7%和100%, 蛋白水平上的相似度也分别达到99.5%和100% (表6和表7)。利用全长蛋白序列构建进化树结果表明Pi47与Pi1明显地聚集到一起(图2)。综上说明, Pi47与Pi1可能是同一个基因。
Table 6
表6
表6Pi47-1与Pik位点对应的已克隆基因在DNA和蛋白水平上的序列相似性
Table 6
| 基因 Gene | Pik1-KA | Pi7-1 | Piks-1 | Pik-2 | Pikp-1 | Pikm1-TS | Pi1-5 |
|---|---|---|---|---|---|---|---|
| Pi47-1 DNA | 0.997 | 0.975 | 0.998 | 0.997 | 0.975 | 0.998 | 0.997 |
| Pi47-1蛋白 Pi47-1 protein | 0.987 | 0.947 | 0.993 | 0.989 | 0.946 | 0.994 | 0.995 |
新窗口打开|下载CSV
Table 7
表7
表7Pi47-2与Pik位点对应的已克隆基因在DNA和蛋白水平上的序列相似性
Table 7
| 基因 Gene | Pik2-KA | Pi7-2 | Piks-2 | Pik-1 | Pikp-2 | Pikm2-TS | Pi1-6 |
|---|---|---|---|---|---|---|---|
| Pi47-2 DNA | 0.999 | 0.998 | 0.999 | 0.999 | 0.998 | 0.999 | 1.000 |
| Pi47-2蛋白 Pi47-2 protein | 0.999 | 0.996 | 0.999 | 0.999 | 0.996 | 0.999 | 1.000 |
新窗口打开|下载CSV
图2
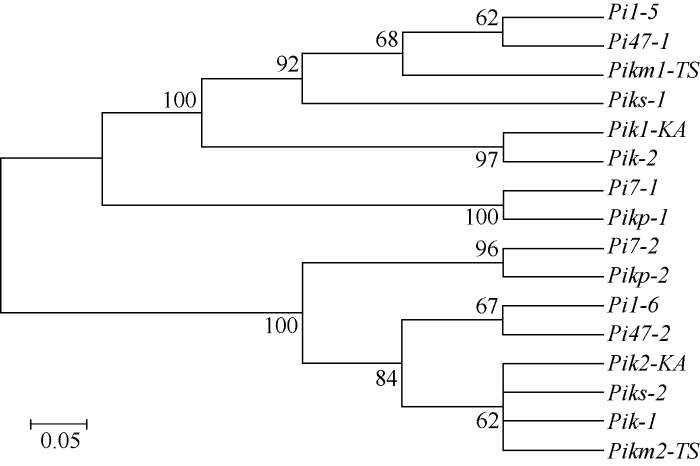 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Pi47与Pik位点已克隆基因的系统进化分析
Fig. 2Phylogenetic analysis of Pi47 and cloned genes at Pik locus
2.5 Pi47与其等位基因的抗谱比较
在Pi47定位区间RM224~RM5926区段, 已有Pikm[37]、Pi46[38]、Pik[39]、Pikp[40]、Pikh[41] 5个抗病基因被精细定位或者克隆, 表明Pi47可能为这些基因的等位基因。为进一步明确Pi47与上述基因的关系和抗谱的差异, 利用来自不同地区的17个稻瘟病菌小种, 对Pi47基因的单基因系CX18和Pik、Pikm、Pikh及Pikp 4个基因CO39背景的近等基因系(NIL)[42]进行接种鉴定。结果发现, 6份材料均对日本小种KOH表现高感, Pik与Pikp对来自福建的小种RB18表现感病, 而Pi47、Pikm和Pikh对其表现抗性。Pi47和Pikp对来自湖南的小种195-2-2表现感病, 而其余3个基因对该小种表现抗性(表8), 表明Pi47与这4个基因抗谱不同。Table 8
表8
表8Pi47及其附近位置抗稻瘟病基因的抗谱分析
Table 8
| 稻瘟菌小种 Isolate | 来源 Origin | 水稻品种 Rice cultivar | |||||
|---|---|---|---|---|---|---|---|
| CO39 | CX18 (Pi47) | IRBLk-Ka (Pik) | IRBLkm-Ts (Pikm) | IRBLkh-K3 (Pikh) | IRBLkp-K60 (Pikp) | ||
| 318-2 | 中国湖南 Hunan, China | S | R | R | R | R | R |
| CHL473 | 中国湖南 Hunan, China | S | R | R | R | R | R |
| X2007A-3 | 中国湖南 Hunan, China | S | R | R | R | R | S |
| CHL440 | 中国湖南 Hunan, China | S | R | R | R | R | S |
| CHL438 | 中国湖南 Hunan, China | S | R | R | R | R | R |
| X2007A-7 | 中国湖南 Hunan, China | S | R | R | R | R | R |
| 195-2-2 | 中国湖南 Hunan, China | S | S | R | R | R | S |
| 87-4 | 中国湖南 Hunan, China | S | R | R | R | R | R |
| 193-1-1 | 中国湖南 Hunan, China | S | R | R | R | R | R |
| 110-2 | 中国湖南 Hunan, China | S | R | R | R | R | R |
| 236-1 | 中国湖南 Hunan, China | S | R | R | R | R | R |
| RB4 | 中国广东 Guangdong, China | S | R | R | R | R | S |
| RB6 | 中国广东 Guangdong, China | S | R | R | R | R | S |
| RB18 | 中国福建 Fujian, China | S | R | S | R | R | S |
| RB19 | 中国福建 Fujian, China | S | R | R | R | R | R |
| RB12 | 中国 China | MR | MR | MR | MR | MR | MR |
| KOH | 日本 Japan | S | S | S | S | S | S |
新窗口打开|下载CSV
3 讨论
稻瘟病是水稻生产中危害最严重的真菌病害之一。由于稻瘟菌自然群体的快速变异导致致病性不断改变的特点, 利用单一的抗病基因很难从根本上持久解决稻瘟病防治的问题, 必须不断挖掘和克隆新的抗病基因, 以适应稻瘟菌群致病性的不断变化。本实验室在前期研究中从世界各地收集的种质资源中鉴定了多份广谱、持久高抗稻瘟病的新种质, 并通过构建F2和重组自交系群体对其中湘资3150[43]、天津野生稻[44]、魔王谷[45]、龙S[46]和Jefferson[47] 5份种质中的抗病基因进行了定位。Huang等[32]利用重组自交系群体从湘资3150中初定位了分别位于水稻第11、第12染色体上的2个抗稻瘟病基因Pi47和Pi48, 本研究进一步对Pi47基因进行了精细定位和候选基因的克隆。从重组自交系群体筛选的4个Pi47单基因系构建的F2分离群体用于精细定位(表2)表明, 3个群体(CX18/CO39、CX52/CO39与CX279/CO39)抗感比例符合3∶1单基因分离规律(表2), 可以用于进一步的精细定位。而CX155/CO39F2群体抗感分离不符合单基因分离规律(表2), 不适用于下一步精细定位; 造成这个结果的原因可能一是接种条件不适导致发病不充分, 二是该株系实际可能不是单基因系, 因为用于鉴定单基因系的连锁标记与目的基因还有较大的遗传距离, 遗传重组导致鉴定不准确。
在利用3个分离群体中1687个感病单株对Pi47进行精细定位的过程中, 我们在目标区段内筛选了56个SSR标记才得到1个多态标记, 表明抗病亲本湘资3150和感病亲本CO39在目标区段的多态性极低, 这可能是群体双亲都为籼稻导致基因组序列差异小。我们进一步在该区段开发了11个新标记, 包括4个CAPS标记(S4、S10、S11和S32)和7个STS标记(S6、SC1、SC12、SC3、SC6、SC7和SC12)(表3和图1), 最终将Pi47基因精细定位于标记SC12和K33之间约67.8 kb的区域内(图1)。这些标记不仅可用于Pi47基因的分子标记辅助选择育种, 还可以用于其他相关的水稻种质资源基因型鉴定、基因定位和克隆。
通过比对日本晴参考基因组在Pi47精细定位区间的基因组信息, 构建了Pi47基因在RM224和K134标记间约490 kb的BAC克隆重叠群(图1), 并对SC12和K33间67.8 kb区间内日本晴基因组序列进行了注释, 发现该区间编码8个结构基因, 其中2个编码NBS-LRR抗病蛋白类似基因(表5), 可能为Pi47的候选基因。利用同源PCR克隆, 测定了抗病亲本湘资3150中这2个NBS-LRR基因的序列, 比对和注释发现这2个基因是由同一个启动子驱动的方向相反的串联基因。进一步对数据库比对发现, Pi47两个候选基因(Pi47-1和Pi47-2)与已鉴定的Pik位点多个等位基因在DNA序列和蛋白序列上相似度非常高(表6、表7和图2)。同时, 发现Pi47-2与Pi1-6基因在DNA和蛋白序列上相似度达到100% (表7), 而Pi47-1与Pi1-5在DNA和蛋白序列上相似度也达到了99.7%和99.5% (表6), 且Pi47和Pi1编码蛋白在系统进化树中聚集在一起(图2), 表明Pi47与Pi1可能为同一功能基因。此前研究表明Pi1只在来自西非利比里亚的LAC23及其衍生品种C101LAC和IRBL1-CL、以及来自东南亚国家越南的Tetep等极少数籼稻品种中存在[16]。本研究克隆的Pi47基因供体材料湘资3150是中国湖南的一个地方品种, 这为拓展Pi1基因的起源进化提供了重要的材料。然而, 在地理位置相距甚远的3个栽培稻品种中均具有同一个抗病基因, Pi47/Pi1基因在自然选择和人工培育过程中的演化进程有待进一步研究。
目前定位和克隆的抗稻瘟病基因中, 多数抗病基因往往成簇分布在少数几个染色体位点[48], 如第6染色体靠近着丝粒附近的Pi2/Pi9位点, 至少已发现了10个具有广谱抗性的等位基因[48,49]。本研究中Pi47所在的第11染色体长臂末端Pik位点也定位了至少9个等位基因, 如Pik、Pikh、Piks、Pikm、Pikp、Pi1、Pi7、Pi47、Pi49等, 其中Pik、Pikh、Pikm、Pikp、Pi1等5个基因已被克隆[8-11,16]。我们比较了Pi47单基因系与Pik、Pikm、Pikh、Pikp 4个基因近等基因系的抗谱, 发现Pi47与这4个基因的抗谱有差异(表8)。这些不同抗谱的等位基因是在对抗不断变异的稻瘟菌的过程中进化而成, 如Pik对应的无毒基因Avr-Pik中鉴定出5种变异类型Avr-Pik-A、B、C、D和E, 可由Pik位点的5个等位基因Pik、Pikp、Pikm、Piks和Pikh分别识别[50]。这些抗御不同无毒基因的抗病等位基因, 为不同地区利用抗性基因培育抗病品种提供了重要资源。本研究发掘的Pik位点的等位基因Pi47, 在湖南地区连续多年的病圃鉴定中表现出高水平持久抗性, 可望作为重要的抗性基因应用于抗病育种。此外, 我们在携带Pi47基因的水稻品种湘资3150中还定位了Pi48基因[34], 2个基因Pi47和Pi48在湘资3150广谱、持久抗性中的作用机制有待进一步研究。
4 结论
采用3个Pi47单基因系与感病亲本CO39构建的F2群体的1687个感病单株, 将Pi47精细定位在S32与K33间0.24 cM区域的171.2 kb物理区间内, 背景分析进一步缩小至SC12和K33间67.8 kb的区间内。该区间编码8个结构基因, 其中2个编码NBS-LRR抗病类似蛋白基因, 为Pi47的候选功能基因。Pi47可能与Pi1为同一基因。Pi47与Pik的4个等位基因Pik、Pikm、Pikh及Pikp的抗谱不同。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
DOI:10.1146/annurev.phyto.39.1.285URL [本文引用: 1]
[本文引用: 1]
DOI:10.1111/j.1365-313X.2010.04348.xURLPMID:20807214 [本文引用: 1]

Summary Top of page Summary Introduction Results Discussion Experimental Procedures Acknowledgements References Supporting Information Rice blast is one of the most widespread and destructive plant diseases worldwide. Breeders have used disease resistance ( R ) genes that mediate fungal race-specific ‘gene-for-gene’ resistance to manage rice blast, but the resistance is prone to breakdown due to high pathogenic variability of blast fungus. Panicle blast 1 ( Pb1 ) is a blast-resistance gene derived from the indica cultivar ‘Modan’. Pb1 -mediated resistance, which is characterized by durability of resistance and adult/panicle blast resistance, has been introduced into elite varieties for commercial cultivation. We isolated the Pb1 gene by map-based cloning. It encoded a coiled-coil–nucleotide-binding-site–leucine-rich repeat (CC–NBS–LRR) protein. The Pb1 protein sequence differed from previously reported R-proteins, particularly in the NBS domain, in which the P-loop was apparently absent and some other motifs were degenerated. Pb1 was located within one of tandemly repeated 60-kb units, which presumably arose through local genome duplication. Pb1 transcript levels increased during the development of Pb1+ cultivars; this expression pattern accounts for their adult/panicle resistance. Promoter:GUS analysis indicated that genome duplication played a crucial role in the generation of Pb1 by placing a promoter sequence upstream of its coding sequence, thereby conferring a Pb1 -characteristic expression pattern to a transcriptionally inactive ‘sleeping’ resistance gene. We discuss possible determinants for the durability of Pb1 -mediated blast resistance.
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
DOI:10.1007/s11032-011-9638-yURL [本文引用: 2]
Abstract) specificities exhibited by allelic genes at the rice blast resistance locus gene from rice cultivar Kanto51 and compared its molecular features with those of and of another gene cloned from cv. Kusabue. Like , is composed of two adjacent NBS-LRR (nucleotide-binding site, leucine-rich repeat) genes: the first gene, -, and the second gene, -. from Kanto51 and from Kusabue were not identical; although the predicted protein sequences of the second genes were identical, the sequences differed by three amino acids within the NBS domain of the first genes. The Pik proteins from Kanto51 and Kusabue differed from Pikm in eight and seven amino acids, respectively. Most of these substituted amino acids were within the coiled-coil (CC) and NBS domains encoded by the first gene. Of these substitutions, all within the CC domain were conserved between the two Pik proteins, whereas all within the NBS domain differed between them. Comparison of the two Pik proteins and Pikm suggests the importance of the CC domain in determining the resistance specificities of and . This feature contrasts with that of most allelic or homologous NBS-LRR genes characterized to date, in which the major specificity determinant is believed to lie in the highly diverged LRR domain. In addition, our study revealed high evolutionary flexibility in the genome at the locus, which may be relevant to the generation of new specificities at this locus.
[本文引用: 1]
DOI:10.1534/genetics.108.095034URLPMID:18940787 [本文引用: 2]
Abstract The rice blast resistance gene Pikm was cloned by a map-based cloning strategy. High-resolution genetic mapping and sequencing of the gene region in the Pikm-containing cultivar Tsuyuake narrowed down the candidate region to a 131-kb genomic interval. Sequence analysis predicted two adjacently arranged resistance-like genes, Pikm1-TS and Pikm2-TS, within this candidate region. These genes encoded proteins with a nucleotide-binding site (NBS) and leucine-rich repeats (LRRs) and were considered the most probable candidates for Pikm. However, genetic complementation analysis of transgenic lines individually carrying these two genes negated the possibility that either Pikm1-TS or Pikm2-TS alone was Pikm. Instead, it was revealed that transgenic lines carrying both of these genes expressed blast resistance. The results of the complementation analysis and an evaluation of the resistance specificity of the transgenic lines to blast isolates demonstrated that Pikm-specific resistance is conferred by cooperation of Pikm1-TS and Pikm2-TS. Although these two genes are not homologous with each other, they both contain all the conserved motifs necessary for an NBS-LRR class gene to function independently as a resistance gene.
[本文引用: 2]
[本文引用: 1]
DOI:10.1007/s00122-010-1393-7URLPMID:20589366 [本文引用: 1]
DNA markers that allow for identification of resistance genes in rice germplasm have a great advantage in resistance breeding because they can assess the existence of the genes without laborious inoculation tests. Functional markers (FMs), which are designed from functional polymorphisms within the sequence of genes, are unaffected by nonfunctional allelic variation and make it possible to identify an individual gene. We previously showed that the resistance function of the rice blast resistance gene Pit in a resistant cultivar, K59, was mainly acquired by up-regulated promoter activity through the insertion of a long terminal repeat (LTR) retrotransposon upstream of Pit. Here, we developed PCR-based DNA markers derived from the LTR-retrotransposon sequence and used these markers to screen worldwide accessions of rice germplasm. We identified 5 cultivars with the LTR-retrotransposon insertion out of 68 rice accessions. The sequence and expression pattern of Pit in the five cultivars were the same as those in K59 and all showed Pit-mediated blast resistance. The results suggest that the functional Pit identified using the markers was derived from a common progenitor. Additionally, comparison of the Pit coding sequences between K59 and susceptible cultivars revealed that one nucleotide polymorphism, which caused an amino acid substitution, offered another target for a FM. These results indicate that our DNA markers should enhance prediction of Pit function and be applicable to a range of rice varieties/landraces cultivated in various regions worldwide and belonging to the temperate japonica, tropical japonica, and indica groups.
DOI:10.2307/3871103URLPMID:11090207 [本文引用: 1]
Abstract The rice blast resistance (R) gene Pi-ta mediates gene-for-gene resistance against strains of the fungus Magnaporthe grisea that express avirulent alleles of AVR-Pita. Using a map-based cloning strategy, we cloned Pi-ta, which is linked to the centromere of chromosome 12. Pi-ta encodes a predicted 928-amino acid cytoplasmic receptor with a centrally localized nucleotide binding site. A single-copy gene, Pi-ta shows low constitutive expression in both resistant and susceptible rice. Susceptible rice varieties contain pi-ta(-) alleles encoding predicted proteins that share a single amino acid difference relative to the Pi-ta resistance protein: serine instead of alanine at position 918. Transient expression in rice cells of a Pi-ta(+) R gene together with AVR-Pita(+) induces a resistance response. No resistance response is induced in transient assays that use a naturally occurring pi-ta(-) allele differing only by the serine at position 918. Rice varieties reported to have the linked Pi-ta(2) gene contain Pi-ta plus at least one other R gene, potentially explaining the broadened resistance spectrum of Pi-ta(2) relative to Pi-ta. Molecular cloning of the AVR-Pita and Pi-ta genes will aid in deployment of R genes for effective genetic control of rice blast disease.
DOI:10.1094/MPMI-19-1216URLPMID:17073304 [本文引用: 3]
Abstract The rice blast resistance (R) genes Pi2 and Piz-t confer broad-spectrum resistance against different sets of Magnaporthe grisea isolates. We first identified the Pi2 gene using a map-based cloning strategy. The Pi2 gene is a member of a gene cluster comprising nine gene members (named Nbs1-Pi2 to Nbs9-Pi2) and encodes a protein with a nucleotide-binding site and leucine-rich repeat (LRR) domain. Fine genetic mapping, molecular characterization of the Pi2 susceptible mutants, and complementation tests indicated that Nbs4-Pi2 is the Pi2 gene. The Piz-t gene, a Pi2 allele in the rice cultivar Toride 1, was isolated based on the Pi2 sequence information. Complementation tests confirmed that the family member Nbs4-Piz-t is Piz-t. Sequence comparison revealed that only eight amino-acid changes, which are confined within three consecutive LRR, differentiate Piz-t from Pi2. Of the eight variants, only one locates within the xxLxLxx motif. A reciprocal exchange of the single amino acid between Pi2 and Piz-t did not convert the resistance specificity to each other but, rather, abolished the function of both resistance proteins. These results indicate that the single amino acid in the xxLxLxx motif may be critical for maintaining the recognition surface of Pi2 and Piz-t to their respective avirulence proteins.
DOI:10.1007/s00122-012-1894-7URLPMID:22643901 [本文引用: 4]
We report the isolation of Pi1, a gene conferring broad-spectrum resistance to rice blast (Magnaporthe oryzae). Using loss- and gain-of-function approaches, we demonstrate that Pi1 is an allele at the Pik locus. Like other alleles at this locus, Pi1 consists of two genes. A functional nucleotide polymorphism (FNP) was identified that allows differentiation of Pi1 from other Pik alleles and other non-Pik genes. A extensive germplasm survey using this FNP reveals that Pi1 is a rare allele in germplasm collections and one that has conferred durable resistance to a broad spectrum of pathogen isolates.
[本文引用: 1]
DOI:10.1534/genetics.105.044891URLPMID:16387888 [本文引用: 1]
Abstract The broad-spectrum rice blast resistance gene Pi9 was cloned using a map-based cloning strategy. Sequencing of a 76-kb bacterial artificial chromosome (BAC) contig spanning the Pi9 locus led to identification of six tandemly arranged resistance-like genes with a nucleotide-binding site (NBS) and leucine-rich repeats (LRRs) (Nbs1-Pi9-Nbs6-Pi9). Analysis of selected Pi9 deletion mutants and transformation of a 45-kb fragment from the BAC contig into the susceptible rice cultivar TP309 narrowed down Pi9 to the candidate genes Nbs2-Pi9 and Nbs3-Pi9. Disease evaluation of the transgenic lines carrying the individual candidate genes confirmed that Nbs2-Pi9 is the Pi9 gene. Sequence comparison analysis revealed that the six paralogs at the Pi9 locus belong to four classes and gene duplication might be one of the major evolutionary forces contributing to the formation of the NBS-LRR gene cluster. Semiquantitative reverse transcriptase (RT)-PCR analysis showed that Pi9 was constitutively expressed in the Pi9-resistant plants and was not induced by blast infection. The cloned Pi9 gene provides a starting point to elucidate the molecular basis of the broad-spectrum disease resistance and the evolutionary mechanisms of blast resistance gene clusters in rice.
DOI:10.1534/genetics.107.075465URLPMID:17507669 [本文引用: 1]
The indica rice variety Kasalath carries Pi36, a gene that determines resistance to Chinese isolates of rice blast and that has been located to a 17-kb interval on chromosome 8. The genomic sequence of the reference japonica variety Nipponbare was used for an in silico prediction of the resistance (R) gene content of the interval and hence for the identification of candidate gene(s) for Pi36. Three such sequences, which all had both a nucleotide-binding site and a leucine-rich repeat motif, were present. The three candidate genes were amplified from the genomic DNA of a number of varieties by long-range PCR, and the resulting amplicons were inserted into pCAMBIA1300 and/or pYLTAC27 vectors to determine sequence polymorphisms correlated to the resistance phenotype and to perform transgenic complementation tests. Constructs containing each candidate gene were transformed into the blast-susceptible variety Q1063, which allowed the identification of Pi36-3 as the functional gene, with the other two candidates being probable pseudogenes. The Pi36-encoded protein is composed of 1056 amino acids, with a single substitution event (Asp to Ser) at residue 590 associated with the resistant phenotype. Pi36 is a single-copy gene in rice and is more closely related to the barley powdery mildew resistance genes Mla1 and Mla6 than to the rice blast R genes Pita, Pib, Pi9, and Piz-t. An RT-PCR analysis showed that Pi36 is constitutively expressed in Kasalath.
DOI:10.1534/genetics.107.080648URLPMID:17947408 [本文引用: 1]
The resistance (R) gene Pi37, present in the rice cultivar St. No. 1, was isolated by an in silico map-based cloning procedure. The equivalent genetic region in Nipponbare contains four nucleotide binding site- leucine-rich repeat (NBS-LRR) type loci. These four candidates for Pi37 (Pi37-1, -2, -3, and -4) were amplified separately from St. No. 1 via long-range PCR, and cloned into a binary vector. Each construct was individually transformed into the highly blast susceptible cultivar Q1063. The subsequent complementation analysis revealed Pi37-3 to be the functional gene, while -1, -2, and -4 are probably pseudogenes. Pi37 encodes a 1290 peptide NBS-LRR product, and the presence of substitutions at two sites in the NBS region (V239A and 1247M) is associated with the resistance phenotype. Semiquantitative expression analysis showed that in St. No. 1, Pi37 was constitutively expressed and only slightly induced by blast infection. Transient expression experiments indicated that the Pi37 product is restricted to the cytoplasm. Pi37-3 is thought to have evolved recently from -2, which in turn was derived from an ancestral -1 sequence. Pi37-4 is likely the most recently evolved member of the cluster and probably represents a duplication of -3. The four Pi37 paralogs are more closely related to maize rp1 than to any of the currently isolated rice blast R genes Pita, Pib, Pi9, Pi2, Piz-t, and Pi36.
DOI:10.1007/s00122-012-2031-3URLPMID:23400829 [本文引用: 1]
The major quantitative trait locus qBR9.1 confers broad-spectrum resistance to rice blast, and was mapped to a ~69.1/kb region on chromosome 9 that was inherited from resistant variety Sanhuangzhan No 2 (SHZ-2). Within this region, only one predicted disease resistance gene with nucleotide binding site and leucine-rich repeat (NBS-LRR) domains was found. Specific markers corresponding to this gene cosegregated with blast resistance in F 2 and F 3 populations derived from crosses of susceptible variety Texianzhan 13 (TXZ-13) to SHZ-2 and the resistant backcross line BC-10. We tentatively designate the gene as Pi56(t). Sequence analysis revealed that Pi56(t) encodes an NBS-LRR protein composed of 743 amino acids. Pi56(t) was highly induced by blast infection in resistant lines SHZ-2 and BC-10. The corresponding allele of Pi56(t) in the susceptible line TXZ-13 encodes a protein with an NBS domain but without LRR domain, and it was not induced by Magnaporthe oryzae infection. Three new cosegregating gene-specific markers, CRG4-1, CRG4-2 and CRG4-3, were developed. In addition, we evaluated polymorphism of the gene-based markers among popular varieties from national breeding programs in Asia and Africa. The presence of the CRG4-2 SHZ-2 allele cosegregated with a blast-resistant phenotype in two BC 2 F 1 families of SHZ-2 crossed to recurrent parents IR64-Sub1 and Swarna-Sub1. CRG4-1 and CRG4-3 showed clear polymorphism among 19 varieties, suggesting that they can be used in marker-assisted breeding to combine Pi56(t ) with other target genes in breeding lines.
DOI:10.1007/s11032-014-0067-6URL [本文引用: 1]
Pikahei - 1 (t) is the strongest quantitative trait locus (QTL) for blast resistance in upland rice cv. Kahei, which has strong field resistance to the rice blast disease. A high-quality bacterial artificial chromosome library was used to fine-map Pikahei - 1 (t) within ~300/kb on the 31-Mb region on rice chromosome 4. Of the 42 predicted open reading frames, seven resistance gene analogs ( RGAs ) with the nucleotide-binding site and leucine-rich repeat (NBS-LRR) domain were identified. Among these, RGA1, 2, 3, 5, and 7, but not RGA4 and 6, were found to be expressed in Kahei and monogenic lines containing Pikahei - 1 (t). Blast inoculation of transgenic rice lines carrying the genomic fragment of each RGA revealed that only RGA3 was associated with blast resistance. On the basis of these results, we concluded that RGA3 is the Pikahei - 1 (t) and named it Pi63 . Pi63 encoded a typical coiled-coil-NBS-LRR protein and showed isolate-specificity. These results suggest that Pi63 behaves like a typical Resistance ( R ) gene, and the strong and broad-spectrum resistance of Kahei is dependent on natural pyramiding of multiple QTLs. The blast resistance levels of Pi63 were closely correlated with its gene expression levels, indicating a dose-dependent response of Pi63 function in rice resistance. Pi63 is the first cloned R gene in the R gene cluster on rice chromosome 4, and its cloning might facilitate genomic dissection of this cluster region.
[本文引用: 1]
DOI:10.1007/s00122-015-2579-9URLPMID:26183036 [本文引用: 1]
Key message We characterized a novel blast resistance gene Pi50 at the Pi2/9 locus; Pi50 is derived from functional divergence of duplicated genes. The unique features of Pi50 should facilitate its use in rice breeding and improve our understanding of the evolution of resistance specificities. Abstract Rice blast disease, caused by the fungal pathogen Magnaporthe oryzae , poses constant, major threats to stable rice production worldwide. The deployment of broad-spectrum resistance ( R ) genes provides the most effective and economical means for disease control. In this study, we characterize the broad-spectrum R gene Pi50 at the Pi2/9 locus, which is embedded within a tandem cluster of 12 genes encoding proteins with nucleotide-binding site and leucine-rich repeat (NBSRR) domains. In contrast with other Pi2/9 locus, the Pi50 cluster contains four duplicated genes ( Pi50_NBS4_1 to 4 ) with extremely high nucleotide sequence similarity. Moreover, these duplicated genes encode two kinds of proteins (Pi50_NBS4_1/2 and Pi50_NBS4_3/4) that differ by four amino acids. Complementation tests and resistance spectrum analyses revealed that Pi50_NBS4_1/2 , not Pi50_NBS4_3/4 , control the novel resistance specificity as observed in the Pi50 near isogenic line, NIL-e1. Pi50 shares greater than 96% amino acid sequence identity with each of three other R proteins, i.e., Pi9, Piz-t, and Pi2, and has amino acid changes predominantly within the LRR region. The identification of Pi50 with its novel resistance specificity will facilitate the dissection of mechanisms behind the divergence and evolution of different resistance specificities at the Pi2/9 locus.
DOI:10.1007/s00122-016-2681-7URLPMID:26883042 [本文引用: 1]
Key message A novel R gene was mapped to a locus on chromosome 11 from 30.42 to 30.8502 Mb, which was proven to be efficient in the improvement of rice blast resistance.
DOI:10.1126/science.aai8898URLPMID:28154240 [本文引用: 1]
Abstract Crop breeding aims to balance disease resistance with yield, however single resistance (R) genes can lead to resistance breakdown and R gene pyramiding may impact growth fitness. Here we report that the rice Pigm locus contains a cluster of genes encoding nucleotide-binding leucine-rich repeat (NLR) receptors that confer durable resistance to the fungus Magnaporthe oryzae without yield penalty. In the cluster, PigmR confers broad-spectrum resistance, whereas PigmS competitively attenuates PigmR homodimerization to suppress resistance. PigmS expression, and thus PigmR-mediated resistance, are subjected to tight epigenetic regulation. PigmS increases seed production to counteract the yield cost induced by PigmR Therefore, our study reveals a mechanism balancing high disease resistance and yield through epigenetic regulation of paired antagonistic NLRs, providing a tool to develop elite crop varieties. Copyright 2017, American Association for the Advancement of Science.
DOI:10.1111/j.1365-313X.2006.02739.xURLPMID:16709195 [本文引用: 1]
Summary Top of page Summary Introduction Results Discussion Experimental procedures Acknowledgements References Rice blast, caused by the fungal pathogen Magnaporthe grisea , is one of the most devastating diseases in rice worldwide. The dominant resistance gene, Pi-d2 [previously named Pi-d(t)2 ], present in the rice variety Digu, confers gene-for-gene resistance to the Chinese blast strain, ZB15. Pi-d2 was previously mapped close to the centromere of chromosome 6. In this study, the Pi-d2 gene was isolated by a map-based cloning strategy. Pi-d2 encodes a receptor-like kinase protein with a predicted extracellular domain of a bulb-type mannose specific binding lectin (B-lectin) and an intracellular serine hreonine kinase domain. Pi-d2 is a single-copy gene that is constitutively expressed in the rice variety Digu. Transgenic plants carrying the Pi-d2 transgene confer race-specific resistance to the M.grisea strain, ZB15. The Pi-d2 protein is plasma membrane localized. A single amino acid difference at position 441 of Pi-d2 distinguishes resistant and susceptible alleles of rice blast resistance gene Pi-d2 . Because of its novel extracellular domain, Pi-d2 represents a new class of plant resistance genes.
DOI:10.1126/science.1175550URLPMID:19696351 [本文引用: 1]
Blast disease is a devastating fungal disease of rice, one of the world's staple foods. Race-specific resistance to blast disease has usually not been durable. Here, we report the cloning of a previously unknown type of gene that confers non--race-specific resistance and its successful use in breeding. Pi21 encodes a proline-rich protein that includes a putative heavy metal--binding domain and putative protein-protein interaction motifs. Wild-type Pi21 appears to slow the plant's defense responses, which may support optimization of defense mechanisms. Deletions in its proline-rich motif inhibit this slowing. Pi21 is separable from a closely linked gene conferring poor flavor. The resistant pi21 allele, which is found in some strains of japonica rice, could improve blast resistance of rice worldwide.
DOI:10.1094/PHYTO-10-12-0260-RURLPMID:23384860 [本文引用: 1]
Abstract The rice blast resistance gene Pid3 encodes a nucleotide-binding-site leucine-rich repeat (NBS-LRR) protein. This gene was cloned from the rice 'Digu' (indica) by performing a genome-wide comparison of the NBS-LRR gene family between two genome-sequenced varieties, '9311' (indica) and 'Nipponbare' (japonica). In this study, we performed functional analysis of Pid3-A4, an ortholog of Pid3 revealed by allele mining in the common wild rice A4 (Oryza rufipogon). The predicted protein encoded by Pid3-A4 shares 99.03% sequence identity with Pid3, with only nine amino-acid substitutions. In wild rice plants, Pid3-A4 is constitutively expressed, and its expression is not induced by Magnaporthe oryzae isolate Zhong-10-8-14 infection. Importantly, in transgenic plants, Pid3-A4, as compared with Pid3, displays a distinct resistance spectrum to a set of M. oryzae isolates, including those that prevail in the rice fields of Sichuan Province. Therefore, Pid3-A4 should be quite useful for the breeding of rice blast resistance, especially in southwestern China.
[本文引用: 1]
[本文引用: 1]
DOI:10.3724/SP.J.1005.2009.00999URL [本文引用: 2]
抗病性是水稻遗传改良的主要目标,是水稻高产、优质的重要保证。近年来,水稻的抗病机制研究取得实质性进展,鉴定并克隆了一批重要的抗性基因。文章综述了这些基因的鉴定、克隆和生物学功能研究进展,探讨了抗病基因的有效利用。
DOI:10.3724/SP.J.1005.2009.00999URL [本文引用: 2]
抗病性是水稻遗传改良的主要目标,是水稻高产、优质的重要保证。近年来,水稻的抗病机制研究取得实质性进展,鉴定并克隆了一批重要的抗性基因。文章综述了这些基因的鉴定、克隆和生物学功能研究进展,探讨了抗病基因的有效利用。
[本文引用: 1]
DOI:10.1094/PHYTO-08-10-0209URLPMID:21171885 [本文引用: 4]
The indica rice cultivar Xiangzi 3150 (XZ3150) confers a high level of resistance to 95% of the isolates of Magnaporthe oryzae (the agent of rice blast disease) collected in Hunan Province, China. To identify the resistance (R) gene(s) controlling the high level of resistance in this cultivar, we developed 286 F(9) recombinant inbred lines (RILs) from a cross between XZ3150 and the highly susceptible cultivar CO39. Inoculation of the RILs and an F(2) population from a cross between the two cultivars with the avirulent isolate 193-1-1 in the growth chamber indicated the presence of two dominant R genes in XZ3150. A linkage map with 134 polymorphic simple sequence repeat and single feature polymorphism markers was constructed with the genotype data of the 286 RILs. Composite interval mapping (CIM) using the results of 193-1-1 inoculation showed that two major R genes, designated Pi47 and Pi48, were located between RM206 and RM224 on chromosome 11, and between RM5364 and RM7102 on chromosome 12, respectively. Interestingly, the CIM analysis of the four resistant components of the RILs to the field blast population revealed that Pi47 and Pi48 were also the major genetic factors responsible for the field resistance in XZ3150. The DNA markers linked to the new R genes identified in this study should be useful for further fine mapping, gene cloning, and marker-aided breeding of blast-resistant rice cultivars.
[本文引用: 1]
DOI:10.1038/nature03895URLPMID:16100779 [本文引用: 1]
Abstract Rice, one of the world's most important food plants, has important syntenic relationships with the other cereal species and is a model plant for the grasses. Here we present a map-based, finished quality sequence that covers 95% of the 389 Mb genome, including virtually all of the euchromatin and two complete centromeres. A total of 37,544 non-transposable-element-related protein-coding genes were identified, of which 71% had a putative homologue in Arabidopsis. In a reciprocal analysis, 90% of the Arabidopsis proteins had a putative homologue in the predicted rice proteome. Twenty-nine per cent of the 37,544 predicted genes appear in clustered gene families. The number and classes of transposable elements found in the rice genome are consistent with the expansion of syntenic regions in the maize and sorghum genomes. We find evidence for widespread and recurrent gene transfer from the organelles to the nuclear chromosomes. The map-based sequence has proven useful for the identification of genes underlying agronomic traits. The additional single-nucleotide polymorphisms and simple sequence repeats identified in our study should accelerate improvements in rice production.
DOI:10.1007/s11032-007-9118-6URL [本文引用: 1]
The Pik m gene in rice confers a high and stable resistance to many isolates of Magnaporthe oryzae collected from southern China. This gene locus was roughly mapped to the long arm of rice chromosome 11 with restriction fragment length polymorphic (RFLP) markers in the previous study. To effectively utilize the resistance, a linkage analysis was performed in a mapping population consisting of 659 highly susceptible plants collected from four F 2 populations using the publicly available simple sequence repeat (SSR) markers. The result showed that the locus was linked to the six SSR markers and defined by RM254 and RM144 with ≈13.4 and ≈1.202cM, respectively. To fine map this locus, additional 10 PCR-based markers were developed in a region flanked by RM254 and RM144 through bioinformatics analysis (BIA) using the reference sequence of cv. Nipponbare. The linkage analysis with these 10 markers showed that the locus was further delimited to a 0.3-cM region flanked by K34 and K10, in which three markers, K27, K28, and K33, completely co-segregated with the locus. To physically map the locus, the Pik m -linked markers were anchored to bacterial artificial chromosome clones of the reference cv. Nipponbare by BIA. A physical map spanning ≈27802kb in length was constructed by alignment of sequences of the clones anchored by BIA, in which only six candidate genes having the R gene conserved structure, protein kinase, were further identified in an 84-kb segment.
DOI:10.1007/s11032-010-9481-6URL [本文引用: 1]
Finding novel sources of resistance (R) to rice blast disease should facilitate breeding for improved resistance. The objectives of the present study were to evaluate reactions to blast and identify in a space-induced mutant an R gene to a representative isolate of rice blast pathogen. The mutant H4, its parent and twelve monogenic lines were evaluated for their responses to 35 isolates collected from Guangdong Province, China. H4 was found to be resistant to more isolates than its parent and the twelve monogenic lines, suggesting newly acquired resistance may be a function of one or more R genes. A representative isolate GD0193 was used to identify and map the R gene from H4. Genetic analysis revealed that resistance to the isolate GD0193 was controlled by a single dominant gene, designated Pi46 ( t ). Linkage analysis using susceptible F2 individuals showed that Pi46 ( t ) was mapped between the markers RM224 and RM27360 within 1.04 and 1.2cM on the long arm of chromosome 11. Subsequently, Pi46 ( t ) was delimited to an interval of approximately 183.7kb flanked by the markers K67 and T94. These results provide essential information for the cloning of the Pi46 ( t ) gene and will facilitate marker-assisted selection in rice breeding.
DOI:10.1111/j.1469-8137.2010.03462.xURLPMID:21118257 [本文引用: 1]
Abstract 090004 The rice-rice blast pathosystem is of great interest, not only because of the damaging potential of rice blast to the rice crop, but also because both the pathogen and its host are experimentally amenable. The rice blast resistance gene Pik, which is one of the five classical alleles located at the Pik locus on the long arm of chromosome 11, confers high and stable resistance to many Chinese rice blast isolates. 090004 The isolation and functional characterization of Pik were performed in the present study through genetic and genomic approaches. 090004 A combination of Pik-1 and Pik-2 is required for the expression of Pik resistance. Both Pik-1 and Pik-2 encode coiled-coil nucleotide binding site leucine-rich repeat (NBS-LRR) proteins, and each shares a very high level of protein identity with corresponding proteins encoded by the Pik-m and Pik-p alleles. Pik could be distinguished from other Pik alleles, including Pik-m and Pik-p, by the allele-specific, single-nucleotide polymorphism T1-2944G. 090004 The coupled genes probably did not evolve as a result of a duplication event, and are far from any NBS-LRR R gene characterized. Pik is a younger allele at the locus that probably emerged after rice domestication. 0008 The Authors (2010). Journal compilation 0008 New Phytologist Trust (2010).
DOI:10.1094/PHYTO-99-8-0900URLPMID:19594308 [本文引用: 1]
Pik-p is carried by cv. K60, which is one of the Japanese differentials widely used in both Japan and China since the 1980s. Its utility and specificity was evaluated with a total of 612 isolates of Magnaporthe oryzae collected from various regions in China in combination with 16 main resistance genes being used in the breeding programs. Pik-p is an independently and dominantly acting gene in the Pik cluster, which conditions differential reactions against many isolates and contains higher resistance in Guangdong, Jiangsu, and Sichuan provinces, China, indicating that this gene could be still used in these regions. A high-resolution genetic map of Pik-p was constructed using genomic position-ready markers. A set of 47 recombinants out of 681 F(2) plants derived from the crosses cv. K60 (resistant) x cv. AS20-1 (susceptible) and x cv. Kasalath (susceptible) was identified in the genetic interval defined by the markers RM5926 and K37 which flank the Pik gene cluster. This set was then genotyped with seven markers known to reside within the interval. The closest markers to Pik-p were K28 (approximately 0.60 centimorgans [cM]) and K39 (approximately 0.07 cM). A further four markers in the K28-K39 interval were developed from an in silico analysis based on the cv. Nipponbare genome sequence, and these all co-segregated with Pik-p. This 0.67-cM region is equivalent to a physical separation in cv. Nipponbare of approximately 126 kb, plus an as-yet-unfilled genomic gap of unknown length. Four nucleotide-binding site leucine-rich repeat-type resistance genes are present in this interval, and these represent good candidates for Pik-p.
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
由Magnaporthe oryzae引起的稻瘟病是水稻主要病害之一,选育和推广水稻抗病品种(组合)是控制这一病害最安全、经济、环保的方法。湘资3150是湖南省的一个地方品种,经多年的试验证明,该品种对稻瘟病表现出广谱持久抗性。为了定位和克隆湘资3150中的抗瘟基因,本研究以湘资3150和感病品种C039杂交(C/X)后代F2为基础,采用单粒传法,经8代自交构建了包含286个株系的F10重组自交系群体(recombinant inbred line, RIL)对湘资3150中的抗瘟基因进行了定位,主要结果如下: 1.选用来自湖南省各地采集的303个稻瘟病菌菌株,对湘资3150进行接种鉴定,结果表明,湘资3150对测试菌株的抗菌频率达到了95.38%,对其中30个生理小种,包括湖南省的优势生理小种ZB13的抗菌频率都为100%。这说明湘资3150的抗谱极广,并且在湖南本省具有良好的应用价值。 2.检测了914对引物(SSR引物848对,SFP引物66对)在湘资3150和CO39之间的多态性,得到多态性引物220对,其中SSR引物200对,SFP引物20对,平均每条染色体约18对,品种间分子标记的多态性检出率为24.10%。 3.选用强致病力和产孢稳定的稻瘟病菌小种193-1-1(ZC11)接种C/X Flo代RIL,抗感比为3:1,表明湘资3150对该小种的抗性由2对主效基因控制。 4.大约150对多态性引物(SSR和SFP)被用于RIL株系的基因型鉴定,并利用Mapmaker/Exp3.0作图软件构建了覆盖全基因组的遗传连锁图谱,134个多态标记(136个位点)被分成19个连锁群,图谱总长2097.1cM,相邻位点平均遗传距离为15.42 cM。 5.复合区间作图(composite interval mapping, CIM)结果表明,两个主效抗性基因分别位于第11号染色体和第12号染色体,分别命名为Pi47(t)和Pi48(t)。Pi47(t)位于第11号染色体长臂近端体的RM206~RM224之间33.0cM的区域,与RM224距离大约为4cM;LOD最大值为14.62,对湘资3150的抗性贡献率为23.81%;Pi48以(t)位于第12号染色体近着丝粒的RM5364~RM7102之间4.2cM的区域,LOD最大值为17.95,对湘资3150的抗性贡献率为23.39%。。这些连锁标记RM206、RM224、RM5364和RM7102的发现,将有利于这2个基因的克隆和分子标记辅助育种(marker assisted breeding, MAS). 6.通过连锁标记的辅助选择,选择了12个和28个分别含有Pi47(t)和Pi48(t)的RI株系与CO39回交,进一步构建BCF2回交群体或者近等基因系,用于2个基因的精细定位。
URL [本文引用: 1]
由Magnaporthe oryzae引起的稻瘟病是水稻主要病害之一,选育和推广水稻抗病品种(组合)是控制这一病害最安全、经济、环保的方法。湘资3150是湖南省的一个地方品种,经多年的试验证明,该品种对稻瘟病表现出广谱持久抗性。为了定位和克隆湘资3150中的抗瘟基因,本研究以湘资3150和感病品种C039杂交(C/X)后代F2为基础,采用单粒传法,经8代自交构建了包含286个株系的F10重组自交系群体(recombinant inbred line, RIL)对湘资3150中的抗瘟基因进行了定位,主要结果如下: 1.选用来自湖南省各地采集的303个稻瘟病菌菌株,对湘资3150进行接种鉴定,结果表明,湘资3150对测试菌株的抗菌频率达到了95.38%,对其中30个生理小种,包括湖南省的优势生理小种ZB13的抗菌频率都为100%。这说明湘资3150的抗谱极广,并且在湖南本省具有良好的应用价值。 2.检测了914对引物(SSR引物848对,SFP引物66对)在湘资3150和CO39之间的多态性,得到多态性引物220对,其中SSR引物200对,SFP引物20对,平均每条染色体约18对,品种间分子标记的多态性检出率为24.10%。 3.选用强致病力和产孢稳定的稻瘟病菌小种193-1-1(ZC11)接种C/X Flo代RIL,抗感比为3:1,表明湘资3150对该小种的抗性由2对主效基因控制。 4.大约150对多态性引物(SSR和SFP)被用于RIL株系的基因型鉴定,并利用Mapmaker/Exp3.0作图软件构建了覆盖全基因组的遗传连锁图谱,134个多态标记(136个位点)被分成19个连锁群,图谱总长2097.1cM,相邻位点平均遗传距离为15.42 cM。 5.复合区间作图(composite interval mapping, CIM)结果表明,两个主效抗性基因分别位于第11号染色体和第12号染色体,分别命名为Pi47(t)和Pi48(t)。Pi47(t)位于第11号染色体长臂近端体的RM206~RM224之间33.0cM的区域,与RM224距离大约为4cM;LOD最大值为14.62,对湘资3150的抗性贡献率为23.81%;Pi48以(t)位于第12号染色体近着丝粒的RM5364~RM7102之间4.2cM的区域,LOD最大值为17.95,对湘资3150的抗性贡献率为23.39%。。这些连锁标记RM206、RM224、RM5364和RM7102的发现,将有利于这2个基因的克隆和分子标记辅助育种(marker assisted breeding, MAS). 6.通过连锁标记的辅助选择,选择了12个和28个分别含有Pi47(t)和Pi48(t)的RI株系与CO39回交,进一步构建BCF2回交群体或者近等基因系,用于2个基因的精细定位。
DOI:10.3724/SP.J.1238.2013.00040URL [本文引用: 1]
为了更好地揭示天津野生稻对稻瘟病抗性的遗传机理,以天津野生稻 与感病品种CO39杂交构建的F2群体为研究对象,利用稻瘟病菌株CHL 1743、110-2和318-2进行室内接种鉴定,F2群体的抗感单株分离比符合3∶1,表明其对3个稻瘟病菌株的抗性均由1个主效基因控制,命名为 Pi2-1.利用群体分离分析法和隐性群体分离法进行初步定位分析,将Pi2-1基因定位在水稻第6号染色体着丝粒附近的SSR标记AP5659-5到 RM7213之间,与2个标记的遗传距离分别为0.9 cM和1.4 cM.
DOI:10.3724/SP.J.1238.2013.00040URL [本文引用: 1]
为了更好地揭示天津野生稻对稻瘟病抗性的遗传机理,以天津野生稻 与感病品种CO39杂交构建的F2群体为研究对象,利用稻瘟病菌株CHL 1743、110-2和318-2进行室内接种鉴定,F2群体的抗感单株分离比符合3∶1,表明其对3个稻瘟病菌株的抗性均由1个主效基因控制,命名为 Pi2-1.利用群体分离分析法和隐性群体分离法进行初步定位分析,将Pi2-1基因定位在水稻第6号染色体着丝粒附近的SSR标记AP5659-5到 RM7213之间,与2个标记的遗传距离分别为0.9 cM和1.4 cM.
URL [本文引用: 1]
稻瘟病严重影响水稻生产,是由子囊菌Magnaporthe oryzae引起的一种真菌性病害。传统的化学防治方法容易带来安全、环保、经济性等方面的问题。利用稻瘟病抗性基因培育水稻抗稻瘟病品种是目前最有效、最经济的稻瘟病防治方法。水稻主效抗稻瘟病基因的定位和克隆将为培育水稻稻瘟病抗病品种提供优良基因资源,对理解水稻和稻瘟病菌分子互作机理解析具有重要作用。 魔王谷是云南省的一个地方粳稻品种,长期的研究和田间种植证明其具有广谱、持久的稻瘟病抗性。本实验室利用魔王谷与感病籼稻品种C039杂交构建重组自交系F8和F9,通过QTL分析初步定位了一个位于11号染色体的主效抗稻瘟病基因。在此基础上我们对该基因进行了精细定位,并命名为Pi49,并进行了BAC文库筛选,主要结果如下: 1.利用208株感病单株将Pi49初步定位于SSR标记RM224与RM144之间。 2.通过再开发位于标记RM224与RM144之间分子标记,最终将Pi49定位于CAPS标记K27与SSR标记K10之间,Pi49与这两个分子标记的遗传距离都是0.16cM与0.16cM. 3.构建了魔王谷的BAC文库,并用目的基因两侧的标记K10和K134,通过富集PCR筛选的方法对该BAC文库进行了筛选,得到了含有目的基因片段的单克隆。
URL [本文引用: 1]
稻瘟病严重影响水稻生产,是由子囊菌Magnaporthe oryzae引起的一种真菌性病害。传统的化学防治方法容易带来安全、环保、经济性等方面的问题。利用稻瘟病抗性基因培育水稻抗稻瘟病品种是目前最有效、最经济的稻瘟病防治方法。水稻主效抗稻瘟病基因的定位和克隆将为培育水稻稻瘟病抗病品种提供优良基因资源,对理解水稻和稻瘟病菌分子互作机理解析具有重要作用。 魔王谷是云南省的一个地方粳稻品种,长期的研究和田间种植证明其具有广谱、持久的稻瘟病抗性。本实验室利用魔王谷与感病籼稻品种C039杂交构建重组自交系F8和F9,通过QTL分析初步定位了一个位于11号染色体的主效抗稻瘟病基因。在此基础上我们对该基因进行了精细定位,并命名为Pi49,并进行了BAC文库筛选,主要结果如下: 1.利用208株感病单株将Pi49初步定位于SSR标记RM224与RM144之间。 2.通过再开发位于标记RM224与RM144之间分子标记,最终将Pi49定位于CAPS标记K27与SSR标记K10之间,Pi49与这两个分子标记的遗传距离都是0.16cM与0.16cM. 3.构建了魔王谷的BAC文库,并用目的基因两侧的标记K10和K134,通过富集PCR筛选的方法对该BAC文库进行了筛选,得到了含有目的基因片段的单克隆。
DOI:10.3724/SP.J.1006.2012.00408URL [本文引用: 1]
龙S是一个广谱抗稻瘟病的水稻两用核不育系,利用分子标记技术精细定位其主效抗性基因,对于培育抗稻瘟病水稻新品种具有重要意义。采用来自国内外的41个稻瘟病菌系通过接种鉴定方式对龙S进行了稻瘟病抗谱分析,结果显示龙S的抗性频率为100%,对其中39个菌系表现高水平抗性,与Pi9的携带品种75-1-127抗性频率和抗病级别基本相当。群体遗传分析表明龙S的抗性基因表现为显性遗传方式,对于不同菌系龙S表现出不同的抗病遗传模式,其中龙S对稻瘟菌系318-2的抗性由单基因控制。通过抗病亲本龙S与感病亲本日本晴构建F2分离群体,采用BSA(bulk segregant analysis)及RCA(recessive class analysis)分析方法,将龙S的主效抗病基因精细定位于第9染色体上的SSR标记M1-M2所在的1.31cM区间,与已克隆的广谱抗稻瘟病基因Pi5位于相邻的染色体区域。抗谱分析表明,龙S与Pi5、Pii单基因系的抗性频率差异明显,抗谱较后二者更广。龙S主效抗性基因的精细定位,为进一步揭示其与Pi5、Pii的等位关系以及通过分子标记辅助选择培育抗病水稻新品种奠定了基础。
DOI:10.3724/SP.J.1006.2012.00408URL [本文引用: 1]
龙S是一个广谱抗稻瘟病的水稻两用核不育系,利用分子标记技术精细定位其主效抗性基因,对于培育抗稻瘟病水稻新品种具有重要意义。采用来自国内外的41个稻瘟病菌系通过接种鉴定方式对龙S进行了稻瘟病抗谱分析,结果显示龙S的抗性频率为100%,对其中39个菌系表现高水平抗性,与Pi9的携带品种75-1-127抗性频率和抗病级别基本相当。群体遗传分析表明龙S的抗性基因表现为显性遗传方式,对于不同菌系龙S表现出不同的抗病遗传模式,其中龙S对稻瘟菌系318-2的抗性由单基因控制。通过抗病亲本龙S与感病亲本日本晴构建F2分离群体,采用BSA(bulk segregant analysis)及RCA(recessive class analysis)分析方法,将龙S的主效抗病基因精细定位于第9染色体上的SSR标记M1-M2所在的1.31cM区间,与已克隆的广谱抗稻瘟病基因Pi5位于相邻的染色体区域。抗谱分析表明,龙S与Pi5、Pii单基因系的抗性频率差异明显,抗谱较后二者更广。龙S主效抗性基因的精细定位,为进一步揭示其与Pi5、Pii的等位关系以及通过分子标记辅助选择培育抗病水稻新品种奠定了基础。
URL [本文引用: 1]
水稻是全世界最重要的粮食作物之一。稻瘟病(rice blast)由子囊菌灰色大角间座壳菌[Magnaporthe grisea]引起,是水稻生产中的最严重病害之一。实践证明,选育和推广抗病水稻品种(组合)是控制这一病害最安全、经济、环保的方法,而抗性基因的发掘与利用又是抗病育种的基础和核心。因此挖掘更多新的抗瘟基因,对于我们了解水稻抗病的分子机制具有重要的意义,同时对水稻抗病育种也具有巨大的实践应用价值。本研究对多个国家和地区收集到的十份抗源材料进行了抗谱分析和等位性测定,并利用分子标记对其中的两份候选抗源的抗瘟基因进行了定位,具体结果如下: 1.选取了来自4个国家的10份水稻抗源材料,利用从国内外7个稻瘟病发病区分离的32个稻瘟病菌生理小种进行室内接种,鉴定筛选出了Amber、谷梅2号、Jefferson、魔王谷、湘资3150和天津野生稻6份广谱抗瘟资源。 2.选取Amber、Jefferson、谷梅2号三份抗源材料与Pi9基因的供体水稻品系75-1-127杂交获得F2遗传群体,利用稻瘟病菌生理小种318-2接种,观察该群体的抗感分离情况从而判断候选抗源材料的主效抗性基因与Pi9的等位性关系。结果表明谷梅2号和Jefferson含有与Pi9等位或紧密连锁的抗瘟基因,而Amber的抗瘟基因与Pi9不等位。 3.将谷梅2号、Jefferson两份抗源材料与感病亲本CO39构建F2遗传群体,使用稻瘟病菌生理小种318-2接种,群体大小分别为102株和127株,抗感单株分离比均为3:1,表明两份材料对318-2小种的抗性由一对显性基因控制。利用分布在水稻第6号染色体的SSR标记对抗感群体进行基因型分析,筛选到与谷梅2号抗病基因共分离的4个SSR标记,与Jefferson抗病基因共分离的7个SSR标记,进而在分子水平证实了Jefferson、谷梅2号中对318-2的主效抗性基因与Pi9基因的等位性关系。其中Jefferson抗性与分子标记RM7311、RM7178、RM6836、AP4791、AP5659-5、AP5695-1、AP5413共分离;谷梅2号的抗性与AP4791、AP5930、AP5659-5、AP4007共分离。 4.利用已克隆的广谱抗瘟基因Pi2、Pi9、Piz-t的序列信息设计引物,通过RT-PCR方法在Jefferson、谷梅2号中扩增Pi9的同源cDNA序列,均获得了约3.1kb的片段。测序结果显示二者与已克隆的三个基因具有较高的同源性,差别主要集中在LRR结构域中,NBS结构域相对更为保守。谷梅2号的序列与Pi9相比共有46个氨基酸差异,其中在NBS结构域有6个氨基酸差别,在LRR结构域有31个氨基酸的差异;Jefferson的序列与Pi9相比共有18个氨基酸差别,且全在LRR结构域中。
URL [本文引用: 1]
水稻是全世界最重要的粮食作物之一。稻瘟病(rice blast)由子囊菌灰色大角间座壳菌[Magnaporthe grisea]引起,是水稻生产中的最严重病害之一。实践证明,选育和推广抗病水稻品种(组合)是控制这一病害最安全、经济、环保的方法,而抗性基因的发掘与利用又是抗病育种的基础和核心。因此挖掘更多新的抗瘟基因,对于我们了解水稻抗病的分子机制具有重要的意义,同时对水稻抗病育种也具有巨大的实践应用价值。本研究对多个国家和地区收集到的十份抗源材料进行了抗谱分析和等位性测定,并利用分子标记对其中的两份候选抗源的抗瘟基因进行了定位,具体结果如下: 1.选取了来自4个国家的10份水稻抗源材料,利用从国内外7个稻瘟病发病区分离的32个稻瘟病菌生理小种进行室内接种,鉴定筛选出了Amber、谷梅2号、Jefferson、魔王谷、湘资3150和天津野生稻6份广谱抗瘟资源。 2.选取Amber、Jefferson、谷梅2号三份抗源材料与Pi9基因的供体水稻品系75-1-127杂交获得F2遗传群体,利用稻瘟病菌生理小种318-2接种,观察该群体的抗感分离情况从而判断候选抗源材料的主效抗性基因与Pi9的等位性关系。结果表明谷梅2号和Jefferson含有与Pi9等位或紧密连锁的抗瘟基因,而Amber的抗瘟基因与Pi9不等位。 3.将谷梅2号、Jefferson两份抗源材料与感病亲本CO39构建F2遗传群体,使用稻瘟病菌生理小种318-2接种,群体大小分别为102株和127株,抗感单株分离比均为3:1,表明两份材料对318-2小种的抗性由一对显性基因控制。利用分布在水稻第6号染色体的SSR标记对抗感群体进行基因型分析,筛选到与谷梅2号抗病基因共分离的4个SSR标记,与Jefferson抗病基因共分离的7个SSR标记,进而在分子水平证实了Jefferson、谷梅2号中对318-2的主效抗性基因与Pi9基因的等位性关系。其中Jefferson抗性与分子标记RM7311、RM7178、RM6836、AP4791、AP5659-5、AP5695-1、AP5413共分离;谷梅2号的抗性与AP4791、AP5930、AP5659-5、AP4007共分离。 4.利用已克隆的广谱抗瘟基因Pi2、Pi9、Piz-t的序列信息设计引物,通过RT-PCR方法在Jefferson、谷梅2号中扩增Pi9的同源cDNA序列,均获得了约3.1kb的片段。测序结果显示二者与已克隆的三个基因具有较高的同源性,差别主要集中在LRR结构域中,NBS结构域相对更为保守。谷梅2号的序列与Pi9相比共有46个氨基酸差异,其中在NBS结构域有6个氨基酸差别,在LRR结构域有31个氨基酸的差异;Jefferson的序列与Pi9相比共有18个氨基酸差别,且全在LRR结构域中。
[本文引用: 2]
DOI:10.1094/MPMI-20-0063URLPMID:17249423 [本文引用: 1]
Abstract The Pi2/9 locus contains at least four resistance specificities to Magnaporthe grisea and belongs to a gene complex comprised of multiple genes that encode highly homologous nucleotide binding site (NBS) and leucine rich repeat (LRR) proteins. To investigate the genetic events involved in the evolution of the Pi2/9 locus, we analyzed the Pi2/9 locus at the inter- and intralocus levels in five rice cultivars. The NBS-LRR genes in the five cultivars belong to the same phylogenetic clade among rice NBS-LRR genes, and all have a phase-2 intron at the N-terminus. However, the paralogs within each haplotype show a significant sequence divergence and their N-terminal intron and 5' regulatory regions are very different. On the contrary, the orthologs from different haplotypes are highly similar, indicating an obvious orthologous relationship has been maintained during the evolution of the Pi2/9 locus. These results suggest that sequence diversification in the 5' regulatory regions and N-terminal introns of the paralogs may have led to suppression of meiotic recombination between the paralogs within each haplotype, facilitating the maintenance of the orthologous relationship among rice cultivars. Our observations provide valuable insight into the genomic dynamics and evolutionary mechanism of an NBS-LRR resistance-gene complex in rice.
URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}