关键词:全基因组关联分析; 上位性; 混合线性模型; 多位点模型 Advances on Methodologies for Genome-wide Association Studies in Plants FENG Jian-Ying1, WEN Yang-Jun1, ZHANG Jin1, ZHANG Yuan-Ming2,* 1 State Key Laboratory of Crop Genetics and Germplasm Enhancement, Nanjing Agricultural University, Nanjing 210095, China
2 College of Plant Science and Technology, Huazhong Agricultural University, Wuhan 430070, China
Fund:This work was supported by National Natural Science Foundation of China (31301004) and Fundamental Research Funds for the Central Universities (KJQN201422) AbstractGenome-wide association studies (GWAS) have been widely used in human, animal and plant genetics, and many new approaches and their softwares have been developed in recent years. To make a better use of the GWAS methods in applied research, in this study we summarized the advances on methodologies and softwares for GWAS. First, LD score regression was introduced to investigate the effect of population structure on GWAS. Then, the main approaches and their softwares for GWAS in plants were reviewed, including a single-locus model, a multi-locus model, epistasis, and multiple correlated traits. Finally, we prospected the future developments in GWAS. It should be noted that, in real data analysis at present, the methodologies for genome-wide single-marker scan under polygenic background and population structure controls are widely used, and the corresponding results are complementary to those derived from non-parameter approaches with high false discovery rate. However, the future approaches for GWAS should be based on the multi-locus genetic model, QTN-by-environment interaction, epistatic detection and multivariate analysis. Our purpose was to provide beneficial information in theoretical and applied researches.
Keyword:Genome-wide association study; Epistasis; Mixed linear model; Multi-locus model Show Figures Show Figures
RischN, MerikangasK. The future of genetic studies of complex human diseases. Science, 1996, 273: 1516-1517[本文引用:1]
[2]
PurcellS, NealeB, Todd-BrownK, ThomasL, FerreiraM, BenderD, MallerJ, SklarP, De BakkerP, DalyM, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 2007, 81: 559-575[本文引用:2]
[3]
WanX, YangC, YangQ, XueH, Fan XD, Tang N L S, Yu W C. BOOST: a fast approach to detecting gene-gene interactions in genome-wide case-control studies. Am J Hum Genet, 2010, 87: 325-340[本文引用:1]
[4]
TakeuchiF, SerizawaM, YamamotoK, FujisawaT, NakashimaE, OhnakaK, IkegamiH, SugiyamaT, KatsuyaT, MiyagishiM, NakashimaN, NawataH, NakamuraJ, KonoS, TakayanagiR, KatoN. Confirmation of multiple risk loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes, 2009, 3: 1690-1699[本文引用:1]
[5]
MichailidouK, BeesleyJ, LindstromS, CanisiusS, DennisJ, Lush MJ, Maranian MJ, Bolla MK, WangQ, ShahM, Perkins BJ, CzeneK, ErikssonM, DarabiH, Brand JS, Bojesen SE, Nordestgaard BG, FlygerH, Nielsen SF, RahmanN, TurnbullC, BOCS, FletcherO, Peto J, Gibson L, dos-Santos- Silva I, Chang-Claude J, Flesch-Janys D, Rudolph A, Eilber U, Behrens S, Nevanlinna H, Muranen T A, Aittomäki K, Blomqvist C, Khan S, Aaltonen K, Ahsan H, Kibriya M G, Whittemore A S, John E M, Malone K E, Gammon M D, Santella R M, Ursin G, Makalic E, Schmidt D F, Casey G, Hunter D J, Gapstur S M, Gaudet M M, Diver W R, Haiman C A, Schumacher F, Henderson B E, Le Marchand L, Berg C D, Chanock S J, Figueroa J, Hoover R N, Lambrechts D, Neven P, Wildiers H, van Limbergen E, Schmidt M K, Broeks A, Verhoef S, Cornelissen S, Couch F J, Olson J E, Hallberg E, Vachon C, Waisfisz Q, Meijers-Heijboer H, Adank M A, van der Luijt R B, Li J, Liu J, Humphreys K, Kang D, Choi J Y, Park S K, Yoo K Y, Matsuo K, Ito H, Iwata H, Tajima K, Guénel P, Truong T, Mulot C, Sanchez M, Burwinkel B, Marme F, Surowy H, Sohn C, Wu A H, Tseng C C, Van Den Berg D, Stram D O, González-Neira A, Benitez J, Zamora M P, Perez J I, Shu X O, Lu W, Gao Y T, Cai H, Cox A, Cross S S, Reed M W, Andrulis I L, Knight J A, Glendon G, Mulligan A M, Sawyer E J, Tomlinson I, Kerin M J, Miller N, kConFab Investigators, AOCS Group, Lindblom A, Margolin S, Teo S H, Yip C H, Taib N A, Tan G H, Hooning M J, Hollestelle A, Martens J W, Collée J M, Blot W, Signorello L B, Cai Q, Hopper J L, Southey M C, Tsimiklis H, Apicella C, Shen C Y, Hsiung C N, Wu P E, Hou M F, Kristensen V N, Nord S, Alnaes G I, NBCS, Giles G G, Milne R L, McLean C, Canzian F, Trichopoulos D, Peeters P, Lund E, Sund M, Khaw K T, Gunter M J, Palli D, Mortensen L M, Dossus L, Huerta J M, Meindl A, Schmutzler R K, Sutter C, Yang R, Muir K, Lophatananon A, Stewart- Brown S, Siriwanarangsan P, Hartman M, Miao H, Chia K S, Chan C W, Fasching P A, Hein A, Beckmann M W, Haeberle L, Brenner H, Dieffenbach A K, Arndt V, Stegmaier C, Ashworth A, Orr N, Schoemaker M J, Swerdlow A J, Brinton L, Garcia- Closas M, Zheng W, Halverson S L, Shrubsole M, Long J, Goldberg M S, Labrèche F, Dumont M, Winqvist R, Pylkäs K, Jukkola-Vuorinen A, Grip M, Brauch H, Hamann U, Brüning T; GENICA Network, Radice P, Peterlongo P, Manoukian S, Bernard L, Bogdanova N V, Dörk T, Mannermaa A, Kataja V, Kosma V M, Hartikainen J M, Devilee P, Tollenaar R A, Seynaeve C, Van Asperen C J, Jakubowska A, Lubinski J, Jaworska K, Huzarski T, Sangrajrang S, Gaborieau V, Brennan P, McKay J, Slager S, Toland A E, Ambrosone C B, Yannoukakos D, Kabisch M, Torres D, Neuhausen S L, Anton-Culver H, Luccarini C, Baynes C, Ahmed S, Healey C S, Tessier D C, Vincent D, Bacot F, Pita G, Alonso M R, Álvarez N, Herrero D, Simard J, Pharoah P P, Kraft P, Dunning A M, Chenevix-Trench G, Hall P, Easton D F. Genome-wide association analysis of more than 120 000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet, 2015, 47: 373-380[本文引用:1]
[6]
ScuteriA, SannaS, Chen WM, UdaM, AlbaiG, StraitJ, NajjarS, NagarajaR, OrrúM, UsalaG, DeiM, LaiS, MaschioA, BusoneroF, MulasA, Ehret GB, Fink AA, Weder AB, Cooper RS, GalanP, ChakravartiA, SchlessingerD, CaoA, LakattaE, Abecasis GR. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet, 2007, 3(7): e115[本文引用:1]
[7]
Thornsberry JM, Goodman MM, DoebleyJ, KresovichS, NielsenD, Buckler ES. Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet, 2001, 28: 286-289[本文引用:1]
[8]
HansenM, KraftT, GanestamS, SällT, Nilsson NO. Linkage disequilibrium mapping of the bolting gene in sea beet using AFLP markers. Genet Res, 2001, 77: 61-66[本文引用:1]
[9]
Zhang YM, Mao YC, Xie CQ, SmithH, LuoL, XuS. Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L. ). Genetics, 2005, 169: 2267-2275[本文引用:2]
[10]
YuJ, PressoirG, Briggs WH, Vroh BiI, YamasakiM, Doebley JF, McMullen M D, Gaut B S, Nielsen D M, Holland J B, Kresovich S, Buckler E S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet, 2006, 38: 203-208[本文引用:4]
[11]
Kang HM, Zaitlen NA, Wade CM, KirbyA, HeckermanD, Daly MJ, EskinE. Efficient control of population structure in model organism association mapping. Genetics, 2008, 178: 1709-1723[本文引用:2]
[12]
ZhangZ, ErsozE, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu JM, Arnett DK, Ordovas JM, Buckler ES. Mixed linear model approach adapted for genome-wide association studies. Nat Genet, 2010, 42: 355-360[本文引用:2]
[13]
AtwellS, Huang YS, Vilhjálmsson BJ, WillemsG, HortonM, LiY, MengD, PlattA, Tarone AM, Hu TT, JiangR, Muliyati NW, ZhangX, Amer MA, BaxterI, BrachiB, ChoryJ, DeanC, Debieu M, de Meaux J, Ecker J R, Faure N, Kniskern J M, Jones J D, Michael T, Nemri A, Roux F, Salt D E, Tang C, Todesco M, Traw M B, Weigel D, Marjoram P, Borevitz J O, Bergelson J, Nordborg M. Genome-wide association study of 10phenotypes in Arabidopsis Thaliana inbred lines. J Am Soc Mass Spectrom, 2010, 465: 627-631[本文引用:1]
[14]
Wang SB, Feng JY, Ren WL, HuangB, ZhouL, Wen YJ, ZhangJ, Jim MD, Xu SZ, Zhang YM. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci Rep, 2016, 6: 19444[本文引用:3]
[15]
Liu XL, HuangM, FanB, Buckler ES, Zhang ZW. Iterative usage of fixed and rand om effect models for powerful and efficient genome-wide association studies. PLoS Genet, 2016, 12(2): e1005767[本文引用:2]
[16]
Zhang FT, Zhu ZH, Tong XR, Zhu ZX, QiT, ZhuJ. Mixed linear model approaches of association mapping for complex traits based on omics variants. Sci Rep, 2015, 5: 10298[本文引用:3]
[17]
DevlinB, RoederK. Genomic control for association studies. Biometrics, 1999, 55: 997-1004[本文引用:1]
[18]
SongM, HaoW, Storey JD. Testing for genetic associations in arbitrarily structured populations. Nat Genet, 2015, 47: 550-556[本文引用:1]
[19]
Pritchard JK, StephensM, DonnellyP. Inference of population structure using multilocus genotype data. Genetics, 2000, 155: 945-959[本文引用:1]
[20]
Wilson LM, Whitt SR, Ibáez AM, Rocheford TR, Goodman MM, Buckler ES. Dissection of maize kernel composition and starch production by cand idate gene associations. Plant Cell, 2004, 16: 2719-2733[本文引用:1]
[21]
SabattiC, Service SK, Hartikainen AL, PoutaA, RipattiS, BrodskyJ, Jones CG, Zaitlen NA, VariloT, KaakinenM, SovioU, RuokonenA, LaitinenJ, JakkulaE, CoinL, HoggartC, CollinsA, TurunenH, GabrielS, ElliotP, McCarthy M I, Daly M J, Järvelin M R, Freimer N B, Peltonen L. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet, 2009, 41: 35-46[本文引用:1]
[22]
Price AL, Pattersom NJ, Plenge RM, Weinblatt ME, Shadick NA, ReichD. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet, 2006, 38: 904-909[本文引用:1]
[23]
Lee AB, LucaD, KleiL, DevlinB, RoederK. Discovering genetic ancestry using spectral graph theory. Genet Epidemiol, 2010, 34: 51-59[本文引用:1]
[24]
Bulik-Sullivan B K, Loh P R, Finucane H K, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N, Daly M J, Price A L, Neale B M. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet, 2015, 47: 291-295[本文引用:2]
[25]
Bu SH, Zhao XW, YiC, WenJ, Tu JX, Zhang YM. Interacted QTL mapping in partial NCII design provides evidences for breeding by design. PLoS One, 2015, 10(3): e0121034[本文引用:2]
[26]
LiM, Liu XL, BradburyP, Yu JM, Zhang YM, Todhunter RJ, Buckler ES, Zhang ZW. Enrichment of statistical power for genome-wide association studies. BMC Biol, 2014, 12: 73-82[本文引用:1]
[27]
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, SabattiC, EskinE. Variance component model to account for sample structure in genome-wide association studies. Nat Genet, 2010, 42: 348-354[本文引用:1]
[28]
Svishcheva GR, Axenovich TI, Belonogova N M, van Duijn C M, Aulchenko Y S. Rapid variance components-based method for whole-genome association analysis. Nat Genet, 2012, 44: 1166-1170[本文引用:1]
[29]
ZhouX, StephensM. Genome-wide efficient mixed-model analysis for association studies. Nat Genet, 2012, 44: 821-826[本文引用:1]
[30]
LippertC, ListqartenJ, LiuY, Kadie CM, Davidson RI, HeckermanD. Fast linear mixed models for genome-wide association studies. Nat Methods, 2011, 8: 833-835[本文引用:1]
[31]
ListgartenJ, LippertC, Kadie CM, Davidson RI, EskinE, HeckermanD. Improved linear mixed models for genome- wide association studies. Nat Methods, 2012, 9: 525-526[本文引用:1]
[32]
Loh PR, TuckerG, Bulik-Sullivan B K, Vilhjálmsson B J, Finucane H K, Salem R M, Chasman D I, Ridker P M, Neale B M, Berger B, Patterson N, Price A L. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet, 2015, 47: 284-290[本文引用:1]
[33]
WangQ, TianF, PanY, Buckler ES, ZhangZ. A SUPER powerful method for genome wide association study. PLoS One, 2014, 9: e107684[本文引用:1]
[34]
ZhaoK, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, ReynoldsA, MezeyJ, McClung A M, Bustamante C D, McCouch S R. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun, 2011, 2: 467-476[本文引用:1]
[35]
Wen ZX, Tan RJ, Yuan JZ, BalesC, Du WY, Zhang SC, Chilvers MI, SchmidtC, Song QJ, Cregan PB, Wang DC. Genome-wide association mapping of quantitative resistance to sudden death syndrome in soybean. BMC Genomics, 2014, 15: 809-819[本文引用:1]
[36]
YangN, Lu YL, Yang XH, HuangJ, ZhouY, AliF, Wen WW, LiuJ, Li JS, Yan JB. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet, 2014, 10(9): e1004573[本文引用:2]
[37]
McCullagh P, Nelder J A. Generalized Linear Models, 2nd edn. London: Chapman and Hall, 1989[本文引用:1]
[38]
YiN, Liu NJ, Zhi DG, LiJ. Hierarchical generalized linear models for multiple groups of rare and common variants: jointly estimating group and individual-variant effects. PLoS Genet, 2011, 7(12): e1002382[本文引用:1]
[39]
Feng JY, ZhangJ, Zhang WJ, Wang SB, Han SF, Zhang YM. An efficient hierarchical generalized linear mixed model for mapping QTL of ordinal traits in crop cultivars. PLoS One, 2013, 8(4): e59541[本文引用:1]
[40]
WangL, JiaP, Wolfinger RD, ChenX, Grayson BL, Aune TM, ZhaoZ. An efficient hierarchical generalized linear mixed model for pathway analysis of genome-wide association studies. BMC Bioinformatics, 2011, 27(5): 686-692[本文引用:1]
[41]
IwataH, UgaY, YoshiokaY, EbanaK, HayashiT. Bayesian association mapping of multiple quantitative trait loci and its application to the analysis of genetic variation among (Oryza sativa L. ) germplasms. Theor Appl Genet, 2007, 114: 1437-1449[本文引用:1]
[42]
IwataH, EbanaK, FukuokaS, Jannink JL, HayashiT. Bayesian multilocus association mapping on ordinal and censored traits and its application to the analysis of genetic variation among (Oryza sativa L. ) germplasms. Theor Appl Genet, 2009, 118: 865-880[本文引用:1]
[43]
Zhang YM, XuS. A penalized maximum likelihood method for estimating epistatic effects of QTL. Heredity, 2005, 95: 96-104[本文引用:1]
[44]
Hoggart CJ, Whittaker JC, De IorioM, Balding DJ. Simultaneous analysis of all SNPs in genome-wide and resequencing association studies. PLoS Genet, 2008, 4: e1000130[本文引用:1]
[45]
SeguraV, Vilhjálmsson BJ, PlattA, KorteA, SerenÜ, LongQ, NordborgM. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet, 2012, 44: 825-830[本文引用:1]
[46]
YangJ, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet, 2011, 88: 76-82[本文引用:2]
[47]
Goddard ME, Wray NR, VerbylaK, Visscher PM. Estimating effects and making predictions from genomewide marker data. Stat Sci, 2009, 24: 517-529[本文引用:1]
[48]
ZhouX, CarbonettoP, StephensM. Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet, 2013, 9(2): e1003264[本文引用:1]
[49]
MoserG, Lee SH, Hayes BJ, Goddard ME, Wray NR, Visscher PM. Simultaneous discovery, estimation and prediction analysis of complex traits using a Bayesian mixture model. PLoS Genet, 2015, 11(4): e1004969[本文引用:1]
[50]
ZhangY, Liu JS. Bayesian inference of epistatic interactions in case-control studies. Nat Genet, 2007, 39: 1167-1173[本文引用:2]
[51]
Tang WW, Wu XB, JiangR. Epistatic module detection for case-control studies: a Bayesian model with a Gibbs sampling strategy. PLoS Genet, 2009, 5(5): e1000464[本文引用:1]
[52]
ChoS, KimH, OhS, KimK, ParkT. Elastic-net regularization approaches for genome-wide association studies of rheumatoid arthritis. BMC Proc, 2009, 3(suppl 7): S25[本文引用:1]
[53]
HanB, ParkM, Chen XW. A Markov blanket-based method for detecting causal SNPs in GWAS. BMC Bioinformatics, 2010, 11(suppl 3): S5[本文引用:1]
[54]
HanB, Chen XW, TalebizadehZ. FEPI-MB: identifying SNPs-disease association using a Markov blanket-based approach. BMC Bioinformatics, 2011, 12(Suppl 12): S3[本文引用:1]
[55]
LiJ, DanJ, Li CL, Wu RL. A model-free approach for detecting interactions in genetic association studies. Brief Bioinform, 2014, 15: 1057-1068[本文引用:1]
[56]
WangD, Eskridge KM, CrossaJ. Identifying QTLs and epistasis in structured plant populations using adaptive mixed LASSO. J Agric Biol Environ Stat, 2011, 16: 170-184[本文引用:1]
[57]
Lü HY, Liu XF, Wei SP, Zhang YM. Epistatic association mapping in homozygous crop cultivars. PLoS One, 2011, 6(3): e17773[本文引用:1]
[58]
WenJ, Zhao XW, Wu GR, XiangD, LiuQ, Bu SH, YiC, Song QJ, Dunwell JM, Tu JX, Zhang TZ, Zhang YM. Genetic dissection of heterosis using epistatic association mapping in a partial NCII mating design. Sci Rep, 2015, 5: 18376[本文引用:2]
[59]
AschardH, Vilhjálmsson BJ, GrelicheN, Morange PE, Trégouët DA, KraftP. Maximizing the power of principal- component analysis of correlated phenotypes in genome-wide association studies. Am J Hum Genet, 2014, 94: 662-676[本文引用:1]
[60]
Ferreira MA, Purcell SM. A multivariate test of association. Bioinformatics, 2009, 25: 132-133[本文引用:1]
BolormaaS, Pryce JE, ReverterA, ZhangY, BarendseW, KemperK, TierB, SavinK, Hayes BJ, Goddard ME. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet, 2014, 10: e1004198[本文引用:1]
[63]
XuY, Hu WM, Yang ZF, Xu CW. A multivariate partial least squares approach to joint analysis for multiple correlated traits. Crop J, 2016, 4(1): 21-29[本文引用:1]
[64]
KorteA, Vilhjálmsson BJ, SeguraV, PlattA, LongQ, NordborgM. A mixed-model approach for genome-wide association studies of correlated traits in structured populations. Nat Genet, 2012, 44: 1066-1071[本文引用:1]
[65]
ZhouX, StephensM. Efficient algorithm for multivariate linear mixed models in genome-wide association studies. Nat Methods, 2014, 11: 407-409[本文引用:1]
[66]
Casale FP, RakitschB, LippertC, StegleO. Efficient set tests for the genetic analysis of correlated traits. Nat Methods, 2015, 12: 755-758[本文引用:1]
[67]
Furlotte NA, EskinE. Efficient multiple-trait association and estimation of genetic correlation using the matrix-variate linear mixed model. Genetics, 2015, 200: 59-68[本文引用:1]
[68]
WanX, YangC, YangQ, XueH, Tang N L S, Yu W C. Predictive rule inference for epistatic interaction detection in genome-wide association studies. Bioinformatics, 2010, 26: 30-37[本文引用:1]
[69]
Bradbury PJ, ZhangZ, Kroon DE, Casstevens TM, RamdossY, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. BMC Bioinformatics, 2007, 23: 2633-2635[本文引用:1]
[70]
TangY, LiuX, WangJ, LiM, WangQ, TianF, SuZ, PanY, LiuD, Lipka AE, Buckler ES, ZhangZ. GAPIT Version 2: Enhanced integrated tool for genomic association and prediction. Plant Genome, 2016, 9(2): doi: DOI:10.3835/plantgenome2015.11.0120[本文引用:1]
[71]
张福涛. 遗传分析方法的GPU并行计算与优化研究. 浙江大学博士学位论文, 浙江杭州, 2014. pp 89-97Zhang FT. Parallelization and Optimization of GPU Computation for Genetic Analysis Methods. PhD Dissertation of Zhejiang University, Hangzhou, China, 2014. pp 89-97 (in Chinese with English abstract)[本文引用:1]
[72]
Sul JH, BilowM, Yang WY, KostemE, FurlotteN, HeD, EskinE. Accounting for population structure in gene-by- environment interactions in genome-wide association studies using mixed models. PLoS Genet, 2016, 12(3): e1005849[本文引用:1]
[73]
ZhangW, DaiX, WangQ, XuS, Zhao PX. PEPIS: a pipeline for estimating epistatic effects in quantitative trait locus mapping and genome-wide association studies. PLoS Comput Biol, 2016, 12(5): e1004925[本文引用:1]
[74]
Collard B C Y, Mackill D J. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci, 2008, 363(1491): 557-572[本文引用:1]
杨小红, 严建兵, 郑艳萍, 余建明, 李建生. 植物数量性状关联分析研究进展. 作物学报, 2007, 33: 523-530Yang XH, Yan JB, Zheng YP, Yu JM, Li JS. Reviews of association analysis for quantitative traits in plants. Acta Agron Sin, 2007, 33: 523-530 (in Chinese)[本文引用:1]
[77]
谭贤杰, 吴子恺, 程伟东, 王天宇, 黎裕. 关联分析及其在植物遗传学研究中的应用. 植物学报, 2011, 46: 108-118Tan XJ, Wu ZK, Cheng WD, Wang TY, LiY. Association analysis and its application in plant genetic research. Chin Bull Bot, 2011, 46: 108-118 (in Chinese)[本文引用:1]
[78]
布素红. 多亲本群体QTL定位和优异杂交组合预测. 南京农业大学博士学位论文, 江苏南京, 2015. pp 57-68Bu SH. Mapping of Quantitative Trait Loci and Prediction of Elite Hybrid Combination in Multi-parental Populations. PhD Dissertation of Nanjing Agricultural University, Nanjing, China, 2015. pp 57-68 (in Chinese with English abstract)[本文引用:1]
[79]
Chan E K F, Rowe H C, Kliebenstein D J. Understand ing the evolution of defense metabolites in Arabidopsis thaliana using genome-wide association mapping. Genetics, 2010, 185: 991-1007[本文引用:1]
[80]
RiedelsheimerC, LisecJ, Czedik-EysenbregA, SulpiceR, FlisA, GriederC, AltmannT, StittM, WillmitzerL, Melchinger AE. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc Natl Acad Sci USA, 2012, 109: 8872-8877[本文引用:1]
[81]
Wen WW, LiD, LiX, Gao YQ, Li WQ, Li HH, LiuJ, Liu HJ, ChenW, LuoJ, Yan JB. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat Commun, 2014, 5: 3438-3447[本文引用:1]
 , 温阳俊
, 温阳俊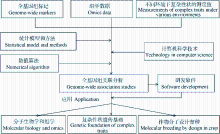
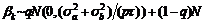
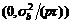
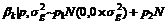



{kind=link}