全文HTML
--> --> -->在现有各种OLEDs蓝光材料中, 聚芴(PFO)是一种刚性平面联苯结构的化合物, 具有高稳定性, 非常容易成膜[6,7]. 其能隙大于2.95 eV, 薄膜的荧光量子效率可达80%以上, 是一种备受产业界期待的蓝色发光材料[8]. Mark等[9]利用聚芴制作的有机电致发光器件, 在开启电压仅为6 V时, 器件亮度高达10000 cd/m2. Niu等[10]详细研究了PFO与Ir-67之间的激子能量传递机制. 但是, Gong等[11-13]经过数十年的系统研究发现, PFO有一个非常重大的缺点: PFO的电致发光器件, 发光峰出现长波偏移, 会产生本不该出现的异常绿色发光带. 这严重影响了PFO器件的饱和色纯度, 也严重制约了其产业化进程[14].
自旋交叉材料(spin crossover, SCO)作为一类典型的分子基磁性材料, 通常具有电子构型3d4–3d7, 经常存在高自旋态(high spin, HS)和低自旋态(low spin, LS) 两种电子自旋态排布. 如Fe(II)配合物的配位原子为N原子时, 就会存在


本文首次报道了使用Fe(NH2trz)3·(BF4)2自旋交叉材料掺杂有机聚合物PFO, 解决了困扰业界多年的聚芴异常绿光难题. 成功制备出结构为ITO/PEDOT:PSS(30 nm)/PFO:Fe(NH2trz)3·(BF4)2(65 nm)/CsCl(0.6 nm)/Al(120 nm)的有机电致发光器件. 这种新型SCO器件抑制了PFO的发光峰向长波偏移, 实现了器件的强烈蓝光纯正发射. 运用光电磁一体化测量技术, 进一步研究了PFO掺杂Fe(NH2trz)3.(BF4)2器件的磁发光(MEL)和磁电导(magneto-conductivity, MC)效应.
2.1.实验方法
本实验制作了结构是ITO/PEDOT: PSS (30 nm)/PFO:Fe(NH2trz)3·(BF4)2 (65 nm)/CsCl (0.6 nm)/Al(120 nm)的电致发光器件. ITO/PEDOT: PSS是器件的复合阳极, CsCl/Al作为器件的复合阴极, PFO:Fe(NH2trz)3·(BF4)2是器件的发光层. 器件结构如图1(a)所示. PFO:Fe(NH2trz)3·(BF4)2用氯仿在无水无氧的条件下共掺溶解48 h.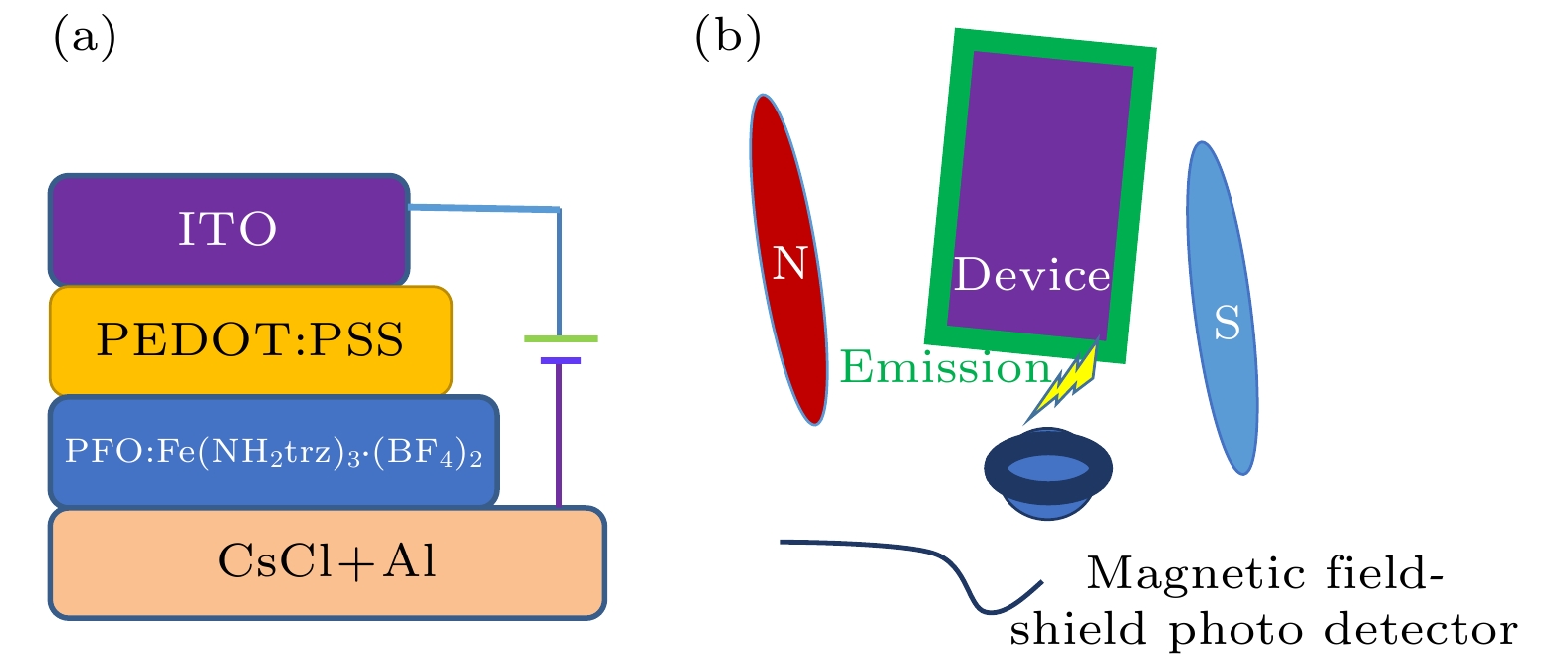 图 1 (a)器件结构; (b)磁效应测试原理示意图
图 1 (a)器件结构; (b)磁效应测试原理示意图Figure1. Schematic description of the device structure (a) and the device fabricated for the MEL measurements (b).
器件制备过程如下:
将ITO玻璃衬底用丙酮、酒精、去离子水分别按照顺序依次超声15 min, 然后放入干燥柜于115 ℃下烘干45 min. 接着将基片放入臭氧发生器进行10 min处理; 然后把基片放入水氧隔绝的手套箱按照顺序分别进行PEDOT:PSS和PFO: Fe(NH2trz)3·(BF4)2 (掺杂质量比10∶1)旋涂成膜, 干燥30 min后, 再把样品基片从手套箱直接传递至真空蒸镀室, 待真空度抽到9 × 10–5 Pa时开始蒸镀CsCl和Al的薄膜. 用石英振荡仪监测各薄膜层的厚度和成膜速率. 在各层薄膜生长的过程中, 系统的真空度维持在2 × 10–4 Pa左右. 电子缓冲层CsCl的厚度和成膜速率分别是0.6 nm和0.01 nm/s, Al电极的厚度和成膜速率分别是120 nm和0.15 nm/s. 所有的测试都在室温大气环境下进行. 测试方法如图1(b)所示. 被测器件均未被封装, 也未使用光耦合输出等附属装置.
2
2.2.材料与仪器
本实验所用的有机材料PFO购自西安宝莱特有限公司; Fe(NH2trz)3·(BF4)2为本实验室自行合成, 实验所用材料分子结构和Fe(NH2trz)3·(BF4)2合成路线如图2所示. 向盛有4-氨基三氮唑(0.5 g, 5.947 mmol)的200 mL圆底烧瓶中加入20 mL甲醇, 得到无色透明溶液. 在超声条件下, 向该溶液逐滴加入含Fe(BF4)2·6H2O(0.6691 g, 1.982 mmol)的甲醇溶液(80 mL), 此时, 该溶液立即产生大量白色沉淀, 超声1 h后将反应液离心, 接着倾倒上清液, 然后再将白色粉末状固体用无水乙醇洗涤、离心三次, 真空干燥, 得到Fe(NH2trz)3·(BF4)2. 其合成路线和材料分子结构如图2所示. 器件镀膜系统使用重庆师范大学和沈阳真空研究所联合研制的Future-2019型有机电致发光器件制造专用镀膜系统. 器件的电压、亮度、电流、电致发光光谱等参数由电脑全自动控制的Keithley-2400数字源表和PR-655扫描光谱仪组成的测量系统检测. Lakeshore-475电磁铁提供测试磁效应(MEL和MC)所需磁场[10]. 图 2 Fe(NH2trz)3·(BF4)2合成路线和材料分子结构示意图
图 2 Fe(NH2trz)3·(BF4)2合成路线和材料分子结构示意图Figure2. Schematic description of synthetic route and molecular structures of the materials studied.
 图 3 不同电压下, 器件ITO/PEDOT: PSS/PFO/CsCl/Al的归一化电致发光EL谱
图 3 不同电压下, 器件ITO/PEDOT: PSS/PFO/CsCl/Al的归一化电致发光EL谱Figure3. Normalized EL spectra of the device with ITO/PEDOT: PSS/PFO/CsCl/Al.
由图3可知, 器件发出非常强烈的异常绿光, 中心峰值波长为553 nm, 色坐标是(0.33, 0.45). 并且无论电压增大或减小, 其绿色发光带始终远远大于其蓝色发光带.
图4是PFO薄膜归一化的光致发光PL谱和电致发光EL谱(8 V)的对比图. 图4的插图是PFO薄膜的吸收谱, 在378 nm处呈现出最大吸收. 在380 nm氙灯光源的激发下, PFO薄膜的光致发光PL谱表现出高灵敏度的振动发射特性, 0-0电子跃迁中心在438 nm处. 与氯仿稀溶液PFO的光致发光PL谱相似, 但有一定的红移. 这种红移是由构象变化、聚集体、激子形成等共同作用引起[6,7,18].
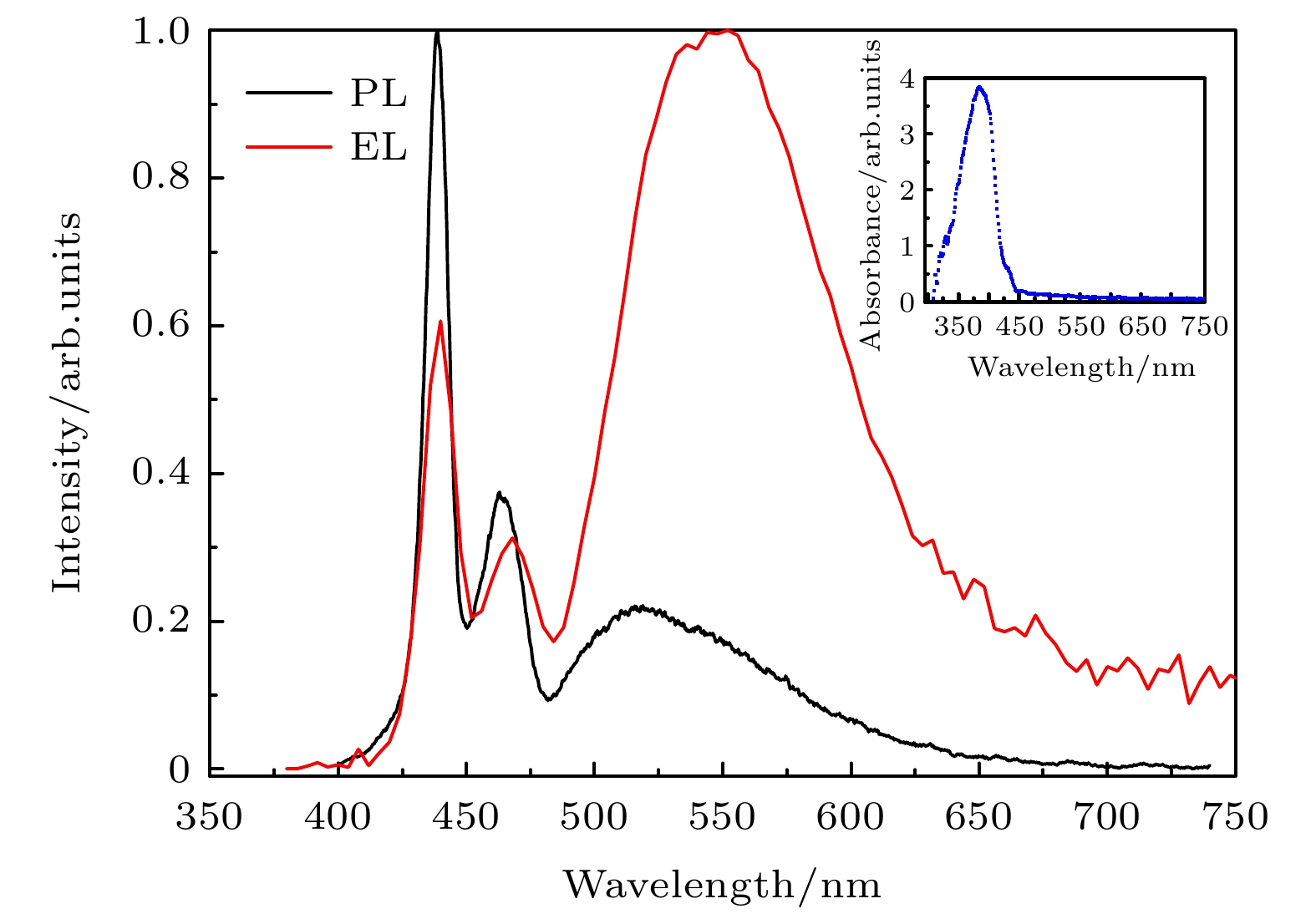 图 4 PFO薄膜的PL谱(红色)和8 V偏置电压的 EL谱(黑色)
图 4 PFO薄膜的PL谱(红色)和8 V偏置电压的 EL谱(黑色)Figure4. PL(red)and EL(black)spectra of the PFO film at 8 V.
与PFO薄膜的PL谱相比, 其EL谱的绿光带发射非常强. 这种异常绿光带并不是首次被观察到, 此前也被很多科学家观察到并进行过非常系统的研究[11-13]. 芴类绿光带可能的形成机制主要包括: 1)激基缔合物机制[19,20]; 2)芴酮缺陷机制[21-23]; 3)聚芴端基界面氧化机制[24,25]. 并且, 到目前为止, 关于PFO异常绿光的产生原因还争论较大. Heeger等[11]认为, PFO单体中含少量未烷基化芴, 在光、热、电等外界条件作用下, 这种残留物被氧化后生成芴酮. 这种新产生的芴酮导致了PFO器件电致发光绿光带的异常发射. Emil等[12]合成了一组含少量单烷基化芴的聚合物发光材料, 利用荧光光谱、红外吸收光谱和紫外吸收光谱等表征手段, 深入研究了在电场作用下这些聚合物薄膜的物性变化. 他们认为: 器件在空气氛围运行时, 聚合物链上产生了芴酮. 因此, 在电致发光过程中, 很容易发生由寡聚芴到芴酮的能量传递, 与这种能量传递对应的辐射跃迁导致异常绿光发射. 然而, Mathieu等[13]的观点与上述主张完全不同, 借助计算模拟和原子力显微镜(AFM)技术, 他们探讨了不同侧基对共轭高分子链段之间π-π堆积的作用, 发现烷基取代的聚芴更容易形成长程有序的聚集, 这种聚集会产生激基缔合物从而引起这些聚合物材料的绿光发射.
从图5(a)可知, Fe(NH2trz)3·(BF4)2掺杂PFO后, 在不同电压下, 器件ITO/PEDOT:PSS/PFO:Fe(NH2trz)3·(BF4)2/CsCl/Al发出强烈的蓝光, 峰值波长为438 nm, 与PFO薄膜的PL谱峰值波长完全一致. 这表明器件的蓝光发射由PFO产生, 掺杂没有引起PFO发光内在本征性质的改变. 并且, 与未掺杂Fe(NH2trz)3·(BF4)2器件相比, 绿色发光带被成功压制. 这种掺杂器件, 完全实现了PFO电致发光器件的本征蓝色发射, 标准三基色坐标是(0.23, 0.22). 并且, 随着器件偏置电压的改变, 器件光谱的蓝光部分在整个EL谱所占比例几乎没有发生改变. 图5(b)是器件的电流-电压-亮度曲线. 在4.0 V时, 器件开始明显发亮. 随着电压的不断增大, 器件的电流呈现指数式上升; 器件的亮度也在不断增大. 如在10 V时, 器件的亮度达到782 cd/m2; 电流效率为1.85 cd/A. 由于本工作是为了研究Fe(NH2trz)3·(BF4)2掺杂的作用, 尽量避免其它因素的干扰, 本器件未添加可以提高器件亮度和效率的电子注入层和空穴缓冲层, 也没有对器件进行特别的优化, 因而器件的整体光电性能不高. 未掺杂Fe(NH2trz)3·(BF4)2的相同结构的器件, 在10 V时, 器件的亮度达到739 cd/m2; 电流效率为1.87 cd/A. 由此可知, 掺杂Fe(NH2trz)3·(BF4)2的器件, 亮度有所提升, 但电流效率基本相同.
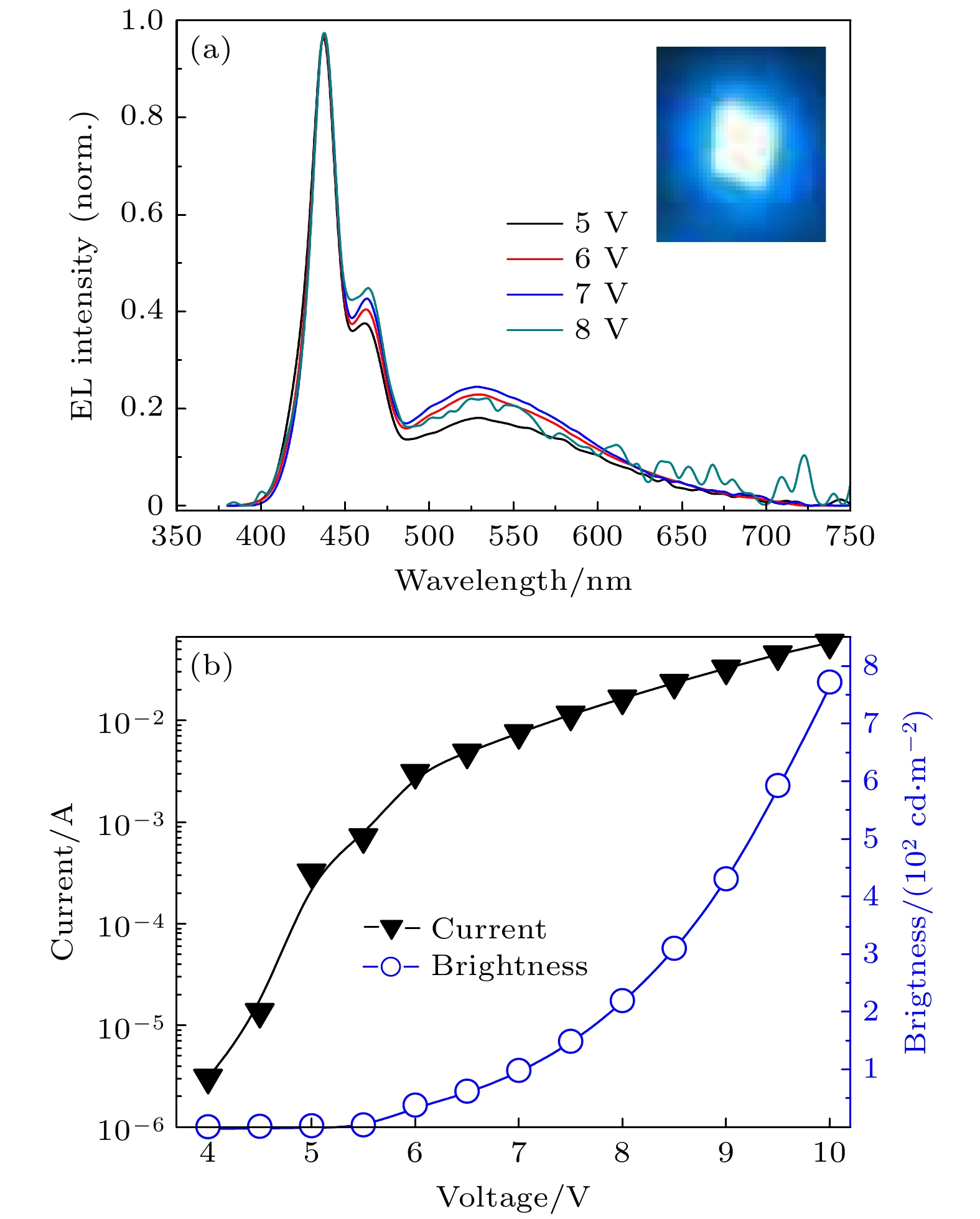 图 5 (a)器件ITO/PEDOT: PSS/PFO: Fe(NH2trz)3·(BF4)2/CsCl/Al的电致发光EL谱; (b)该器件的电流-电压-亮度曲线
图 5 (a)器件ITO/PEDOT: PSS/PFO: Fe(NH2trz)3·(BF4)2/CsCl/Al的电致发光EL谱; (b)该器件的电流-电压-亮度曲线Figure5. (a) EL spectra of the device with ITO/PEDOT: PSS/PFO: Fe(NH2trz)3·(BF4)/CsCl/Al; (b) I-V-L characteristics response of the device
图6是PFO:Fe(NH2trz)3·(BF4)2薄膜归一化的光致发光PL谱和电致发光EL谱(7 V)的对比图. 与PFO:Fe(NH2trz)3·(BF4)2薄膜的PL谱相比, 其EL谱的形状变化很小. 二者的主峰蓝光带几乎完全一致; 右边两个肩峰强度明显下降, 位置有2 nm的红移. 由此可知, PFO:Fe(NH2trz)3·(BF4)2薄膜的光致发光和电致发光的内在能级跃迁过程非常类似.
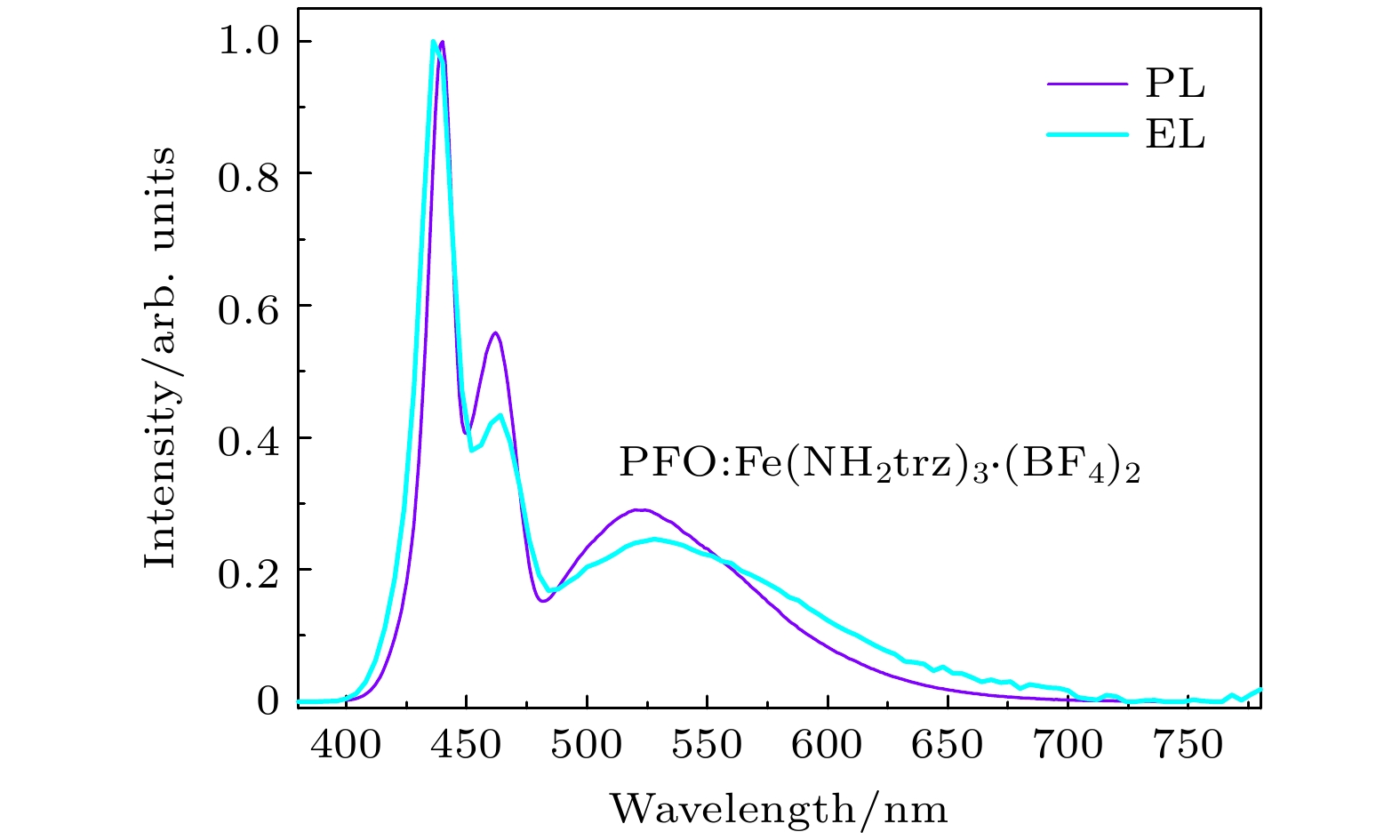 图 6 PFO: Fe(NH2trz)3·(BF4)2薄膜的PL谱(紫色)和7 V偏置电压的 EL谱(青色)
图 6 PFO: Fe(NH2trz)3·(BF4)2薄膜的PL谱(紫色)和7 V偏置电压的 EL谱(青色)Figure6. PL(purple)and EL(ching)spectra of the PFO: Fe(NH2trz)3·(BF4)2 film at 7 V.
对于Fe(NH2trz)3·(BF4)2而言, Fe离子处于正八面体强配位场, 在这些配位场强作用下, 电子存在LS或HS这两种不同的排布方式[15,26]. 此时, Fe离子的5个简并d轨道能级将分别分裂为能级较高的轨道能级和能级较低的轨道能级. 最重要的是, d电子的排布方式遵循能量最低原理还是洪特规则, 这由配位场分裂能和电子成对能的相对大小所决定. 如果配位场分裂能大于电子成对能, d电子排布就遵循能量最低原理, 其先填满能量较低的轨道, 形成LS的电子排布方式. 如果配位场分裂能大于电子成对能, 其排布方式遵循洪特规则, 此时尽可能保持最多的未成对电子数, 形成HS的电子排布方式. 这些不同的自旋态很容易与其周围的PFO分子的电子自旋态和光子态密度进行耦合. 光子态密度是单位体积内某频率附近单位频带宽度上的光子本征态的数目, 对于一个具体的系统每个光子态内的光子数目可能不相同. 改变光子态密度, 可以抑制或增强原子的自发辐射. 对照文献[11,13]的研究结果, 结合图5(a)的EL谱, 推断电子自旋态和光子态密度之间的耦合作用或者降低了PFO薄膜中激基缔合物的形成, 或者减弱了PFO薄膜中芴酮的产生.
激基缔合物是指同种分子之间形成的一种激发态缔合物[27]. 1954年, Kasper等[28]就在芘的正己烷溶液中发现了激基缔合物. 从此, 激基缔合物的研究就引起了人们的广泛关注. 许多科学家对不同的芳环有机化合物进行了大量的研究, 在苯、蒽及其衍生物荧光发射光谱中都发现了激基缔合物的谱带. 在聚合物溶液中, 形成激基缔合物有两种比较典型的情况: 1)分子内近邻生色团之间或分子链内远程的生色团之间形成激基缔合物; 2)不同分子链上的生色团彼此靠近时形成激基缔合物. 就其物理外在本质特征而言, 激基缔合物荧光光谱相对于自身的单体分子会出现明显的红移和展宽. 这种特征是激发态的分子与基态分子之间的相互缔合作用引起. 由于分子间的这种相互缔合作用会降低激发态的能量, 改变原来分子的性质, 因此激基缔合物的PL光谱与原来形成它的分子的PL光谱相比, 会出现红移和展宽. 然而, 通过利用PL光谱的红移和展宽现象来确定是否有激基缔合物的存在还是非常片面[29].
近年来, 有机磁场效应(organic magnetic field effect, OMFE)因其与有机半导体薄膜的载流子自旋属性高度相关, 可以作为一种直接而且高效的方法来研究OLEDs中激发态的动力学过程, 受到了人们的广泛关注[4,29]. 有机磁场效应主要是利用外加磁场, 一般是几到几百mT的磁场, 然后通过测试磁场下流过器件的电流强度和发光强度等参数随外磁场的变化, 根据出现的数据曲线特征, 揭示器件相应的内在动力学过程, 如三重态-三重态湮灭(triplet-triplet annihilation, TTA)[30]和系间窜跃(intersystem crossing, ISC)[10]等. 有机磁场效应主要包括器件的磁发光效应(magneto-electroluminescence, MEL)和MC效应. 2019年, 熊祖红等[31]通过OMFE方法研究发现, 激基缔合物型OLED 的MEL数据曲线特征非常明显: 曲线分为低场部分和高场部分两段. 随着磁场强度增大, 低场部分的MEL曲线快速上升, 其幅值随注入电流增大而减小; 然而, 高场部分MEL曲线快速下降, 并且随注入电流增大下降特别明显.
对于耦合作用是否降低或消除了PFO薄膜中激基缔合物的形成, 我们利用有机磁场效应(OMFE)方法, 研究了PFO:Fe(NH2trz)3·(BF4)2器件的MEL和MC效应:
在不同的偏置电压下, 器件ITO/PEDOT:PSS/PFO:Fe(NH2trz)3·(BF4)2/CsCl/Al的磁发光MEL的值均为正值. 在0—200 mT磁场范围内, 随着器件电压的不断增大, 器件的电流也不断增大, 磁发光MEL的值随注入电流的增加而几乎不变; 在磁场超过200 mT时, MEL值有所下降, 但下降非常缓慢.
如图7(a)所示, 在4—8 V不同的偏置电压下, 器件ITO/PEDOT:PSS/PFO:Fe(NH2trz)3·(BF4)2/CsCl/Al的磁发光MEL的值均为正值, 5 V时达2%. 在相同磁场下, 随着电压的增大(5—8 V), MEL缓慢下降. 但是, 在相同磁场强度下, 5 V的MEL值略大于4 V的MEL值. 这是由于在OLED中, 电流-电压特性可分为: 欧姆接触区间(Ohmic)、陷阱填充空间电荷限制电流区间(TF-SCLC)和陷阱填满限制电流区间(TFL-SCLC)[32,33]. 4 V时器件刚刚启亮, 属于欧姆接触区间; 5 V时属于陷阱填充空间电荷限制电流区间. 这两个不同区间之间的跃变, 导致了在相同磁场强度下, 5 V的MEL值略大于4 V的MEL值. 在同一电压下, 在0—50 mT磁场范围内, 磁发光MEL的值随注入磁场的增大而迅速增大; 但在磁场超过45 mT时, MEL值增大的斜率有所放缓, 但其始终在增大. 特别需要注意的是, 在相同电压下, MEL值随着磁场的增大而增大, 并未出现激基缔合物的MEL“指纹”特征[31](低场部分的MEL快速上升, 其幅值随注入电流增大而减小; 然而, 高场部分MEL快速下降. 并且随注入电流增大下降更明显). 图7(b)是这种掺杂器件的MC曲线. 从图可知, 在较小电压(4 V)下, 器件的MC值为2.08%. 随着电压的增大, 器件的MC值微弱下降. 在同一电压下, 随着磁场的增大, 器件的MC不断增大. 这种曲线变化规律与已经报道的很多激子(exciton)型器件的MC特征[4,34]很类似: 随着磁场的增大, 曲线先上升后下降, 幅值有变化但变化程度很微弱. 由此可以断定, PFO: Fe(NH2trz)3·(BF4)2薄膜内没有激基缔合物的产生. 那么, 未掺杂的纯PFO薄膜内是否有激基缔合物的产生? 如图8所示, 我们测试了器件ITO/PEDOT:PSS/ PFO/CsCl/Al的MEL与MC, 也未出现激基缔合物的MEL“指纹”特征. 由此可见, 并不是掺杂Fe(NH2trz)3·(BF4)2降低或消除了PFO薄膜中激基缔合物的形成, 而是PFO:Fe(NH2trz)3·(BF4)2和纯PFO薄膜内本就没有激基缔合物的产生.
 图 7 不同电压下器件ITO/PEDOT: PSS/PFO: Fe(NH2trz)3·(BF4)2/CsCl/Al的磁发光曲线(a)和MC曲线(b)
图 7 不同电压下器件ITO/PEDOT: PSS/PFO: Fe(NH2trz)3·(BF4)2/CsCl/Al的磁发光曲线(a)和MC曲线(b)Figure7. MEL (a) and MC (b) of the device with ITO/PEDOT: PSS/ PFO: Fe(NH2trz)3·(BF4)2/CsCl/Al under different voltage
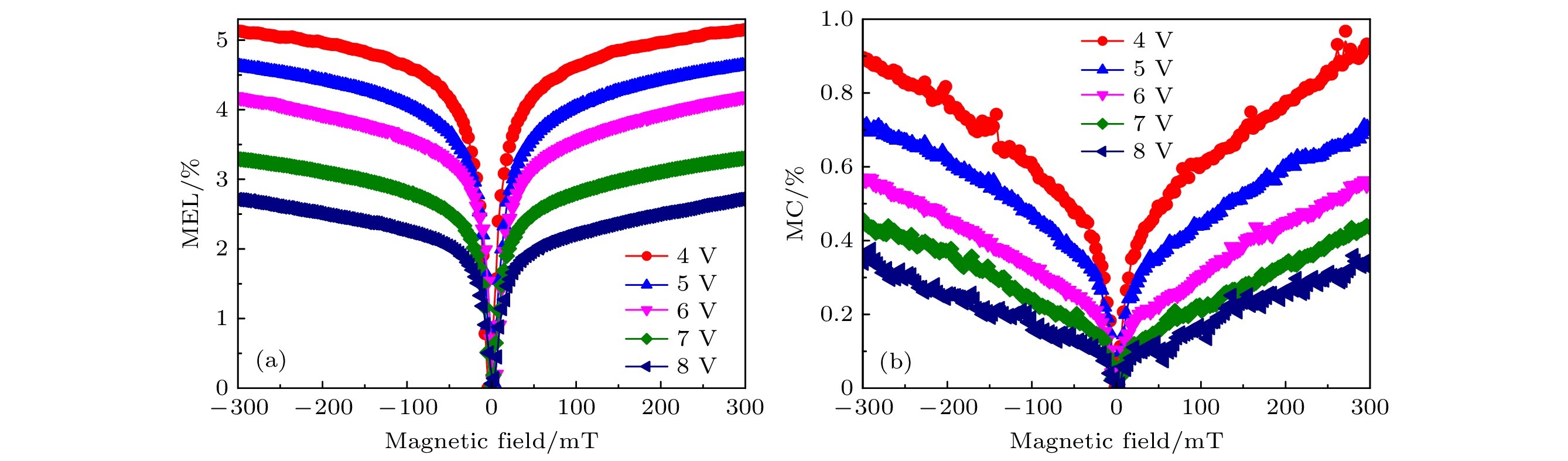 图 8 不同电压下器件ITO/PEDOT: PSS/ PFO/CsCl/Al的磁发光曲线 (a)和MC曲线(b)
图 8 不同电压下器件ITO/PEDOT: PSS/ PFO/CsCl/Al的磁发光曲线 (a)和MC曲线(b)Figure8. MEL (a) and MC (b) of the device with ITO/PEDOT: PSS/ PFO/CsCl/Al under different voltage.
既然PFO:Fe(NH2trz)3·(BF4)2薄膜内没有激基缔合物的产生, 现在就需要特别考虑这种薄膜内的能量传递过程[35]. 根据发光动力学理论, 聚合物的电致发光过程就是一个伴随电子得失的辐射跃迁过程. 电子的不断得失过程, 也就是一个连续的氧化还原过程. 对于没有掺杂Fe(NH2trz)3·(BF4)2的PFO薄膜, 一定比例的PFO在电致发光过程中很容易被氧化为芴酮. 另外, 通过对纯PFO薄膜器件浸入液氮测试, 纯PFO薄膜器件也被放入CCS-350 s真空罩进行测试, 均观察到PFO绿光带明显减弱. 由此也反证, PFO被氧化产生了芴酮是其产生绿光带的重要原因. PFO到芴酮的能量传递对应的辐射跃迁导致553 nm异常绿光发射. 对于掺杂Fe(NH2trz)3·(BF4)2的PFO薄膜, HS的电子排布方式很容易与其周围的PFO分子的电子自旋态和光子态密度进行耦合. 如图9所示, 这种耦合可以“剪断”PFO在电致发光过程中被氧化为芴酮的通道[12]. 还需要特别考虑的是: Fe(NH2trz)3·(BF4)2是一种自旋交叉配合物, 在氯仿溶剂中的溶解性不高, 只能是部分溶解. 这样, Fe(NH2trz)3·(BF4)2与PFO链之间极易发生交联. 分子链间的交联结构使聚合物链之间的距离变大, 这样PFO到芴酮之间的能量传递不容易发生. 因而, Fe (NH2trz)3·(BF4)2掺杂PFO器件中, 异常绿光带被成功压制, 观察到了纯正的蓝光发射.
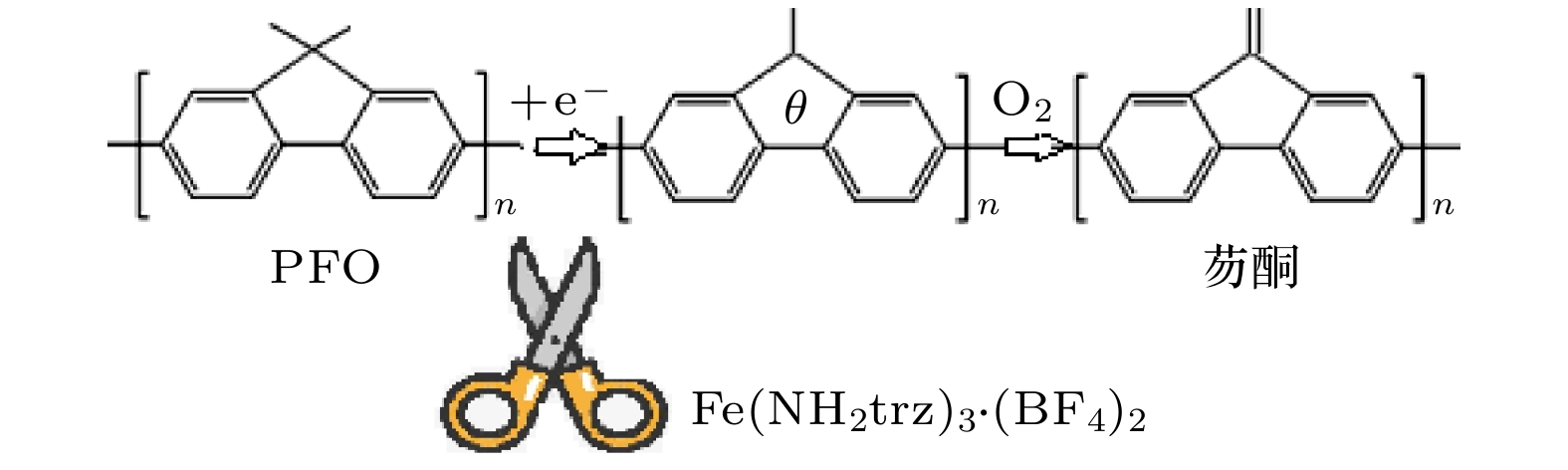 图 9 Fe(NH2trz)3·(BF4)2耦合“剪断”PFO在电致发光过程中被氧化为芴酮机理图
图 9 Fe(NH2trz)3·(BF4)2耦合“剪断”PFO在电致发光过程中被氧化为芴酮机理图Figure9. Mechanism diagram of Fe(NH2trz)3·(BF4)2 coupling cutting PFO oxidation to fluorenone in electroluminescence.
