全文HTML
--> --> -->大气压非平衡等离子体由于其独特的非平衡特性, 即电子温度一般为0—10 eV (1 eV ≈ 11605 K ≈ 96.32 kJ/mol), 而重粒子的温度仅为几百K, 电子温度远高于重粒子温度. 高能电子的能量高于温室气体分子的化学键能, 能够扮演化学“催化剂”的角色, 碰撞解离温室气体分子, 从而实现常温常压下温室气体分子的活化. 但不得不指出, 虽然大气压非平衡等离子体能够为CH4和CO2分子活化提供独特的非热平衡和活化环境, 但也存在反应物转化率低, 产物种类复杂, 目标产物选择性低等缺点. 能量利用率和副产物是制约大气压非平衡等离子体催化工业化的两个最主要因素. 这是因为在等离子体中的化学反应可控性较差, 导致最终目标产物的选择性和产率不够理想. 目前, 学术界主流观点认为, 可通过大气压非平衡等离子和利于目标产物生成的化学催化剂组合, 实现大气压非平衡等离子体催化的大规模生产使用.
目前, 大气压非平衡等离子体作为一种新兴的放电技术, 作为“反应载体”用以实现二氧化碳和甲烷重组, 受到越来越多的科研人员关注, 主要通过实验诊断[10-17]和数值模拟[18-24]对甲烷放电和DRM反应机理进行了详细的研究. 例如2020年, Zhang等[10]通过发射光谱和化学气相色谱等表征手段, 研究了电子碰撞等离子体化学反应和温度热效应化学反应, 在温度可控的大气压介质阻挡反应腔中甲醇和二氧化碳重组制取合成气所扮演的角色. Brune等[11]研究了介质阻挡放电(dielectric barrier discharge, DBD)反应器和催化剂对大气压甲烷DRM过程的影响, 研究表明填充了多孔γ-Al2O3载体填充式DBD反应器, 能够改变放电模式和化学活性, 从而实现CH4/CO2协同催化重组. 此外, 催化剂为Cu时, CH4的转化率高达90%, 这是由于Cu催化剂对于O原子和CO分子有更好的吸附和解吸附能力. Maqueo等[12]研究了反应气体能量输入, 单脉冲能量和脉冲频率对大气压纳秒脉冲放电甲烷重组的影响, 实验结果论证了大气压纳秒脉冲反应器可作为燃料的重整腔室. 脉冲频率是最为重要工作参数, 增加脉冲频率有助于提高反应气体的转化率和能量效益. Alawi等[13]研究了掺杂气体N2和Ar流量, 微波功率和CO2/CH4体积比, 对大气压低功率微波等离子体辅助DRM的影响. 实验结果表明, 在N2和Ar氛围内的DRM过程有着相同的反应性能. 在相同的工作参数下, 如恒定微波功率700 W, CO2/CH4体积比为2∶1, 掺杂气体(N2或Ar), CO2, CH4的流量分别为1.5, 0.4和0.2 L/min, Ar氛围内的DRM能产生更高的合成气体选择性和产率. 王晓玲等[14]通过详细的电学和化学气相色谱, 对微秒脉冲电源驱动的DBD甲烷干重整反应过程进行了诊断, 主要研究了脉冲DBD放电特性及重复频率和脉宽对气体转化特性的影响. 研究结果表明, 增大脉冲重复频率有利于提高CH4和CO2的转化率, 同时获得较高的合成气产率. Wu等[15]利用耦合了磁场和切线流的旋转滑动弧等离子体, 实现了等离子体辅助的DRM, 主要研究了电压和CH4/CO2体积比对等离子体基本放电参数(电子激发温度, 电子密度和转动温度)及放电产物分布的影响. 在论文综述中, 详细地介绍了DBD等离子体DRM的研究现状、取得的研究进展以及遇到的研究瓶颈[16]. 现有的研究主要集中于DBD反应结构, 如电极形貌、放电间隙、放电体积及填充的催化剂对DRM过程的影响. 研究结果表明, 反应气体的空间流动速率、能量密度以及反应气体的体积比对放电产物的分布起着至关重要的作用.
在数值模拟方面, 前人也做了很多有意义的研究工作. 赵曰峰等[18]建立了二维大气压甲烷针-板放电流体模型, 并进行了相关实验验证, 研究得到了主要粒子浓度的时空分布和反应路径. Slaets等[19]采用零维(zero-dimensional, 0D)化学反应动力学模型研究了滑动弧等离子管中, 掺入少量的氮气和氧气对大气压甲烷的干重整的影响. 研究结果表明, 氮气有助于提高二氧化碳的转化率, 但是部分输入能量会耗散在氮气分子转动激发上. 此外, 氧气有助于提高甲烷的转化率, 降低能量损耗, 但二氧化碳的转化率基本保持不变. 王伟宗等[20]总结出一套详细完备的等离子体化学反应集, 可实现CH4, CO2, N2, O2和H2O等反应气体任意组合所涉及的等离子体化学反应模拟, 从而构建出这一研究领域中, 大气压DBD等离子体更为宽广的等离子体化学图像. Snoeck等[21]采用了零维化学反应动力学模型, 研究了等离子体放电脉冲及其余辉中的等离子体行为和化学反应机理, 计算给出了反应气体的转化率、高价值含氧化合物的选择性和产率, 以及DBD等离子体的能量效益. Liu等[22]对等离子体催化CH4/CO2转化成三种不同的含氧化合物(CH3OH, HCHO和CH3COOH) 潜在的反应机理, 从ns-ms和nm-mm的时空尺度进行了详细的总结. 比利时安特卫普大学Bogaerts教授课题组[23,24]分别采用0D和1D等离子体化学反应动力学模型, 模拟了大气压CH4/CO2 DBD等离子体复杂的等离子体化学转化反应机理, 计算给出了DBD活性粒子数密度, 放电产物的转化率、产率和反应气体的转化率, 及整体反应流程图等.
综上所述, 目前大气压非平衡等离子体DRM的研究主要通过实验诊断(发射光谱、电学特性、化学气相色谱等)和流体模型数值模拟, 研究DBD反应器工作参数, 如CO2/CH4体积比, 反应停留时间, 催化剂类型和填充方式, 掺杂气体, 电源工作参数, 电极结构等对等离子体DRM过程中产物分布的影响, 从而分析出DRM过程中潜在的反应机理. 前人的研究结果表明, 初始甲烷摩尔分数是等离子体DRM过程的一个很重要的影响参数. 本文采用零维等离子体化学反应动力学模型, 研究了初始甲烷摩尔分数对等离子体DRM过程的影响, 重点关注合成气(CO和H2)以及高价值的含氧化合物(CH2O和CH3OH)主导的生成和损耗反应路径, 反应气体的转化率和重要产物的选择性. 最后总结归纳出了主要等离子体粒子之间的整体化学反应流程图, 直观地展示出等离子体粒子之间的化学转化关系.
本模型中考虑的等离子体化学反应集, 主要参考了比利时安特卫普大学Bogaerts教授课题组[21,23,24]相关的大气压非平衡CH4/CO2等离子体零维模型数值模拟工作. 在此基础上, 增加了15个C和H相关的中性粒子碰撞反应[29,31]. 最终本模型所考虑的CH4/CO2等离子体化学反应集, 包含82种等离子体粒子和885个等离子体化学反应(60个电子碰撞反应, 334个离子反应和491个中性粒子反应).
3.1.电子时间演化特性
对于大多数反应气体, 大气压DBD通常处在所谓的丝状状态, 包含大量的独立微放电丝状通道, 其持续时间为纳秒量级, 通常为1—100 ns[21]. 在微放电丝状通道中, 电子的能量通过电子直接碰撞的形式, 用于反应气体分子的激发、电离和解离等反应过程, 从而产生大量的电子、离子、激发态粒子和中性自由基等. 这也就是这些丝状放电对于流体模型描述化学反应至关重要的原因. 在本文的0D模型中, 并不能刻画描述丝状放电的空间演化过程, 但可以描述微放电脉冲及其余辉的等离子体反应过程. 因此, 施加到等离子体反应器的为三角脉冲, 其持续时间为30 ns(约为一个丝状放电持续时间), 图1虚线是脉冲功率的波形图, 其峰值功率密度为6.0 × 105 W/cm3. 0 ns时脉冲功率开始线性增加, 15 ns时达到峰值功率, 在随后的15 ns时间内线性下降至0, 因此三角形脉冲功率的持续时间为30 ns. 一个脉冲周期中余辉期间为60 ns, 即脉冲的周期为90 ns. 图1实线给出的是甲烷摩尔分数为10%, 30%, 50%, 70%和90%时, 在五个周期内电子温度随时间的演化曲线, 从图1中可以清楚地看出, 电子温度随着脉冲功率增加而线性快速上升, 然后上升速率略微变缓, 在脉冲功率未达到最大值时, 电子温度已达到峰值约为2.0 eV, 这主要是由于电子快速响应电场加速获得的能量. 随后, 在脉冲功率达到最大值之前, 电子温度又快速线性下降至0左右, 并在余辉中也保持在0附近. 我们也注意到, 在参考文献[21]中, 也观察到类似的电子温度随时间的演化特性. 数值模拟结果表明, 甲烷的摩尔分数越大, 电子温度的增加速率越快, 电子温度的峰值也越高. 例如, 甲烷摩尔分数为10%, 30%, 50%, 70%和90%时, 电子温度的峰值分别为1.85, 1.98, 2.18, 2.38和2.55 eV. 电子密度随时间演化特性与电子温度不尽相同. 如图2所示, 随着脉冲功率的线性增加并未达到峰值前, 电子密度也线性增加至峰值(1014—1015 cm–3), 随后快速小幅度地下降, 再随着时间缓慢下降. 不同甲烷的摩尔分数时, 电子密度随时间的整体演化规律相类似, 但较高的甲烷含量放电产生的电子密度更高. De Bie等[24]的研究也发现, 在CH4/CO2反应气体等离子体体系中, 较高的甲烷占比能使得电子密度维持在一个更高的水平.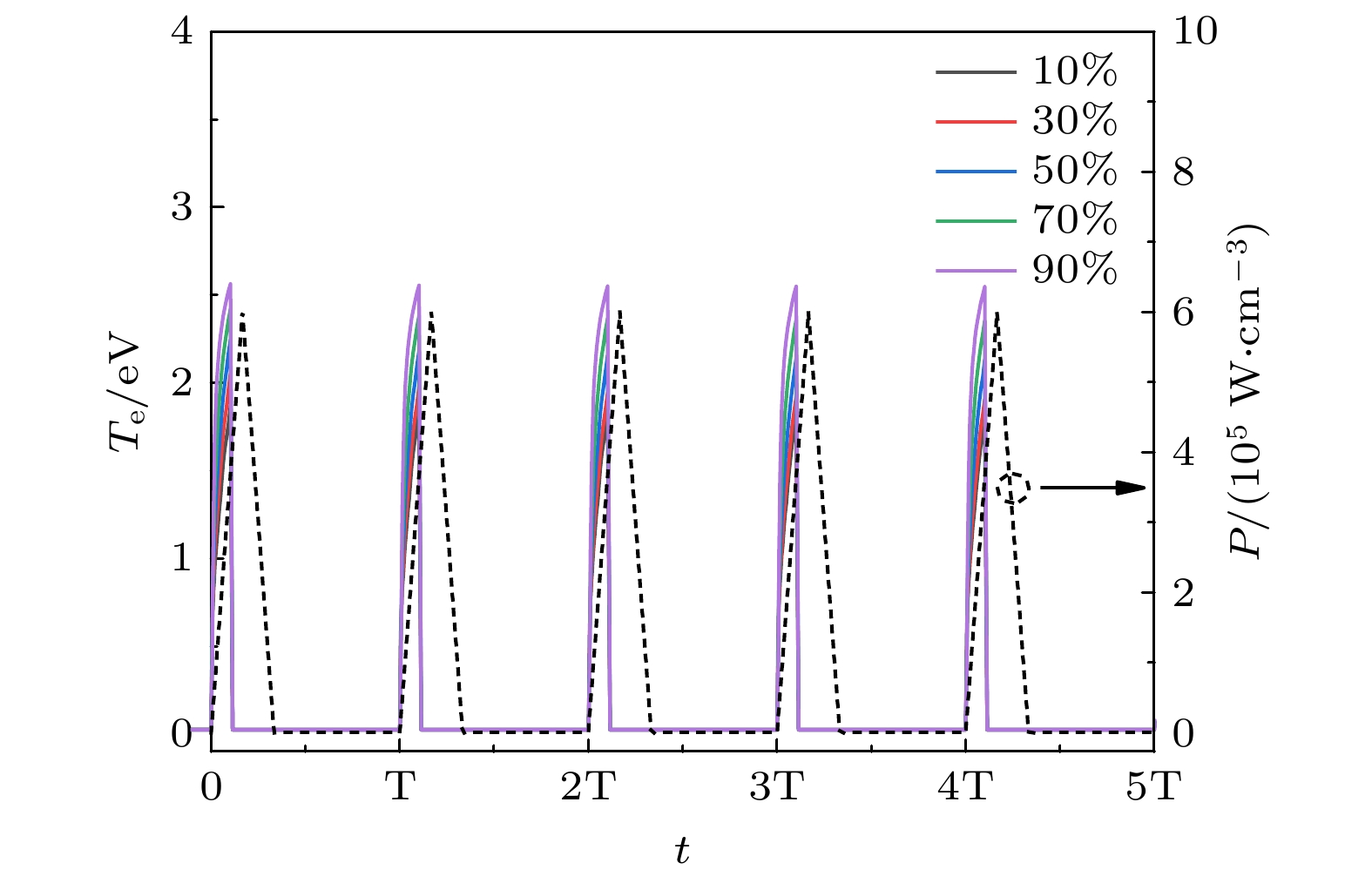 图 1 甲烷摩尔分数为10%, 30%, 50%, 70%和90%时的电子温度随时间演化规律
图 1 甲烷摩尔分数为10%, 30%, 50%, 70%和90%时的电子温度随时间演化规律Figure1. Electron temperature as a function of time for methane mole fractions of 10%, 30%, 50%, 70% and 90%.
 图 2 甲烷摩尔分数为10%, 30%, 50%, 70%和90%时的电子密度随时间变化趋势
图 2 甲烷摩尔分数为10%, 30%, 50%, 70%和90%时的电子密度随时间变化趋势Figure2. Electron density as a function of time for methane mole fractions of 10%, 30%, 50%, 70% and 90%.
2
3.2.粒子时间演化特性
图3展示了不同甲烷含量时主要自由基数密度随时间的变化趋势以及自由基值周期平均值随甲烷摩尔分数的变化规律. 如图3所示, 大气压非平衡等离子体DRM的主要自由基为H, CH2, CH3, O, OH, C, CH3O和CH2OH等, 其中H原子的密度最高维持在1015—1016 cm–3数量级, 其次是CH2, CH3等. 随着甲烷含量的增加, H, CH2, CH3自由基密度会随着增加, 但由于CO2含量在减小, 因而缺乏足够的O原子参与反应, 导致最终生成的含氧化合物的密度会降低, 如图3(d)所示. 例如甲烷摩尔分数为10%时, H原子的数密度达到了1015 cm–3的数量级, O原子的数密度达到1014 cm–3的数量级, 而当甲烷摩尔分数增加到90%时, O原子的数密度显著降低为1013 cm–3的数量级, H原子的数密度小幅度增加接近1016 cm–3的数量级. 这主要是由于O原子和H原子只能分别来源于进料气体CO2和CH4, CH4含量的增加(同时CO2的含量在减小), 解离产生的O原子更少, 从而生成的含氧化合物密度会降低. 从图3(a)—(c)中可看出, CH3自由基的数密度随时间的变化呈现出较为明显周期性波动, 上下波动范围可达 1个数量级. 值得一提的是, 图3中所展示的只是仿真起始一段脉冲周期内(2 μs, 约22个周期)的中性自由基数密度随时间的演化趋势, 这段起始时间内CH3自由基的密度比CH2密度略低, 但经过一段反应时间后, CH3自由基的密度会高于CH2自由基. 一方面, CH3和CH2自由基的主要生成反应路径均是电子和CH4分子的直接碰撞解离反应, 即E + CH4→E + CH3 + H和E + CH4→E + CH2 + H2, 但CH3自由基主要生成反应的反应速率比CH2自由基主要生成反应的反应速率至少高了1个数量级(数据未给出). 另一方面, 本模型中三角形脉冲周期内余辉持续时间仅为60 ns, 导致起始一段周期内CH3自由基的密度会略低于CH2自由基, 且CH3自由基随时间的变化呈现出较为明显的周期性波动, 而CH2自由基未能及时呈现周期性波动. 图 3 (a) 10%, (b) 50%和(c) 90%甲烷摩尔分数时主要自由基的数密度随时间变化趋势, 以及(d) 主要自由基的周期平均值随甲烷摩尔分数的变化
图 3 (a) 10%, (b) 50%和(c) 90%甲烷摩尔分数时主要自由基的数密度随时间变化趋势, 以及(d) 主要自由基的周期平均值随甲烷摩尔分数的变化Figure3. The number densities of main radicals as a function of time for methane mole fractions of (a) 10%, (b) 50%, (c) 90%, and (d) time averaged number densities of main radicals as a function of initial CH4 fraction.
图4展示了不同甲烷含量时主要离子的数密度随时间的变化规律以及离子周期平均值随甲烷摩尔分数的变化规律. 如图4所示, 主要离子为









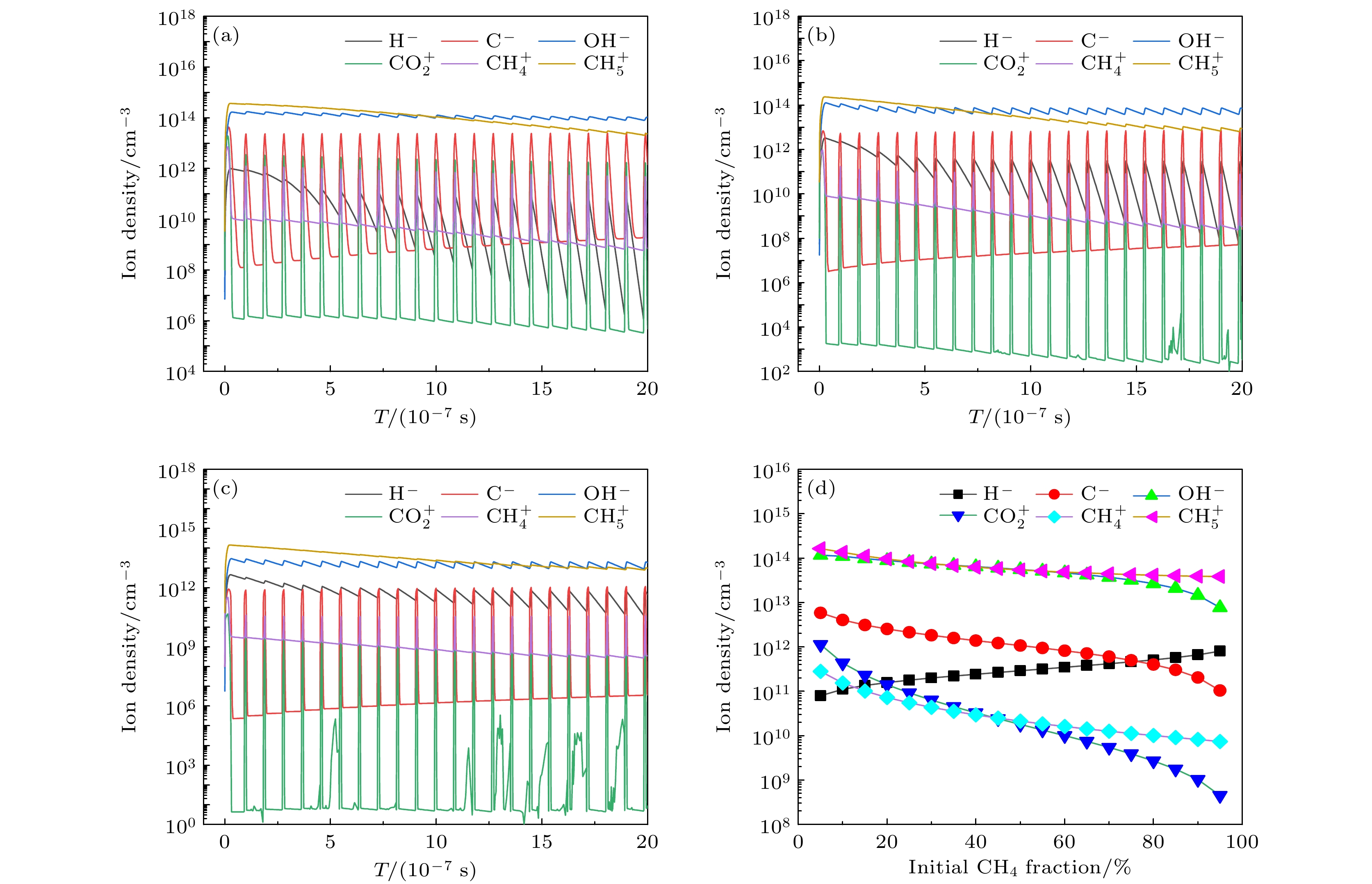 图 4 (a) 10%, (b) 50%和(c) 90%甲烷摩尔分数时主要离子的数密度随时间的变化趋势, 以及(d)主要离子的周期平均值随甲烷摩尔分数的变化
图 4 (a) 10%, (b) 50%和(c) 90%甲烷摩尔分数时主要离子的数密度随时间的变化趋势, 以及(d)主要离子的周期平均值随甲烷摩尔分数的变化Figure4. The number densities of main ions as a function of time for methane mole fractions of (a) 10%, (b) 50%, (c) 90%, and (d) time averaged densities of main ions as a function of initial CH4 fraction.
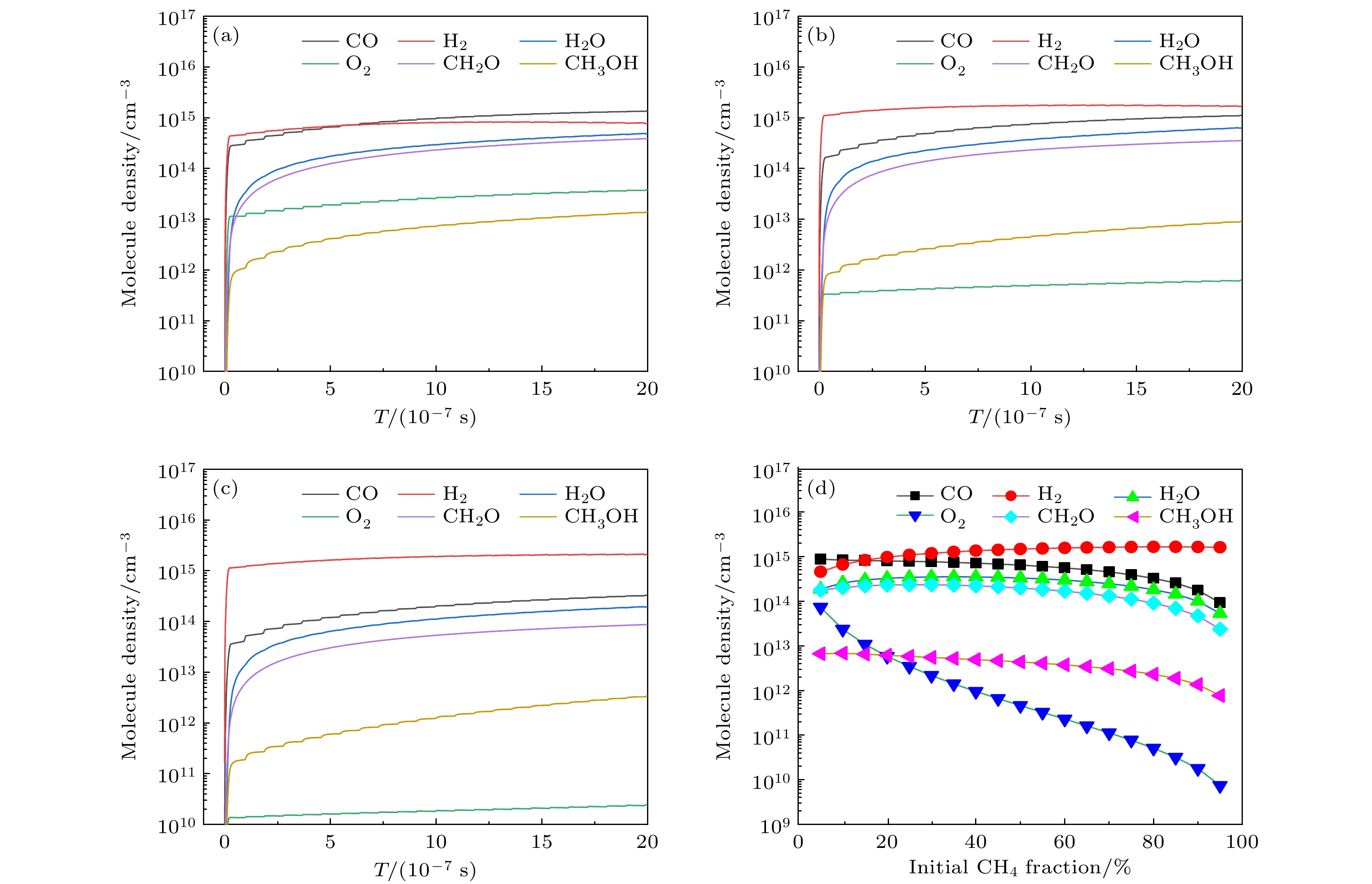 图 5 (a) 10%, (b) 50%和(c) 90%甲烷摩尔分数时主要分子数密度随时间的变化规律, 及(d)主要分子的周期平均值随甲烷摩尔分数的变化
图 5 (a) 10%, (b) 50%和(c) 90%甲烷摩尔分数时主要分子数密度随时间的变化规律, 及(d)主要分子的周期平均值随甲烷摩尔分数的变化Figure5. The number densities of main molecules as a function of time for methane mole fractions of (a) 10%, (b) 50%, (c) 90%, and (d) time averaged densities of main molecules as a function of initial CH4 fraction.
2
3.3.反应气体转化率和主要产物选择性
反应气体的转化率X, 和主要产物选择性S的计算公式分别为: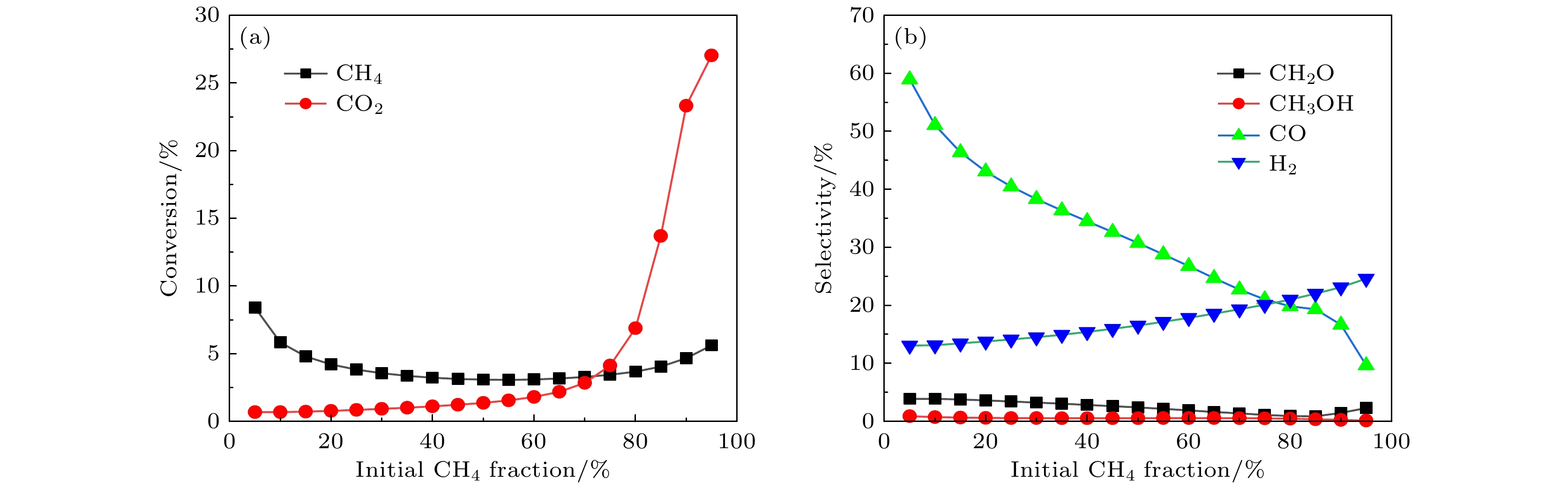 图 6 (a)进料气体的转化率和(b)合成气和重要含氧化合物的选择性随着甲烷摩尔分数的变化趋势
图 6 (a)进料气体的转化率和(b)合成气和重要含氧化合物的选择性随着甲烷摩尔分数的变化趋势Figure6. Time-averaged (a) conversion, (b) selectivity as a function of initial CH4 mole fraction.
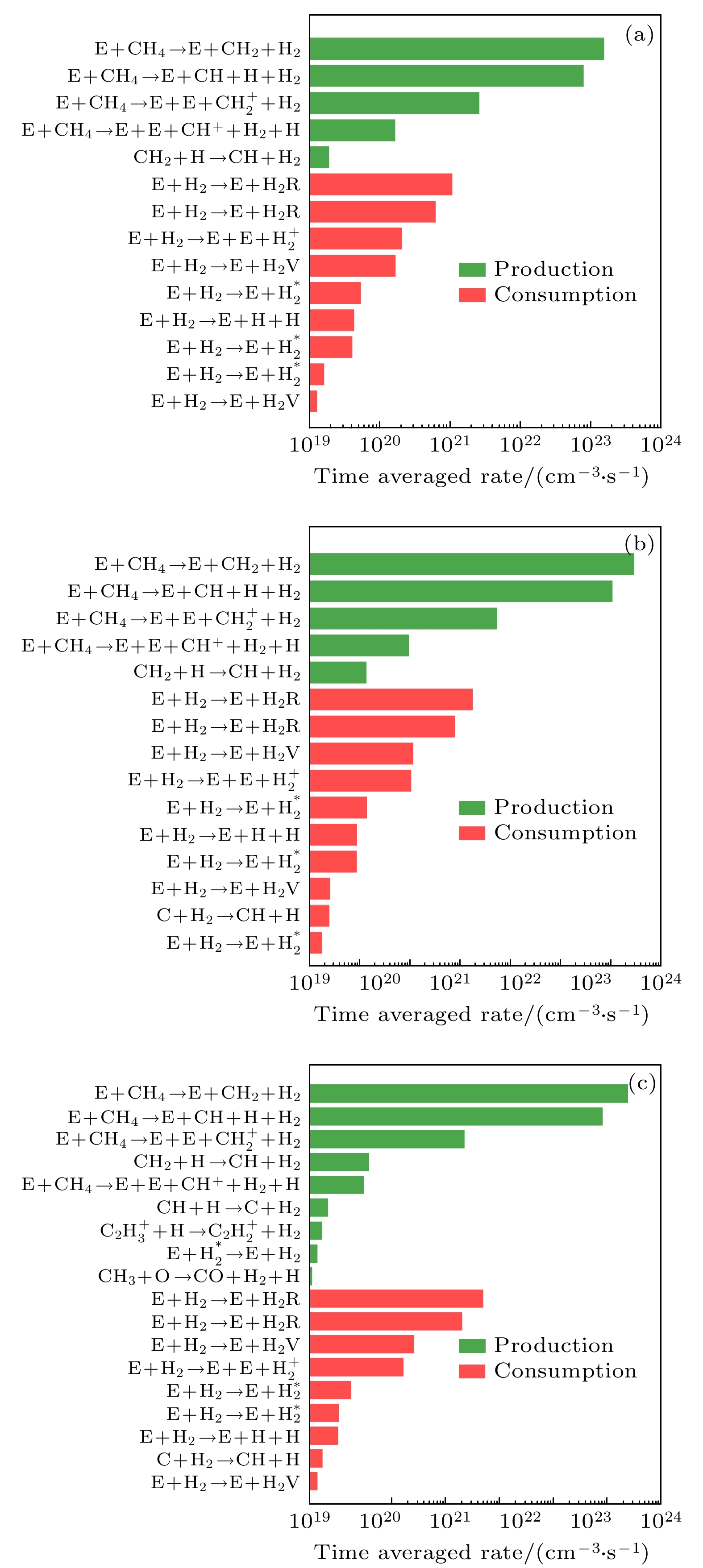 图 10 H2的主要生成和损耗反应的时间平均反应速率随甲烷摩尔分数的变化柱状图 (a) 10%, (b) 50%, (c) 90%
图 10 H2的主要生成和损耗反应的时间平均反应速率随甲烷摩尔分数的变化柱状图 (a) 10%, (b) 50%, (c) 90%Figure10. Time-averaged reaction rates of the dominant reaction pathways for the production and consumption of H2 as a function of methane mole fraction (a) 10%, (b) 50%, (c) 90%.
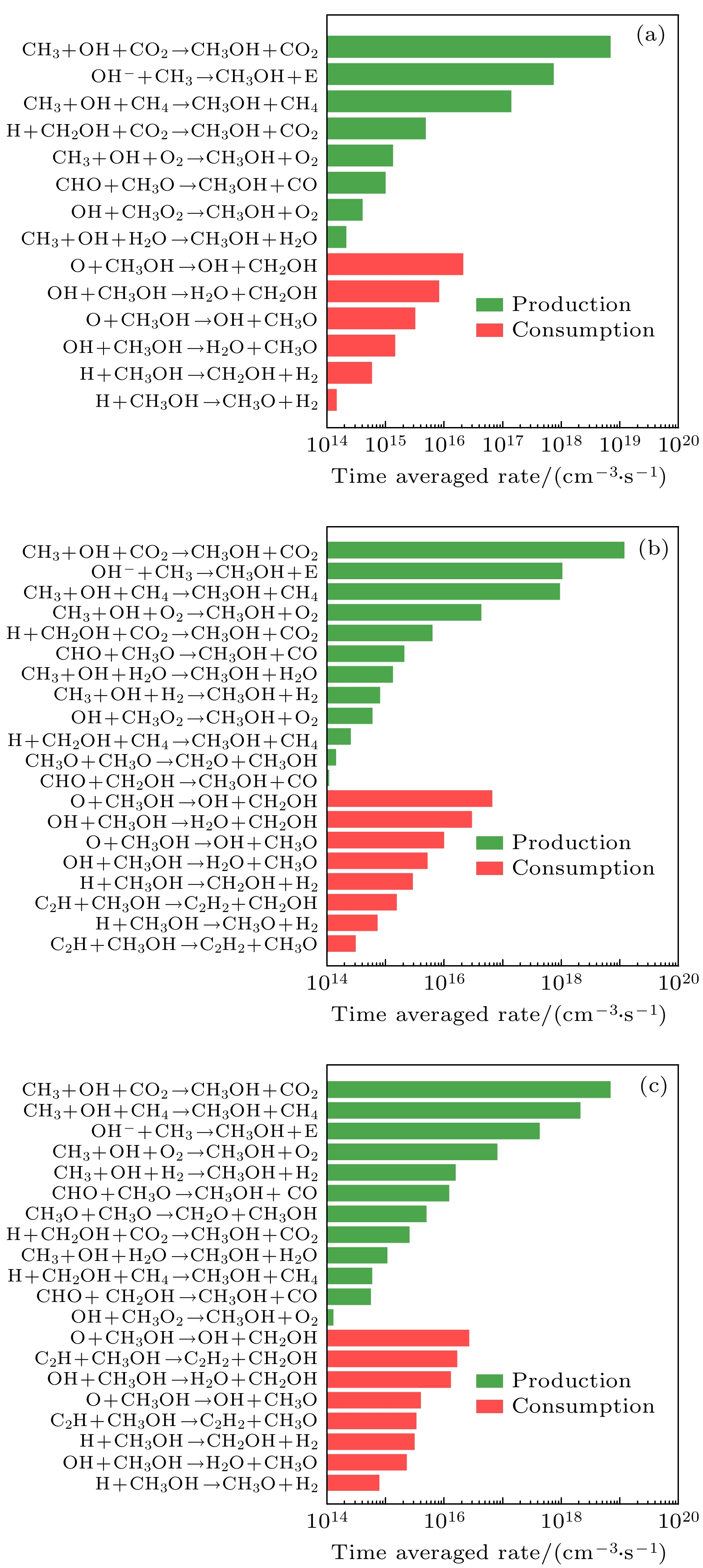 图 8 CH3OH的主要生成和损耗反应的时间平均反应速率随甲烷摩尔分数的变化柱状图 (a) 10%, (b) 50%, (c) 90%
图 8 CH3OH的主要生成和损耗反应的时间平均反应速率随甲烷摩尔分数的变化柱状图 (a) 10%, (b) 50%, (c) 90%Figure8. Time-averaged reaction rates of the dominant reaction pathways for the production and consumption of CH3OH as a function of methane mole fraction: (a) 10%, (b) 50%, (c) 90%.
 图 9 CO的主要生成和损耗反应的时间平均反应速率随甲烷摩尔分数的变化柱状图 (a) 10%, (b) 50%, (c) 90%
图 9 CO的主要生成和损耗反应的时间平均反应速率随甲烷摩尔分数的变化柱状图 (a) 10%, (b) 50%, (c) 90%Figure9. Time-averaged reaction rates of the dominant reaction pathways for the production and consumption of CO as a function of methane mole fraction (a) 10%, (b) 50%, (c) 90%.
2
3.4.主导反应路径
为了进一步探究合成气和重要含氧化合物的生成和损耗机理, 图7—图10依次给出了CH2O, CH3OH, CO, H2主要的生成和损耗反应的时间平均反应速率随甲烷摩尔分数(10%, 50%, 90%)的柱状图. 如图7所示, 当甲烷摩尔分数较低时, CH2和CO2的反应是主要的CH2O生成反应路径, 而当甲烷摩尔分数增加时, CH3和O之间的反应变成了主要的CH2O生成反应路径. CH2O的主要损耗反应路径是CH2O分别和O和OH生成CHO的反应. 如图8所示, CH3OH主要的生成化学反应路径是CH3, OH和CO2的三体复合反应(CH3+OH+CO2→CH3OH+CO2), 这是因为这两种自由基的高浓度和活性, 各自孤立的电子恰好形成一对共价键. 值得注意的是, 随着CH4摩尔分数增加为90%时, CH3, OH和CH4的三体复合反应速率比OH–和CH3之间的反应速率更大, 成为CH3OH分子生成的次要反应. 这是因为甲烷摩尔分数从10%增加到90%时, 一方面OH–密度下降了至少一个量级(如图4(c)), 另一方面是CH4的含量远大于CO2. CH3OH的主要损耗反应路径是CH3OH分子分别与O和OH反应生成CH2OH的反应. 此外, O原子可分别从CH3OH的甲基(-CH3)或者羟基(-OH)中夺取H原子分别生成CH2OH和CH3O. 数值模拟结果表明, O和CH3OH反应生成CH2OH的反应速率远大于O和CH3OH反应生成CH3O的反应速率, 这表明CH3OH中的甲基比羟基更容易失去氢原子. 图 7 CH2O的主要生成和损耗反应的时间平均反应速率随甲烷摩尔分数的变化柱状图 (a) 10%, (b) 50%, (c) 90%
图 7 CH2O的主要生成和损耗反应的时间平均反应速率随甲烷摩尔分数的变化柱状图 (a) 10%, (b) 50%, (c) 90%Figure7. Time-averaged reaction rates of the dominant reaction pathways for the production and consumption of CH2O as a function of methane mole fraction: (a) 10%, (b) 50%, (c) 90%.
如图9所示, CO主要来源于CO2的电子碰撞反应, 该电子碰撞反应在所有的CO生成反应中反应速率最大. 当甲烷含量较低为10%时, E + CO2→O– + CO的反应速率比其他CO生成反应大了近三个数量级. 而当甲烷摩尔分数逐渐升高时, E + CO2→O–+CO的反应速率逐渐减小, 而其他的CO生成反应的反应速率逐渐增加, 如C+O2→O + CO, E + CO2+→O + CO, 但E + CO2→O–+CO始终是CO的主导生成反应路径. 电子和CO碰撞解离和电离反应, 是CO的主要消耗反应路径. 图10给出的是H2的主要生成和损耗反应路径. 结果表明, CH4的电子碰撞解离反应(E + CH4→E + CH2 + H2, E + CH4→E + CH + H + H2)是H2分子最重要的生成反应路径. 而电子和H2碰撞电离反应则是H2主要的损耗反应路径. 如图9和图10所示, H2主要生成反应的反应速率远大于CO的生成反应速率, 而且H2主要损耗反应的反应速率却小于CO的主要损耗反应速率, 这也就是生成物中H2的密度高于CO的原因(如图5所示).
图11总结给出了CH4/CO2摩尔分数比为1∶1时大气压非平衡等离子体DRM(CH4/CO2 = 1∶1)生成合成气和CH2O, CH3OH等含氧化合物的总体反应流程图. 为了流程图的清晰和紧凑起见, 仅考虑时间平均的净反应速率超过1018 cm3/s的反应路径, 且图中箭头的宽度与时间平均的净反应速率成线性正比. 电子和甲烷分子的碰撞解离反应生成CH3, CH2, H2, 其中生成CH3自由基的净反应速率最大. 形成的甲基中, 一部分CH3自由基将重新结合成高级烃(C2H5, C2H6和C3H8等), 一部分参与反应形成含氧化合物如CH2O和CH3OH等, 还有一部分反应生成H2. 含氧化合物中的O来源于CO2分子, 主要通过电子和CO2分子的碰撞解离形成CO, O, O2等, 其中绝大部分形成了CO, CO进一步生成O原子, 因此O原子主要是由CO2解离形成的. O原子进一步转移到OH等粒子, 并与CH3反应参与形成CH2O与CH3OH等含氧化合物. 此外, CO2也可和甲烷解离产物CH2反应形成CH2O. 如图11所示, 反应气体CH4/CO2的电子碰撞解离反应生成CO和H2的净反应速率明显高于其他含氧化合物, 此外甲基和羟基对于含氧化合物CH2O与CH3OH的生成至关重要. 总体反应流程图进一步揭示了大气压非平衡等离子体DRM的反应机理, 为大气压非平衡等离子体DRM提供了理论参考.
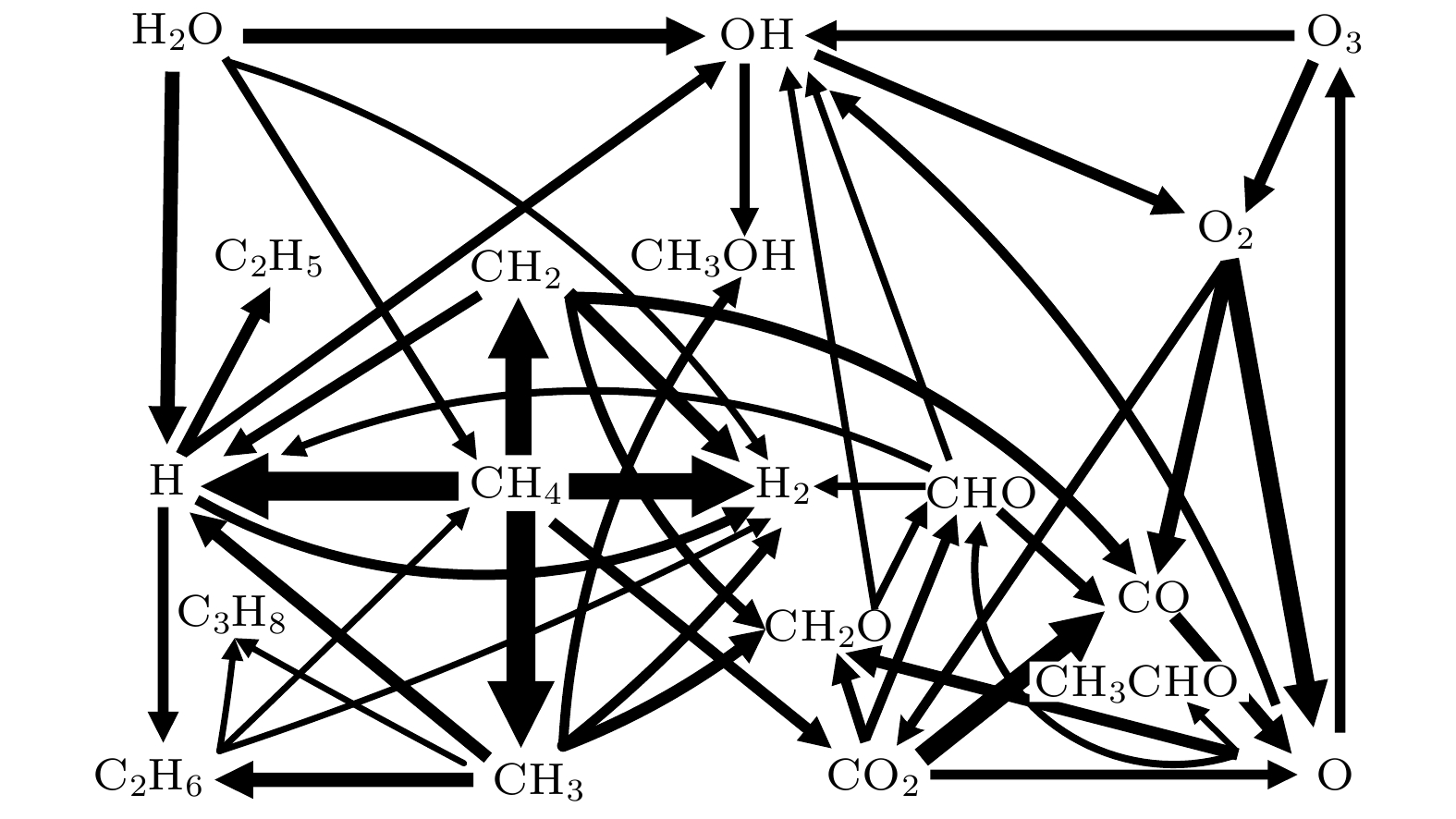 图 11 CH4/CO2摩尔分数比为1:1的大气压非平衡等离子体DRM反应总体流程图. 箭头的粗细与时间平均的净反应速率成线性正比
图 11 CH4/CO2摩尔分数比为1:1的大气压非平衡等离子体DRM反应总体流程图. 箭头的粗细与时间平均的净反应速率成线性正比Figure11. Schematic overview of the dominant reaction pathways for the conversion of CH4 and CO2 into representative higher oxygenates and syngas in atmospheric non-equilibrium plasma for a 1:1 CH4/CO2 gas mixture. The thickness of the arrows is linearly proportional to time-averaged rate of net reaction.

