Fund Project:Project supported by the Jiangxi Provincial Natural Science Foundation, China (Grant Nos. 20202BAB204004, 20171BAB216001), the Scientific Research Project of Jiangxi Provincial Education Department, China (Grant Nos. GJJ191114, GJJ161242), the Qinglan Scholars Program of Nanchang Normal University, China, and the National Natural Science Foundation of China (Grant Nos. 51871096, 11664028)
Received Date:06 February 2020
Accepted Date:10 July 2020
Available Online:10 October 2020
Published Online:20 October 2020
Abstract:By using large-scale atomic/molecular massively parallel simulator (LAMMPS) code, a molecular dynamics simulation is performed in the NPT ensemble at zero pressure to investigate the influence of melting rates γ on the evolutional characteristics of vanadium atomic structure such as body-centered cubic (BCC), hexagonal close-packed structure (HCP), face centered cubic (FCC), simple cubic (SC) and icosahedra (ICO) during the rapid melting of solid vanadium crystal at five different melting rates (γ1 = 1 × 1011 K/s, γ2 = 1 × 1012 K/s, γ3 = 1 × 1013 K/s, γ4 = 1 × 1014 K/s , γ5 = 1 × 1015 K/s), in which 16000 atoms in a cubic box under the periodic boundary condition are considered, and their motion equations are solved by Verlet’s algorithm in the velocity form in time steps of 1 fs. Constant pressure P and temperature T are imposed by a modified Nose-Hoover method for both P and T variables, and an embedded-atom model (EAM) potential is utilized. For identifying the local atomic structures of liquid and solid vanadium at different temperatures, a polyhedral template matching method (PTMM) is used by measuring the root-mean square deviation (RMSD), in which clusters are classified as the topology of the local atomic environment without any ambiguity in the classification. Subsequently, the variation of the potential energy, entropy and Gibbs free energy of FCC, HCP, BCC and ICO vanadium clusters are calculated through ab initio MD simulation in the canonical ensemble (NVT) at selected temperatures, and the lowest-energy dynamic structure and its corresponding static heating structure are also shown in this paper. Based on the above calculated results, it is found that the melting point of refractory metal vanadium increases obviously with the increase of heating rate, but the heating rate only presents a limited effect on the population of atomic structure for each of BCC, HCP, FCC, SC and ICO. Namely, the temperature still plays a dominant role in the rapid melting process of V rather than heating rate. Moreover, the ab initio MD simulation and thermodynamics analysis further reveal that lots of ICO clusters of vanadium can exist stably in the liquid region rather than in solid crystal, which is not only due to its higher stability and longer lifetime than those of crystalline atomic clusters, but also because ICO possesses higher entropy and lower Gibbs free energy in high temperature liquid region. Keywords:molecular dynamics/ vanadium/ melting/ cluster
针对含有16000个钒原子的BCC结构, 图1中首先给出了5种不同熔化速率下势能曲线随温度的演变过程. 从图1(a)与(b)中可以看出, 4种熔化速率(γ1 = 1 × 1011 K/s, γ2 = 1 × 1012 K/s, γ3 = 1 × 1013 K/s, γ4 = 1 × 1014 K/s)的能量曲线存在明显的突变, 其熔化转变温度分别为Tm1 = 3246 K, Tm2 = 3309 K, Tm3 = 3384 K以及Tm4 = 3553 K. 而随着熔化速率进一步升高至$1\!\times \!10^{15}$ K/s, 体系势能曲线的突变消失, 在T = 3250 K附近呈现渐变现象, 直至4236 K与另外4种熔化曲线重合. 即熔化速率越高, Tm也越高. 导致这一现象产生的原因是过高的熔化速率使得原子体系在极短的时间内来不及达到稳定的动力学与热力学平衡. 与此同时, 图2中也同时给出了5种不同熔化速率下体积随温度的演变过程. 从图2知, 在熔化速率γ ≤ γ4 (1 × 1014 K/s)时, 不同熔化速率下体系的体积存在明显的突变, 说明有一级相变产生, 其突变所对应的温度点与图1中势能曲线的转变点呈现完美的一一对应关系, 反映出固态钒的体积明显小于液态钒的体积. 当考虑不同熔化速率下体系的压强随温度的波动变化时, 图3中明确显示熔化速率最低时(γ1 = 1 × 1011 K/s), 压强波动最大, 而当熔化速率越来越高时, 压强的波动幅度则逐渐减小, 当速率升高至1 × 1015 K/s时, 压强的变化幅度在图3中是最小的. 该现象说明包含16000个钒原子的周期性模拟体系在熔化速率较低时, 研究对象有相对充分的时间来达到相对稳定的亚稳或者稳定平衡状态, 而速率较高时, 体系则经历剧烈的热力学过程. 图 1 5种不同熔化速率下金属钒体系的势能变化与温度之间的演变关系, (b)是(a)的局部放大图 Figure1. The potential energy of the vanadium metal as a function of temperature at various melting rates γ, (b) is a partial enlarged view of (a).
图 2 5种不同熔化速率下金属钒体系的体积变化与温度之间的演变关系, (b)是(a)的局部放大图 Figure2. The volume of the vanadium metal as a function of temperature at various melting rates γ, (b) is a partial enlarged view of (a).
图 3 不同熔化速率下体系压强随温度的演变关系 Figure3. The pressure of the vanadium metal as a function of temperature at various melting rates γ.
23.2.径向分布函数与微结构的演化 -->
3.2.径向分布函数与微结构的演化
针对熔化过程中系统微结构的演变, 径向分布函数(radial distribution function, RDF)是一种简单有效的方法, 与实验上经常使用的结构因子互为傅里叶变换, 是表征结构的一个重要参数, 数学定义如下[1]:
其中$l( {{B}}) = \dfrac{1}{N}\displaystyle\sum\nolimits_{i = 1}^N {\left\| {{b_i} - {{B}}} \right\|}$. 之后确定一个中心原子和其近邻原子之间的结构, 就只需先给出标准模板团簇构型(如图5中的ICO, BCC, HCP, FCC及SC多面体), 然后通过RMSD来判断该标准团簇模板与目标团簇构型之间的结构相似性. RMSD数值越小, 条件越发苛刻, 目标结构与模板的相似度也就越高, 所表征的团簇结构也就越接近于模板结构, 该方法称之为PTMM, 目前已被广泛应用于对非晶与玻璃态结构的表征, 接下来将用其分析不同类型团簇的原子结构. 众所周知, 金属钒在常温下是BCC结构, 图5中已经初步给出了FCC, HCP, BCC, ICO以及SC团簇原子随温度的变化示意图, 在此基础上, 图6给出了5种不同熔化速率(γ1 = 1 × 1011 K/s, γ2 = 1 × 1012 K/s, γ3 = 1 × 1013 K/s, γ4 = 1 × 1014 K/s以及γ5 = 1 × 1015 K/s)下FCC, HCP, BCC, ICO以及SC团簇原子数随温度变化的具体情况. 从图6总的来看, 在低温区间内, 不论是高熔化速率还是低熔化速率, 当温度由室温逐渐升高时, BCC团簇在起始阶段呈现缓慢减少的趋势, 但当温度逐渐逼近Tm时, BCC原子急剧减少, HCP则迅速增加, 之后ICO, FCC以及SC等原子团簇也在T = Tm这一瞬间陡然增加. 而一旦当温度T > Tm时, HCP与ICO则由最大值开始急剧下降, 同时FCC也开始减少. 与此同时虽然SC的数量在高温阶段较少, 但其数目并未随温度的增加而发生明显变化. 简言之, 图6中隐藏着如下的物理规律: 即在BCC急剧消失的时候, 在熔化温度附近有部分原子团簇快速转化为HCP, ICO, FCC以及SC团簇, 这一熔化规律并不随熔化速率的升高而发生改变. 之后为了进一步了解晶体钒在熔化过程中, 不同熔化速率对同一类型团簇原子数目的影响, 图7分别给出了FCC, HCP, BCC, ICO, SC以及其他类型团簇的原子分布随温度的演变关系. 从图7可见, 不论是FCC, HCP, ICO还是SC类型的原子团簇, 其数目都在熔化温度Tm附近明显增加, 与此同时伴随着BCC的急剧减少(图7(c)). 对同一团簇而言, 考虑熔化速率效应, 发现在速率γ ≤ 1 × 1012 K/s时, 速率的改变对团簇数目的影响并不显著. 当γ大于1 × 1012 K/s时, 熔化速率越高, BCC类型团簇减少得越慢, 而FCC, HCP, ICO以及SC则增长得越慢. 之后随着温度逐渐升高, FCC, HCP以及ICO的数目在达到顶峰后才逐渐减少. 图 6 利用多面体模板匹配法分析得到的5种不同熔化速率下FCC, HCP, BCC, ICO以及SC类型团簇原子随温度的演变关系 Figure6. The population of the FCC, HCP, BCC, ICO and SC types atoms in V system as a function of temperature obtained from the polyhedral template matching at various rates, respectively.
图 7 利用多面体模板匹配法分析得到的5种不同熔化速率下各种类型团簇原子分布随温度的演变关系 (a) FCC; (b) HCP; (c) BCC; (d) ICO; (e) SC; (f)其他类型团簇 Figure7. The fraction of the various types atoms in V system as a function of temperature obtained from the polyhedral template matching at five various rates: (a) FCC; (b) HCP; (c) BCC; (d) ICO; (e) SC; (f) other types atoms counts.
针对图6与图7中 ICO原子团簇在高温阶段持续存在的物理现象, 图8进一步利用ab initio MD模拟了BCC(15个原子)、FCC(13个原子)、HCP(13个原子)以及ICO(13个原子)分别在300, 500 K和1100 K时的结构稳定性与团簇寿命. 结果显示在300 K和500 K时, FCC与HCP均在模拟结束后转变为了ICO, BCC也在模拟结束后转变为了含有ICO团簇核心的几何结构, 而ICO则几乎保持其初始结构而未发生变化. 当温度升高至1100 K时, BCC演变为一个含有畸变ICO的结构, 而FCC与HCP则经过一系列不同的亚稳结构最终演变为非规则构型. 唯独ICO仍然保持其基本构型, 且未发生明显改变, 这说明在高温阶段ICO的稳定性的确高于晶体型原子团簇, 这也为ICO在温度高于熔化温度后的大量出现提供了团簇层面的解释. 图 8 FCC, HCP, BCC以及ICO 团簇平均每原子势能随模拟步长的演变趋势 (a) 300 K; (b) 500 K; (c) 1100 K Figure8. The variation of the potential energy of per atom of FCC, HCP, BCC and ICO cluster as a function of time step, respectively: (a) 300 K; (b) 500 K; (c) 1100 K
此外, 考虑到熔化过程中的MD模拟[35]与实验[37,40]均是在有限温度下进行的, 所以本文通过计算团簇的原子振动频率[52]后, 进一步从熵与吉布斯自由能的角度来研究各团簇的热力学稳定性[52], 期望以此找到具有5次对称性而缺乏平移对称性的ICO能够在高温区稳定存在的原始因素. 因为熵与吉布斯自由能均是描述热力学系统的重要态函数, 其大小均可清晰反映系统所处状态的稳定情况, 指明热力学过程进行的方向, 可以为研究对象提供定量表述, 即熵越高, 吉布斯自由能越低, 孤立体系越稳定. 基于此, 图9中分别给出了FCC, HCP, ICO及BCC(为了与其他三种标准团簇具有可比性,此处是仅含13原子的标准BCC碎片)原子团簇的熵与吉布斯自由能随温度的演变关系. 可知, 虽然各团簇的熵与自由能随温度的演变趋势恰好相反, 但其所指示的物理意义[52]却完全一致, 即与FCC, HCP, BCC相比, ICO的熵最高, 吉布斯自由能最低, 而且随着温度逐渐升高, 该现象愈加显著. 这充分表明高温液态金属区域ICO的热力学稳定性远远优于各晶体型原子团, 为液态金属中ICO原子团的大量存在提供了热力学层面的理论解释. 图 9 FCC, HCP, BCC及ICO 原子团簇的熵与吉布斯自由能随温度的演变关系 (a) 熵; (b) 吉布斯自由能 Figure9. The variation of the entropy and Gibbs free energy of FCC, HCP, BCC and ICO cluster as a function of temperature, respectively: (a) Entropy; (b) Gibbs free energy.

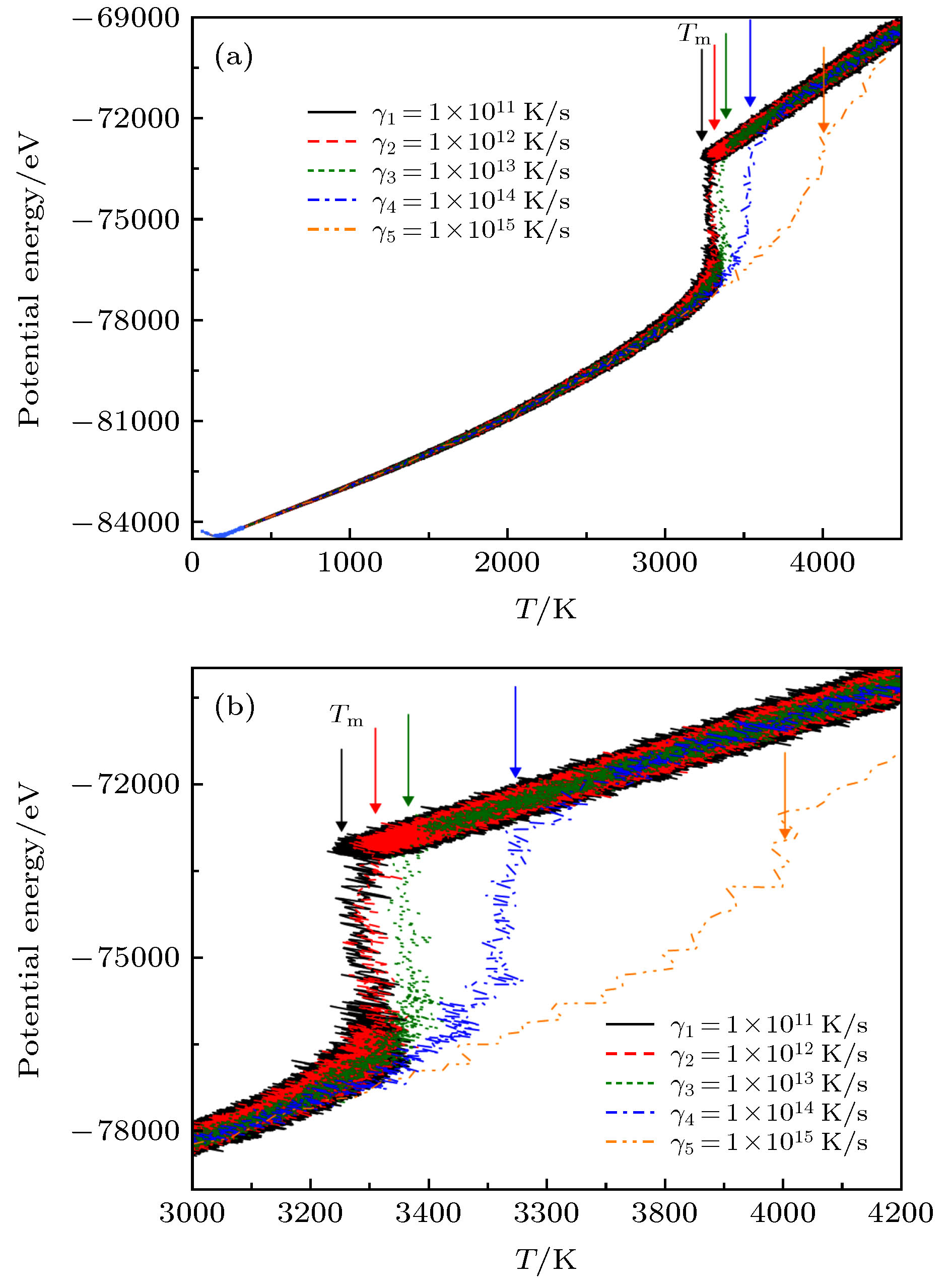 图 1 5种不同熔化速率下金属钒体系的势能变化与温度之间的演变关系, (b)是(a)的局部放大图
图 1 5种不同熔化速率下金属钒体系的势能变化与温度之间的演变关系, (b)是(a)的局部放大图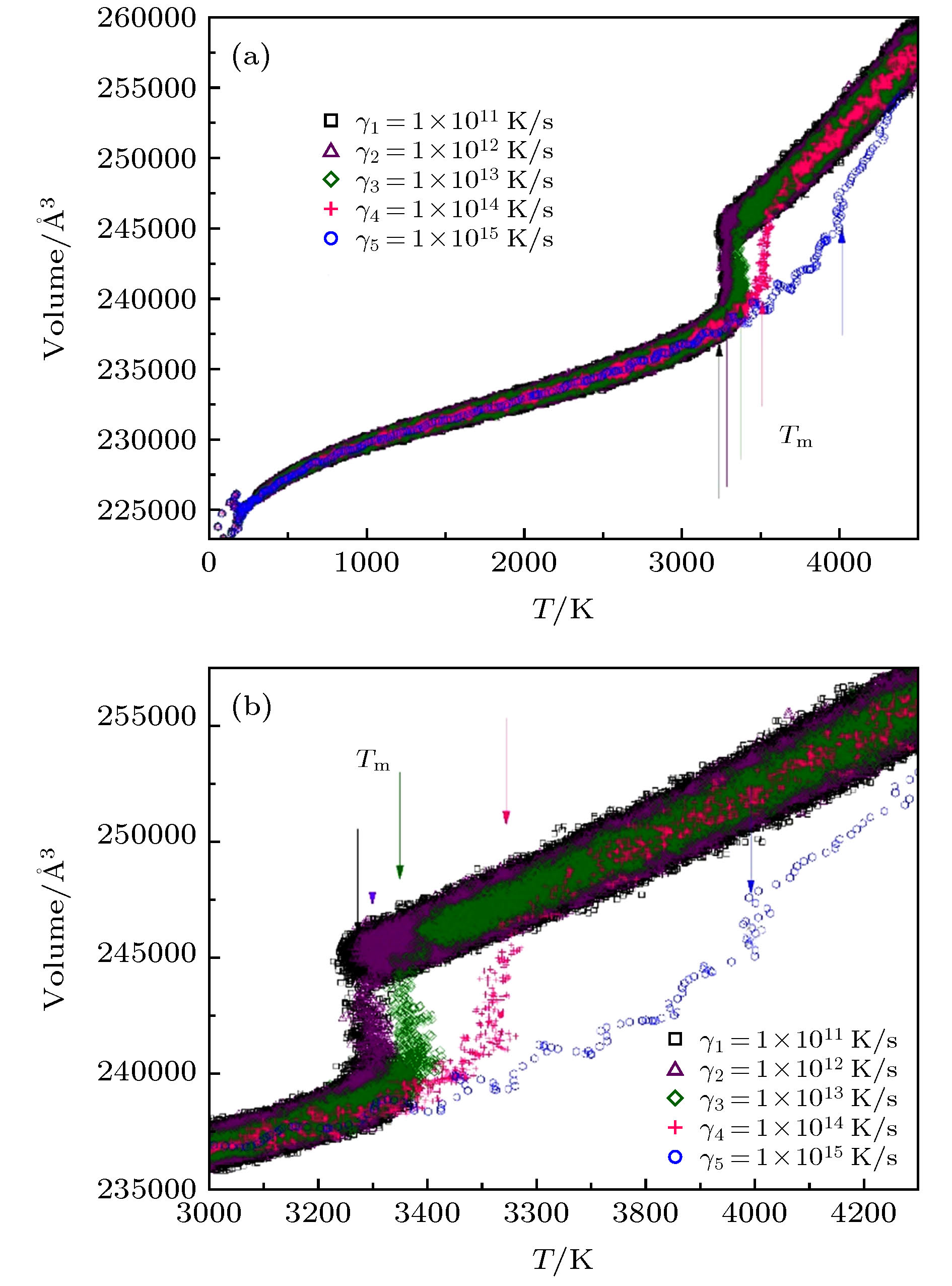 图 2 5种不同熔化速率下金属钒体系的体积变化与温度之间的演变关系, (b)是(a)的局部放大图
图 2 5种不同熔化速率下金属钒体系的体积变化与温度之间的演变关系, (b)是(a)的局部放大图 图 3 不同熔化速率下体系压强随温度的演变关系
图 3 不同熔化速率下体系压强随温度的演变关系










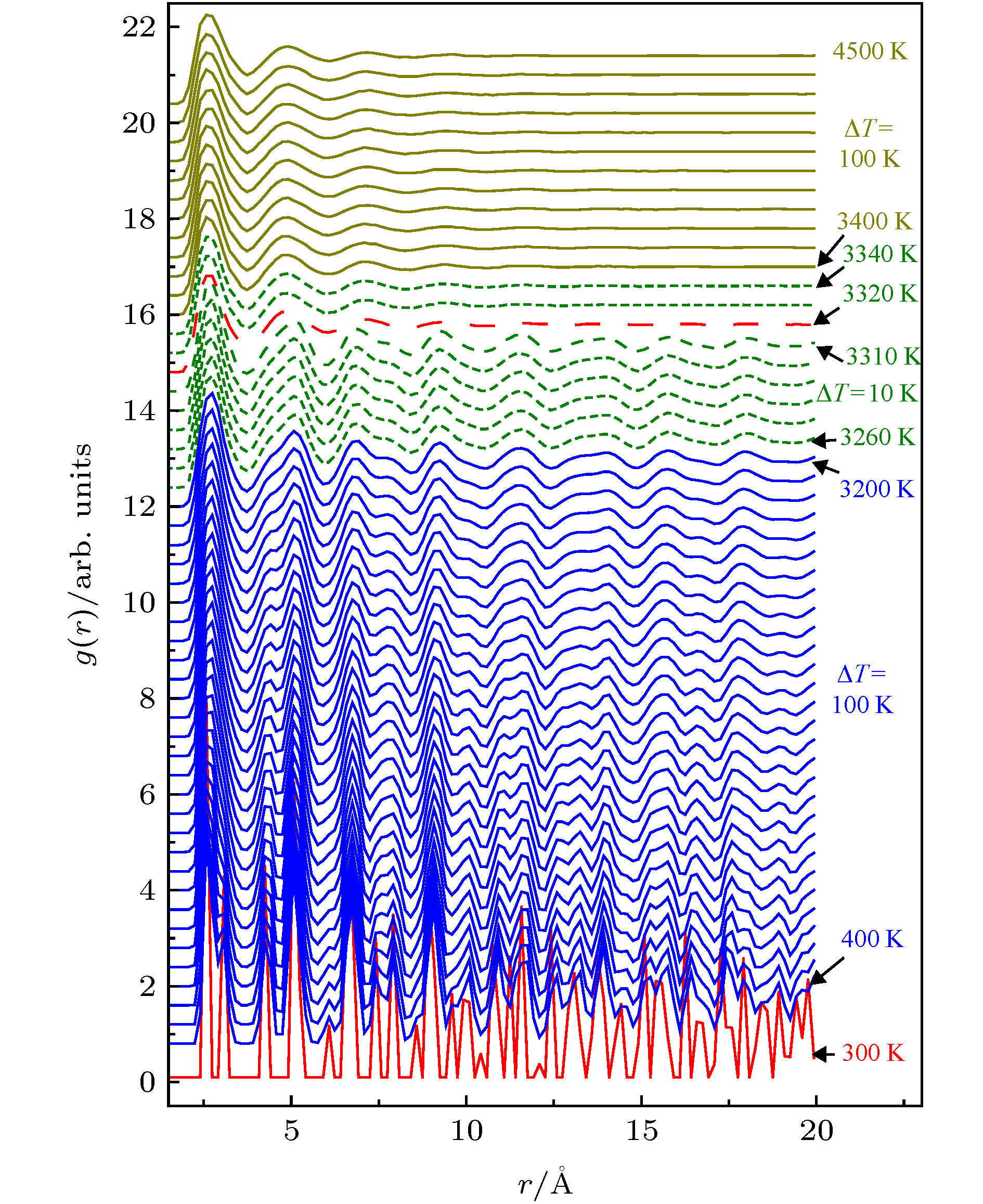 图 4 熔化速率为γ2 = 1 × 1012 K/s时RDF在不同温度区间的演变关系
图 4 熔化速率为γ2 = 1 × 1012 K/s时RDF在不同温度区间的演变关系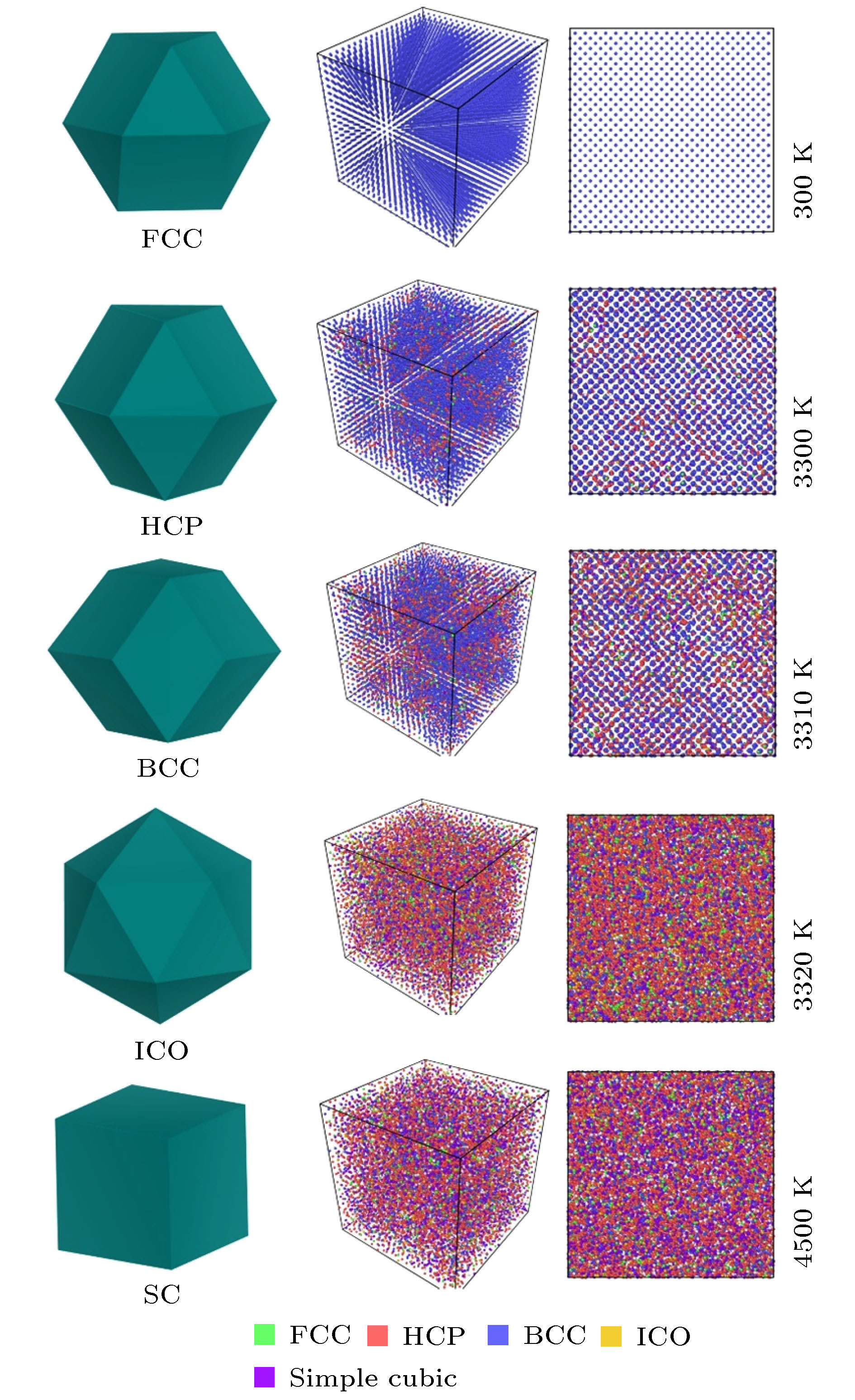 图 5 晶体钒熔化过程不同类型团簇的演变示意图(γ2 = 1 × 1012 K/s)
图 5 晶体钒熔化过程不同类型团簇的演变示意图(γ2 = 1 × 1012 K/s)




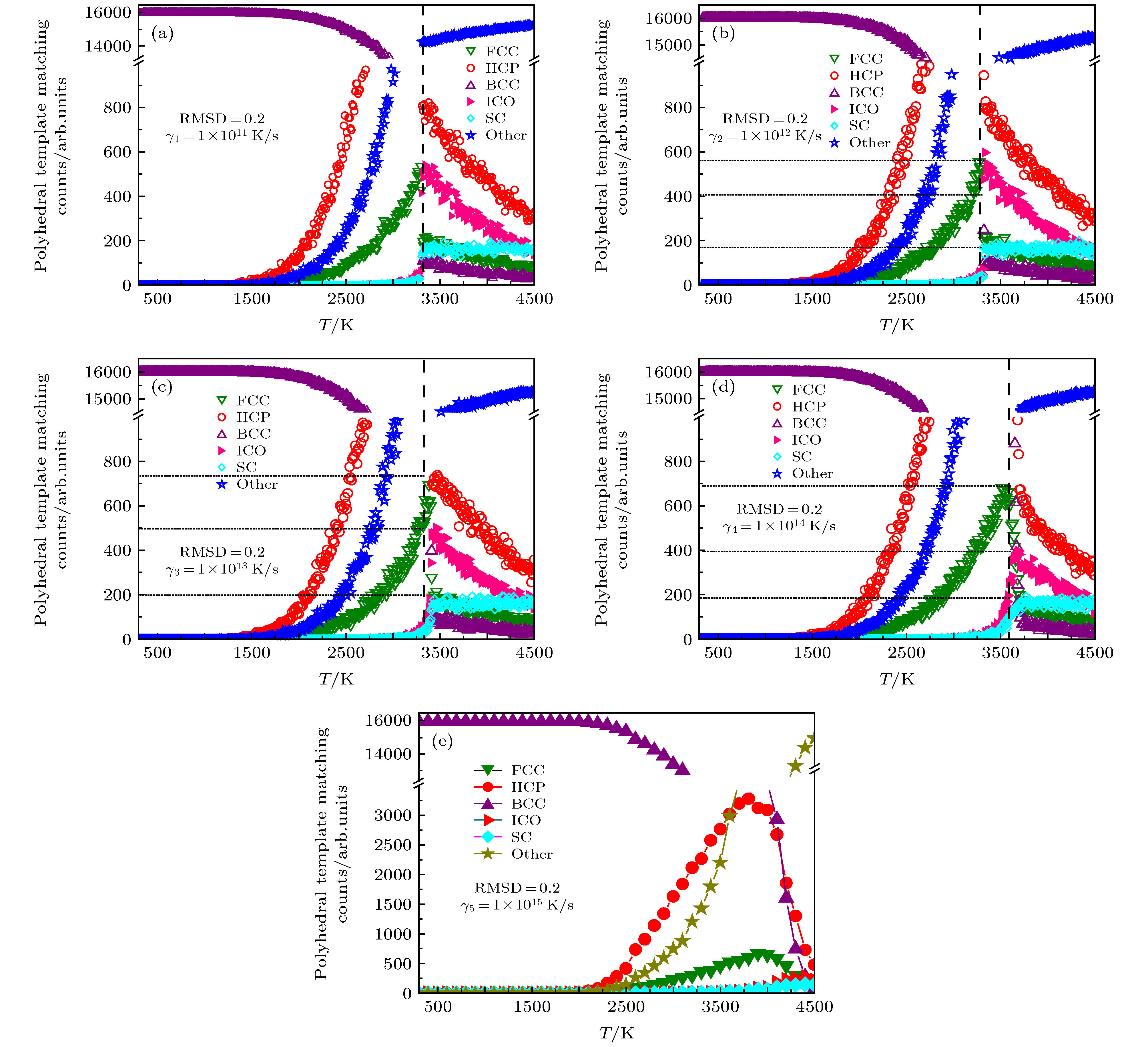 图 6 利用多面体模板匹配法分析得到的5种不同熔化速率下FCC, HCP, BCC, ICO以及SC类型团簇原子随温度的演变关系
图 6 利用多面体模板匹配法分析得到的5种不同熔化速率下FCC, HCP, BCC, ICO以及SC类型团簇原子随温度的演变关系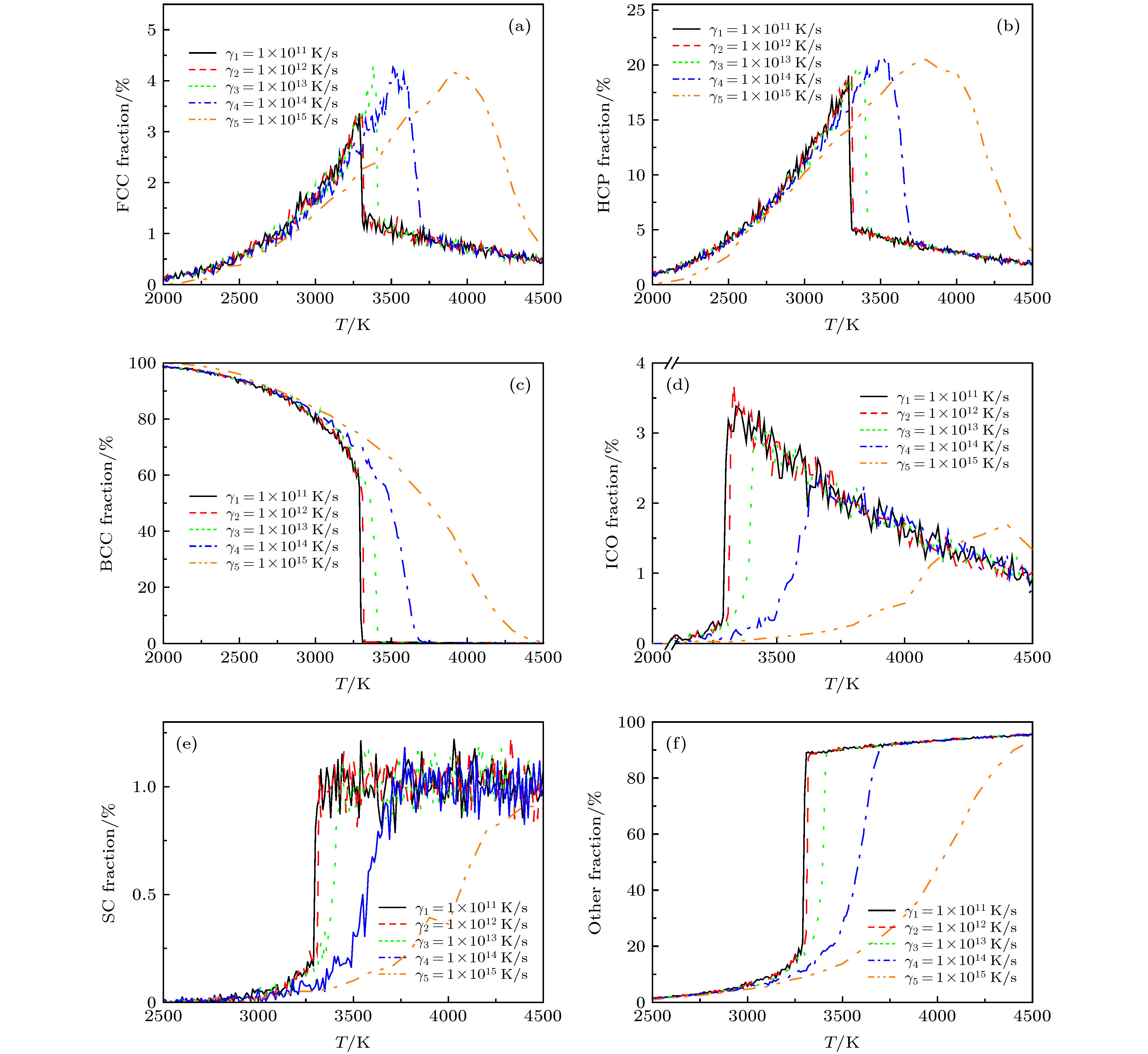 图 7 利用多面体模板匹配法分析得到的5种不同熔化速率下各种类型团簇原子分布随温度的演变关系 (a) FCC; (b) HCP; (c) BCC; (d) ICO; (e) SC; (f)其他类型团簇
图 7 利用多面体模板匹配法分析得到的5种不同熔化速率下各种类型团簇原子分布随温度的演变关系 (a) FCC; (b) HCP; (c) BCC; (d) ICO; (e) SC; (f)其他类型团簇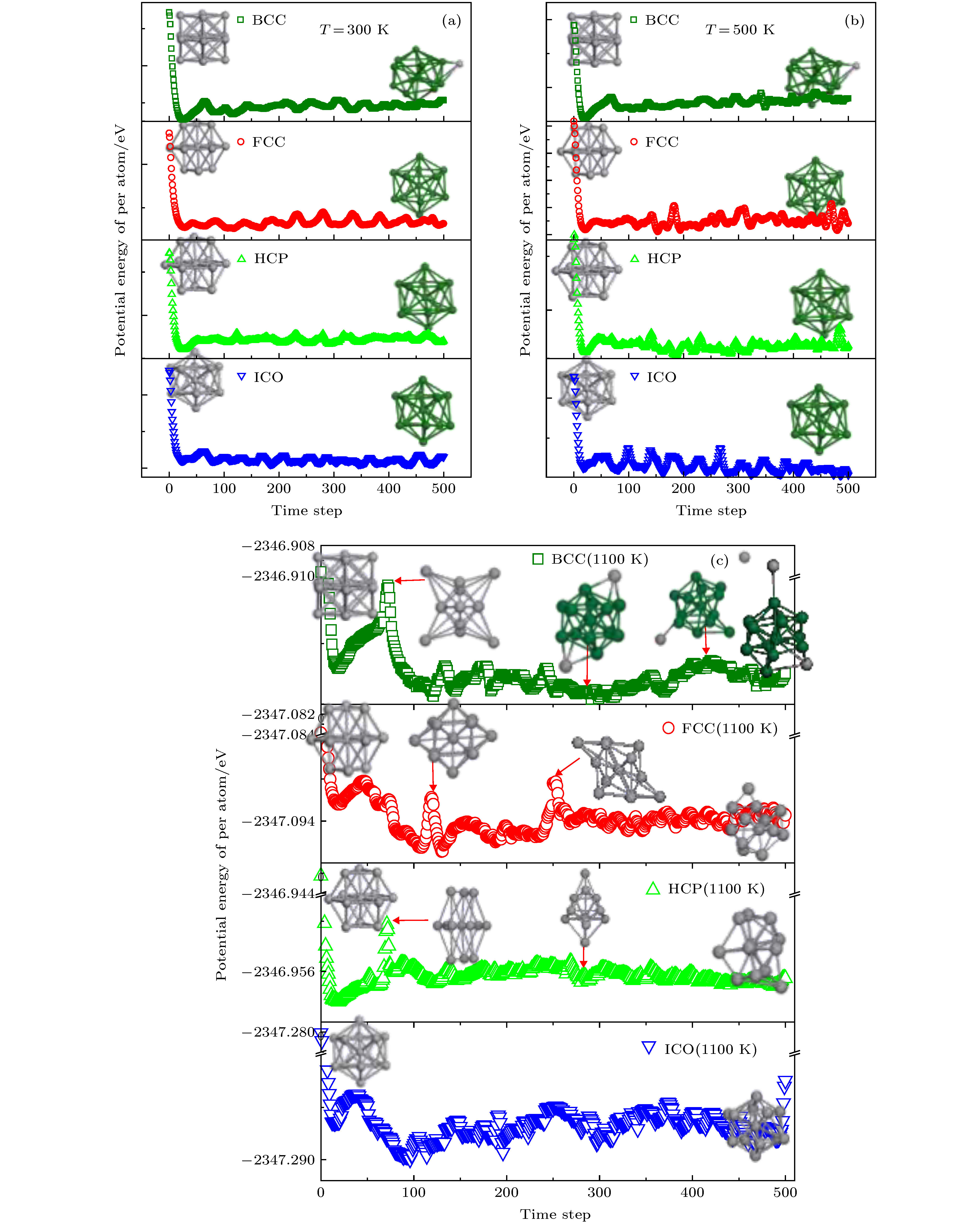 图 8 FCC, HCP, BCC以及ICO 团簇平均每原子势能随模拟步长的演变趋势 (a) 300 K; (b) 500 K; (c) 1100 K
图 8 FCC, HCP, BCC以及ICO 团簇平均每原子势能随模拟步长的演变趋势 (a) 300 K; (b) 500 K; (c) 1100 K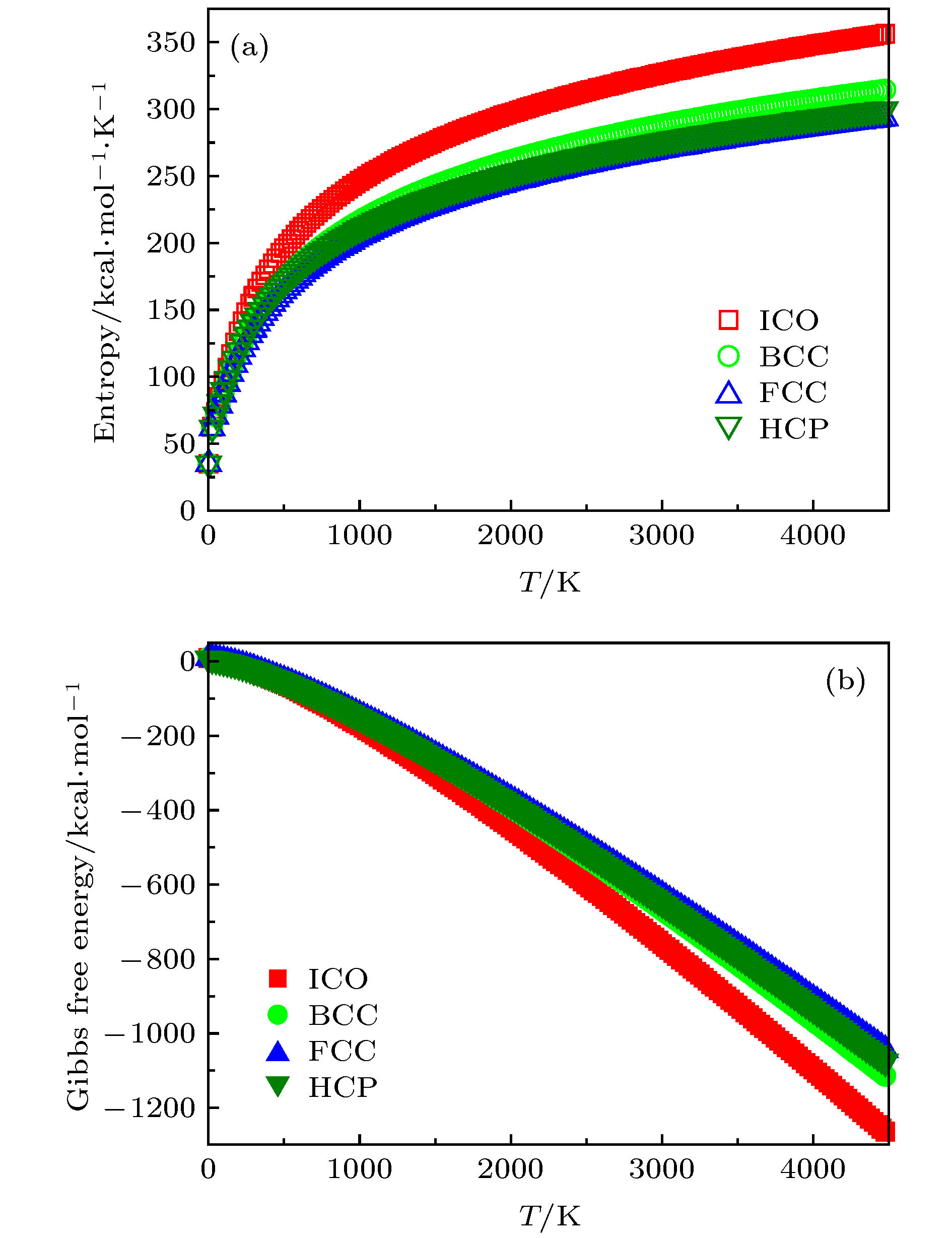 图 9 FCC, HCP, BCC及ICO 原子团簇的熵与吉布斯自由能随温度的演变关系 (a) 熵; (b) 吉布斯自由能
图 9 FCC, HCP, BCC及ICO 原子团簇的熵与吉布斯自由能随温度的演变关系 (a) 熵; (b) 吉布斯自由能