全文HTML
--> --> -->无膜细胞器中通常含有大量的内禀无序蛋白质或者固有无序区域(intrinsically disordered proteins or regions, IDPs/IDRs)[4,12,16-25], 因此目前对蛋白质相分离的研究大多数主要集中在内禀无序蛋白及其相分离的过程[26,27]. 通过简单地突变/修饰内禀无序蛋白质或者无序多肽中的氨基酸序列, 就可以完全打乱蛋白质的热响应特性[28]. 然而, 蛋白质序列的复杂性和构象的多样性, 给内禀无序蛋白质的计算和实验研究带来了重大挑战. 虽然内禀无序蛋白质有很多种类, 但是仍可以将其归类为低复杂度序列 (low complexity sequences, LCS) [29]. 这种序列仅由二十种天然氨基酸中的小部分构成. 对这样的序列构成的蛋白质而言, 其能量面形貌通常有很多个极小点并且各点的势垒很低[30,31]. 此外, 这一类分子内部相互作用的简并表现为分子间相互作用的多价性而导致自组装. 由于序列的低复杂度、单分子水平上状态的简并性以及聚集和/或相分离的倾向性, 使得揭示这些序列对应的分子的详细结构和机理的实验研究变得十分困难[32-35]. 理论研究和计算机模拟方法能够提供理论预测和显示分子细节, 这不但有助于解释实验现象[13,36], 同时可以用来补充和指导蛋白质的实验研究而展现出其特有的优势.
关于蛋白质分子的“液-液相分离”的多尺度理论模型和多分辨率计算方法很多[32,37,38] (图1). 由于蛋白质分子序列和构象的多样性和复杂性, 给蛋白质分子的理论研究、计算机模拟和实验研究带来了巨大的挑战[39,40]. 本文将对蛋白质分子“液-液相分离”的理论基础和计算机模拟方法进行简要的综述, 目的是为进一步研究蛋白质等生物大分子的“液-液相分离”的物理化学机制和过程提供理论和方法借鉴.
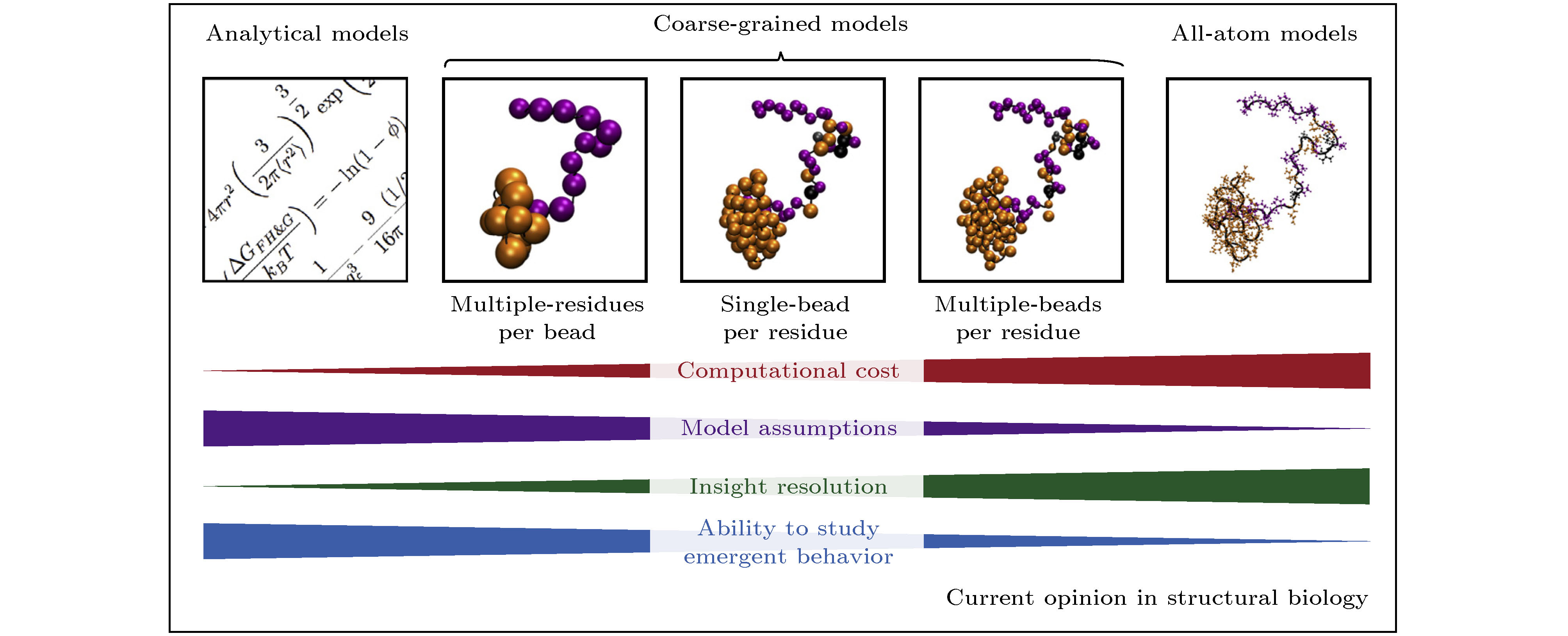 图 1 研究蛋白质“液-液相分离”的计算方法: 从左至右分别是解析模型、粗粒化模型(包括多个残基单个粒子、单个残基单个粒子、单个残基多个粒子的模型)和全原子模型, 分辨率的提升对计算资源的耗费程度急剧增高, 而模型假设数的减少会减弱研究涌现行为的能力[35]
图 1 研究蛋白质“液-液相分离”的计算方法: 从左至右分别是解析模型、粗粒化模型(包括多个残基单个粒子、单个残基单个粒子、单个残基多个粒子的模型)和全原子模型, 分辨率的提升对计算资源的耗费程度急剧增高, 而模型假设数的减少会减弱研究涌现行为的能力[35]Figure1. Computational approaches utilized to study protein liquid-liquid phase separation (LLPS). From left to right are the analytical model, the coarse-grained model (multiple-residues per bead, single-bead per residue and multiple-beads per residue coarse-grained models), and all-atom model. High-resolution descriptions increase the computational cost of the simulations. All-atom model is often impractical for the study of emergent behavior of LLPS[35].
2.1.理论研究
32.1.1.Flory-Huggins理论
从热力学角度看, 相分离的发生降低了体系整体的自由能[11]. 体系的自由能包括焓与熵变化的贡献, 其中焓变包含蛋白质分子与溶剂之间、蛋白质分子之间以及溶剂分子之间势能的变化, 而熵变衡量了体系自由度的变化. Flory[41]和Huggins[42]在20世纪40年代提出了高分子溶液的似晶格模型, 用高分子的体积分数





3
2.1.2.其他统计热力学理论
在Flory和Huggins的焓变公式推导过程中只考虑了近程的相互作用, Overbeek和Voorn 在他们的工作基础上考虑了长程静电相互作用, 应用于带Z个正电和Z个负电而整体不带电的聚电解质高分子溶液. 他们假设这些电荷随机的分布于溶液中, 与盐溶液类似可以用Debye-Hückel理论假设计算体系中的电荷间相互作用的能量. 虽然他们的平均场理论模型在高分子聚合物及其相分离领域提供了十分简洁的形式, 但是该种模型对内禀无序蛋白内部电荷排列及关联性没有成功的解决. 随后, 随机相近似(random phase approximation, RPA)方法被应用于的聚电解质高分子溶液静电作用的计算[45], 该方法可用于任意电荷分布样式的聚电解质高分子溶液体系. 虽然RPA理论模型可以给任何形式电荷排列的无序多肽的相分离研究提供了基本的理论框架, 但是在涨落很大的情况下, RPA模型在IDPs构象特性和相分离行为的预测可能会失败. Lin等[21,45]将该方法应用于蛋白质分子相分离体系的计算, 解释了同种电荷在序列上集中分布对于相分离的驱动作用, 以及离子强度对于静电作用为主的相分离体系的影响. 场理论模拟(field theory simulation, FTS)方法是一种更细致处理聚电解质高分子溶液体系的方法[46,47], 通过对场理论中的共轭变量进行模拟采样, 来获得体系的相图. McCarty等[1]用离散的链模型研究了不同谷氨酸(E)和赖氨酸(K)二组分构成的多肽链, 用场理论模拟方法获得了它们的盐浓度-蛋白浓度相图, 并得到跟RPA理论相近的结果.2
2.2.计算机模拟方法
蛋白质分子“液-液相分离”的研究中用到最多的是粗粒化模拟方法, 全原子模型的计算机模拟方法偶尔也会用到. 这里的粗粒化模拟方法是指将多个原子简化为单个粒子来降低分辨率提高计算效率的计算机模拟方法[35]. 该方法是通过显著地减少粒子的数量的模型, 并为理论研究和全原子模拟之间的沟通提供了一个桥梁. 下面将重点综述近几年发展起来的粗粒化模型.3
2.2.1.非格点模型的粗粒化模拟方法
1) 多个残基简化为单个球的粗粒化模型根据蛋白质中氨基酸序列的特征, 可将一条多肽简化为一个粗粒化的粒子或将一个重复的短肽片段处理成一个粗粒化的粒子. 对于低复杂度序列组成的无序蛋白质, 其序列的性质有利于粗粒化建模, 对同聚蛋白质尤为方便[13,48,49]. 该种类型的蛋白质指的是由单种氨基酸组成的短肽或是短肽重复片段组成的蛋白质. 为了研究重复序列长度和序列关联性对相分离的影响, 可以将重复序列构成的一条多肽简化为一个粒子或将多个重复的片段简化为一个粒子. 例如, 将huntingtin蛋白可视为二嵌段聚合物而得到了广泛的粗粒化研究[50]. huntingtin蛋白的外显子包含一个多聚谷氨酰胺束, 可以看作是一个仅由多聚谷氨酰胺和脯氨酸构成的二嵌段共聚物[50]. Burke等[48]利用多个氨基酸残基简化为一个粒子的粗粒化模型探索了多聚谷氨酰胺束长度和构象倾向性对其自组装过程的影响. 研究表明, 其自组装的驱动力主要取决于多聚谷氨酰胺束的长度、蛋白质浓度和两个区块的相对疏水性, 而不是多聚谷氨酰胺束的构象偏好[48]. Condon等[51]研究了(elastin-like polypeptide, ELP)弹性蛋白, 通过用一个粒子表示1~2个重复单元, 这些模拟能够达到与实验研究相当的长度和时间尺度, 并可重现与实验测量的涌现行为一致的特征.
2) 单个残基简化为单个球的粗粒化模型
根据蛋白质中氨基酸序列的特征, 可以将单个氨基酸简化为包含疏水性和电荷性质特征的球, 也就是通常所说的残基化水平的粗粒化模型. 残基化水平的粗粒化模型可以用来理解驱动分子间相互作用和相分离行为序列的重要特征[36,52,53]. 单个氨基酸被简化为单个粒子, 通常用疏水性指数来参数化成对氨基酸之间的相互作用, 并且重复出了实验上观察到的构象. 例如, Dignon等[17,52]基于Kapcha和Rossky[54]氨基酸的疏水性指数和Ashbaugh-Hatch[55]势函数的形式 开发出一种将单个残基简化为单个球的粗粒化模型来研究蛋白质的相分离的计算方法. 在这种相互作用势函数形式中, 通过比较粗粒化模拟与实验测定得到的回转半径, 确定了成对氨基酸之间短程相互作用的最佳数值. Ghavami等[56]利用类似的方法研究了酵母核孔(nuclear pore complex, NPC)中苯丙氨酸-甘氨酸序列组成多肽的自组装结构. 通过实验测定的斯托克斯半径和疏水性指数来确定粗粒化模拟参数, 粗粒化模拟表明, 在NPC的中心区域重复序列形成圆饼状的结构. 序列突变研究发现, 这种组织结构依赖于带电残基的组分和排列. Borgia等[57]使用同样的方法研究了相同数目的谷氨酸和天冬氨酸以及赖氨酸和精氨酸的短肽. 模拟结果与核磁共振光谱数据吻合得很好, 表明相互作用纯粹是静电驱动的, 不同类型带电氨基酸之间的结构差异对于这个系统来说并不显著[58].
单个残基水平的粗粒化模型不仅可以提供相分离过程中蛋白质分子间何种相互作用驱动相分离和序列特征, 而且弥补了理论模型的局限性. 例如, 基于RPA理论[45]和粗粒化模拟研究表明, (EK)50序列中不同电荷排列模式导致相分离结果定性相似, 但定量不同的现象. 同样地, 通过使用一个氨基酸一个粒子的显式粗粒化模型, Song等[59]的研究表明, (EK)50序列的回转半径和链末端距离由于构象异质性而不相关, 这是均聚物模型通常缺少的特征. 综合起来, 单个残基水平的粗粒化模型提供了一个可以与解析理论进行比较的途径, 有助于识别系统的局限性, 同时也为模拟低复杂度序列提供了一个简单而强大的模拟框架.
3) 单个残基简化为多个球的粗粒化模型
单个残基简化为多个球的粗粒化模型是更精细的粗粒化模型. 这里每个残基可视为由多个粒子组成的. 该模型有两种类型: 可移植的模型和系统特定模型(在所有被研究的系统中永久化是固定的)[60]. 常见的可移植多个粒子组成单个残基的粗粒化模型有PRIME[61-65], PLUM[66-68], AWSEM[69]和MARTINI[70-73]模型. 这些模型被用于模拟同源蛋白的原纤化形成和重复序列短肽的聚集过程[62-64], 且可用来研究NPC蛋白的门控和运输机制和类蚕丝弹性蛋白热应答行为的分子机制[71]. 其中PLUM模型被用来研究类蚕丝弹性蛋白单体的转变温度, 与实验研究结果是一致的[66].
尽管上述的多粒子的粗粒化模型具有明显的普遍性, 可移植模型并不总是适合于研究低复杂度的蛋白质序列. 一个给定模型的相关性通常取决于模型最初参数化. 例如, 原始的PLUM模型可能导致一些IDPs的二级结构过度稳定, 而MARTINI模型, 可能不会重现出蛋白质的结构特征并且禁止了结构转变[70]. 由于可移植模型的局限性, 系统特定的粗粒化模型仅仅被频繁地用于研究低复杂度序列. 这些模型经常使用从全原子模拟收集的信息来参数化特定的研究对象. 从全原子模拟到粗粒化模型的建立有两种主要方法: 基于结构的方法和力匹配方法[74]. 在基于结构的方法中, 目标是从全原子模拟中再现特定的构象分布. 而在力匹配方法从全原子模拟映射到粗粒化模型中更简单的经验力场(已经被用来研究与疾病相关的同源蛋白的聚集). 由Hills和Voth[75]开创的力匹配方法, 该方法被用来研究了酵母菌核孔复合体中的由苯丙氨酸和甘氨酸组成的低复杂度序列(phenylalanine-glycine LCS within the yeast nuclear pore complex, FG-nups)的构象特性. 这种方法捕捉到了几个较长的FG-nups的两相无序态, 并且表明序列组分以及序列特异性对纤维蛋白核糖核酸酶的构象偏差有强烈的影响[76].
4) 长条形盒子动力学模拟(slab simulation)的粗粒化方法
该模拟方法将所有分子放在长条形的周期性盒子(三个方向边长: x = y

粗粒化模型对多肽早期聚集事件和聚集形态可以提供有价值的信息. 这些模型可以明确地捕获序列特异性, 这是解析模型中通常缺乏的特征. 然而粗粒化模型有其自身的局限性. 粗粒化模型的参量化往往基于全原子模拟的模型, 因此高度依赖于全原子模拟的准确性, 并且多体相互作用项可以通过对较小数量分子的模拟得到. 此外, 粗粒化模型的相关性依赖于这样的假设: 模型能够恰当地捕获到感兴趣的区域的特性. 对于能够在高阶自组装过程中经历涌现构象转变的低复杂度序列, 粗粒化模型在其捕获自联结的热力学和动力学的能力方面可能会从根本上受到限制. 综上所述, 虽然粗粒化模型提供了一条重要的途径来评估个别分子的序列依赖性偏差, 但在可以得出的结论的确定性方面, 应始终考虑到它们的局限性.
3
2.2.2.格点模型的粗粒化模拟
格点模型也可以被用来研究生物大分子的“液-液相分离”过程. 例如, Pappu课题组和Chan课题组分别开发了不同的格点模型用来研究无序多肽或者生物大分子的“液-液相分离”. 其中美国华盛顿大学的Pappu实验组专注于该方法的研究, 他们开发了LASSI (Lattice simulation of Sticker and Spacer Interactions, LASSI)具体实现了格点模型的聚集模拟[79]. 在格点模型模拟过程中, 一般将生物大分子的结构域或者功能模块整体作为单个粗粒化的粒子, 粒子只能处于离散立方网格的格点中, 不同粒子所在格点不能重合, 一般假设只有相邻格点粒子有相互作用并且粒子对的相互作用能参数化为一个确定值, 体系用蒙特卡洛方法演化采样. Pappu实验室用此模拟方法研究了核仁FIB1-NPM1-RNA三元体系中结构域相互作用模式影响形成分层液滴的机制, 其中用一个分枝的结构模拟NPM1分子五聚体的结构[13]. 他们也用此模拟方法做了(SH3)m + (PRM)n体系中无序连接链对相分离的影响的研究[18]. 模拟结果显示连接链的有效溶剂体积影响该体系聚集态的性质, 连接链有效溶剂体积大、主要为较刚性的伸展构象时体系倾向于聚集成凝胶, 相反的, 连接链以压缩状态为主时体系倾向于聚集成液滴.Das等[53]应用格点模型上的粗粒化模拟方法研究了(EK)25序列的不同电荷排布情况下的多肽序列的相分离特点. 在他们的研究中, 重点考察了正负电荷混合均匀和完全分离的两种典型的序列. 他们认为每一条多肽链以自回避的形式占据三维空间点. 将每一个氨基酸残基简化为单个粒子并只占有一个格点, 两个相邻的粒子之间通过共价键来链接, 此外所有的粒子静电作用为屏蔽的库伦静电相互作用. 通过大量的蒙特卡洛模拟研究发现, 可以发现两种不同的序列相分离的临界温度有明显的差异, 对于正负电荷分离形成大的电荷模块的序列来说, 这一类序列显示出明显的相分离特性. 他们在格点模型上的大规模蒙特卡洛模拟研究与RPA理论研究的结果是一致的.
3
2.2.3.全原子模型
虽然粗粒化模拟已被用于研究低复杂度序列的聚集和组装, 全原子模拟对于在单个多肽水平上提取详细的构象特征具有更高的价值. 鉴于其普遍的序列重复性和高聚集倾向性, 这些蛋白质的研究往往给实验带来了巨大的挑战性. 全原子模拟提供了一个方便的解决方案, 提供了无限稀释下的“无限分辨率”[80-87]. 对于易于聚集的同源蛋白质(例如, 聚谷氨酰胺、聚甘氨酸、聚丙烯酸)的全原子模拟已经对单体的构象偏差有了相当深入的了解. 单分子荧光共振实验补充了全原子模拟的研究. 例如, Warner等他们能够提取huntingtin外显子1单体的构象系综. 全原子模拟也可以与其它实验相结合技术, 如核磁共振波谱法和小角度X射线散射产生互补信息. 例如TDP-43的LCS结构域、hnRNPA2的LCS结构域和RNA聚合酶Ⅱ的碳末端等系统得到了全原子模拟和上述实验技术的研究.尽管全原子模拟提供了高分辨率的解析精度, 但它们并非没有局限性. 考虑到它们的计算成本, 研究大量分子的分子间结合和聚集仍然令人望而却步, 得出的结论的准确性依赖于力场的可靠性. 在低复杂度序列的研究中力场的局限性往往被放大. 特别是在全原子分子动力学的框架下, 构型状态的高度简并性使得构型抽样成为一个主要的挑战. 因此, 尽管全原子模拟已经为理解LCS序列提供了较高的精度, 但是依然要像粗粒化方法一样要谨慎地考虑全原子模拟方法的局限性.
3
2.2.4.各种模型的优缺点概述
上述不同的理论方法和计算模型有着各自的优势和用处, 也有着不足和缺陷, 在使用的时候不能全盘否定. 例如, 平均场理论十分简洁地给出了高分子聚合物及其相分离的理论表达式, 但是在针对具体体系和涉及电荷关联性较强的情况下, 理论预测可能显得不是太准确. RPA理论模型的优点在于能够给出电荷相互作用如何调控IDPs相分离过程的清晰的物理图像, 缺点在于针对具体体系常常预测能力显得不是十分充足. 格点模型的优势就是抓住了整个问题中的最核心要素, 简化了构象空间, 使得计算简单并且快速. 而非格点模型描述构象相对比较准确, 各种相互作用也可以考虑得更精细, 粗粒化模型的参量化往往是基于全原子模拟的模型, 因此高度依赖于全原子模拟的准确性. 全原子模型的优点在于计算精确, 能够给出具体的相互作用细节, 能够更为精细地描述结构及其特征, 但是由于原子数目的显著增加, 导致计算太耗费时间, 目前只能研究小体系或者相分离的早期聚集等行为特征. 总之, 在实际的研究过程中, 要根据所研究对象的需求和具体的特征, 来选择合适的方法恰当的描述系统的相互作用及其集体运动行为.生物大分子相分离过程的研究当前处于起步和发展阶段. 研究者可以将上述的各种方法巧妙地应用于生物系统的相分离中来. 也可以发展新的理论方法以及模拟手段研究生物体系的相分离. 最终为深入地理解蛋白质等生物大分子的序列、结构以及相分离产生的临界条件之间的基本关系提供恰当而快速的理论方法和模拟手段. 此外, 也可以对上述的研究方法进行混合来解决生物大分子的相分离问题. 相信在未来, 生物大分子涉及的相分离将会吸引越来越多的科学研究人员的关注, 并且应用上述的研究手段或者发展新的方法来解决相分离相关的问题.
