关键词:普通小麦; dsDNA; 荧光定量; 显性标记; 分子育种 dsDNA Fluorescent Quantification and Genotyping in Common Wheat by FLUOstar System XIAO Yong-Gui1, Susanne DREISIGACKER2, Claudia NUÑEZ-RÍOS2, HU Wei-Guo3, XIA Xian-Chun1, HE Zhong-Hu1,4,* 1 Institute of Crop Science, Chinese Academy of Agricultural Sciences (CAAS) / National Wheat Improvement Center, Beijing 100081, China
2 International Maize and Wheat Improvement Center (CIMMYT), 06600 México, DF México;
3 Wheat Research Institute, Henan Academy of Agricultural Sciences, Zhengzhou 450002, China
4 CIMMYT China Office, c/o CAAS, Beijing 100081, China
Fund:This study was supported by the Key Project of the National Research and Development Program (2016YFD0101804-6), the National Natural Science Foundation of China (31671691), the National Key Technology R&D Program of China (2014BAD01B05), and the International Science & Technology Cooperation Program of Ministry of Science and Technology (2013DFG30530). AbstractQuantitative analysis on double-stranded DNA (dsDNA) lays a foundation in molecular biology research in plants, particularly important for genotyping in molecular breeding. The objective of this study was to establish standard curve for fluorescence quantitative analysis by lambda DNA, to compare the difference between dsDNA value in fluorescence system and ultraviolet spectrophotometry, and to identify the allelic variations of rust resistance genes in wheat. The fluorescent dye could be efficiently performed in the quantitative analysis with micro dsDNA concentration (< 1.1 ng μL-1). However, the fluorescent dye could lead to uncertainty of original concentrations of wheat leaf and grain genome DNA, due to more fold serial dilutions for higher DNA concentration. A downward tendency was happened in fluorescent intensity when fluorescent reaction volume was tapered, which influenced the accuracy of DNA concentration. The volume of reaction system mixed nucleic acid and fluorescent dye should be more than 200 μL for accurate determination of micro dsDNA. For genotyping on PCR products, the volume of fluorescent reaction system should be more than 40 μL. FLUOstar could be used for identifying the dominant marker, for instance csSr32#1 ( Sr32) and IB-267 ( Sr50), its accuracy was 100% in correspondence with that from agarose gel electrophoresis. Co-dominant marker with the characteristic of peculiarity and major difference in amplified fragment length ( ≥100 bp), such as We173( Yr26), could also be identified by fluorescent analysis. Compared with agarose gel electrophoresis method, fluorescent method have a simple, convenient, and rapid oparetion with high repeatability, and can be used for segregating generations in marker-assisted breeding.
Keyword:Common wheat; dsDNA; Fluorescent quantitative analysis; Dominant marker; Molecular breeding Show Figures Show Figures
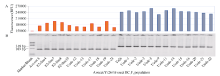
图1 λ 噬菌体dsDNA样品A和B制作的标准曲线及不同反应体系间的荧光差异Fig. 1 Lambda dsDNA standard curve in sample A and sample B and the fluorescence differences among four reaction systems
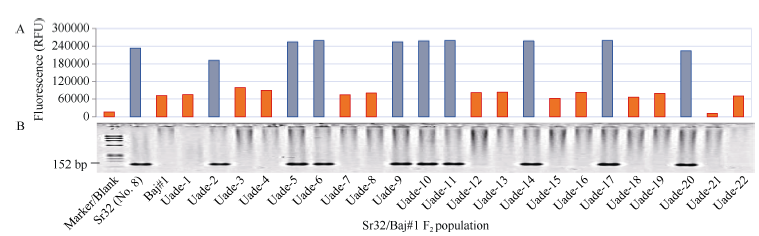
图4 显性标记csSr32#1扩增产物的荧光定量分析(A)和琼脂糖凝胶电泳检测(B)Fig. 4 Amplification products of the dominant csSr32#1marker determined by QuantiFluor dye (A) and agarose gel electrophoresis (B)
图5 共显性标记We173扩增产物的荧光定量分析(A)和凝胶电泳检测结果(B)Fig. 5 Amplification products of the co-dominant We173marker determined by QuantiFluor dye (A) and agarose gel electrophoresis (B)
表2 琼脂糖电泳与荧光染料的经济效益差异分析 Table 2 Comparing the efficiency difference of genetic typing between agarose gel and QuantiFluor dye reagents
试剂 Reagent
包装量 Amount per kit
价格 Price (USD)
96孔样品量 96 samples per microplate
标记类别 Marker type
使用量 Amount
费用 Cost (USD)
耗时 Time (min)
琼脂糖 Agarose
100 g
152
2 g
3.0
90
CD/D
荧光染料 QuantiFluor
1000 μ L (200× )
408
1920 μ L
4.1
8
CD/D
The prices of reagents were from Promega (Madison, WI, USA). Time estimation for the agarose gel electrophoresis method is 10 min of agarose gel preparation, 5 min of sample loading, 60 min of electrophoresis, 10 min of ethidium bromide staining, and 5 min of fluorescence imaging. Time estimation for the QuantiFluor dye method is 3 min of reaction reagents preparation, 3 min of sample loading, and 3 min of fluorescence detection. Marker types CD and D stand for co-dominant and dominant markers, respectively. The QuantiFluor dye method with co-dominant marker is limited by the amplified fragment differences between alleles. 试剂报价来自Promega (Madison, WI, 美国)。耗时计算, 琼脂糖胶电泳方法包括制胶10 min、点样5 min、电泳60 min、EB染色10 min、成像5 min; 荧光定量分析法包括配置反应试剂3 min、分样2 min、检测3 min。标记类别, CD和D分别表示共显性和显性标记, 荧光定量分析法检测共显性标记受等位基因片段差异大小的限制。
表2 琼脂糖电泳与荧光染料的经济效益差异分析 Table 2 Comparing the efficiency difference of genetic typing between agarose gel and QuantiFluor dye reagents
BhatS, CurachN, MostynT, Bains GS, Griffiths KR, Emslie KR. Comparison of methods for accurate quantification of DNA mass concentration with traceability to the international system of units. Anal Chem, 2010, 82: 7185-7192[本文引用:2]
[2]
聂晓静, 赵筱萍, 王毅. 基于荧光图像的抗肿瘤细胞迁移药物筛选方法. 药学学报, 2011, 46: 793-797Nie XJ, Zhao XP, WangY. Development of fluorescence imaging based assay for screening compounds with anti-migration activity. Acta Pharm Sin, 2011, 46: 793-797 (in Chinese with English abstract)[本文引用:3]
[3]
王宇, 刘景晶. 核酸的定量技术研究进展. 药学进展, 2006, 30: 385-390WangY, Liu JJ. Recent advances in research on quantification of nucleic acids. Prog Pharmac Sci, 2006, 30: 385-390 (in Chinese with English abstract)[本文引用:4]
[4]
Lyu ZZ, Liu JC, ZhouY, GuanZ, Yang SM, LiC, Chen AL. Highly sensitive fluorescent detection of small molecules, ions, and proteins using a universal label-free aptasensor. Chem Commun(Camb), 2013, 49: 5465-5467[本文引用:2]
[5]
RengarajanK, Cristol SM, MehtaM, Nickerson JM. Quantifying DNA concentrations using fluorometry: a comparison of fluorophores. Mol Vis, 2002, 8: 416-421[本文引用:2]
[6]
Lucena-AguilarG, Sánchez-López A M, Barberán-Aceituno C, Carrillo-Ávila J A, López-Guerrero J A, Aguilar-Quesada R. DNA source selection for downstream applications based on DNA quality indicators analysis. Biopreserv Biobank, 2016, 14: 264-270[本文引用:2]
[7]
Haque KA, Pfeiffer RM, Beerman MB, Struewing JP, Chanock SJ, Bergen AW. Performance of high-throughput DNA quantification methods. BMC Biotechnol, 2003, 3: 20[本文引用:4]
[8]
Georgiou CD, PapapostolouI. Assay for the quantification of intact/fragmented genomic DNA. Anal Biochem, 2006, 358: 247-256[本文引用:1]
[9]
MarieD, VaulotD, PartenskyF. Application of the novel nucleic acid dyes YOYO-1, YO-PRO-1 and PicoGreen for flow cytometric analysis of marine prokaryotes. Appl Environ Microb, 1996, 62: 1649-1655[本文引用:2]
[10]
桂海娈, 金庆日, 张亚军, 王晓杜, 杨永春, 邵春艳, 程昌勇, 卫芳芳, 杨扬, 杨梦华, 宋厚辉. 基于荧光染料PicoGreen 和核酸适配体的伏马毒素B1检测方法. 生物工程学报, 2015, 31: 1393-1400Gui HL, Jin QR, Zhang YZ, Wang XD, Yang YC, Shao CY, Cheng CY, Wei FF, YangY, Yang MH, Song HH. Development of an aptamer/fluorescence dye PicoGreen-based method for detection of fumonisin B1. Chin J Biotechnol, 2015, 31: 1393-1400 (in Chinese with English abstract)[本文引用:2]
[11]
曾国平, 向东山, 何治柯. 基于Hoechst33258荧光染料检测单链DNA的方法研究. 化学学报, 2011, 69: 1450-1456Zeng GP, Xiang DS, He ZK. Fluorimetric method for the determination of sequence-specific DNA with the fluorescent dye Hoechst 33258. Acta Chim Sin, 2011, 69: 1450-1456 (in Chinese with English abstract)[本文引用:2]
[12]
MagoR, VerlinD, ZhangP, BansalU, BarianaH, JinY, EllisJ, HoxhaS, DundasI. Development of wheat- Aegilops speltoidesrecombinants and simple PCR-based markers for Sr32and a new stem rust resistance gene on the 2S#1 chromosome. Theor Appl Genet, 2013, 126: 2943-2955[本文引用:1]
[13]
MagoR, Bariana HS, Dundas IS, SpielmeyerW, Lawrence GL, Prior AJ, Ellis JG. Development of PCR markers for the selection of wheat stem rust resistance genes Sr24 and Sr26 in diverse wheat germplasm. Theor Appl Genet, 2005, 111: 496-504[本文引用:1]
[14]
ZhangX, HanD, ZengQ, DuanY, YuanF, ShiJ, WangQ, WuJ, HuangL, KangZ. Fine mapping of wheat stripe rust resistance gene Yr26 based on collinearity of wheat with Brachypodium distachyonand rice. PLoS One, 2013, 8: e57885[本文引用:1]
[15]
HuangH, ShiS, GaoX, GaoR, ZhuY, WuX, ZangR, YaoT. A universal label-free fluorescent aptasensor based on Ru complex and quantum dots for adenosine, dopamine and 17 β-estradiol detection. Biosens Bioelectron, 2016, 79: 198-204[本文引用:1]
[16]
LeungK, HeB, YangC, Leung CH, Wang HD, MaD. Development of an aptamer-based sensing platform for metal ions, proteins, and small molecules through terminal deoxynucleotidyl transferase induced G-Quadruplex formation. ACS Appl Mater Interfaces, 2015, 7: 24046-24052[本文引用:1]
[17]
LisantiS, Omar W A W, Tomaszewski B, De Prins S, Jacobs G, Koppen G, Mathers J C, Langie S A S. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS One, 2013, 8: e79044[本文引用:1]
[18]
Fraga MF, UriolE, Borja DL, BerdascoM, EstellerM, Cañal MJ, RodriguezR. High-performance capillary electrophoretic method for the quantification of 5-methyl 2’-deoxycytidine in genomic DNA: application to plant, animal and human cancer tissues. Electrophoresis, 2002, 23: 1677-1681[本文引用:2]
[19]
KarimiM, JohanssonS, StachD, CorcoranM, Grand erD, SchallingM, BakalkinG, LykoF, LarssonC, Ekstrõm TJ. LUMA (LUminometric Methylation Assay): a high throughput method to the analysis of genomic DNA methylation. Exp Cell Res, 2006, 312: 1989-1995[本文引用:2]
[20]
Kuo KC, McCune R A, Gehrke C W, Midgett R, Ehrlich M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucl Acids Res, 1980, 8: 4763-4776[本文引用:1]
[21]
Burns MJ, Nixon GJ, Foy CA, HarrisN. Stand ardization of data from real-time quantitative PCR methods-evaluation of outliers and comparison of calibration curves. BMC Biotechnol, 2005, 5: 31[本文引用:1]
[22]
Wilfinger WW, MackeyK, ChomczynskiP. Effect of pH and ionic strength on the spectrophotometric assessment of nucleic acid purity. Biotechniques, 1997, 22: 474-481[本文引用:2]
[23]
SedlackovaT, RepiskaG, CelecP, SzemesT, MinankG. Fragmentation of DNA affects the accuracy of the DNA quantitation by the commonly used methods. Biol Proced Online, 2013, 15: 5[本文引用:1]
[24]
Hollegaard MV, GroveJ, GrauholmJ, Kreiner-MøllerE, BønnelykkeK, NørgaardM, Benfield TL, Nørgaard-PedersenB, Mortensen PB, MorsO, Sørensen HT, Harboe ZB, Børglum AD, DemontisD, Ørntoft TF, BisgaardH, Hougaard DM. Robustness of genome-wide scanning using archived dried blood spot samples as a DNA source. BMC Genet, 2011, 12: 58[本文引用:1]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}