全文HTML
--> --> -->近年来, 在电气工程领域, 天然酯以其可再生、环保及优良的绝缘性能受到了人们的关注[3-7]. 天然酯绝缘油通常以大豆油、菜籽油或山茶籽油为原料, 并添加抗氧化剂而制成, 其主要成分为支链上含有碳碳双键的不饱和甘油三酯分子. 将天然酯代替矿物油应用于变压器中, 能够有效地避免因变压器油泄露而造成的环境污染. 然而, 天然酯绝缘油的运动黏度在20 ℃测试条件下为85 mm2/s, 远高于矿物绝缘油在同等测试条件下22 mm2/s 的运动黏度[8]. 天然酯绝缘油这种高运动黏度的性质将明显影响其散热性能. 短-中链甘油三酯的运动黏度在20 ℃测试条件下约为10—40 mm2/s, 且其理化性质稳定[9]. 可将短-中链甘油三酯添加到天然酯绝缘油中, 以扩大天然酯绝缘油中短-中链甘油三酯的占比, 从而改善绝缘油的散热性能. 若将短-中链甘油三酯添加到天然酯绝缘油中, 其必然将承受相应等级电场强度的作用. 已有的研究结果表明, 绝缘油中放电起始与发展的电场等级在108 V/m的量级, 绝缘油以快速流注击穿时的电场等级可达109 V/m以上[10-12]. 在高场强作用下分子结构与性质将会发生明显变化, 而目前对饱和甘油三酯在相应的电场强度等级下的分子特性变化规律还不清楚.
密度泛函理论(density functional theory, DFT)可用于绝缘材料分子特性的研究[13-16]. 文献[13]研究了碳碳双键对不饱和甘油三酯分子电离势与亲和势的影响; 文献[14]对比研究了甘油三油酸酯与芳香烃类分子的电子结构及电离势, 解释了天然酯与矿物油两种绝缘油在放电现象上所表现的差异; 文献[15]研究了电场对芳香烃及烷烃分子电离势与激发能的影响. 众所周知, 天然酯是以不饱和长支链甘油三酯分子为主的液体电介质. 短-中链甘油三酯不含碳碳双键, 在电场下将表现出不同于不饱和甘油三酯的分子性质, 因此还有待进一步研究.
本文使用密度泛函与含时密度泛函方法, 通过设置与绝缘油中放电相应的电场强度等级, 计算了电场下三乙酸甘油酯(triacetin, C2:0)、三丁酸甘油酯(tributyrin, C4:0)、三己酸甘油酯(tricaproin, C6:0)、三辛酸甘油酯(tricaprylin, C8:0)和三癸酸甘油酯(tricaprin, C10:0)共5种不同碳链长度的饱和甘油三酯分子的总能量、偶极矩、红外光谱、分子轨道能量与激发特性, 探索了不同碳链长度的饱和甘油三酯在电场影响下分子特性的变化规律. 研究结果有助于提高对短-中链饱和甘油三酯绝缘介质中放电机理的认识, 并为天然酯绝缘油的性能改进提供一定的理论支撑.

根据含时密度泛函理论(time dependent density functional theory, TD-DFT), 分子激发态激发能Eex可表示为[20,21]



图1给出了C6∶0分子的构型, 其三个支链上分别包含6个碳原子. 图1中红色、黑色与蓝色位置分别表示氧、碳与氢三种原子, 并用数字进行标识, Z轴贯穿1C与2C两原子. C2∶0, C4∶0, C8∶0, C10:0等4种分子的构型与C6:0分子相比仅存在支链上碳原子个数的差异, 限于篇幅不在此展示. 本文的研究步骤如下. 首先, 对C2∶0, C4∶0, C6∶0, C8∶0与C10∶0这5种分子在无电场条件下进行结构优化, 获得其稳定构型. 图1中的黄色箭头指示了C6∶0分子在结构优化后的固有偶极矩的方向, 其数值为6.66 Debye, 在X, Y与Z轴上的分量分别为4.05, –4.72及–2.38 Debye. 然后, 在Z轴方向上分别施加–0.0075, –0.005, –0.0025, –0.001, –0.0005, +0.0005, +0.001, +0.0015, +0.0025, +0.005与+0.0075 a.u.的电场, 其中“–”与“+”分别表示施加电场的方向沿Z轴负方向和正方向. 1 a.u.电子场强相当于5.1











 图 1 C6∶0的分子构型
图 1 C6∶0的分子构型Figure1. Molecular configuration of C6∶0.
在具体方法与基组选择方面, 比较了HF/631+G*, B3LYP/631G*, B3LYP/631+G*和B3LYP/6311++G**4种方法对无电场条件下分子的主要官能团振动频率计算结果, 如表1所列, 其中0.90与0.96为修正系数[24]. 可以发现, 相比HF方法, B3LYP方法的计算结果更接近实验值; 3种B3LYP方法中, B3 LYP/631+G*和B3LYP/6311++G**的计算结果与实验值更为接近[25]. 考虑精确度与时效性, 同时为了便于比较, 本文其他计算都选择了B3LYP方法及631+G*基组.
| 方法 | 甘油 三酯 | 波数 v/ cm–1 | |||
| C—O—O | C=O | C—H | |||
| C2:0 | 1235.8 | 1816.3 | 2938.4 | ||
| C4:0 | 1185.9 | 1809.5 | 2938.3 | ||
| HF/631+ G*/0.90 | C6:0 | 1162 | 1809.2 | 2926.4 | |
| C8:0 | 1179.5 | 1809.1 | 2926.2 | ||
| C10:0 | 1160.2 | 1808.9 | 2908.9 | ||
| C2:0 | 1173.9 | 1787.1 | 2602.1 | ||
| C4:0 | 1131.2 | 1778.3 | 2993.7 | ||
| B3LYP/631G*/0.96 | C6:0 | 1126.8 | 1778.2 | 2983.1 | |
| C8:0 | 1125.2 | 1778.2 | 2981.7 | ||
| C10:0 | 1089.3 | 1778.2 | 2907.7 | ||
| C2:0 | 1181.6 | 1756.8 | 2956.7 | ||
| C4:0 | 1123.3 | 1750.7 | 2987.5 | ||
| B3LYP/631+G*/0.96 | C6:0 | 1119.1 | 1750.5 | 2975.6 | |
| C8:0 | 1117.6 | 1750.4 | 2940.5 | ||
| C10:0 | 1116.5 | 1750.4 | 2943.9 | ||
| C2:0 | 1164.1 | 1751.3 | 2936.3 | ||
| C4:0 | 1107.6 | 1745.4 | 2966.1 | ||
| B3LYP/6311++G**/0.96 | C6:0 | 1104.2 | 1745.1 | 2959.5 | |
| C8:0 | 1103 | 1745 | 2895.9 | ||
| C10:0 | 1102.3 | 1745 | 2929.9 | ||
| C2:0 | 1208.0 | 1738.6 | 2950.0 | ||
| 实验值 | C4:0 | 1159.0 | 1736.5 | 2962.5 | |
| C6:0 | 1159.8 | 1744.4 | 2950.4 | ||
| C8:0 | 1155.5 | 1747.5 | 2958.3 | ||
| C10:0 | ? | ? | ? | ||
表1不同计算方法的比较
Table1.Comparison of different calculation methods
3.1.键长与红外光谱
键长是分子的基本结构参数, 键长的变化对分子特性的影响起到关键性作用. 甘油三酯分子所包含的化学键数量很多, 限于篇幅, 表2给出了C6∶0分子在不同电场下部分主要化学键的键长. 从表2可以发现, 键长与电场强度存在明显的依赖关系. 例如, 当电场在–3.8



| E/(108 V·m–1) | 2C—9O/? | 12C—9O/? | 12C=13O/? | 5C—10O/? | 14C—10O/? | 14C = 15O/? | 1C—11O/? | 16C=17O/? |
| –38 | 1.435 | 1.389 | 1.205 | 1.434 | 1.379 | 1.209 | 1.438 | 1.214 |
| –25 | 1.434 | 1.388 | 1.207 | 1.433 | 1.379 | 1.207 | 1.432 | 1.211 |
| –13 | 1.434 | 1.384 | 1.207 | 1.431 | 1.376 | 1.207 | 1.430 | 1.209 |
| –5.1 | 1.434 | 1.381 | 1.207 | 1.431 | 1.373 | 1.206 | 1.429 | 1.208 |
| –2.6 | 1.434 | 1.380 | 1.207 | 1.432 | 1.372 | 1.208 | 1.428 | 1.208 |
| 0 | 1.434 | 1.380 | 1.207 | 1.433 | 1.371 | 1.207 | 1.428 | 1.207 |
| 2.6 | 1.434 | 1.379 | 1.207 | 1.433 | 1.370 | 1.208 | 1.428 | 1.207 |
| 5.1 | 1.434 | 1.378 | 1.207 | 1.434 | 1.369 | 1.208 | 1.428 | 1.207 |
| 13 | 1.435 | 1.375 | 1.208 | 1.436 | 1.366 | 1.210 | 1.428 | 1.206 |
| 25 | 1.434 | 1.373 | 1.209 | 1.439 | 1.360 | 1.213 | 1.428 | 1.205 |
| 38 | 1.434 | 1.370 | 1.211 | 1.443 | 1.355 | 1.216 | 1.425 | 1.203 |
表2电场下C6∶0分子的键长
Table2.Bond length of C6∶0 molecule under electric field.
键长变化与电场下电荷的转移有关[17,26]. 化学键两端的原子在引力与斥力组成的内应力作用下, 在平衡距离处达到稳定状态. 当施加电场时, 电子发生与电场方向相反的运动, 电荷转移, 原子间的内应力发生变化, 平衡距离改变, 键长因此而变化.
图2给出了C6:0分子在–3.8






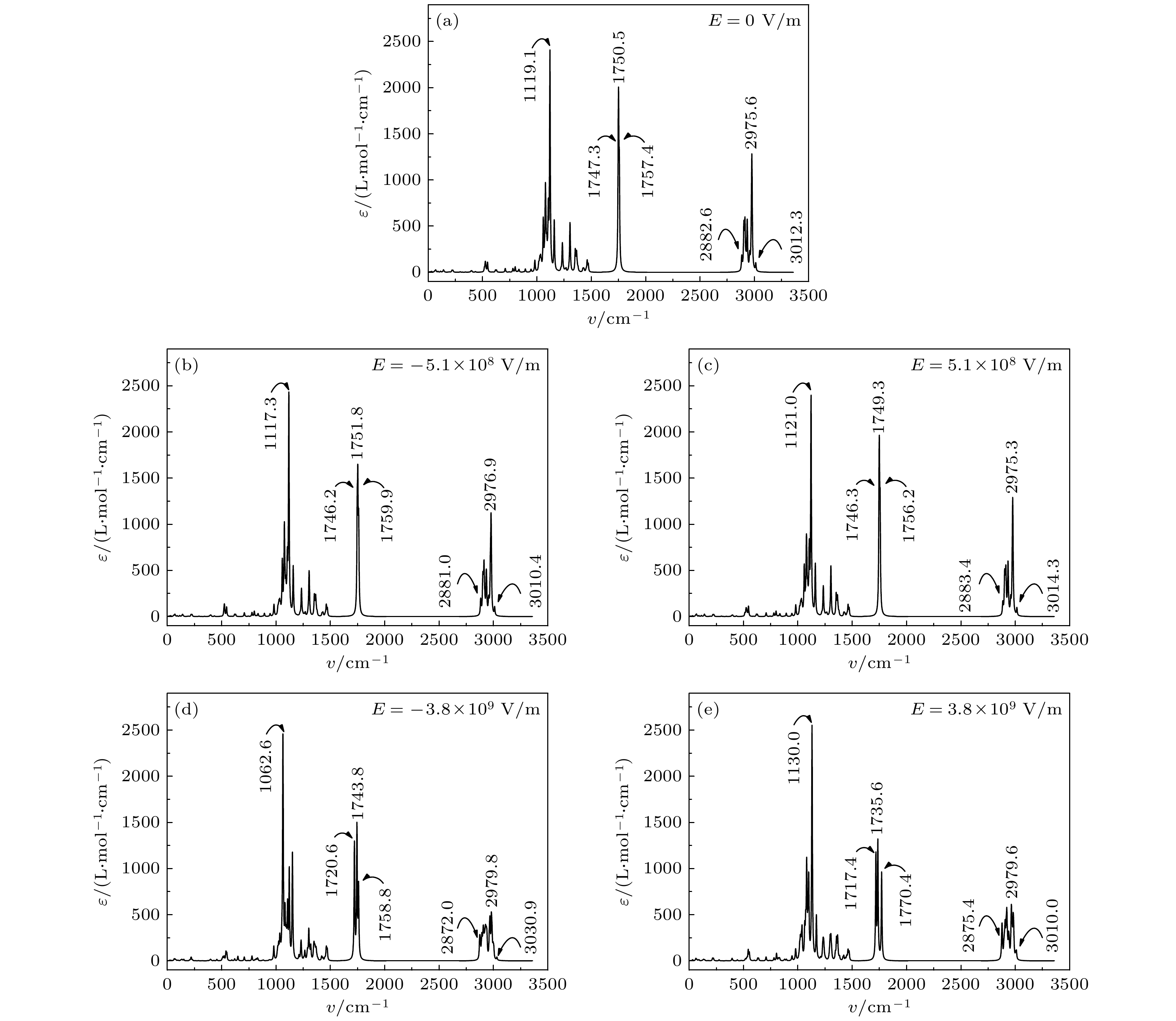 图 2 电场下C6∶0的分子的红外光谱
图 2 电场下C6∶0的分子的红外光谱Figure2. The infrared spectra of C6∶0 molecule under electric field.
波数峰的移动与键长变化有关[22]. 当电场在–3.8


2
3.2.偶极矩与总能量
正负电荷中心不重合的分子称作极性分子, 分子极性与其偶极矩呈正相关. 图3(a)给出了C2∶0, C4∶0, C6∶0, C8∶0与C10∶0这5种甘油三酯分子在不同电场下的偶极矩. 从图3(a)可以发现, 在无电场条件下, 这5种分子皆为极性分子, 其偶极矩差距不大, 约为6.65—6.71 Debye, 即其固有偶极矩. 当在Z轴负方向上增大施加电场强度时, 这5种分子的偶极矩都是先减小后增大. 如本文第2节所述, 分子的固有偶极矩在–Z轴上有分量. 当沿着Z轴负方向上施加电场时, 由电场引起的偶极矩的方向沿Z轴正方向, 即电场引起的偶极矩的方向与电场方向相反, 由此导致了上述分子偶极矩先减小后增大的现象.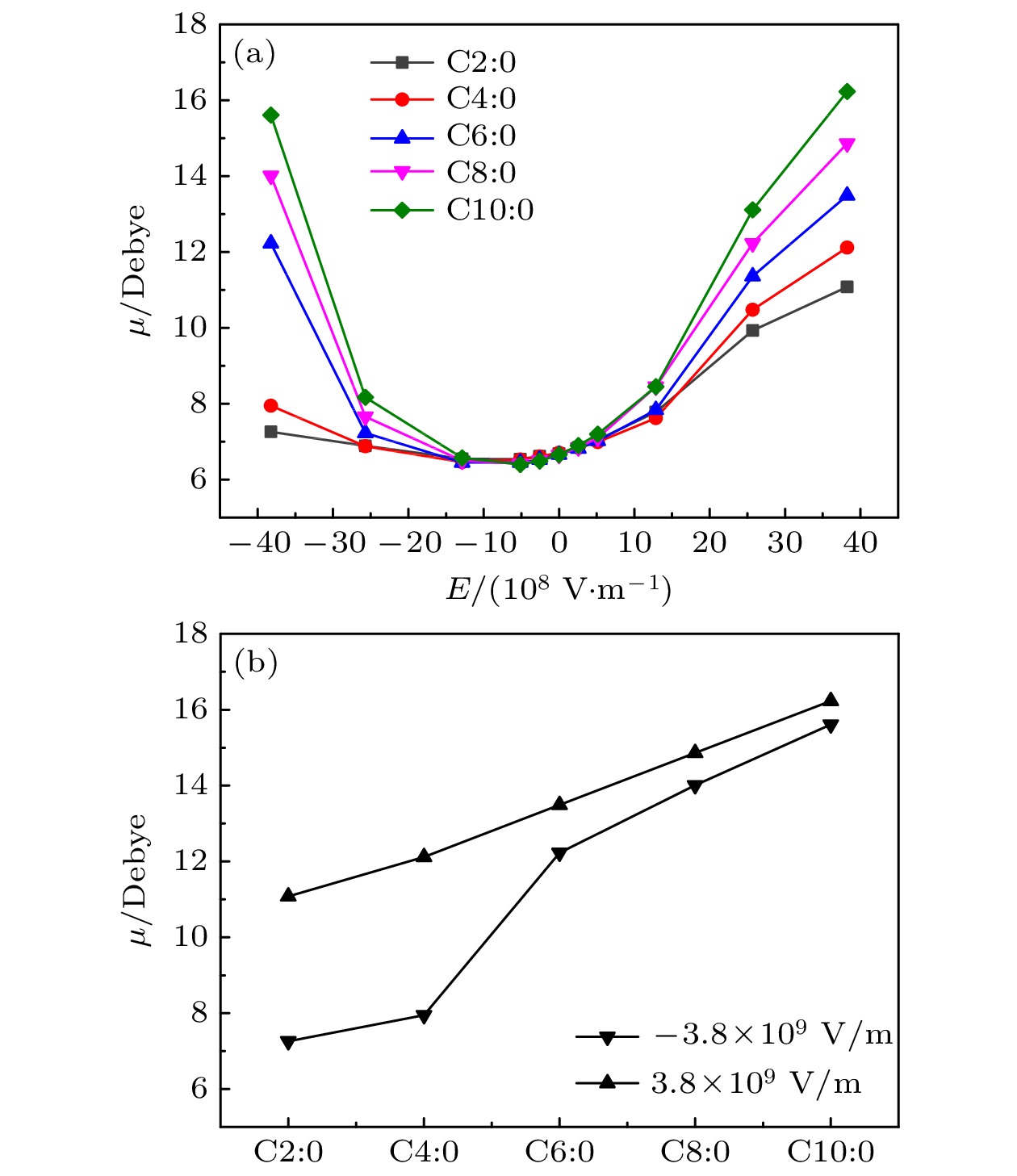 图 3 电场下分子的偶极矩
图 3 电场下分子的偶极矩Figure3. The molecular dipole moment under electric field.
另外值得注意的是, 在相同电场下, 随着碳链长度的增大, 甘油三酯分子的偶极矩也同时增大. 例如, 施加电场–3.8

图4给出了C2∶0, C4∶0, C6∶0, C8∶0与C10∶0这5种甘油三酯分子在不同电场下的总能量. 从图4可以看出, 当电场在–3.8


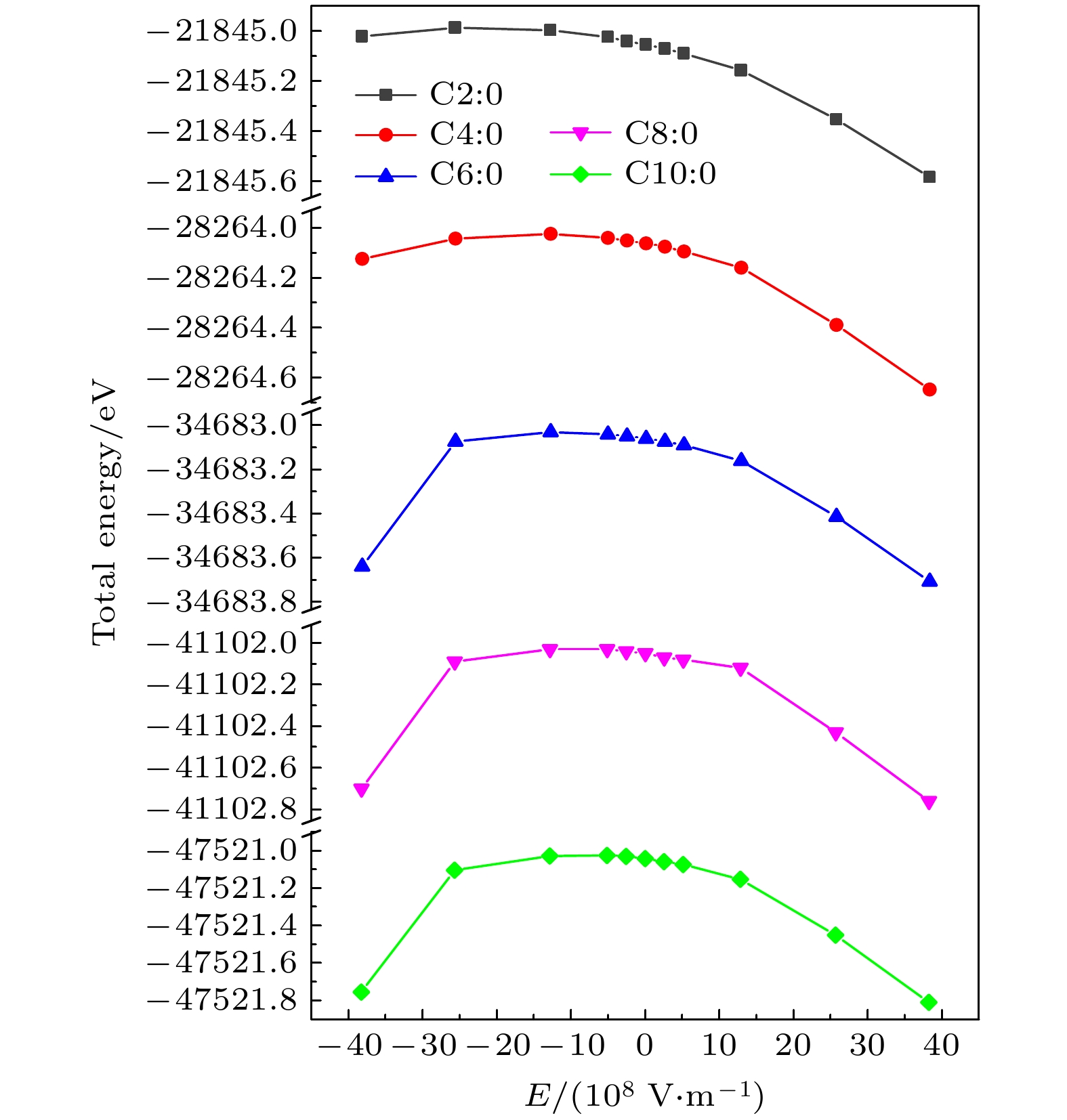 图 4 电场下分子的总能量
图 4 电场下分子的总能量Figure4. The molecular total energy under electric field.
2
3.3.前线轨道与电离势
分子前线轨道包括最高占据态分子轨道(the highest occupied molecular orbital, HOMO)和最低非占据态分子轨道(the lowest unoccupied molecular orbital, LUMO). 前线分子轨道对分子性质具有重要影响. 图5给出了C2∶0, C4∶0, C6∶0, C8∶0与C10:0这5种甘油三酯分子在不同电场下的前线轨道能量. 从图5可以看出, 这5种分子的LUMO能量随电场强度的变化趋势与变化幅度基本相同. 在+Z轴方向上, 随着电场强度大小的增加, 这5种分子的LUMO能量均减小. LUMO能量的减小预示着分子得电子能力的增强. 电场下这5种分子的HOMO能量随电场强度的变化趋势基本相同, 但其变化幅度差异较大. 在相同电场强度下, 碳链越长分子的HOMO能量越高, 这预示着分子越容易失去轨道电子.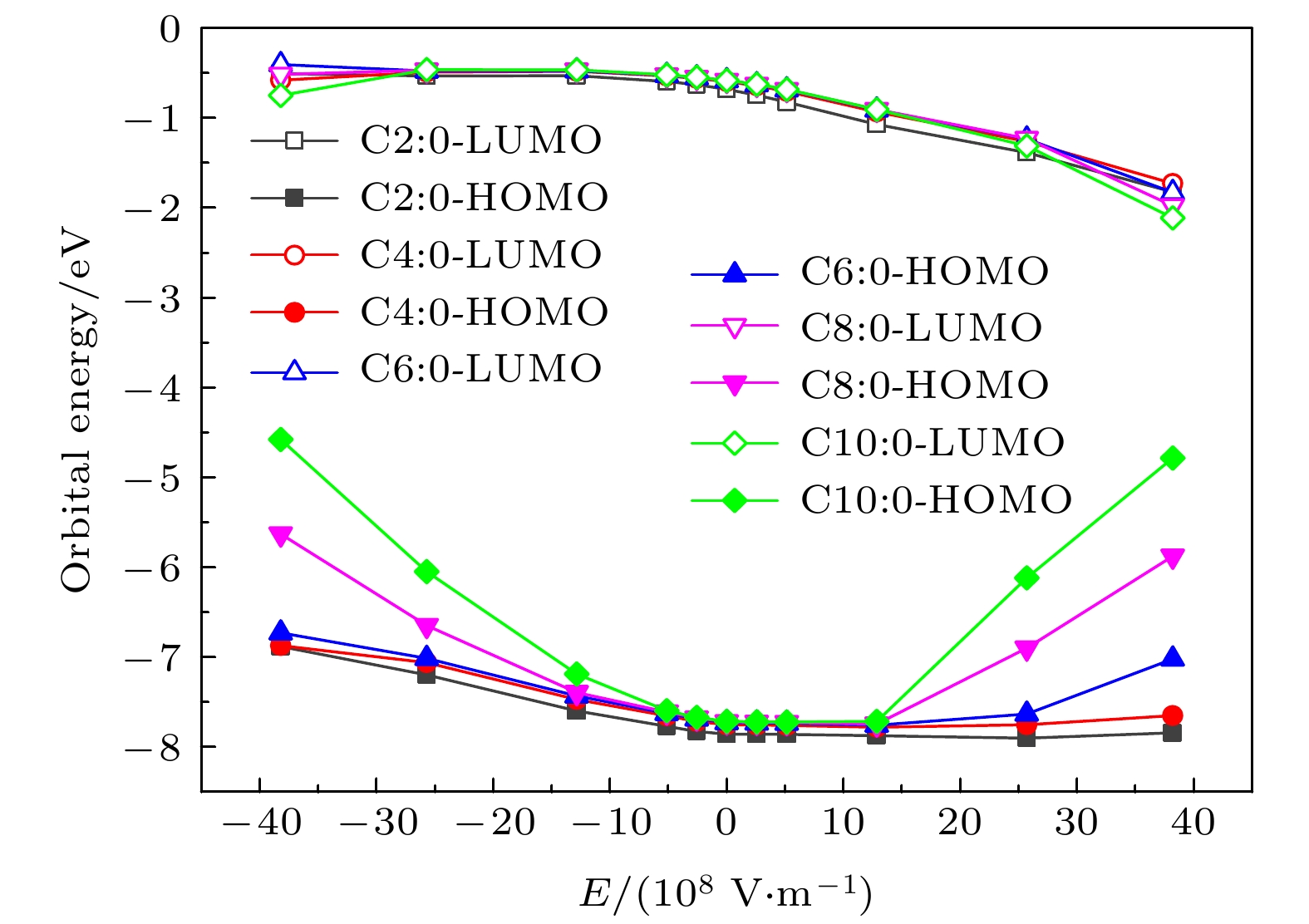 图 5 电场下分子的前线轨道能量
图 5 电场下分子的前线轨道能量Figure5. The Frontier molecular orbital energy under electric field.
分子能隙定义为HOMO与LUMO的能量差, 能隙在一定程度上反映了分子参与化学反应的活性[28,29]. 图6给出了C2∶0, C4∶0, C6∶0, C8∶0与C10∶0这5种甘油三酯分子在不同电场下的能隙. 由图6可以看出, 在–Z轴与+Z轴方向上, 随着电场强度大小的增加, 这5种分子的能隙均减小. 相同电场强度下, 随着碳链长度的增加, 分子能隙呈减小趋势.
 图 6 电场下分子的能隙
图 6 电场下分子的能隙Figure6. The molecular energy gap under electric field.
图7给出了C6∶0分子在–3.8





 图 7 电场下C6∶0分子的前线轨道云图
图 7 电场下C6∶0分子的前线轨道云图Figure7. The cloud image of C6∶0 molecular frontier orbital under electric field.
| E/(108 V·m–1) | Composition of the HOMO and LUMO (>3%) | |
| –38 | HOMO | 5C∶3.73, 12C∶4.49, 37C∶3.35, 40C∶11.88, 43H∶4.08, 45C∶18.02, 46C∶13.24, 47H∶4.2, 48H∶10.08, 50H∶6.99 |
| LUMO | 1C∶18.58, 2C∶18.88, 5C∶31.84, 18C∶6.56, 19C∶10.27, 22C∶6.87 | |
| –5.1 | HOMO | 2C∶3.07, 12C∶3.36, 13O∶34.35, 36C∶25.08, 37C∶5.25, 40C∶4.97 |
| LUMO | 1C∶7.12, 2C∶7.25, 14C∶5.28, 18C∶25.35, 19C∶33.40, 27C∶6.48, 28C3.29, 31C∶3.17 | |
| 0 | HOMO | 2C∶5.86, 5C∶5.03, 11O∶7.80, 17O∶31.35, 18C∶17.39, 19C∶5.15, 22C∶5.58 |
| LUMO | 1C∶5.77, 2C∶6.75, 14C∶4.68, 18C∶26.92, 19C∶32.22, 27C∶7.36, 28C∶4.81, 31C∶3.87 | |
| 5.1 | HOMO | 2C∶5.62, 5C∶4.92, 11O∶8.19, 12C∶3.31, 16C∶3.17, 17O∶32.67, 18C∶17.61, 19C∶5.21, 22C∶5.88 |
| LUMO | 1C∶8.10, 2C∶9.33, 14C∶3.39, 18C∶7.20, 19C∶29.10, 27C∶6.93, 28C∶4.89, 31C∶4.08 | |
| 38 | HOMO | 1C∶15.52, 2C∶31.37, 18C∶5.46, 19C∶7.80, 52C∶8.47, 53C∶6.22, 54H∶4.66, 55H∶5.03, 57H∶3.53, 58H∶3.32 |
| LUMO | 27C∶9.74, 28C∶26.00, 31C∶33.47, 59C∶26.02 |
表3电场下C6∶0分子的前线轨道组分
Table3.The frontier orbital composition of C6∶0 molecule under electric field.
液体电介质中的放电过程十分复杂. 在强电场下, 电介质的中性分子发生电离形成正离子与电子. 正离子和电子在电场力作用下分别移向阴极与阳极, 在这一过程中, 电子被中性分子捕获可形成负离子. 整个放电过程中还存在正离子与负离子复合及正离子与电子复合, 并伴随声光电现象. 中性分子的电离贯穿整个放电过程, 对放电的发展起到重要作用. 分子电离势能够代表分子被电离的难易程度. 库普曼定理认为分子电离势等于HOMO能量的相反数[31], 根据此定理可估算分子的电离势. 需要指出的是, 库普曼定理假定分子在失去一个电子时, 其分子结构及轨道能级不变, 因此根据此定理所获得的电离势可能并不十分精确. 然而在由多种分子组分组成的液体电介质中, 放电过程通常由低电离势分子决定[32], 因此比较分子间电离势的相对大小仍然具有实际意义.
图8给出了不同电场强度下不同甘油三酯分子的电离势. 从图8可以看出, 随着碳链长度的增加, 分子电离势均减小. 这是因为随着碳链长度的增加, 分子体积变大, 分子对最外层的电子的引力减弱, 分子更容易失掉电子. 值得注意的是, 在109 V/m量级的电场强度下, C8∶0与C10∶0分子的电离势急剧减小. 例如, 施加电场–3.8




 图 8 电场下不同分子的电离势
图 8 电场下不同分子的电离势Figure8. The ionization potential of different molecules under electric field.
2
3.4.激发态
本文使用含时密度泛函方法计算了短-中链饱和甘油三酯的激发态. 计算结果显示, 甘油三酯分子间的激发特性差异较小, 表4列出了C2∶0, C6∶0与C10:0三种甘油三酯分子在0 , 5.1

| 分子 | E /(108 V·m–1) | m = 1 | m = 2 | m = 3 | m = 4 | m = 5 | m = 6 | m = 7 | m = 8 | m = 9 | |
| 0 | Eex /eV | 5.6219 | 5.6422 | 5.6657 | 6.6449 | 6.6683 | 6.7503 | 6.7754 | 6.8532 | 6.8874 | |
| λ /nm | 220.54 | 219.74 | 218.83 | 186.59 | 185.93 | 183.67 | 182.99 | 180.91 | 180.02 | ||
| f | 0.0009 | 0.0009 | 0.0004 | 0.0056 | 0.0015 | 0.0177 | 0.0096 | 0.0019 | 0.0032 | ||
| 5.1 | Eex /eV | 5.6149 | 5.6418 | 5.6619 | 6.6358 | 6.6848 | 6.7356 | 6.7626 | 6.8506 | 6.8776 | |
| C2∶0 | λ /nm | 220.81 | 219.76 | 218.98 | 186.84 | 185.47 | 184.07 | 183.34 | 180.98 | 180.27 | |
| f | 0.0009 | 0.0009 | 0.0005 | 0.0071 | 0.0009 | 0.0169 | 0.0098 | 0.0041 | 0.0022 | ||
| 26 | Eex /eV | 5.5859 | 5.6366 | 5.643 | 6.3958 | 6.4734 | 6.5507 | 6.7110 | 6.8013 | 6.8396 | |
| λ /nm | 221.96 | 219.96 | 219.71 | 193.85 | 191.53 | 189.27 | 184.75 | 182.30 | 181.27 | ||
| f | 0.0008 | 0.0009 | 0.0004 | 0.0040 | 0.0024 | 0.0070 | 0.0005 | 0.0227 | 0.0023 | ||
| 0 | Eex /eV | 5.6609 | 5.6751 | 5.7014 | 6.6073 | 6.6248 | 6.7205 | 6.7478 | 6.8553 | 6.8990 | |
| λ /nm | 219.02 | 218.47 | 217.46 | 187.65 | 187.15 | 184.49 | 183.74 | 180.86 | 179.71 | ||
| f | 0.0004 | 0.0005 | 0.0002 | 0.0017 | 0.0115 | 0.0113 | 0.0004 | 0.0013 | 0.0055 | ||
| 5.1 | Eex /eV | 5.6551 | 5.6747 | 5.6991 | 6.6070 | 6.6271 | 6.7076 | 6.7391 | 6.8510 | 6.8983 | |
| C6∶0 | λ /nm | 219.24 | 218.49 | 217.55 | 187.66 | 187.09 | 184.84 | 183.98 | 180.97 | 179.73 | |
| f | 0.0004 | 0.0005 | 0.0002 | 0.0127 | 0.0006 | 0.0110 | 0.0002 | 0.0051 | 0.0017 | ||
| 26 | Eex /eV | 5.6314 | 5.6697 | 5.6808 | 6.3806 | 6.4593 | 6.5178 | 6.7284 | 6.7854 | 6.8576 | |
| λ /nm | 220.17 | 218.68 | 218.25 | 194.31 | 191.95 | 190.22 | 184.27 | 182.72 | 180.80 | ||
| f | 0.0004 | 0.0004 | 0.0001 | 0.0062 | 0.0011 | 0.0037 | 0.0004 | 0.0123 | 0.0011 | ||
| 0 | Eex /eV | 5.6594 | 5.6735 | 5.7005 | 6.6005 | 6.6186 | 6.7133 | 6.7432 | 6.8516 | 6.8955 | |
| λ /nm | 219.08 | 218.53 | 217.5 | 187.84 | 187.33 | 184.68 | 183.87 | 180.96 | 179.80 | ||
| f | 0.0004 | 0.0005 | 0.0002 | 0.0017 | 0.0124 | 0.0117 | 0.0004 | 0.0014 | 0.0056 | ||
| 5.1 | Eex /eV | 5.6538 | 5.6734 | 5.6984 | 6.5991 | 6.6206 | 6.7002 | 6.7336 | 6.8443 | 6.8952 | |
| C10∶0 | λ /nm | 219.29 | 218.54 | 217.58 | 187.88 | 187.27 | 185.05 | 184.13 | 181.15 | 179.81 | |
| f | 0.0004 | 0.0005 | 0.0002 | 0.0136 | 0.0005 | 0.0115 | 0.0002 | 0.0052 | 0.0019 | ||
| 26 | Eex /eV | 5.6303 | 5.6676 | 5.6812 | 6.3752 | 6.4561 | 6.5099 | 6.7246 | 6.7745 | 6.8546 | |
| λ /nm | 220.21 | 218.76 | 218.24 | 194.48 | 192.04 | 190.45 | 184.37 | 183.01 | 180.88 | ||
| f | 0.0004 | 0.0004 | 0.0001 | 0.0067 | 0.0010 | 0.0040 | 0.0005 | 0.0132 | 0.0010 |
表4电场下分子的激发态
Table4.The molecular excited state under electric field.
感谢国家超级计算深圳中心提供计算资源.
