全文HTML
--> --> -->目前已有大量关于金刚石/铝复合材料界面改性的实验研究, 包括金刚石颗粒表面处理[4-8]、基体金属合金化[9,10]以及复合材料制备工艺优化[11,12]等. Wu等[13]认为Cu元素的加入能够降低铝基体的熔点, 界面相Al2Cu使金刚石/铝界面得到强化. Monje等[14]通过气压浸渗和原位化学表面改性制备了金刚石/铝复合材料, 指出清洁或重构的金刚石表面有利于界面热传导. Li等[15]提出了一种在金刚石表面低温合成SiC的新方法, SiC界面层的引入使复合材料的导热系数提高了12.9%, 且在潮湿环境中表现出良好的导热稳定性.
近年来, 基于密度泛函理论的第一性原理计算也成为材料界面及导热研究的有效手段[16-21], 促进了物理学、化学和材料科学等学科的发展. 利用第一性原理对界面性质进行分析, 可以在理论上对实验进行更深入阐述. Qi和Hector[22]利用VASP软件包研究了氢吸附对金刚石/铝界面结合和转移的影响, 计算表明未重构金刚石{111}是清洁金刚石与铝的有利表面. 吴孔平等[23]由原子轨道态密度分析金属诱导带隙态产生于金刚石一侧, 并根据平均静电势计算结果得到金刚石/铝界面的肖特基势垒的高度为1.03 eV. Zhao等[18]利用第一性原理计算解释了α-Al核倾向于在C终端中心位置的Al4C3 (0001)上形成的原因. 上述计算均与实验符合良好, 但有关金刚石不同晶面与铝界面性质的系统研究尚未报道.
本文利用第一性原理计算金刚石/铝的界面性质, 根据粘附功、界面电子结构分析了金刚石/铝体系中Al, C相互作用及Al4C3的引入对金刚石/铝界面性质的影响. 采用真空气压浸渗法制备金刚石/铝复合材料, 对其界面反应进行研究; 通过湿热实验研究界面反应对金刚石/铝复合材料性能的影响规律, 阐明界面反应调控对于复合材料热性能稳定性的重要意义.
2.1.样品制备
选用纯铝及金刚石颗粒作为实验原材料. 其中, 质量分数为99.99%的工业纯铝作为基体, 厂家为东北轻合金有限责任公司; MBD4型, 粒径为100 μm的人造单晶金刚石颗粒为增强体, 厂家为河南飞孟金刚石股份有限公司. 采用真空气压浸渗法制备金刚石/铝复合材料, 浸渗温度为700 ℃, 浸渗时间为30 min. 制备金刚石/铝复合材料中增强体体积分数约为60%.2
2.2.表征方法
使用Helios Nanolab600i扫描电镜对金刚石形貌及复合材料微观组织进行观察. 采用型号为 Dimension Icon的原子力扫描探针显微镜对金刚石不同晶面形貌及粗糙度进行表征. 使用型号为Talos F200×的场发射透射电子显微镜(trans-mission electron microscope, TEM)观察金刚石/铝复合材料的界面结构, 该试样由美国FEI公司生产的Helios Nanolab600i聚焦离子束扫描电子显微镜(focused ion beam, FIB)制备. 利用德国耐驰公司LFA467Nanoflash热导率测试仪测试复合材料的热扩散系数(k). 根据阿基米德原理测试复合材料的密度(ρ), 混合定律计算比热容(c), 代入公式λ=k × ρ × c即可计算得到金刚石/铝复合材料热导率. 利用INSTRON-5569电子万能试验机测试金刚石/铝复合材料三点弯曲性能, 试样尺寸为3 mm × 4 mm × 36 mm. 湿热环境实验在东莞皓天试验设备有限公司的SMC-150PF恒温恒湿试验箱中进行, 测试温度设定为70 ℃, 湿度设定为90% R.H, 温度波动 ± 2 ℃, 湿度偏差+2%, –3% R.H. 第0, 3, 6, 12, 24, 36, 48和60 d时取出测试其热导率, 湿热前和湿热60 d后分别测试其三点弯曲性能.2
2.3.计算方法
采用基于密度泛函理论的第一性原理计算方法, 对金刚石/铝界面性质进行研究. 使用Materials Studio 2017中的Materials Visualizer模块进行模型搭建及界面匹配; 结构优化、能量计算及电子结构计算在CASTEP模块下完成. 计算选取Ultrasoft超软赝势[24], 交换关联泛函使用GGA-PBE形式[25], 布里渊区K点取样选取Monkhorst-Pack网格[26]. 铝晶胞结构优化及表面计算采用截断能为310 eV, K点网格分别为8 × 8 × 8和18 × 18 × 1; 金刚石晶胞结构优化及表面计算选取截断能为400 eV, K点网格分别为8 × 8 × 8和8 × 8 × 1; 界面模型最佳界面间距测试及结构优化使用截断能为400 eV, K点网格为7 × 7 × 1. 自洽场能量收敛到1 × 10–5 eV/atom, 弛豫到每个原子间的力不超过0.03 eV/?, 应力误差不超过0.05 GPa, 位移误差不超过0.001 ?.3.1.金刚石/铝界面性质第一性原理计算
33.1.1.模型构建与优化
金刚石和铝均属于立方晶系, 面心立方布喇菲点阵. 其中, 人造单晶金刚石为六-八面体, 由{111}与{100}两个晶面族围封而成, 铝密排面为{111}. (111)和(100)晶面分别属于{111}和{100}晶面族, 因此, 计算金刚石(111)/铝(111)和金刚石(100)/铝(111)两种界面模型可为金刚石/铝界面性质研究提供明确参考.为使界面模型中金刚石和铝表面层的厚度足以反映块体性质, 首先计算了金刚石、铝的晶格常数和表面能. 金刚石的晶格常数为3.568 ?, 与文献计算值3.547 ?[27]及实验数据3.560 ?[28]相近; 铝的晶格常数4.047 ?也与Liu等[29]和Xu等[30]的计算结果4.050 ?, 4.044 ?相吻合, 即计算参数选取满足精度要求. 表面能计算结果表明, 六层金刚石(111)、七层金刚石(100)以及五层铝(111)面的表面能趋于稳定, 分别为5.75 J/m2, 9.83 J/m2和0.81 J/m2. 基于此, 确定了构建金刚石(111)/铝(111)、金刚石(100)/铝(111)两种界面模型的表面层数. 为使两种模型具有良好的匹配程度, 选取金刚石(111)




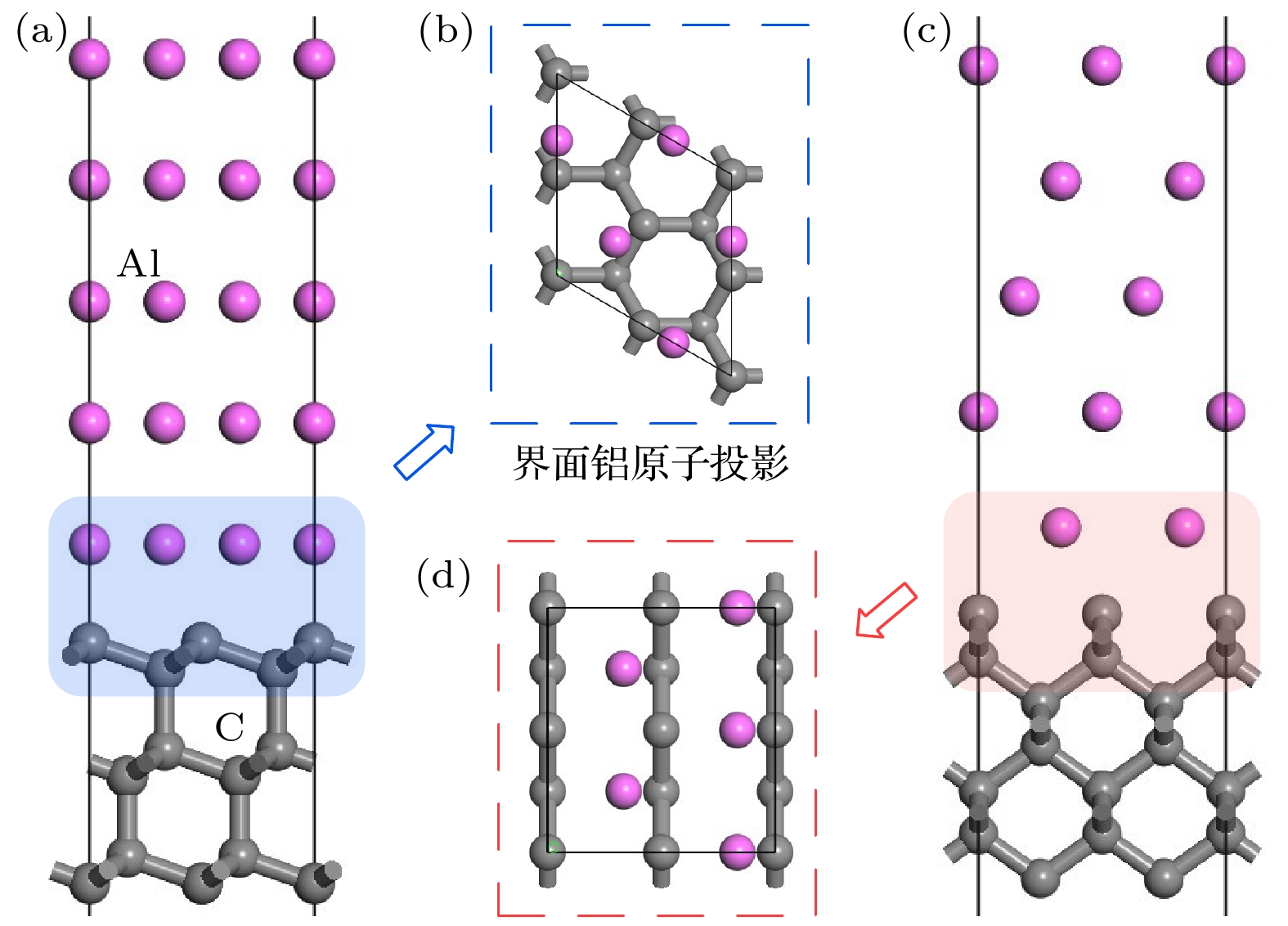 图 1 (a), (b) 金刚石(111)/铝(111)主、俯视图; (c), (d) 金刚石(100)/铝(111)主、俯视图
图 1 (a), (b) 金刚石(111)/铝(111)主、俯视图; (c), (d) 金刚石(100)/铝(111)主、俯视图Figure1. (a), (b) Front and top view of model diamond (111)/Al(111); (c), (d) front and top view of model diamond(100)/Al(111).
采用UBER(universal binding energy relation)曲线测试模型最佳界面间距d0, 使构建界面模型更合理稳定. 测试得到金刚石(111)/铝(111)、金刚石(100)/铝(111)模型最佳界面间距分别为1.95 ?和1.70 ?. 随后, 对两种界面模型进行结构优化, 仅弛豫界面处两层铝原子和两层碳原子, 并利用(1)式计算其界面粘附功Wad. 结果表明, 金刚石(111)/铝(111)的界面粘附功为4.14 J/m2, 金刚石(100)/铝(111)的界面粘附功为5.85 J/m2, 后者较前者的界面粘附功提高了41%. 界面粘附功越大, 界面结合越强, 界面粘附功最大、界面间距最小的界面具有最强的界面结合性能和最佳的稳定性[31]. 因此, 金刚石(100)面与铝(111)面的结合力更强.
3
3.1.2.金刚石/铝界面差分电荷密度分析
进一步分析两种金刚石/铝模型界面处的电荷转移, 模型结构优化后的差分电荷密度如图2所示. 金刚石碳原子之间存在大量电荷聚集, 且为球状, 呈现共价键合的特点. 在金刚石(111)/铝(111)界面处, 碳原子附近呈淡红色, 得到少量电荷, 铝原子在靠近碳原子的一侧存在较多电荷损耗; 相较于金刚石(111)/铝(111), 金刚石(100)/铝(111)中碳原子附近红色区域颜色变深、范围变大, 在碳原子与铝原子之间存在椭球状电荷聚集区域, 高电荷密度分布在碳原子一侧. 电荷转移促进Al—C键合形成, 且电荷转移越多, 形成键合趋势越强. 因而金刚石(100)面的碳原子与铝原子之间相互作用更强, 界面粘附功更大.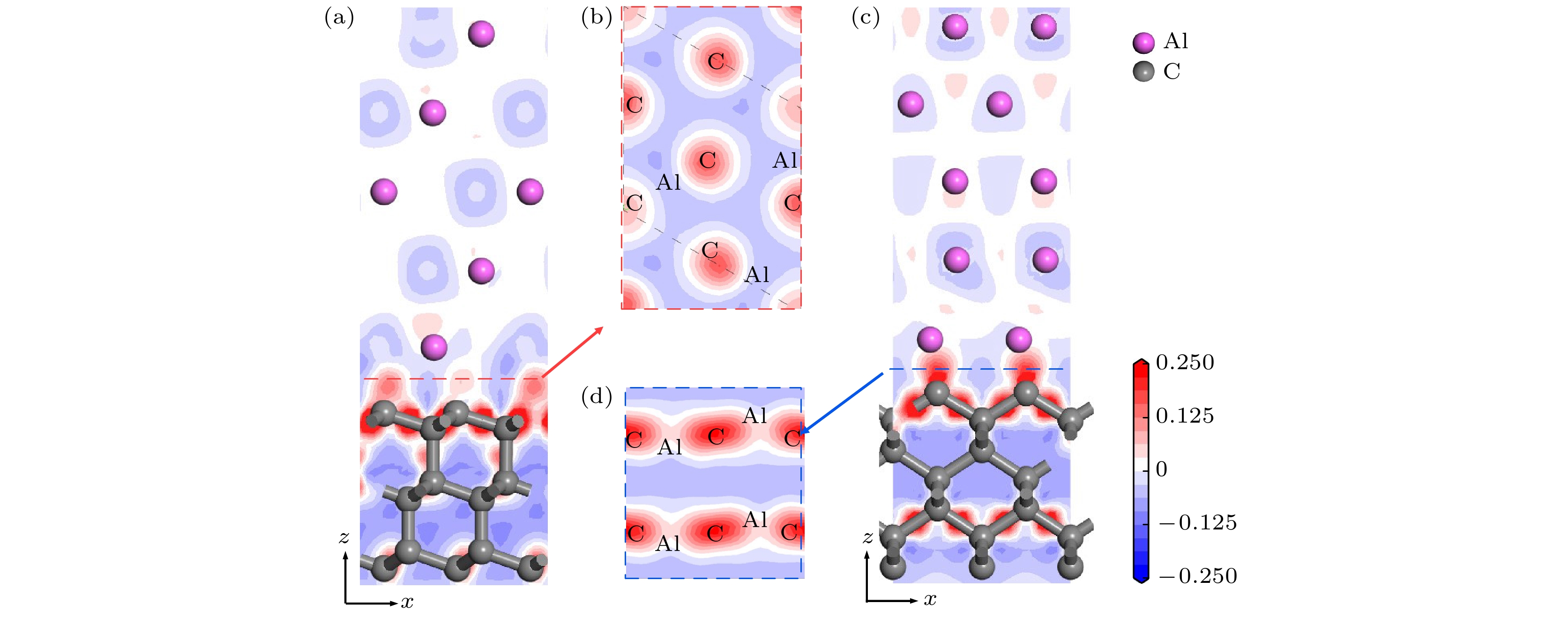 图 2 (a), (b) 金刚石(111)/铝(111); (c), (d) 金刚石(100)/铝(111)差分电荷密度图
图 2 (a), (b) 金刚石(111)/铝(111); (c), (d) 金刚石(100)/铝(111)差分电荷密度图Figure2. Differential charge density image of model: (a), (b) Diamond (111)/Al(111); (c), (d) diamond(100)/Al(111).
3
3.1.3.金刚石/铝界面分波态密度分析
为明确原子间成键本质, 分别对两种模型界面附近两层Al原子及两层C原子的分波态密度(PDOS)进行分析, 如图3所示. 对于金刚石(111)/铝(111)界面, 界面处第一层Al原子的s, p轨道具有更宽的能量范围, 在–12.4—–11.2 eV出现了新的能量状态. 在–4.8—–1.5 eV范围内, 第一层Al原子的电子态密度相对第二层Al的峰强和峰位均发生了变化. 而界面碳原子在该范围内出现了尖锐的杂化峰, 对比可知, Al原子与C原子存在相互作用. 界面附近C原子在费米能级处的态密度不为零, 在金属Al的诱导作用下呈现一定的金属性质. 且在0.8—3.9 eV附近, C原子p轨道与Al原子s轨道的电子态密度出现共振峰, 而大于5.5 eV的范围内, 两层Al, C原子各自态密度无明显差别. 图 3 金刚石/铝界面分波态密度 (a) 金刚石(111)/铝(111); (b) 金刚石(100)/铝(111)
图 3 金刚石/铝界面分波态密度 (a) 金刚石(111)/铝(111); (b) 金刚石(100)/铝(111)Figure3. PDOS of diamond/Al interface: (a) Diamond(111)/Al(111); (b) diamond(100)/Al(111).
金刚石(100)/铝(111)中的电子态密度主要由C原子的p轨道和Al原子的p轨道贡献. 界面处Al原子在–16—12 eV附近出现新的峰值, 表明C的s轨道与Al的s及p轨道产生杂化作用. 在–4.4, –3.5 和–2.8 eV处, 界面Al原子与C原子的p轨道出现多处共振峰, 界面处C原子与Al原子成键特征明显. 界面处C原子的s, p轨道态密度均向费米能级附近移动, 且Al原子在0—13.5 eV范围内具有较大的态密度, 有利于C与Al原子相互作用形成Al—C键. 此外, 界面处C原子的态密度峰形尖锐, 出现2.1 eV的赝能隙, 表明金刚石C原子具有强烈的共价特性. 而第二层碳原子态密度受Al, C界面的影响较小. 综上, 金刚石(100)/铝(111)比金刚石(111)/铝(111)中Al, C分波态密度重叠区域更大, 且尖峰数目更多, 因此模型金刚石(100)/铝(111)中Al与C之间杂化作用更加强烈, 形成离子键(少量共价键)的趋势更大, 与差分电荷密度分析结果一致.
3
3.1.4.金刚石/铝界面原子轨道布居分析
原子轨道布居数可以描述各个原子轨道电子分布情况, 对形成的键合进行定量分析. 金刚石/铝界面模型界面层铝、碳原子轨道布居分析如表1所列, 相应原子编号如图4所示.| Interface model | Position | s | p | Total | Charge |
| Diamond(111)/Al(111) | Al1 | 1.03 | 1.61 | 2.64 | 0.36 |
| Al2 | 1.03 | 1.61 | 2.64 | 0.36 | |
| Al3 | 1.03 | 1.61 | 2.64 | 0.36 | |
| C1 | 1.18 | 3.07 | 4.25 | –0.25 | |
| Diamond(100)/Al(111) | C2 | 1.18 | 3.09 | 4.26 | –0.26 |
| Al1 | 0.88 | 1.73 | 2.61 | 0.39 | |
| Al2 | 0.89 | 1.72 | 2.60 | 0.40 | |
| Al3 | 0.88 | 1.73 | 2.61 | 0.39 | |
| Al4 | 0.89 | 1.72 | 2.61 | 0.39 | |
| C1 | 1.33 | 3.10 | 4.43 | 0.43 | |
| C2 | 1.33 | 3.10 | 4.43 | 0.43 |
表1金刚石/铝界面原子轨道布居分析
Table1.Atomic orbital population analysis of diamond/Al interface.
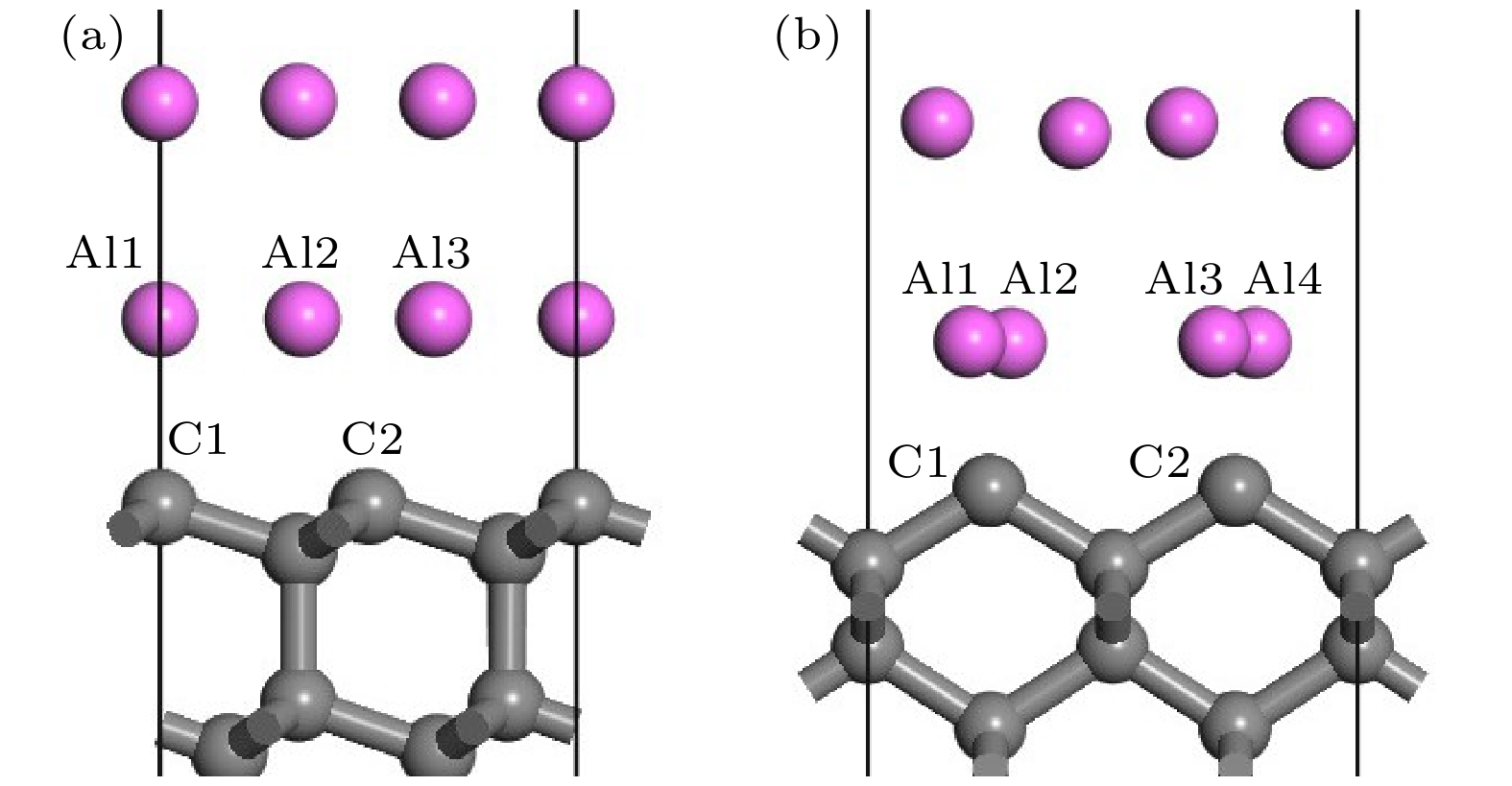 图 4 金刚石/铝界面模型结构优化后局部图 (a) 金刚石(111)/铝(111); (b) 金刚石(100)/铝(111)
图 4 金刚石/铝界面模型结构优化后局部图 (a) 金刚石(111)/铝(111); (b) 金刚石(100)/铝(111)Figure4. Partial image of optimized structure of diamond/Al interface: (a) Diamond(111)/Al(111); (b) diamond(100)/Al(111).
在金刚石/铝两种界面模型中, 界面处铝原子均失去电子, 碳原子得到电子, 铝和碳的价电子均主要集中在p轨道. 对于金刚石(111)/铝(111)界面, 界面处三个铝原子均失去0.36e, 碳原子得到0.25e和0.26e. C和Al之间存在电荷转移, Al的s, p轨道价电子向C的s, p轨道转移, 形成Al—C键合; 金刚石(100)/铝(111)界面附近铝原子向碳原子靠近, 碳原子得到电荷增加为0.43e, 电荷增加主要体现在C的s轨道, Al的s轨道失去更多电荷. 电荷转移越多, 形成键合越强, 所以金刚石(100)/铝(111)界面形成的Al—C键强于金刚石(111)/铝(111). 键重叠布居分析表明, 金刚石(100)/铝(111)中Al—C键长平均为0.208 nm, 小于金刚石(111)/铝(111)中Al—C键长0.211 nm. 因此, 金刚石(100)/铝(111)中形成Al—C键能更高, 界面粘附功更大, 界面更稳定.
2
3.2.考虑界面反应的金刚石(100)/铝(111)界面性质第一性原理计算
由上述计算结果可知, 金刚石(100)面与Al(111)面形成Al—C键合的趋势更强, 更容易形成界面产物Al4C3. 为了进一步研究Al4C3形成对金刚石/铝界面的影响, 将界面反应考虑到金刚石(100)/铝(111)界面模型构建中.采用在金刚石(100)/铝(111)界面引入Al—C键的方式模拟存在界面反应的金刚石(100)/铝(111)界面(简写为修正的金刚石(100)/铝(111)). 首先, 优化了添加C原子的位置和个数. 共考虑了C原子在Al原子正上方(①), C原子在两个Al原子中间的上方(②)和C原子在四个Al原子中间的上方(③)三种位置情况, 如图5(b)所示. 并研究了不同C原子在Al原子表面覆盖率(0.25, 0.50和1.00 个原子层(ML))对吸附能的影响. (2)式为吸附能计算公式, 计算结果列于表2. 参照Al4C3晶体中Al—C键长, 吸附模型中Al—C键长确定为1.94 ?. 由于吸附能越大越稳定, 最终选择C原子吸附在②位置, 覆盖率选择为0.50 ML, 其模型示意图如图5(d)所示.
 图 5 C原子在Al(111)表面吸附位置确定 (a) Al(111)表面模型; (b) C原子在Al(111)表面吸附位置俯视图; (c) Al4C3模型局部图; (d) 优化的C原子在Al(111)表面的位置
图 5 C原子在Al(111)表面吸附位置确定 (a) Al(111)表面模型; (b) C原子在Al(111)表面吸附位置俯视图; (c) Al4C3模型局部图; (d) 优化的C原子在Al(111)表面的位置Figure5. Determination of adsorption position of C atom on Al (111) surface: (a) Al (111) surface model; (b) top view of the adsorption position of C atom on Al (111) surface; (c) the local map of Al4C3 model; (d) the position of the optimized C atom on Al (111) surface.
| Coverage/ML | ① | ② | ③ |
| 0.25 | 4.12 | 7.96 | 7.63 |
| 0.50 | 6.89 | 8.62 | 8.21 |
| 1.00 | 4.27 | 7.41 | 5.76 |
表2不同覆盖率及不同吸附位置时C原子在Al(111)表面的吸附能 (eV·atom–1)
Table2.Adsorption energy of C atom on Al (111) surface with different coverage and adsorption position (eV·atom–1).
基于上述计算结果, 在金刚石(100)/铝(111)模型的界面处添加两个C原子, 引入Al—C键, 研究修正的金刚石(100)/铝(111)模型的界面性质. 图6为结构优化前后的模型示意图. 结构优化后, 铝在界面附近的两层原子均出现较大幅度弛豫. 第二层铝原子产生褶皱. 此外, 金刚石(100)的C原子相互靠近, 具有成键趋势. 模型粘附功计算结果为9.72 J/m2, 显著大于前述两种模型, 即Al—C键合的引入增强了金刚石与铝之间的界面结合.
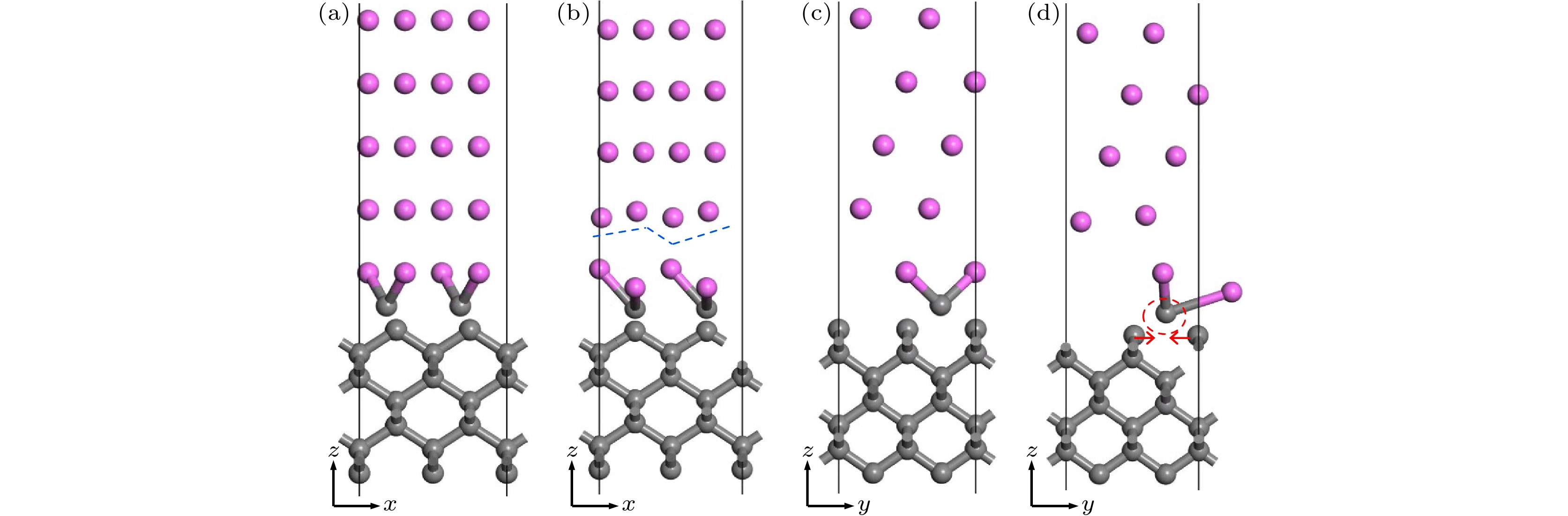 图 6 修正的金刚石(100)/铝(111)模型示意图 (a), (c) 优化前; (b), (d) 优化后
图 6 修正的金刚石(100)/铝(111)模型示意图 (a), (c) 优化前; (b), (d) 优化后Figure6. Schematic diagram of diamond(100)/Al(111) modified model: (a), (c) Before optimization; (b), (d) after optimization.
修正的金刚石(100)/铝(111)模型差分电荷密度如图7所示. Al1与C1间由于存在Al—C键聚集较多电荷. 由图7(b) 可知, C3与其左侧的C原子之间也存在电荷转移, 金刚石(100)表面的C原子在引入Al—C键的影响下具有重构趋势. 此外, C1与C3, C4之间, C2与C5, C6之间聚集大量电荷, 形成共价键特征明显. 因此, Al4C3的引入促进了界面处C原子之间的相互作用, 同时C与Al一侧通过Al—C化学键相连, 有利于金刚石与铝之间的界面结合.
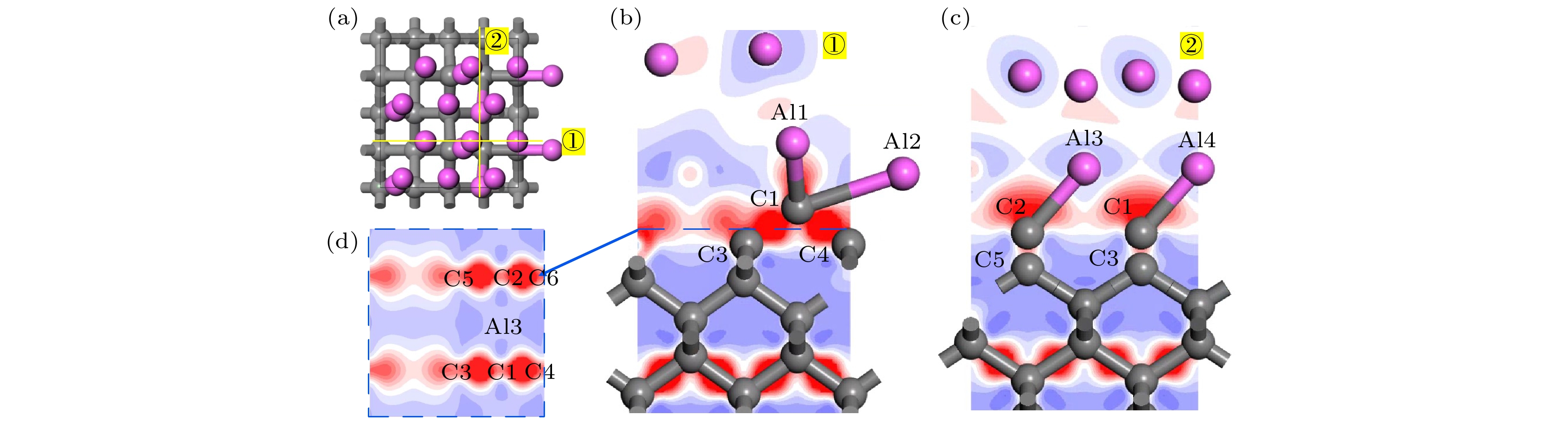 图 7 修正的金刚石(100)/铝(111)模型差分电荷密度分析 (a) 结构优化后模型俯视图; (b) (a)中①截面差分电荷密度图; (c) (a)中②截面差分电荷密度图; (d) (b)中虚线部分的差分电荷密度图
图 7 修正的金刚石(100)/铝(111)模型差分电荷密度分析 (a) 结构优化后模型俯视图; (b) (a)中①截面差分电荷密度图; (c) (a)中②截面差分电荷密度图; (d) (b)中虚线部分的差分电荷密度图Figure7. Differential charge density analysis of diamond(100)/Al(111) modified model: (a) Top view of the model after structure optimization; (b) cross section differential charge density diagram of ① in Fig. (a); (c) cross section differential charge density diagram of ② in Fig. (a); (d) the differential charge density diagram of the dotted line in Fig. (b).
图8为引入Al—C键合后修正的金刚石(100)/铝(111)模型的界面分波态密度. 引入C原子与第一层Al原子存在键合, 在–12.2—–10.1 eV范围内C的p轨道与Al的s, p轨道存在共振峰. 且在–4.3, –3.0和、–1.5 eV附近, C原子的p轨道与Al原子层的p轨道出现多处相似峰值, 费米能级附近添加C原子的能量状态显著增强, 即Al—C键的形成使C原子具有一定金属性质. 在10.3—–15.1 eV附近, C和Al原子也存在相似的能量状态. 同时, C原子的引入使界面处的第一层金刚石碳原子s轨道的的分波态密度在–17.3—–14.9 eV附近出现明显峰值, 且C原子p轨道与第一层金刚石碳原子的p轨道的态密度在–10.2—4.1 eV范围内具有高度一致性, 形成C—C共价键特征明显. 而第二层金刚石碳原子受界面影响作用减弱, 虽然费米能级处的态密度值不为零, 呈现出一定金属性质, 但其值显著低于C原子及第一层金刚石碳原子. 综上, 引入Al—C键能够促进界面处C—C的相互作用, 有利于金刚石与铝的界面结合.
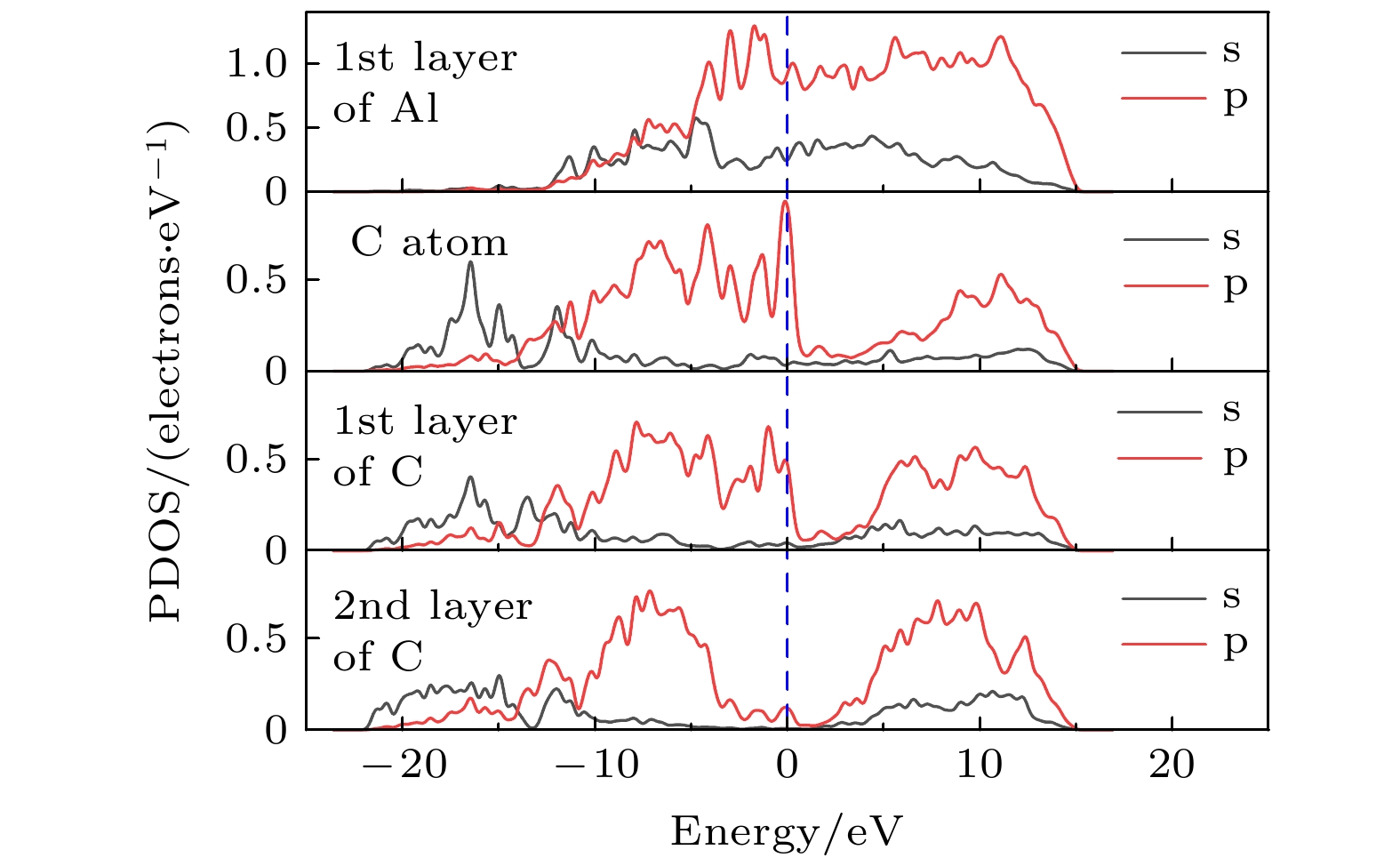 图 8 修正的金刚石(100)/铝(111)模型的分波态密度
图 8 修正的金刚石(100)/铝(111)模型的分波态密度Figure8. PDOS of diamond (100)/Al(111) modified model.
2
3.3.金刚石/铝复合材料界面反应与界面选择性
为研究金刚石/铝复合材料界面反应, 将制备所得复合材料利用碱洗法腐蚀掉铝基体, 提取出金刚石颗粒形貌如图9所示. 图中金刚石颗粒完整, 棱角整齐. 金刚石{100}面存在明显针状蚀坑, 该面与铝基体反应形成界面产物Al4C3; 而金刚石{111}晶面较为光滑, 即金刚石与铝基体间的反应具有界面选择性, Al4C3更容易在金刚石{100}面形成. 图 9 复合材料中提取金刚石的SEM形貌 (a) 提取金刚石颗粒整体形貌; (b) 提取金刚石颗粒局部形貌; (c) 单个金刚石颗粒形貌; (d) {100}晶面上的Al4C3蚀坑
图 9 复合材料中提取金刚石的SEM形貌 (a) 提取金刚石颗粒整体形貌; (b) 提取金刚石颗粒局部形貌; (c) 单个金刚石颗粒形貌; (d) {100}晶面上的Al4C3蚀坑Figure9. SEM morphology of diamond extracted from diamond/Al composites: (a) Overall morphology of extracted diamond particles; (b) local morphology of extracted diamond particles; (c) morphology of single diamond particle; (d) Al4C3 pits on {100} plane.
图10所示为复合材料原子力显微镜照片. 金刚石{111}表面较为平整, 粗糙度Ra为5.53 nm. 而金刚石{100}面观察到大量针状反应物, 表面粗糙度Ra为8.85 nm. 分析结果与前述吻合, 即金刚石{100}面与铝反应生成Al4C3, 使金刚石表面粗糙度增大, 而金刚石{111}面几乎无界面反应发生. Monje等[12]报道在760 ℃、金刚石与液态铝接触时间趋近于零的条件下, 金刚石{100}面生成大量Al4C3, 而{111}面几乎没有任何产物. Che等[32]通过实验认为Al4C3在金刚石{100}面成核速率高于{111}面, 与本文结果一致.
 图 10 复合材料中金刚石原子力显微镜照片 (a) 金刚石{111}面; (b) 金刚石{111}面 10 μm × 10 μm面积; (c) 金刚石{100}面; (d) 金刚石{100}面10 μm × 10 μm面积
图 10 复合材料中金刚石原子力显微镜照片 (a) 金刚石{111}面; (b) 金刚石{111}面 10 μm × 10 μm面积; (c) 金刚石{100}面; (d) 金刚石{100}面10 μm × 10 μm面积Figure10. AFM photographs of diamond surface in composites: (a) Diamond {111}; (b) diamond {111} with 10 μm × 10 μm area; (c) diamond {100}; (d) diamond {100} with 10 μm × 10 μm area.
进一步对复合材料界面反应进行表征. 图11(a)为透射电镜下观察金刚石{100}/铝复合材料界面显微组织. 金刚石{100}面与铝基体之间存在片状及棒状的Al4C3, 其位置用红色数字标出. Al4C3延伸到铝基体中, 另一侧与凹凸不平的金刚石边界相连, 金刚石{100}与铝之间构成反应型界面. 图11(b)为金刚石{111}/铝界面处的高分辨图像, 金刚石与铝之间界面平齐, 无界面产物Al4C3, 未观察到发现含氧非晶层. 此外, 发现了金刚石与铝存在一组位相关系, 即: 金刚石




 图 11 复合材料界面反应表征 (a) 金刚石{100}/铝界面处Al4C3形貌; (b) 金刚石/铝界面处的位相关系
图 11 复合材料界面反应表征 (a) 金刚石{100}/铝界面处Al4C3形貌; (b) 金刚石/铝界面处的位相关系Figure11. Characterization of interface reaction of composites: (a) Al4C3 morphology at the interface of diamond{100}/Al; (b) phase relationship at diamond/Al interface.
金刚石与铝的界面选择性和金刚石表面碳原子成键状态密切相关. 结合上述第一性原理计算结果, 金刚石(100)面与铝界面粘附功更大, 界面结合更强, 因此金刚石(100)面更易与铝粘附; 此外, 金刚石(100)面与铝(111)面在界面处电荷转移更多, 形成Al—C键合趋势更强, 铝与碳反应优先发生在金刚石(100)面. 第一性原理计算从原子键合与电荷转移的角度解释了界面反应在金刚石不同晶面上的差异性.
2
3.4.界面反应对复合材料的影响
33.4.1.界面反应对金刚石/铝复合材料性能的影响
Al4C3对复合材料综合性能的影响受到研究者们广泛关注. 虽然Al4C3可以改善金刚石与铝间的界面结合, 但其在潮湿环境中发生水解, 不利于复合材料性能. 因此, 通过湿热环境处理研究界面反应对金刚石/铝复合材料的影响. 金刚石/铝初始热导率平均为610.2 W/(m·K), 其归一化热导率随湿热处理时间变化曲线如图12所示. 图 12 湿热处理后金刚石/铝复合材料归一化热导率变化
图 12 湿热处理后金刚石/铝复合材料归一化热导率变化Figure12. The variation of normalized thermal conductivity of diamond/Al composites after heat-moisture treatment.
由于真空气压浸渗法制备的复合材料致密度高, 水分率先接触试样表面, 使暴露于表面的Al4C3水解, 而不易进入复合材料内部, 因此初期复合材料热性能下降缓慢. 随湿热时间延长, 水分渗入材料内部, 热导率下降速率变快. 后期Al4C3几乎全部水解, 湿热环境对材料性能影响已达到最低, 热导率变化趋于平缓. 测试复合材料原始及湿热60 d后的力学性能, 其抗弯强度从247.6 MPa降低到138.5 MPa. 由此可见, 界面产物Al4C3水解导致复合材料综合性能急剧下降.
3
3.4.2.界面反应对金刚石/铝复合材料的影响机制
金刚石/铝复合材料湿热前后断口扫描电镜照片如图13所示. 湿热前, 复合材料更倾向于沿金刚石{111}晶面与铝发生界面脱粘, 断口中裸露的金刚石多呈六边形; 而金刚石{100}晶面更容易粘附铝, 呈现出局部塑性断裂, 图13(b)和图13(c)中可以进一步观察到金刚石{100}面粘附大量铝, 而棱边处存在大量针状、岛状的界面产物Al4C3. Al4C3的生成可以将金刚石与铝之间的机械结合转变成化学结合, 当其尺寸较小时, 铝液可以渗入颗粒间隙, 提高复合材料致密度, 有利于界面的紧密结合[33]. 此外, 当形成界面产物Al4C3时, 第一性原理计算表明, Al—C键的形成有利于界面处电荷的重新分布, 金刚石表面的碳原子具有重构的趋势, 同时也促进了共价键的形成, 使界面结合强度得以提高. 然而, 当界面产物Al4C3尺寸较大且生成数量较多时, 铝液的渗入需要更长时间, 冷却后铝液无法完全填充碳化物之间的空隙, 且Al4C3是一种硬脆相, 导致复合材料界面结合变差. 图 13 湿热处理60 d前后金刚石/铝复合材料断口形貌 (a), (b), (c) 湿热前; (d), (e), (f) 湿热后
图 13 湿热处理60 d前后金刚石/铝复合材料断口形貌 (a), (b), (c) 湿热前; (d), (e), (f) 湿热后Figure13. The fracture morphology of diamond/Al composites: (a), (b), (c) before and (d), (e), (f) after 60 days heat-moisture treatment
湿热处理60 d后, 断口中有较多{100}晶面暴露出来, 复合材料界面结合减弱, 金刚石与铝脱粘现象更明显. 由图13(e)和图13(f)可知, {100}晶面粘附铝的能力有所减弱, 原来金刚石表面的Al4C3处出现三角形或梯形蚀坑. 说明大量Al4C3在湿热环境中发生水解, 导致界面结合力降低, 复合材料的抗弯强度下降. 同时, 界面产物Al4C3作为金刚石与铝之间的纽带, 形成连续的导热通道. 其在湿热环境下水解会产生孔洞等缺陷, 增加声子散射的概率, 进而使得复合材料热导率急剧下降. 因此, 虽然金刚石与铝之间细小弥散的Al4C3可以改善界面结合, 提高复合材料热导率. 但Al4C3易水解的特性不利于复合材料在复杂环境中服役稳定性, 改善界面选择性结合、抑制界面产物Al4C3生成对于提高金刚石/铝复合材料性能稳定性至关重要.
2) 采用真空气压浸渗法制备金刚石/铝复合材料, 从多尺度对金刚石/铝的界面结构进行了研究. 实验观察到金刚石{100}面易于粘附铝, 存在大量针状Al4C3且粗糙度较大; 而{111}面不易粘附铝, 表面较光滑. 第一性原理计算从原子键合与电荷转移角度对实验现象进行合理解释.
3) 通过湿热处理研究金刚石/铝复合材料界面反应的影响, 研究发现湿热环境中的性能衰减主要与界面产物Al4C3的水解有关, 湿热60 d后金刚石/铝复合材料的热导率下降了29.9%, 抗弯强度降低了40.1%. 性能的大幅度衰减不利于复合材料在复杂环境的服役稳定性. 因此, 抑制Al4C3形成、改善金刚石与铝选择性结合对于提高金刚石/铝复合材料热性能及其稳定性至关重要. 本文利用第一性原理系统地研究了金刚石/铝的界面性质, 为后续金刚石/金属界面性质的研究及高导热复合材料界面结构设计奠定了理论基础.
