全文HTML
--> --> -->为了实现有效且持久的太阳能到氢气的转化, 光催化材料需要以下特征: 有效且较宽的光吸收范围, 快速的电荷分离以及出色的稳定性[5]. 窄带隙半导体(例如硅和III-IV族复合材料)可以有效地吸收光并分离电子-空穴对[6], 然而它们在氧化还原反应过程中会遭受表面腐蚀和钝化, 因而其寿命往往在数小时之内. TiO2, Fe2O3, BiVO4和 WO3等类型的氧化物半导体具有出色的化学稳定性, 成为光催化应用的热门材料, 但是它们的带隙较宽、电子-空穴对复合率较高. 光催化反应的效率可以通过多种方法提高, 比如: 改变催化材料的物质形态(纳米片、纳米颗粒、纳米花和纳米棒等)提高其比表面积, 通过半导体/金属肖特基结、半导体/半导体异质结等设计弯曲的能带结构和相应的内电场(例如ZnO-TiO2异质结[7]), 通过离子掺杂(例如Fe3+, Ru3+和V4+等掺杂ZnO等[8,9])调控光催化剂的载流子浓度并提高其迁移率.
近年来, 钙钛矿材料在可见光范围内表现出较高的光吸收率和能量转换效率(PCE为输出能量/输入能量)[10,11], 在光伏、光催化等可再生能源生产中显示出很好的应用前景. 钙钛矿材料在光催化方向的研究从SrTiO3材料开始, 但是随着研究的不断深入, Miyauchi等[12]发现SrTiO3的带隙较大(3.2 eV), 导致其只能在紫外光的波长范围内分解水, 进而影响了铁电材料的光催化效率. 在光催化的实际应用中, 人们往往希望得到在可见光范围内发生反应的材料, 因此更多的低带隙材料得到广泛的研究, 如BiFeO3 (2.16—2.7 eV)[13,14], Bi2FeCrO6 (1.4—2.1 eV)[15]和MAPbI3 (1.5 eV)等. 近几年来, 在钙钛矿铁电材料的光催化研究方面取得了一系列进展, 这大体上分为两类: 1)铁电光催化材料, 包括BiFeO3, BaTiO3, NaNbO3, PbZr1–xTixO3 (PZT), Bi2FeCrO6, Bi2MoO6和MAPbI3等; 2)铁电/半导体复合材料, 即通过钙钛矿铁电材料与传统光催化材料构建异质结, 利用铁电体的退极化场(EP)帮助分离光催化材料中光生电子-空穴对并增强光催化性能, 比如 BiFeO3/TiO2, BaTiO3/CdS等[4].
铁电材料由于具有铁电极化(P), 其EP可用于分离光生电子-空穴对, 在光伏和光催化方面具有独特的优势, 这为提高光催化的能量转换效率开辟了一条新的道路. 钙钛矿氧化物铁电体, 即ABO3及其衍生物, 已被应用在电容器、传感器、致动器、存储器、能量存储等方面. 近几年来, 铁电材料成为了光催化、力催化和热催化领域的研究热点. 由于优异的光催化材料对于光催化反应过程中电子-空穴对的分离与运输要求极高, 研究者往往通过外在条件, 例如: 外加电场, 构建电子传输层与空穴传输层等方法, 来增加电子-空穴对的分离与载流子的运输效率. 铁电材料因其EP的存在, 具备天然优势, 材料本身就可以达到增加电子-空穴对分离与运输效率的目的. 铁电极化及其EP在调控光催化半导体材料及其异质结的能带结构和帮助分离光生电子-空穴对等方面具有巨大的发展空间[16-20]. 当铁电材料参与异质结时, 其极化将在相邻的半导体中引起大量的自由载流子重新分布, 因此可以将半导体耗尽区的大小和宽度有效地朝着有利的方向调整[19]. 各种研究已经证明了铁电极化调制半导体器件性能的有效性[21]. 例如, 铁电极化及其EP可以提高光生电子-空穴对的分离效率, 从而提高太阳能电池及光催化材料的PCE, 铁电极化还能够调节BaTiO3, PbZr1–xTixO3 (PZT)和BiFeO3材料表面的电化学反应, 例如罗丹明B、乙烯、刚果红、甲基橙和甲基蓝的降解[22-24]. 当将铁电极化适当地引入到这样的系统中时, 有望显著提高材料的光电转换效率(IPCE)和PCE.
本文阐明了铁电极化及其EP增强光催化性能的物理和电化学机制, 描述了EP帮助分离光生电子-空穴对、分解水产氢、分解污染物或CO2的结构设计和实验过程, 介绍了BiFeO3, BaTiO3, Bi2FeCrO6等氧化物铁电材料和MAPbI3卤素钙钛矿材料在光催化性能研究方面的最新研究进展, 分析了钙钛矿铁电材料在光催化研究和应用方面的挑战和机遇, 讨论了钙钛矿铁电光催化材料的发展方向.
2.1.铁电材料及其特性
铁电材料具有外电场可以翻转的铁电极化[25], 其铁电性源于其晶格结构的不对称性, 晶体内部的正负电荷中心不重合导致其内部存在电偶极子, 也就是说铁电材料的晶体结构具有非中心对称性, 只有单晶和多晶材料具有铁电性, 非晶材料不具有铁电性. 晶体材料有32种不同的点群, 其中11种中心对称点群, 21种非中心对称点群. 在这21种群中, 20种群具有热释电性和压电性[26-28], 其中只有10种具有铁电性, 它们分别是: 三斜晶系1 (C 1)点群、单斜晶系m (Cs)点群和2 (C 2)点群、正交晶系mm 2 (C 2v)点群、三方晶系3 (C 3)点群和3m (C 3v)点群、四方晶系4 (C 4)点群和 图 1 (a) ABX3型钙钛矿铁电材料结构图; (b) P-E电滞回线; (c) 铁电光催化、热释电催化、压电催化机制及其应用
图 1 (a) ABX3型钙钛矿铁电材料结构图; (b) P-E电滞回线; (c) 铁电光催化、热释电催化、压电催化机制及其应用Figure1. (a) Structure diagram of ABX3 type perovskite ferroelectric material; (b) P-E hysteresis loop. (c) photocatalysis, piezocatalysis and pyrocatalysis of a ferroelectric semiconductor and their application
引进铁电畴的概念可以更好地理解铁电体内部极化的情况, 铁电畴是指在铁电晶体内部特定的微小区域内, 所有的电偶极子的取向均为统一取向, 并且该区域中电偶极子的取向不同于其他区域的电偶极子取向, 将这样的小区域称为铁电畴, 铁电畴的存在对于本文所涉及的光催化材料有着重要的影响. 铁电材料通常具有很多的铁电畴, 各个铁电畴也通常具有随机取向的P, 宏观上P通常为零. 因此, 当对材料施加一定方向的外电场E后, 使得材料内部原本混乱的铁电畴出现一定程度的取向, 导致材料沿外电场方向的两个表面富集相反的束缚电荷. 相反的束缚电荷产生退极化场EP, 其方向与外电场E方向相反. EP可以加速材料内部电子-空穴对的分离, 这对于铁电材料在光催化领域的发挥起到了至关重要的作用.
对ABO3型钙钛矿材料来说, A, B和O元素都会对光催化活性有一定的影响. 导带底通常是由O元素的2p轨道和B元素的nd轨道形成的反键轨道决定, 而价带顶则通常是由O元素的非成键2p轨道来决定. 其中B元素对光催化活性有着至关重要的影响; B元素d轨道和O元素2p轨道的相互作用决定着其费米面附近的电子结构. 例如对于Bi2FeCrO6薄膜来说, 通过控制Fe/Cr阳离子序参量及其在薄膜中的分布, 可在1.4—2.1 eV的范围内调控Bi2FeCrO6的带隙, 从而获得更强的光吸收. 对A位离子而言, 其可以间接影响光催化效率, 在晶格内可以起到电荷补偿的作用, 并且A位离子半径的大小影响着BO64–八面体的结构畸变, 较大或者较小半径的A位离子会引起整个晶格的膨胀或者收缩, 使B—O键长发生变化, 从而影响材料的带隙宽度[32].
2
2.2.铁电材料的半导体性质
光催化铁电材料的能带结构(包括带隙大小、直接/间接半导体等)、少数载流子寿命与迁移率、载流子平均扩散长度、内/外量子效率等均对半导体材料的光催化性能有重大的影响. 材料吸收光的能力也是影响光催化材料效率的重要因素. 材料的带隙大小主要影响吸收可见光的范围, 带隙越小, 吸收可见光的范围越大. 少数载流子是指在对应的p型或n型半导体中相对数量最少的载流子, 少数载流子有一定的生存时间, 电子-空穴对并不会直接复合, 其存在的时间有的长有的短, 因此少数载流子寿命就称为τm. 间接带隙半导体往往具有较长的τm, 因此能带的结构包括直接与间接带隙都会影响载流子的分离过程, 对材料的传输效率产生重要的影响. 少数载流子在半导体内部并不是有序的运动, 而是处于无序不规则运动状态, 因此用载流子扩散长度(Lm)来衡量载流子的运动能力. 载流子在半导体内部的传输是一个复杂的过程, 半导体的传输能力主要由少数载流子决定, 半导体材料传输能力的大小与τm和Lm相关联. 铁电材料的铁电极化P及其EP, 可以有效地帮助半导体材料中光生电子-空穴对分离和沿特定方向的运动. 量子效率是指材料内部载流子数目与入射太阳光能量所具有的光子数目的比例. 量子效率分为内量子效率与外量子效率, 内量子效率是指材料的电荷载流子数目与从外部入射到材料内部并被吸收的光子数目之比; 外量子效率是指材料内部载流子数目与入射太阳光能量所具有的光子数目的比例, 一般可以直接测试材料的外量子效率, 内量子效率需要通过测试材料的透射率与反射率来计算. 材料的入射光子/电流转换效率简称光电转换效率(IPCE), 是研究光伏或光催化材料性能的一个重要指标, 其计算公式如下:2
2.3.光催化基本原理
光催化是指利用自然界中最丰富的能量-光能, 通过催化反应, 转变为电能或者化学能, 来分解有机物或者产生化学能. 光催化作为一种化学反应, 属于氧化还原反应, 通过电子的得失得到电能以及化学能. 光催化过程中, 必须具备两个核心要素: 光能与催化剂, 光能承担着启动光催化反应以及维持化学反应进行的任务; 而催化剂承担降低反应活化能, 加快反应进行的任务. 如图2(a)所示, 光催化材料必须保证导带的底部低于H+/H2的氧化电位0 eV; 价带的顶部高于H2O/O2的还原电位1.23 eV. 热力学上驱动该反应所需要的最小光子能量为1.23 eV, 根据Eph(eV) = 1240/λ(nm), 该光子的波长是1008 nm. 波长为λ的光源既可以是自然光, 也可以是人工光源, 为了使半导体的价带电子发生跃迁, 光子能量需要大于等于半导体催化剂的带隙(Eg), 即Eg ≥ 1240/λ(nm). 当激发能量足够时, 电子发生跃迁, 在半导体材料内部与表面产生光生电子-空穴对. H+/H2和O2/H2O的氧化还原电位可能夹在价带的顶部和导带的底部之间[33], 因此需要比光催化剂的带隙大得多的光子能量. 以带隙为3.2 eV的TiO2半导体材料为例, 想要激发此催化剂, 则需要光源的波长小于或等于388 nm. 图 2 (a) 光催化分解水的基本原理; (b) 光催化产氢、析氧反应步骤[33]
图 2 (a) 光催化分解水的基本原理; (b) 光催化产氢、析氧反应步骤[33]Figure2. (a) Basic principle of photocatalytic water-splitting process; (b) photocatalytic reaction steps for hydrogen and oxygen production[33]
在实际光催化反应中大部分光生电子(e–)-空穴(h+)对都会重新复合, 产生热能, 其反应式为
在光催化分解水并生产H2的过程中, 光催化半导体材料称为工作电极, 为了提高电子-空穴对的分离效率, 通常会为催化剂配备参比电极和对电极. 首先, 需要定义理想状态下的标准氢电极(SHE)作为电势参照的参比电极. 将镀有海绵状铂黑的铂片插入到H+浓度是1.0 mol/L的酸性溶液, 再持续输入压强为100 kPa的高纯H2使铂黑吸附H2至饱和, 这时铂片就可以等同于SHE. 我们规定任何温度下标准状态的氢电极的电势为零, 其他电极的“氢标”电势就是其与标准氢电极SHE的电势差. 标准氢电极都是可逆氢电极(RHE), 而RHE不一定会是SHE. 另外, 在常用的三电极光催化反应系统中, Ag/AgCl通常代替标准氢电极SHE作为参比电极. Pt通常是对电极, 当向催化剂施加一定的正向偏压之后, 会迫使光生电子向Pt电极方向移动, 从而使电子-空穴对分离, 减少两者发生复合的机会, 使得光催化效率大大提升. 图2(b)解释了光催化产氢、析氧反应过程, 光催化材料受到光激发后, 电子与空穴分离, 电子参与还原反应产生氢气, 空穴参与氧化反应产生氧气[37-39].
目前, 提高光催化材料的催化活性和能量转换效率PCE, 主要从两点出发: 1)调控光催化材料的能带结构, 增加材料吸收光的波长范围、少数载流子的寿命τm和平均扩散长度Lm; 2)提高光生电子-空穴对的分离效率、外量子效率和PCE. 主要有以下六种方式:
1)离子掺杂改善半导体性能
通过离子掺杂, 尤其是过渡金属离子, 可以有效提高载流子的寿命, 降低电子空穴对的复合速率, 增加半导体内部电子-空穴对的分离程度. 以TiO2为例, Litter和Navio[40]以及Choi等[41]发现对TiO2进行Mo2+, Fe3+, Ru3+, Re5+, Os3+, Rh3+, V4+的掺杂, 可以明显提高TiO2的光催化效率. 但是, 对于金属离子的掺杂存在着一定的极限值, 高于此极限值后, 半导体材料的光催化性能会急剧下降.
2) 半导体/半导体异质结分离电子-空穴对
在半导体催化剂表面复合一层带隙较小的半导体材料, 可以有效地提高半导体内光生载流子的分离效率, 并且可以扩大光谱的吸收范围. 二者的复合必须满足能级匹配的条件, 所复合的半导体材料的带隙必须小于催化剂半导体本身的带隙, 且其导带能级必须高于催化剂半导体的导带能级. 表1列举和比较了各种铁电材料、异质结和肖特基结的光催化性能[14,18,42-59].
| 材料及结构 (铁电材料为粗体) | 铁电 | 带隙/eV | 激励源 | 催化降解物 | 催化活性 | 污染性 | 稳定性(性能/时间) | 文献 |
| BiFeO3纳米粉体 | 强 | 2.18 | 紫外可见光 | 甲基橙 | 8 h降解90% | 中 | — | [14] |
| FTO玻璃/BiVO4/BiFeO3/CuInS2 | 强 | 2.1—2.7 | 可见光 | 对硝基苯酚 | Kobs = 0.02 min–1 | 中 | 相对稳定/5次循环 | [56] |
| NaNbO3纳米棒 | 强 | 3.3 | 光+超声振动 | 甲基蓝 | — | 弱 | 98%/3次循环 | [42] |
| BaTiO3@Ag纳米颗粒 | 强 | 3.2 | 光 | 罗丹明B | Kobs = 0.087 min–1 | 弱 | [43] | |
| BaTiO3/MoO3 | 强 | 3.2 | 紫外-可见光 | 罗丹明B | 60 min降解86% | 弱 | 95%/5次循环 | [44] |
| BaTiO3/Ag2O纳米棒 | 强 | 3.2 | 紫外光+ 超声振动 | 罗丹明B (c = 15 mg·L–1 ) | Kobs = 0.031 min–1 | 弱 | 50%/5次循环 | [18] |
| BaTiO3@非晶BaTiO3–x | 强 | 3.2 | 可见光 | 甲基蓝 | 5 h降解62.4% | 弱 | 97%/5次循环 | [45] |
| PbTiO3/TiO2纳米片 | 强 | 3.6 | 氙灯可见光 | 甲基蓝 | Kobs = 0.057 min–1 132.6 μmol·h–1 ·g–1 产H2 | 强 | — | [46] |
| KNbO3/g-C3N4 | 强 | 3.28 | 氙灯可见光 | — | 180 μmol·h–1 ·g–1 产H2 | 弱 | 95%/4次循环 | [47] |
| {001} Bi3TiNbO9纳米片 | — | 3.3 | 氙灯可见光 | — | 342.6 μmol·h–1 ·g–1 产H2 | 中 | — | [48] |
| KNbO3颗粒 | 强 | 3.28 | 光 | 罗丹明B | Kobs = 0.317 min–1 | 弱 | — | [49] |
| KNbO3纳米片 | 强 | 3.07 | 可见光+超声振动 | 罗丹明B | Kobs = 0.022 min–1 2 h降解92.6% | 弱 | — | [50] |
| FTO玻璃/ZnSnO3纳米线 | 弱 | 3.7 | 光+压力 | 甲基蓝 | Kobs = 0.007 min–1 | 弱 | 90%/1 h | [51] |
| FTO/ZnSnO3–x纳米线 | 弱 | 2.4—3.7 | 光、超声振动、 光和超声振动 | — | 3562, 3453, 3882 μmol·h–1 ·g–1 产H2 | 弱 | 在振动下相对稳定/7 h | [52] |
| FTO/Zn1–xSnO3 纳米线 | 弱 | 2.4—3.7 | 紫外光+振动 | 甲基蓝 | Kobs = 0.015 min–1 | 弱 | — | [53] |
| PZT@TiO2核壳结构 | 强 | 3.6 | 光+搅拌 | 罗丹明B | 80 min完全降解 | 强 | — | [54] |
| BiOI-BaTiO3纳米粒子 | 强 | 3.2 | 可见光 | 甲基橙 | 90 min降解95.4% | 弱 | — | [55] |
| ZnO纳米线 | 压电 | 3.37 | 光+摇摆 | 甲基蓝 | Kobs = 0.025 min–1 | 弱 | 99%/3次循环 | [57] |
| ZnO纳米片/TiO2纳米颗粒 | 压电 | 3.37 | 可见光 | 甲基橙 | Kobs = 0.038 min–1 | 弱 | 相对稳定/11 h | [58] |
| Ag-ZnO纳米线 | 压电 | 3.37 | 光+弯折 | 罗丹明B | Kobs = 0.052 min–1 | 弱 | 90%/8次循环 | [59] |
表1部分压电和铁电材料的光催化降解甲基橙染料或CO2的性能比较
Table1.Photocatalytic degradation of organic compounds using a variety of catalytic methods.
3) 通过半导体/金属的肖特基结分离电子-空穴对
当特定半导体材料与金属材料复合时, 由于两者的费米能级不同, 电子从能级较高的半导体材料向能级较低的金属材料传输时导致载流子重新分布, 直到二者的费米能级相同, 形成肖特基势垒, 相应的内电场可以有效地捕获电子, 阻止电子空穴的重新复合.
4) 通过外电场分离电子-空穴对
外电场对光催化反应有着重要的影响, 外电场帮助催化剂进行电子-空穴对的分离. 同时, 对于不同的催化剂材料, 存在一个特定的最佳电压值, 而且在不同的实验条件下, 此数值也不同. 表2列举和比较了各种铁电材料及其异质结的光电催化性能[15,50,60-73].
| 材料和结构 (铁电材料为粗体) | 铁电 | PCE/% | 带隙/eV | 电解液 | 光源 | 工作电极电势 | 光电流密度/ mA·cm–2 | 污染性 | 稳定性 (性能/时间) | 文献 |
| ITO/BiFeO3/Au | 强 | — | 2.16—2.7 | 0.1 mol/L KCl | AM1.5G | 0 V vs. Ag/AgCl | 0.05 | 弱 | — | [60] |
| SrTiO3/SrRuO3/(111)BiFeO3 | 强 | — | 2.16—2.7 | 0.5 mol/L Na2SO4 | AM1.5G | 0 V vs. Ag/AgCl | 0.08 | 弱 | 100%/700 s | [61] |
| SrTiO3/CaRuO3/(111) Bi2FeCrO6 | 强 | — | 1.9—2.1 | 1 mol/L Na2SO4 | AM1.5G | 0 V vs. Ag/AgCl | –2.02 | 弱 | — | [15] |
| SrTiO3/SrRuO3/Bi2FeCrO6/ NiO | 强 | — | 1.8— –2.7 | 1 mol/L Na2SO4 | AM1.5G | 1.2 V vs. RHE | 0.9 | 弱 | 95%/7 h | [62] |
| TiO2@PbTiO3 核壳结构 | 强 | — | 3.6 | — | 氙灯100 mW·cm–2 | — | 132 μmol·g–1 H2 | 强 | — | [63] |
| FTO/NaNbO3 | 强 | — | 3.37 | 0.5 mol/L Na2SO4 | AM1.5G | 1 V vs. Ag/AgCl | 0.51 | 弱 | — | [64] |
| ITO/KNbO3纳米片 | 强 | — | 2.86 | 0.5 mol/L Na2SO4 | AM1.5G | 0 V vs. Ag/AgCl | 0.82 | 弱 | — | [50] |
| (001) LiNbO3单晶 | 强 | — | 3.26 | mol/LK3PO4 | AM1.5G | 1.23 V vs. RHE | 0.15 | 弱 | — | [65] |
| FTO/TiO2@BaTiO3/Ag2O | 强 | — | 3.2 | 1 mol/LNaOH | AM1.5G | 0.8 V vs. Ag/AgCl | 1.8 | 弱 | 97%/1 h | [66] |
| FTO/TiO2@SrTiO3 (10 nm四方铁电相) | 弱 | — | 3.2 | 1 mol/LNaOH | AM1.5G | 1.23 V vs. RHE | 1.43 | 弱 | — | [67] |
| Glass/FTO/m-TiO2/CH3NH3PbI3/ Spiro-MeOTAD/Au/Ni | 弱 | 14.4 | 1.5 | — | AM1.5G | 1.0 V vs. SHE | 17.4 | 强 | 66%/1 h | [68] |
| FTO/PEDOT:PSS/CH3NH3PbI3/ PCBM/PEIE/Ag/FM | 弱 | 7.7 | 1.5 | — | AM1.5G | 1.2 V vs. RHE | 15.0 | 强 | 80%/1 h | [69] |
| ITO/NiO/CH3NH3PbI3/ PCBM/Ag/Ti/Pt | 弱 | 16.1 | 1.5 | 0.5 mol/L H2SO4 | AM1.5G | 1.2 V vs. RHE | 18 | 强 | 70%/12 h | [70] |
| CH3NH3PbI3 solar cells, a cell for H2O splitting | 弱 | 15.7 | 1.5 | — | AM1.5G | — | 10 | 强 | 75%/10 h | [71] |
| FTO/BiVO4/black-phosphorene/ NiOOH | 无 | — | 2.4—2.5 | 0.5 mol/L KH2PO4 K2HPO4 | AM1.5G | 1.23 V vs. RHE | 4.48 | 强 | 99%/60 h | [72] |
| FTO/H:TiO2 | 无 | 1.63 | 3.2 | 1 mol/LNaOH | AM1.5G | –0.6 V vs. Ag/AgCl | 1.97 | 弱 | 94%/28 h | [73] |
表2近年部分铁电材料光电催化分解水的研究进展(这里ITO, FTO, SrTiO3, Nb-SrTiO3和glass是薄膜基片, PCBM是[6,6]-苯基C61-丁酸甲酯, PEIE是乙氧基化聚乙烯亚胺; PEDOT:PSS是聚苯乙烯磺酸盐(3, 4-乙撑二氧噻吩))
Table2.Photoelectrochemical water splitting of ferroelectric materials in recent years, where ITO, FTO, SrTiO3, Nb-SrTiO3 and glass are substrate of films, PCBM is [6,6]-phenyl-C61-butyric acid methyl ester, PEIE is ethoxylated polyethylenimine, PEDOT:PSS is poly(3, 4-ethylenedioxythiophene) polystyrene sulfonate and FM is In0.51Bi0.325Sn0.165 as protective layer
5)优化反应溶液pH值
溶液pH值的影响原理比较复杂, 在不同的情况以及不同的材料中, pH值表现出了不同的影响, 具体的原理还需要进一步的探究.
6)调控溶液中的氧含量
氧对光催化反应过程的影响主要来自两方面: O2直接参与光催化反应; O2直接影响半导体电极的开路电位光电压响应, O2会影响光生电子向Pt对电极的运动, 催化剂表面的氧成分会大量吸收电子, 阻碍电子-空穴对的转移.
3.1.BiFeO3的光催化
近年来BiFeO3等铁电材料的光电催化效率如表2所示[60-67]. 2008年, 南京大学的Cho等[14]发现BiFeO3纳米粉体的带隙可以降低到2.18 eV, 带隙能量能够很好地与光催化分解水的氧化还原反应匹配, 电子-空穴对在退极化场的作用下容易分离, 有利于光催化效率的提升, 该BiFeO3粉体具有优异的光催化性能, 开启了铁电半导体材料光催化研究的热潮. 为了方便比较, 对于基片/底电极/光催化剂/电解液结构, 指定钙钛矿铁电材料作为阳极使用的为光阳极, 作为阴极使用的为光阴极, 外电场及其电压、铁电极化方向指向电解液为正向电场/正电压(+E/+V )、正向极化(+P), 外电场及其电压、铁电极化方向指向底电极为反向电场/负电压(–E/–V )、反向极化(–P).对于BiFeO3材料而言, 其既可以独自作为光电极使用, 也可以作为辅助性材料与C3N4等经典光催化材料复合, 也可以在BiFeO3材料基础上进行修饰改性(例如, 与Au复合等)[74-76]. 2014年, Cao等[77]在ITO导电玻璃上利用旋涂法制备了高质量的BiFeO3多晶薄膜, ITO薄膜的厚度为100 nm, BiFeO3薄膜厚度为300 nm. 二者的势垒高度为1.24 eV, 为典型的肖特基结. 在进行光催化测试之前, BiFeO3薄膜电极被置于碳酸丙烯脂溶液中进行电化学预处理, 施加10 s的 ± 8 V偏压对BiFeO3薄膜进行不同方向的铁电极化预处理, 控制电荷转移的方向并改变能带结构, 以便提高BiFeO3薄膜的光催化效率. 稳态荧光光谱法是分析光致电荷转移动力学的一种简便方法, 分别对薄膜进行+8 V与–8 V极化之后, 可以发现不同极化方向的BiFeO3薄膜的荧光强度发生了显著的变化. 如图3(a)所示, BiFeO3薄膜经过+8 V外电压极化后, 其铁电极化P方向指向电解液, 其退极化场–EP (即Ebi)指向底电极, 能带在左边缘处向上弯曲、右边缘处向下弯曲, 导致耗尽层宽度增加, 加大电子-空穴对的分离效率, 同时电子与空穴更容易的导出, 它的荧光强度显著大于初始薄膜的荧光强度. 如图3(b)所示, BiFeO3薄膜经过–8 V电压极化后, 其结果与+8 V极化相反, 空穴难以从电解液传输到BiFeO3, 电子难以从BiFeO3传输到电解液. 如图3(c)所示, 经过+8 V极化的BiFeO3薄膜的外量子产生效率是–8 V极化后薄膜外量子效应的10倍, 这说明了BiFeO3的铁电极化方向能够明显的影响材料的外量子效率, 可以通过铁电极化翻转调节外量子效率和光电流密度. 如图3(d)所示, BiFeO3薄膜的铁电极化状态不同, 光催化的电流密度也随之变化. 首先, BiFeO3工作电极为0 V (vs. Ag/AgCl)时, +8 V和–8 V极化后的BiFeO3显示的光电流都为负, 这是因为BiFeO3/ITO界面上存在1.24 eV肖特基势垒, 这会阻碍电子并促进空穴向ITO电极的转移, 从而无论BiFeO3薄膜如何极化, 都只能获得光阴极电流. 其次, BiFeO3薄膜的极化状态可以大幅度调节光电流. 当BiFeO3工作电极为0 V时, –8 V极化后的BiFeO3薄膜的光电流密度为0, +8 V极化后的BiFeO3的光电流密度为–10 μA/cm2.
 图 3 (a), (b) 对BiFeO3薄膜进行+8 V和–8 V 极化后的能带结构示意图; (c) 极化之前和+8 V和–8 V极化之后BiFeO3电极测量的外量子效率; (d) 不同铁电极化状态的BiFeO3工作电极的光电流与电势曲线[24]
图 3 (a), (b) 对BiFeO3薄膜进行+8 V和–8 V 极化后的能带结构示意图; (c) 极化之前和+8 V和–8 V极化之后BiFeO3电极测量的外量子效率; (d) 不同铁电极化状态的BiFeO3工作电极的光电流与电势曲线[24]Figure3. Energy band structure diagram of the BiFeO3 thin film after (a) +8 V and (b) –8 V poling; (c) external quantum yield spectra of BiFeO3 film before poling and after +8 V and –8 V poling; (d) photocurrent–potential characteristics of the photoelectrodes with different polarization states[24]
2018年Song等[61]研究发现: (111)pc BiFeO3单晶外延薄膜具有最大的宏观铁电极化及其EP, 能够显著地提高薄膜样品的光电催化性能. 为了方便比较, 根据ABO3伪立方结构表示BiFeO3的晶面和晶向, 并标记pc. 通过脉冲激光沉积(PLD)薄膜方法在SrTiO3基片生长La0.67Sr0.33MnO3 或SrRuO3底电极. 然后, 分别生长(001)pc, (110)pc, (111)pc三种晶面的外延BiFeO3薄膜. 由于菱面体相BiFeO3的铁电极化方向在ABO3晶胞的体对角线方向, 即沿伪立方体的

Song等[61]构建了Mott-Schottky曲线并计算了BiFeO3薄膜光阳极的带边位置, 阐明了不同晶体取向的BiFeO3薄膜的光催化性能差异. 首先, 根据图4(a)的Mott-Schottky曲线的正斜率可以判断BiFeO3薄膜为n型半导体, 此行为是由氧空位引起的. 另外, Vfb+kT/e, 可通过Mott-Schottky方程中获得
 图 4 (a) 三种50 nm厚外延BiFeO3薄膜光阳极的Mott-Schottky曲线, 相对于Ag/AgCl参比电极的平带电势由曲线斜率与横轴交点决定; (b) 400—800 nm入射光波长范围内的BiFeO3薄膜的吸光度; (c) 三种外延BiFeO3薄膜光阳极的能带位置; (d) BiFeO3薄膜光阳极的电化学阻抗谱[77]
图 4 (a) 三种50 nm厚外延BiFeO3薄膜光阳极的Mott-Schottky曲线, 相对于Ag/AgCl参比电极的平带电势由曲线斜率与横轴交点决定; (b) 400—800 nm入射光波长范围内的BiFeO3薄膜的吸光度; (c) 三种外延BiFeO3薄膜光阳极的能带位置; (d) BiFeO3薄膜光阳极的电化学阻抗谱[77]Figure4. (a) Mott-Schottky plots for the 50-nm-thick epitaxial BiFeO3 thin-film photoanodes with different crystallographic orientations, where the flat-band potentials are obtained from the intercepts of the extrapolated lines; (b) absorbance measurements for these three BiFeO3 thin films with incident light at 400?800 nm wavelength; (c) band positions for the epitaxial BiFeO3 thin-film photoanodes; (d) electrochemical impedance spectroscopy spectra of the BiFeO3 thin-film photoanodes[77]

图5(a)是BiFeO3外延薄膜作为光阳极分解水时的能带图. 当BiFeO3薄膜的铁电极化指向底电极, 在BiFeO3/电解液界面具有负束缚电荷, 能带向上弯曲, 在底电极/BiFeO3界面具有正束缚电荷, 能带向下弯曲. 因此, 电子能够传输到底电极并最终在Pt对电极附近电解液中通过光催化产生H2, 空穴可以传输到电解液并通过光催化产生O2. 当BiFeO3外延薄膜的铁电极化指向电解液时, 电子难以传输到底电极, 空穴难以传输到电解液. 图5(b)的BiFeO3电极的光电流-电势(vs. Ag/AgCl)曲线证实了能带图的理论模型, 在工作电极的电势为正的情况下, 当铁电极化指向底电极时, (111)pc BiFeO3薄膜的光电催化效率表现的最为突出, 光电流密度可达到0.08 mA/cm–2, 当铁电极化指向电解液时, 光电流很小. 图5(c)对比了电势为0情况下, (001)pc和(111)pc薄膜的光电催化效果, 可知铁电极化指向底电极(–P)时(111) BiFeO3薄膜具有最大的光电流密度. –P使(111)pc BiFeO3薄膜的光电流密度增大约5.3倍, 铁电极化翻转使光电流密度变化8000%, 光催化起始电位变化0.33 V. 此外, 当(111)pc BiFeO3薄膜的厚度从20 nm增加到50 nm时, 外延BiFeO3薄膜的光电流密度增加, 并且当厚度进一步从50 nm增加到100 nm时, BiFeO3薄膜的光电流密度减小. 光电流的变化可以用电荷转移模型来解释[87-89]: 随外延BiFeO3薄膜厚度从20 nm增加到50 nm, 光生载流子总数的增加引起光电流密度的增加; 当厚度从50 nm增加到100 nm, 由于载流子扩散长度小于薄膜厚度, 大量的光生电子-空穴对在薄膜内部复合, 光电流密度随薄膜厚度的增加而减小.
 图 5 光催化反应过程中BiFeO3薄膜作为光阳极的能带图 (a) 不同极化状态下, BiFeO3薄膜能带结构的改变; (b) (111)pc BiFeO3薄膜作为光阳极的光电流密度-电势曲线; (c) 在电势为0 V情况下(001)pc和(111)pc BiFeO3薄膜光阳极的光电流密度-时间曲线[61]
图 5 光催化反应过程中BiFeO3薄膜作为光阳极的能带图 (a) 不同极化状态下, BiFeO3薄膜能带结构的改变; (b) (111)pc BiFeO3薄膜作为光阳极的光电流密度-电势曲线; (c) 在电势为0 V情况下(001)pc和(111)pc BiFeO3薄膜光阳极的光电流密度-时间曲线[61]Figure5. Energy band diagrams for BiFeO3 photoanodes in PEC water splitting cells: (a) Changes in the band structure of BiFeO3 thin films under different polarization states; (b) linear sweep voltammetry of 50-nm-thick (111)pc BiFeO3 thin-film photoanodes in different polarization states; (c) photocurrent density versus time curves for (001)pc and (111)pc BiFeO3 thin-film photoanodes with different polarization states under zero bias (0 V vs. Ag/AgCl)[61]
2019年Huang等[89]利用BiFeO3材料涂覆在掺Sn的TiO2纳米棒(Sn:TiO2@BiFeO3)上, 实现了光催化电流密度的增强, 最大达到1.76 mA/cm2. 很多铁电材料具有高介电常数和较低的载流子迁移率, 因此利用铁电薄膜进行能带工程改性时铁电薄膜需要足够薄, 从而尽量避免铁电材料对电荷传输的影响. 首先利用水热法在FTO玻璃衬底上生长规则的Sn:TiO2纳米棒, 在450 ℃退火后, 利用溶胶-凝胶法在FTO玻璃/Sn:TiO2上涂覆一层BiFeO3薄膜, 制备过程及模拟形貌如下图6(a)所示. 通过Sn掺杂与BiFeO3包裹都能够显著提高TiO2纳米棒的光催化性能. 如图6(b)所示, 工作电极电势为1.23 V时, TiO2纳米棒的光电流密度为0.54 mA/cm2, Sn掺杂的TiO2纳米棒的光电流密度提高到0.83 mA/cm2, Sn:TiO2@BiFeO3的光催化电流密度提升到1.51 mA/cm2. 由于BiFeO3 (2.16—2.7 eV)的带隙比TiO2 (3.2 eV)小, 通过复合之后导致Sn:TiO2@BiFeO3结构的可见光吸收范围显著增强. 同时, 如果对该复合结构施加不同方向的外加电场, BiFeO3铁电层的退极化场EP可以调控Sn:TiO2@BiFeO3的光电催化效率. 如图6(c)所示, 外电压极化BiFeO3薄膜后其铁电极化指向电解液, 其退极化场指向Sn:TiO2, Sn:TiO2@BiFeO3界面处的能带向上弯曲, 其耗尽层变宽, 内建电场增加了电子-空穴分离效率, 光阳极的光催化电流密度由1.51 mA/cm2增加至1.76 mA/cm2. 如图6(d)所示, 当外加极化电压指向FTO基片时, Sn:TiO2@BiFeO3界面处的耗尽层变窄, 降低了电子-空穴分离效率, 光阳极的光催化电流密度由1.51 mA/cm2降低至1.02 mA/cm2.
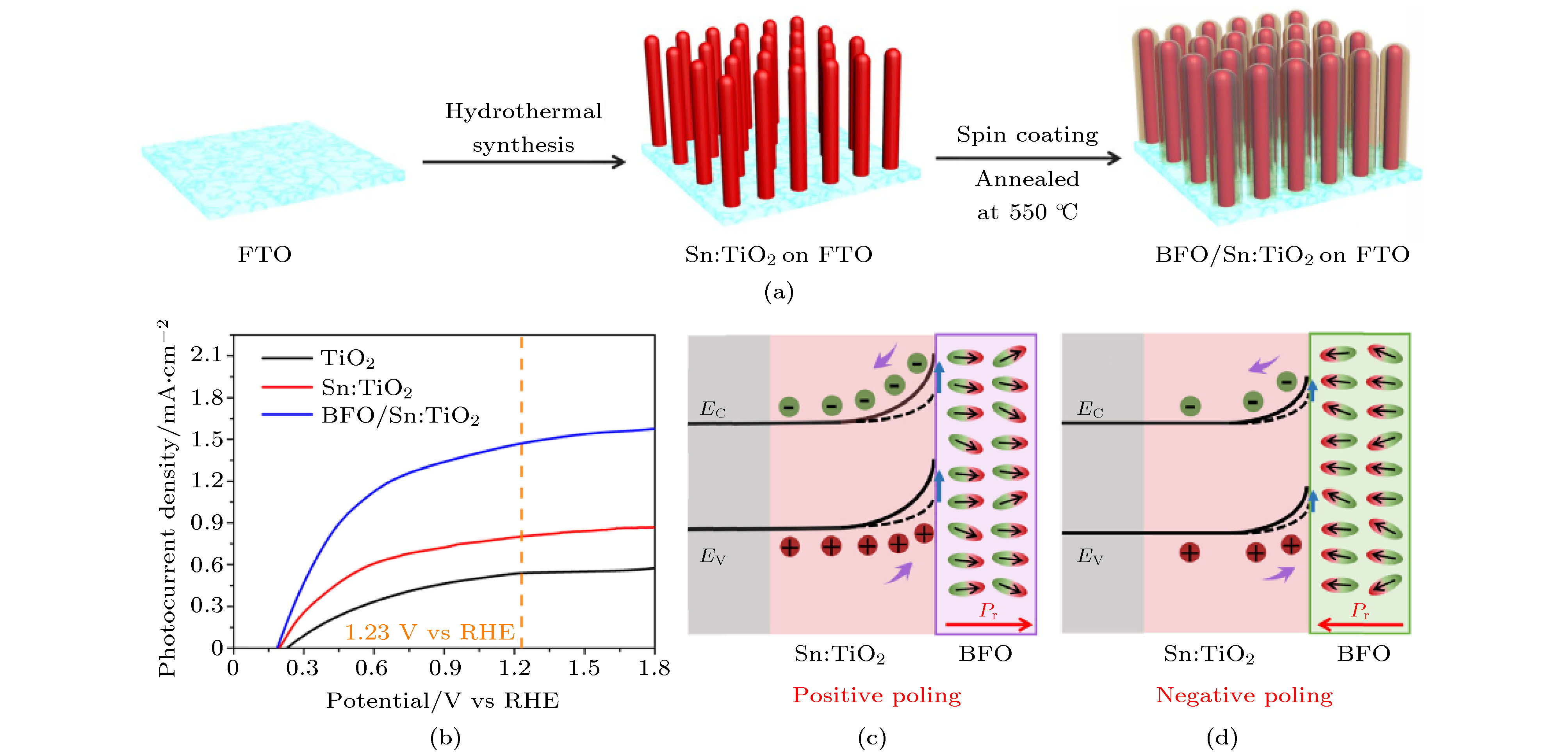 图 6 (a) BiFeO3@Sn:TiO2生长机制示意图; (b) TiO2, Sn:TiO2与Sn:TiO2@BiFeO3作为光阳极时的光电流密度; 铁电极化分别指向(c) 电解液和(d) Sn:TiO2时, Sn:TiO2@BiFeO3的能带示意图[89]
图 6 (a) BiFeO3@Sn:TiO2生长机制示意图; (b) TiO2, Sn:TiO2与Sn:TiO2@BiFeO3作为光阳极时的光电流密度; 铁电极化分别指向(c) 电解液和(d) Sn:TiO2时, Sn:TiO2@BiFeO3的能带示意图[89]Figure6. (a) Schematic representation for the growth mechanism of Sn:TiO2@BiFeO3 nano rods; (b) photocatalysis performance of TiO2, Sn:TiO2 and BiFeO3@Sn:TiO2 nano rods. Schematic electronic band diagram of (c) positive poling BiFeO3 and (d) negative poling BiFeO3[89]
作为n型半导体, BiVO4的带隙约为2.4 eV, 相对较窄的带隙可以吸收大部分可见光[90], 是目前光催化领域中最有希望获得实际应用的光阳极催化材料之一[1,91-96]. 尽管BiVO4的原材料资源丰富, 成本低廉, 但是在AM1.5G标准太阳光照和电势为1.23 V的情况下, BiVO4的光电流密度远小于其理论值7.5 mA/cm2[97,98]. 这主要归因于材料的表面缺陷以及表面腐蚀导致的内部较高的电子-空穴对复合率, 以及析氧反应过程中产生的超电位[99-101]. 通过在BiVO4纳米棒表面包裹BiFeO3铁电层, Xie等[102]显著提高了BiVO4的光催化效率. BiFeO3铁电层不仅可以对BiVO4层进行保护, 防止光腐蚀与化学腐蚀, 同时BiFeO3铁电层的退极化场也可以提高异质结中光生电子-空穴对分离效率. 如图7(a)所示, 通过化学溶剂沉积法在BiVO4纳米棒表面包裹了10 nm以内厚度的BiFeO3铁电层[103]. 由于使用化学方法制备薄膜, 其内部的化学溶剂的蒸发导致薄膜的形貌呈多孔状, 这可以增加材料的吸光性, 进而提高材料的光催化效率. 如图7(b)所示, 在AM1.5G光照和电势为0.6 V条件下, FTO/BiVO4/BiFeO3复合结构光电流密度为0.63 mA/cm2, 比单纯的BiVO4光阳极的光电流密度(即0.14 mA/cm2)提高约4.4倍, 比BiVO4/Co-Pi光阳极的光催化电流密度(即0.50 mA/cm2)提高了24%. 这里Co-Pi为钝化缓冲层, 用于防止溶液腐蚀与光腐蚀. 与普通的BiVO4相比, BiVO4/BiFeO3的电荷复合率也从17 s–1大幅度降低到了0.6 s–1. 如图7(a)和图7(c)所示, BiFeO3的铁电极化方向可以调控BiVO4/BiFeO3的光催化性能, 与未通过外电压极化的BiFeO3相比, BiFeO3铁电极化指向电解液时(+P by positive poling), BiVO4/BiFeO3界面的能带向上弯曲, 增加BiVO4中耗尽层的宽度, 能够促进空穴从BiVO4进入BiFeO3和电解液, BiVO4/BiFeO3光阳极的光电流密度提高了19%. BiFeO3铁电极化指向底电极时(–P by negative poling), BiVO4/BiFeO3光阳极的光电流密度降低了约24.4%, 这是由于BiVO4表面的能带结构向下弯曲降低了电子-空穴对的分离效率. 图7(d)是光电流密度-时间曲线, 在光照12 h之后, BiVO4/BiFeO3光阳极的光电流密度是初始光电流的59%, 与单纯的BiVO4纳米棒相比, BiVO4/BiFeO3的稳定性大大提高, 甚至比BiVO4/Co-Pi更好.
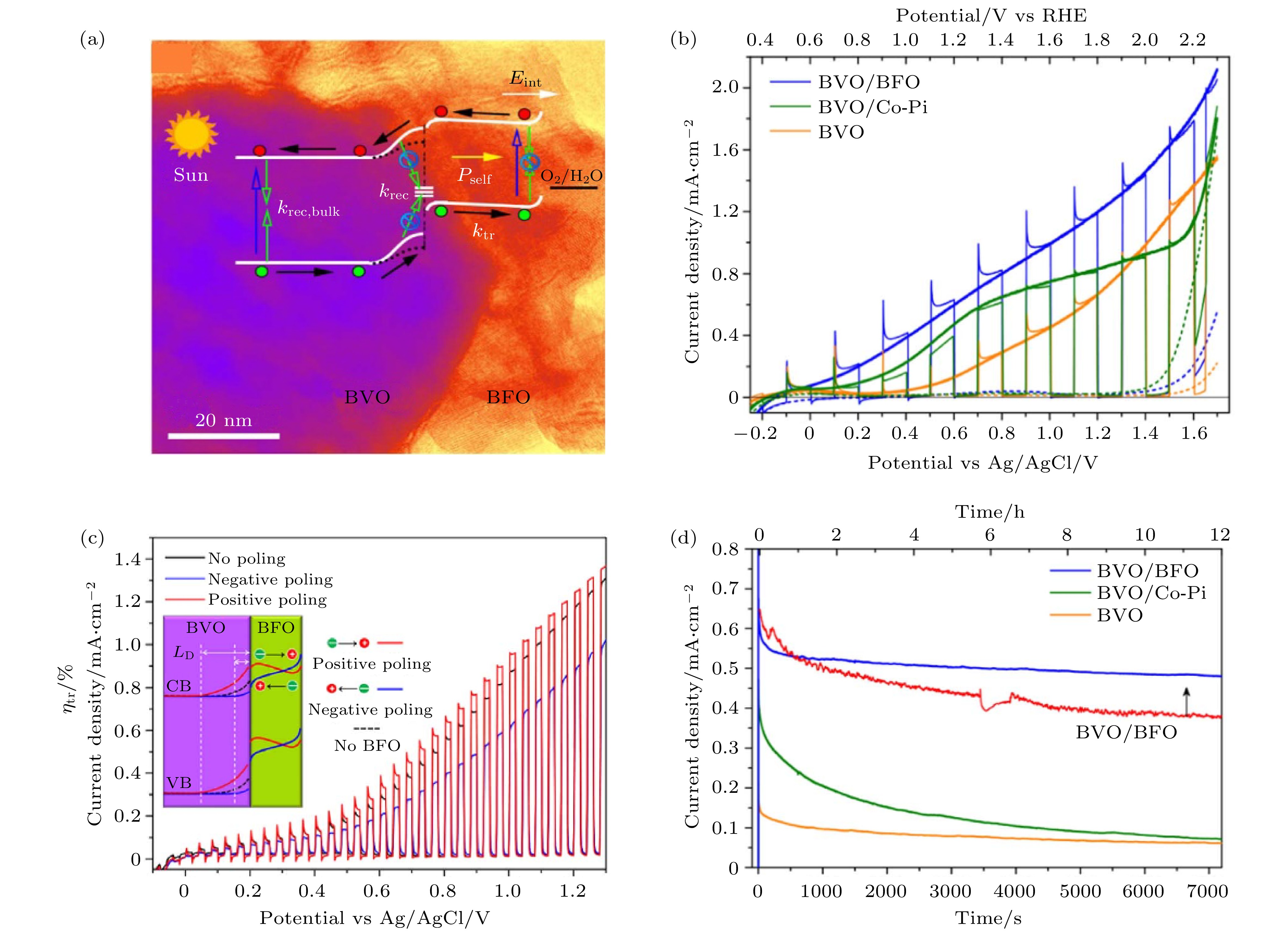 图 7 (a) BiVO4/BiFeO3光阳极的电子能级及结构示意图; (b) BiVO4/Co-Pi, BiVO4和BiVO4/BiFeO3三种结构的光电流密度-电势曲线; (c)不同铁电极化状态下BiVO4/BiFeO3光阳极的光电流密度-电势曲线; (d) 在工作电极的电势为0.6 V时, 三种结构的光电流密度-时间的曲线[102]
图 7 (a) BiVO4/BiFeO3光阳极的电子能级及结构示意图; (b) BiVO4/Co-Pi, BiVO4和BiVO4/BiFeO3三种结构的光电流密度-电势曲线; (c)不同铁电极化状态下BiVO4/BiFeO3光阳极的光电流密度-电势曲线; (d) 在工作电极的电势为0.6 V时, 三种结构的光电流密度-时间的曲线[102]Figure7. (a) Electron energy levels of BiVO4/BiFeO3 photoanode and the structural representation; (b) the photocurrent density curves of three different structures of BiVO4/Co-Pi, BiVO4 and BiVO4/BiFeO3; (c) photocurrent density versus potential curves at three statuses of ferroelectric polarization; (d) long-term photostability of three photoanodes at 0.6 V (V vs. Ag/AgCl)[102]
2
3.2.Bi2FeCrO6的光催化
Bi2FeCrO6具有特殊的双钙钛矿结构, 由于其带隙比较小, 在可见光驱动的氧化还原反应方面具有较大的优势. 2015年Li等[15]使用PLD系统生长了Bi2FeCrO6外延薄膜并研究其光电催化效应. Bi2FeCrO6铁电薄膜材料属于A2BB'O6型, 其中B位Fe/Cr元素的有序度影响到材料的能带结构和光催化性能, 因此调节Bi2FeCrO6的Fe/Cr阳离子有序度可增强光电催化效率. LaAlO3/CaRuO3/Bi2FeCrO6 (120 nm)中Bi2FeCrO6的有序相和无序相所对应的带隙值分别为2.12 eV和2.68 eV. (LaAlO3)0.3(Sr2AlTaO6)0.7(LSAT)/CaRuO3/Bi2FeCrO6 (130 nm)中Bi2FeCrO6的有序相和无序相对应的带隙值为2.05 eV和2.61 eV. SrTiO3/CaRuO3/Bi2FeCrO6 (128 nm)中Bi2FeCrO6的有序相和无序相对应的带隙值为1.94 eV和2.58 eV. 如图8(a)所示, 在AM1.5G标准太阳光照射下、pH = 6.8的1 mol/L Na2SO4电解液中, SrTiO3/CaRuO3/Bi2FeCrO6工作电极在–0.97 V的电势下的光电流密度为–1.02 mA/cm2. 如图8(b)所示, 铁电极化指向电解液(+P)时, 工作电极的电势为–1 V时, 光电流密度为–2.02 mA/cm2, 比没有极化的样品大两倍, 因此+P有利于增强光阴极对水的分解活性. 如图8(c)所示, 相同条件之下, 铁电极化指向底电极时(–P)时, 光催化电流密度显著降低至–0.85 mA/cm2. 这说明Bi2FeCrO6的铁电极化影响了半导体光生载流子的迁移, 通过铁电极化来调控能带结构并设计改进的光电极和太阳能转换智能器件是有前途的. 图 8 SrTiO3/CaRuO3/Bi2FeCrO6样品在Bi2FeCrO6 的(a)初始极化状态、(b) +P (即Pup)、(c) –P(即Pdown)时的结构示意图和在光阴极时Bi2FeCrO6薄膜的光电流-电势(vs. Ag/AgCl)曲线图[15]; (d) 在光阳极时SrTiO3/SrRuO3/Bi2FeCrO6/NiO异质结的光电催化示意图和(e)其光电流-电势曲线[62]
图 8 SrTiO3/CaRuO3/Bi2FeCrO6样品在Bi2FeCrO6 的(a)初始极化状态、(b) +P (即Pup)、(c) –P(即Pdown)时的结构示意图和在光阴极时Bi2FeCrO6薄膜的光电流-电势(vs. Ag/AgCl)曲线图[15]; (d) 在光阳极时SrTiO3/SrRuO3/Bi2FeCrO6/NiO异质结的光电催化示意图和(e)其光电流-电势曲线[62]Figure8. Schematic illumination and variations of the photocurrent density with applied voltage (vs. Ag/AgCl) in 1 mol/L Na2SO4 at pH 6.8 under chopped simulated sunlight illumination (AM1.5G) of SrTiO3/CaRuO3/Bi2FeCrO6 sample: (a) Before, (b) after negative (Pup, –25 V) and (c) and positive poling (Pdown, 25 V)[15]; (d) schematic diagram of the experimental setup and (e) photocurent versus potential (vs. RHE) curves of SrTiO3/SrRuO3/Bi2FeCrO6/NiO[62]
Bi2FeCrO6等半导体与含水电解质接触并发生光电催化时会出现自然腐蚀或钝化的现象, 光阳极/电解质中的电荷复合和有限的电荷转移动力学导致了较大的能量损失, 增加了电子-空穴对的复合速率, 严重影响了半导体材料的光催化效率以及使用寿命, 从而降低了其光催化的性能. 因此通常在使用前需要通过表面处理和改性或通过设置保护层来使其稳定[104-107]. 此外, 通过添加有效的电子传输层来减少光生电子-空穴对的复合, 可以提高n型本征半导体光阳极的光催化反应性能. 对于光阳极材料而言, 此类问题的解决方案在于设计一种既有保护性能也有空穴传输性能的材料, 该材料应该同时具有化学稳定性、抗氧化性、透明性和导电性的优点. 具有这种特性的最合适的材料是p型透明导电氧化物, 也称为p型窗口层. p型半导体氧化镍(NiO)具有立方晶体结构, 其带隙为3.6 eV, 超薄的NiO薄膜具有很高的光学透明性(10 nm时约为80%, 20 nm时为75%), 相对较低的电阻率(120 Ω·cm)和较高导带位置, 因此NiO适合用作保护层与传输层[108]. 如图8(d)所示, 2019年Huang等[62]构建了SrTiO3/SrRuO3/Bi2FeCrO6/NiO异质结. 利用PLD系统在透明的SrTiO3基片上生长了底电极SrRuO3、光阳极层Bi2FeCrO6和空穴传输层NiO, 其中NiO层的厚度为10和20 nm. 在300 K时, 10和20 nm厚度的NiO薄膜的电导率分别约为0.13和0.25 S/cm. 通过霍尔效应测量, 10 nm的NiO空穴浓度和迁移率分别为1.2 × 1018 cm–3与0.7 cm2·V–1·s–1; 而20 nm NiO的空穴浓度和迁移率为1.5 × 1018 cm–3与1.1 cm2·V–1·s–1. 如图8(e)所示, 在这种n-Bi2FeCrO6/p-NiO异质结光电二极管中, 在1.23 V电势下其光电流密度可提高四倍, 达到0.4 mA/cm2, 在4 h内可以保持稳定性, 且在标准太阳光光照下获得了较高的光电转换效率(~3.7%). 此外, 具有+P的Bi2FeCrO6薄膜的光阳极的光电流密度略小于原始状态下的样品, 具有–P的Bi2FeCrO6膜在相同偏压下光电流密度显著增加, 因此可以通过调整铁电极化状态提高电子-空穴分离与运输效率并提高光催化效率.
2
3.3.四方相BaTiO3铁电体的光催化
含有染料、肥料或表面活性剂分子等有机污染物的工业废水的排放是当今社会亟待解决的严重问题[109], 光催化被认为是降解和去除污染物的有效方法[110]. 表1对比了BaTiO3等铁电材料通过光催化或力催化降解甲基橙染料或CO2的性能. BaTiO3是一种广泛使用的铁电材料, 其四方相具有铁电性, 铁电居里温度约为120 °C[111]. 当温度高于其居里温度时或者室温下纳米粉体的表面能足够大时, BaTiO3四方铁电相转变为立方顺电相[112-115]. BaTiO3的带隙为3.18 eV, 可以在光照辐射下降解有机染料, 例如甲基红和甲基橙[116]. 2013年Cui等[43]研究了模拟太阳光照射下四方相BaTiO3铁电纳米粉体对典型染料分子罗丹明B脱色的影响. 在光催化反应过程中, 极性罗丹明B阳离子很可能在铁电材料的极性表面上产生紧密结合的层, 从而导致更大程度的染料分子与催化剂表面结合, 与立方顺电相为主的BaTiO3纳米粉体相比, 四方相为主的BaTiO3纳米粉体的脱色率可以提高3倍. 如下图9(a)和图9(b)所示, 当纳米结构的Ag以电化学的方式沉积在BaTiO3表面时, 催化剂载体表面上的Ag纳米颗粒涂层可提高染料吸附率约1.5倍, 光催化反应速率进一步提高, 染料在45 min左右完全脱色, 相应的催化分解染料的动力学速率常数(Kobs)为0.087 min–1. 通过比较立方顺电相为主的BaTiO3、退火后四方铁电相为主的BaTiO3、Ag-BaTiO3和退火后Ag-BaTiO3的光催化脱色速率, 发现退火和Ag纳米颗粒涂层可以优化BaTiO3的光催化性能. 退火后的样品比未退火样品具有更高的催化活性, 这说明退火后四方铁电性相BaTiO3的比例增加, 从而优化了其光催化性能. 由于铁电半导体退极化场EP的存在, 铁电材料可以有效地分离空穴和电子, EP产生了与典型的PN结中相似的空间电荷层, 并有效分离光生电子-空穴对、加速电子和空穴的运输速率并抑制了电子-空穴对的复合. 此外, 退火后的催化剂表现出更高的染料吸附水平, 这也可以归因于四方相BaTiO3的铁电性质. 图 9 (a) BaTiO3铁电材料的能带结构及其退极化场EP分离光生电子-空穴对的示意图; (b) 太阳光照射下不同催化剂对罗丹明B的光致脱色特性[43]; (c) Glass/500 nm-BaTiO3/67 nm-MoO3异质结的能带结构和载流子分离示意图; (d) 该异质结在紫外可见光和可见光分别照射下分离罗丹明B的效果图[44]; (e) 可见光(λ > 400 nm)照射下BaTiO3-CdS复合材料的光生空穴-电子对分离、载流子迁移以及光催化产生氢气的示意图; (f) 原始CdS, 纯BaTiO3和BaTiO3-CdS (wt 20%)复合材料的光电流-时间曲线(电极0.5 cm × 0.5 cm)[118]
图 9 (a) BaTiO3铁电材料的能带结构及其退极化场EP分离光生电子-空穴对的示意图; (b) 太阳光照射下不同催化剂对罗丹明B的光致脱色特性[43]; (c) Glass/500 nm-BaTiO3/67 nm-MoO3异质结的能带结构和载流子分离示意图; (d) 该异质结在紫外可见光和可见光分别照射下分离罗丹明B的效果图[44]; (e) 可见光(λ > 400 nm)照射下BaTiO3-CdS复合材料的光生空穴-电子对分离、载流子迁移以及光催化产生氢气的示意图; (f) 原始CdS, 纯BaTiO3和BaTiO3-CdS (wt 20%)复合材料的光电流-时间曲线(电极0.5 cm × 0.5 cm)[118]Figure9. (a) Schematic of BaTiO3-Ag composites with the effect of free carrier reorganization on band structure and photoexcited carriers, and (b) photodecolorization profiles of RhB with different catalysts under solar simulator[43]; (c) schematic representation of the 500 nm-BaTiO3/67 nm-MoO3 heterostructure on glass substrate, and (d) its photodecolorization profiles of RhB under UV-visible and visible light (sun light)[44]; (e) schematic of photoinduced hole and electron migration in BaTiO3-CdS composites and photocatalytic hydrogen process under visible light (λ > 400 nm), and (f) its photoelectrochemical properties of pristine CdS, pure BaTiO3 and BaTiO3-CdS (wt 20%) composite[118]
2018年Song等[117]通过原子力显微镜等观察到了BaTiO3的铁电极化调制光催化氧化还原反应的微观过程. 当BaTiO3铁电极化指向电解液, 光催化还原反应在铁电畴表面可以将Ag+还原为Ag, 其效率是铁电极化平行于样品表面时效率的3倍; 铁电极化背向电解液时, 光催化氧化反应可以在铁电畴表面将Pb2+氧化为PbO2, 而铁电极化平行于样品表面时不能将Pb2+氧化为PbO2.
BaTiO3只能吸收紫外光后产生光催化反应. 2018年Li等[45]制备了BaTiO3粉体, 然后在800 °C高真空环境通过Al粉将BaTiO3表面还原为非晶BaTiO3–x, 从而制备了BaTiO3@非晶BaTiO3-x核壳结构. BaTiO3–x薄膜含有大量的氧空位, 显著提高了可见光的吸收率, 该核壳结构样品再可见光照射下, 5 h内可以降解62.4%的甲基蓝染料.
2019年Alex等[44]在玻璃上制备了500 nm-BaTiO3/67 nm-MoO3异质结, 从而显著增强了其光催化效率. 如图9(c)所示, 由于BaTiO3的铁电极化束缚电荷和MoO3半导体的自由电荷存在相互耦合, 也由于BaTiO3和MoO3的能带结构差异, 在BaTiO3/MoO3界面存在指向MoO3层的内电场, 它帮助分离光生电子-空穴对, 其中电子向MoO3迁移, 空穴向BaTiO3迁移. 如图9(d)所示, 在紫外可见光照射下, 异质结在1 h内降解了86%的罗丹明B, 其效率是BaTiO3薄膜的1.7倍, 是MoO3薄膜的3.2倍. 经过5次光催化降解罗丹明B的循环, 异质结的光催化效率仅仅下降了5%. 该实验进一步说明铁电和半导体结合可以显著提高其光催化效果.
为了进一步利用BaTiO3铁电极化提高半导体材料的光催化效率, 2018年Huang等[118]制备了铁电/金属硫化物异质结构, 从而增强了光生电子-空穴对的分离效率并提高了其光催化性能. 如图9(e)所示, CdS吸附在具有四方铁电相的BaTiO3纳米球上, 由于BaTiO3铁电畴及其退极化场的影响, CdS中产生的载流子可以高效地迁移到表面并参与氧化还原反应. 因为BaTiO3的带隙太宽, 可见光无法在BaTiO3纳米球中高效的激发电子-空穴对并进行光催化反应, 它的产H2速率极低. CdS是窄带隙半导体, 光生电子-空穴对容易快速复合, 纯CdS的光催化活性低. 从图9(f)可以看出, BaTiO3-CdS (20 wt%)复合材料比单独的BaTiO3或者CdS表现出更高的光催化产氢活性, 其速率是CdS光催化产氢速率的9.7倍. 在可见光照射下, 复合材料的光催化制氢速率最高可达到483 μmol·h–1·g–1, 在32 h的循环测试中表现出令人满意的制氢性能, 保持良好的稳定性.
2015年Yang等[5]通过利用水热法在单晶TiO2纳米线表面生长了5 nm厚度和具有自发铁电极化的BaTiO3层, 从而获得了TiO2@BaTiO3核壳结构. 如图10(a)所示, 通过铁电极化调控该核壳结构的能带, 大大提高了光电催化性能. 由于BaTiO3退极化场EP的存在, 使得TiO2内部的电子-空穴更加容易分离与传输. 与原始的TiO2纳米线相比, TiO2@BaTiO3的光电流密度提高了67%. 通过计算TiO2@BaTiO3异质结上的电势分布, 研究TiO2和TiO2@BaTiO3纳米线的光吸收、电荷注入和分离特性, 发现BaTiO3的铁电极化显著提高了光生电子-空穴对的分离效率并大幅度增强了TiO2@BaTiO3的光催化性能. 如图10(b)所示, 通过外部电场极化调控铁电极化, 铁电极化指向电解液(即+P)时TiO2-BaTiO3光阳极的光催化电流密度从0.74 mA/cm2提高到0.96 mA/cm2, 增加了29.7%, 相反地, 铁电极化指向TiO2时(即–P) TiO2-BaTiO3光阳极的光催化电流密度降低至0.65 mA/cm2, 降低了12.2%.
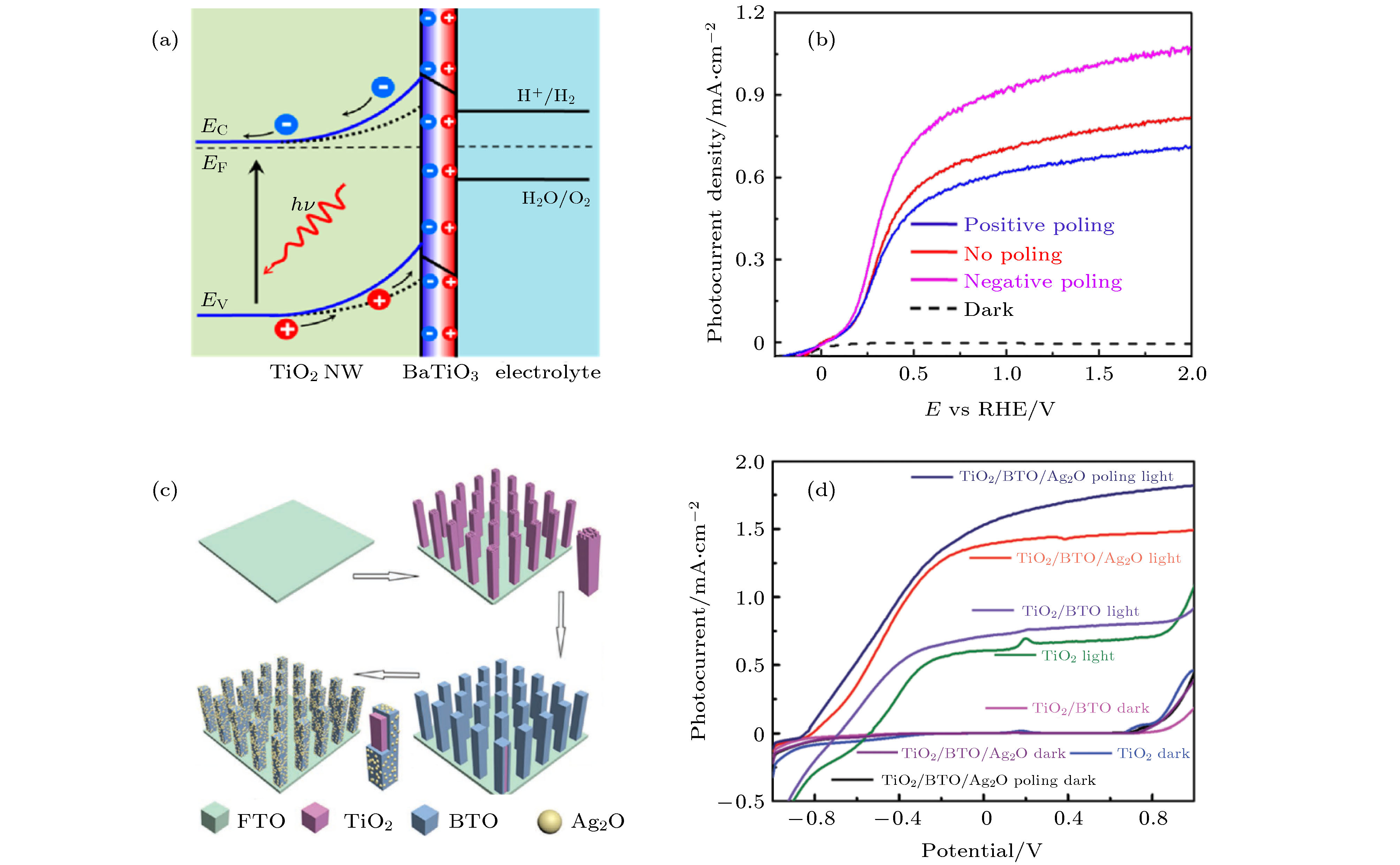 图 10 (a) TiO2@BaTiO3 (BTO)纳米线的能带结构示意图; (b) 铁电极化后TiO2@BaTiO3纳米线光阳极的光电流密度-电势曲线[5]; (c) 在FTO玻璃上制备TiO2@ BTO/Ag2O纳米棒的示意图; (d) 在标准太阳光照射下, TiO2, TiO2@BTO, 初始TiO2@BTO/Ag2O, 铁电极化指向TiO2的TiO2@BTO/Ag2O四种纳米棒阵列的光电流-电势曲线[66]
图 10 (a) TiO2@BaTiO3 (BTO)纳米线的能带结构示意图; (b) 铁电极化后TiO2@BaTiO3纳米线光阳极的光电流密度-电势曲线[5]; (c) 在FTO玻璃上制备TiO2@ BTO/Ag2O纳米棒的示意图; (d) 在标准太阳光照射下, TiO2, TiO2@BTO, 初始TiO2@BTO/Ag2O, 铁电极化指向TiO2的TiO2@BTO/Ag2O四种纳米棒阵列的光电流-电势曲线[66]Figure10. (a) Energy band diagram of nanowire photocatalytic reaction of TiO2@BaTiO3 nanowires; (b) photocurrent density versus potential curve of TiO2@BaTiO3 nanowires at three polarization statuses[5]; (c) scheme of the fabrication process of TiO2@BTO/Ag2O nanorod array, and (d) photocurrent-potential curves in the dark and under Xe lamp irradiation of the different photoanodes[66]
2019年, Liu等[66]在FTO玻璃上制备出了TiO2@BaTiO3核壳结构, 并且在几纳米厚度的BaTiO3薄膜表面生长了Ag2O纳米粒子, TiO2@BaTiO3/Ag2O制备过程如图10(c)所示. 垂直生长的TiO2纳米棒有很好的导电性, 光生电子能够方便快捷地传输到FTO电极上. TiO2属于n型半导体, BaTiO3是绝缘层, Ag2O是p型半导体, 形成n-i-p结构, 在BaTiO3薄膜内部和界面存在指向TiO2的内电场. 如图10(d)所示, 光生电子-空穴对在内电场作用下分离, 电子向FTO迁移, 空穴向Ag2O迁移, 因此TiO2@BaTiO3/Ag2O结构有效地增强了光生电子-空穴对的分离和迁移效率. Ag2O是窄带隙半导体, 可以吸收波长300— 900 nm的太阳光并进行光电催化. 在外电压作用下BaTiO3的铁电极化指向电解液, 其铁电退极化场EP方向和内电场方向一致, 显著增强了光电催化. 在电势为0.8 V (vs. Ag/AgCl)时, TiO2@BaTiO3/Ag2O的光电流密度达到1.8 mA/cm2, 这是TiO2纳米棒光电流密度的2.6倍. 该结构中, BaTiO3铁电薄膜同时用于调整n型半导体TiO2和p型半导体Ag2O的光电催化性能, Ag2O增加了光吸收光谱范围, n-i-p结构内电场和铁电极化退极化场增加了电子-空穴对的分离和迁移效率, 因此, TiO2@BaTiO3/Ag2O具有优异的光电催化性能. 这进一步说明, 铁电极化及其能带工程在改善半导体材料的光电催化性能方面具有广阔的前景.
2019年, Yang等[119]制备了同时具有(100)和(111)晶面的Cu2O纳米颗粒, 发现这两个晶面具有的表面电势差为23 mV, 可产生2.3 kV/cm的内电场. 在Cu2O颗粒的晶面上进一步生长了BaTiO3纳米小球, 形成Cu2O@BaTiO3复合结构, 然后将其与超级吸水的水凝胶复合在一起. 在10 mW/cm2的可见光照射下, 在全封闭的环境中150 mg的Cu2O@BaTiO3和水凝胶复合体可吸收和分解26.5 mg的水汽. 在该过程中, 水凝胶吸收空气中的水汽并提供给Cu2O@BaTiO3, BaTiO3纳米粒子主要帮助分离光生电子-空穴对, Cu2O将分离后的电子和空穴分别迁移到(111)和(100)晶面并产生还原反应和氧化反应. 该工作为光电催化分解水找到了新的应用场景.
2
3.4.四方相PbTiO3和SrTiO3铁电体的光催化
四方相PbTiO3是著名的铁电材料, 带隙大于2.75 eV, 通常条件下是良好的绝缘体, 在紫外光照射下具有一定的光催化效应[120]. 通过 Cu, Fe等离子掺杂, 可以增强该材料的光吸收并改善其光催化效应[121,122]. 同时, 也可以在PbTiO3表面黏附Ag, Pt纳米离子[63], 或者制备TiO2@PbTiO3核壳结构[123], 从而比较显著地提高了该结构的光催化效率. 2020年兰州大学Liu等[124]制备了铁电极化垂直于(001)表面的单晶单畴PbTiO3纳米片, 证实了该样品中分离光生电子-空穴对最主要的驱动力量是铁电退极化场EP. 如图11(a)所示, 由于纳米片上下表面分别聚焦了正负束缚电荷, 纳米片内部存在铁电退极化场EP并导致其能带弯曲; 当屏蔽电荷比较少时, 增加纳米片厚度即可增加纳米片上下表面的电势差ΔV. 如图11(b)所示, 光照条件下纳米片上下表面之间电势随着纳米片厚度的增加而增加, 进一步增强了其光生电子-空穴对的分离效率和纳米片通过光催化制氢的效率. 此外, 在PbTiO3纳米片上下表面之间生长SiO2介电层可以有效抑制纳米片表面自由电荷屏蔽铁电退极化场. 该研究阐明了铁电退极化场及其屏蔽电荷对光催化过程中电子-空穴对分离的影响[124]. 2018年Li等[46]制备了(001)晶面的PbTiO3单晶纳米片, 并在其表面继续生长了TiO2单晶薄膜. 当PbTiO3的铁电极化背向TiO2薄膜时, 该(PbTiO3/TiO2)-纳米片在氙灯的可见光照射下, 其分解甲基蓝的Kobs为0.053 min–1 (图11(c)); 当铁电极化指向TiO2薄膜时, 该(PbTiO3/TiO2)+分解水产生氢气的速率为132.6 μmol·h–1·g –1 (图11(d)).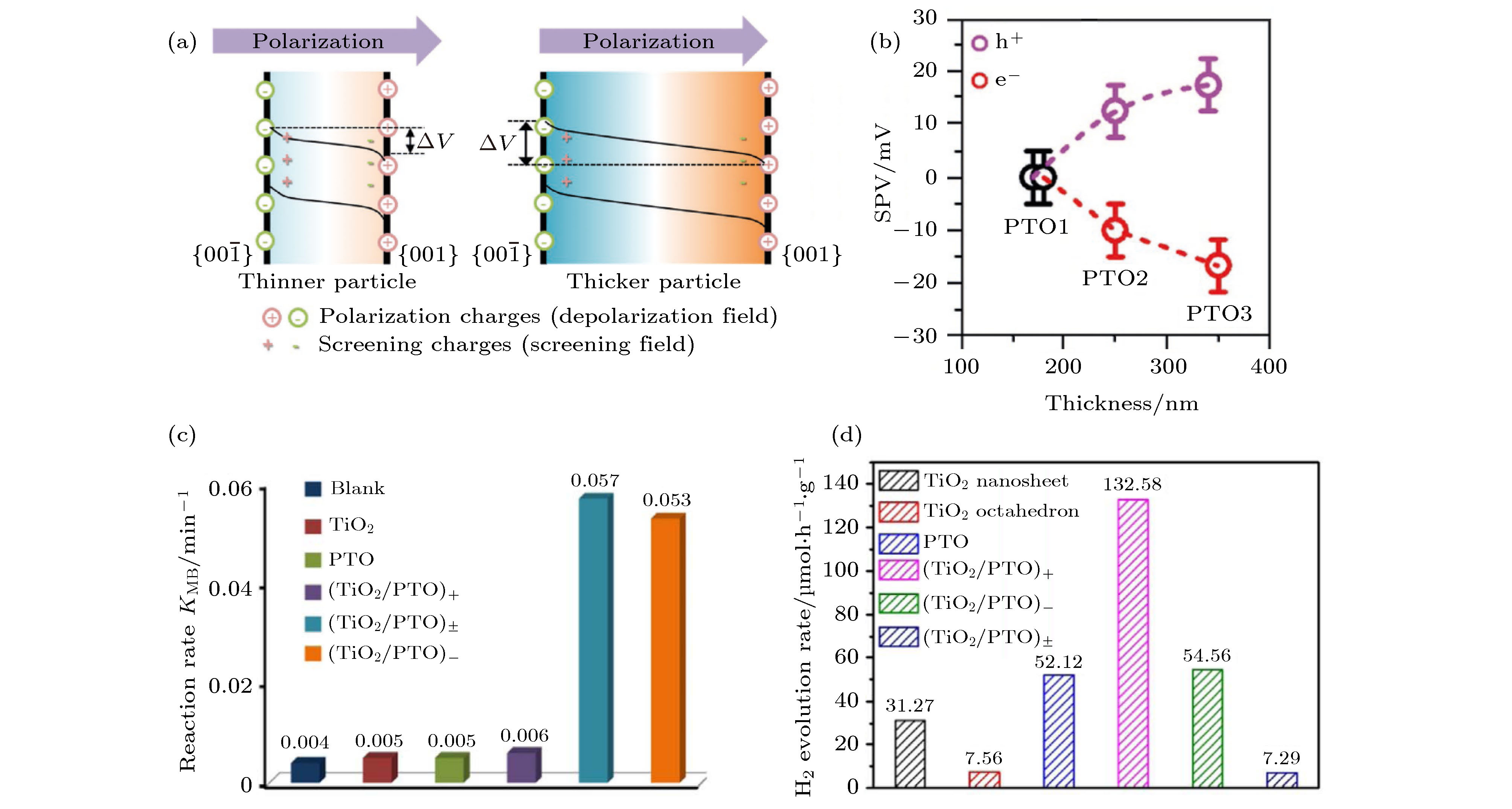 图 11 (a) 铁电极化垂直于表面时, (001) PbTiO3单晶单畴纳米片在200—350 nm不同厚度时的能带图; (b) 通过开尔文探针力显微镜测量的表面光伏电压SPV[124]; (c) (001) PbTiO3单晶纳米片、TiO2粉体、铁电极化指向或背向TiO2层或没有特定指向情况下的PbTiO3/TiO2纳米片的光催化分解甲基蓝的速率常数Kobs (即KMB); (d) 光催化产氢速率[46]
图 11 (a) 铁电极化垂直于表面时, (001) PbTiO3单晶单畴纳米片在200—350 nm不同厚度时的能带图; (b) 通过开尔文探针力显微镜测量的表面光伏电压SPV[124]; (c) (001) PbTiO3单晶纳米片、TiO2粉体、铁电极化指向或背向TiO2层或没有特定指向情况下的PbTiO3/TiO2纳米片的光催化分解甲基蓝的速率常数Kobs (即KMB); (d) 光催化产氢速率[46]Figure11. (a) Schematic of energy band in thinner (001) PbTiO3 (PTO) with smaller built-in voltages (ΔV) and thicker nanosheet with larger ΔV; (b) correlation between surface photovoltaic value measured by Kelvin probe force microscopy and nanosheet thickness[124]; (c) the reaction rate of blank control and photodegradation of MB under visible light (λ ≥ 420 nm) irradiation with (001) PTO, TiO2 and heterostructured TiO2/PTO composites; (d) H2 evolution rate of water splitting under visible light (λ ≥ 420 nm) irradiation[46]
SrTiO3块体材料是立方顺电相, 其在应力作用或者离子掺杂条件下容易发生晶格畸变并转变为四方铁电相. 2004年Haeni等[125]在Natrue上发表的文章表明, 约1%的双轴拉伸应变可以使SrTiO3在室温下转变为四方铁电相, 在DyScO3单晶基片上生长的50 nm厚度的(100) SrTiO3外延单晶薄膜在室温下具有清晰的铁电畴结构. 当然, 与其他典型的铁电材料相变相比, 四方相SrTiO3纳米片的铁电性很弱, 其饱和铁电极化、铁电畴等的研究工作尚不完善. 尽管如此, SrTiO3是著名的光催化材料, 其室温下的电子迁移率为5—8 cm2·V–1·s–1, 远远大于TiO2的电子迁移率0.1—4 cm2·V–1·s–1[4], 其铁电极化增强光催化效应的工作值得我们进一步研究. 2017年Wu 等[67]在FTO玻璃基片上制备了单晶TiO2纳米线, 并进一步包裹了一层10 nm厚度的具有四方铁电相的SrTiO3薄膜, 获得了TiO2@SrTiO3核壳结构. 在TiO2@SrTiO3结构中, SrTiO3同时具有高电子迁移率和四方铁电相两大优势. 与TiO2@BaTiO3 核壳结构类似, SrTiO3的铁电退极化场优化了TiO2/SrTiO3界面的能带结构, 有利于电子-空穴对的分离, SrTiO3的高电子迁移率有利于载流子迁移到FTO电极, 其光生电子-空穴对的分离效率(ηseparation)提高到87.7%. 在AM1.5G的模拟太阳光照射和1.23 V (vs. RHE)电势下, 在1 mol/L的NaOH电解液中, TiO2@SrTiO3光阳极的光电流密度达到1.43 mA/cm2, 比同等条件下TiO2纳米线的光电流密度提高了83%. 此外, 热处理、SrTiO3薄膜的厚度和铁电极化方向都可以显著调控其光电催化性能.
2
3.5.XNbO3 (X = Na, K, Li)和Bi3TiNbO9的光催化
XNbO3 (X = Na, K, Li)也是铁电材料光催化的研究热点之一. NaNbO3是一种n型铁电氧化物半导体, 具有良好的载流子迁移率和有利的水分解能带结构. 2017年Singh和Khare[64]通过水热法制备了NaNbO3纳米颗粒, 涂覆在FTO导电玻璃上, 研究了纳米铁电薄膜的电化学性能, 纳米铁电薄膜的铁电极化可使纳米铁电薄膜的光催化效率提高7%—23%. 如图12(a)所示, NaNbO3纳米薄膜在外加电场铁电极化后, 半导体/电解质界面的能带弯曲, 界面电荷的转移效率提高, 进而增强了材料的光催化效率. 如下图12(b)所示, 与传统的光阳极相比, 在电势为1 V时, 铁电极化指向FTO玻璃基片(–P)的NaNbO3纳米膜光阳极的光电流从0.31 mA/cm2提高到0.51 mA/cm2, 而铁电极化指向电解液(+P)时相应的光电流降低到0.09 mA/cm2. Singh和Khare[126]在PVDF柔性衬底上成功制备了柔性PVDF/Cu/NaNbO3/PVDF光阳极, 如图12(c)所示. 为了研究电场引起的铁电极化对NaNbO3薄膜的光阳极行为的影响, 通过施加5 V直流电压使NaNbO3薄膜产生宏观铁电极化. 如图12(d)所示, 在100 mW/cm2卤钨灯照射下和工作电极电势为1 V时, 铁电极化指向PVDF基片(–P)时光阳极的光催化性能大幅度提高, 其光阳极的光电流高达1 mA/cm2, 而铁电极化指向电解液(+P)时光阳极的光电流密度仅为0.35 mA/cm2. 由于PVDF/Cu/NaNbO3/PVDF光阳极的柔韧性和压电性能, 利用压电效应可进一步提高~26%的光电流.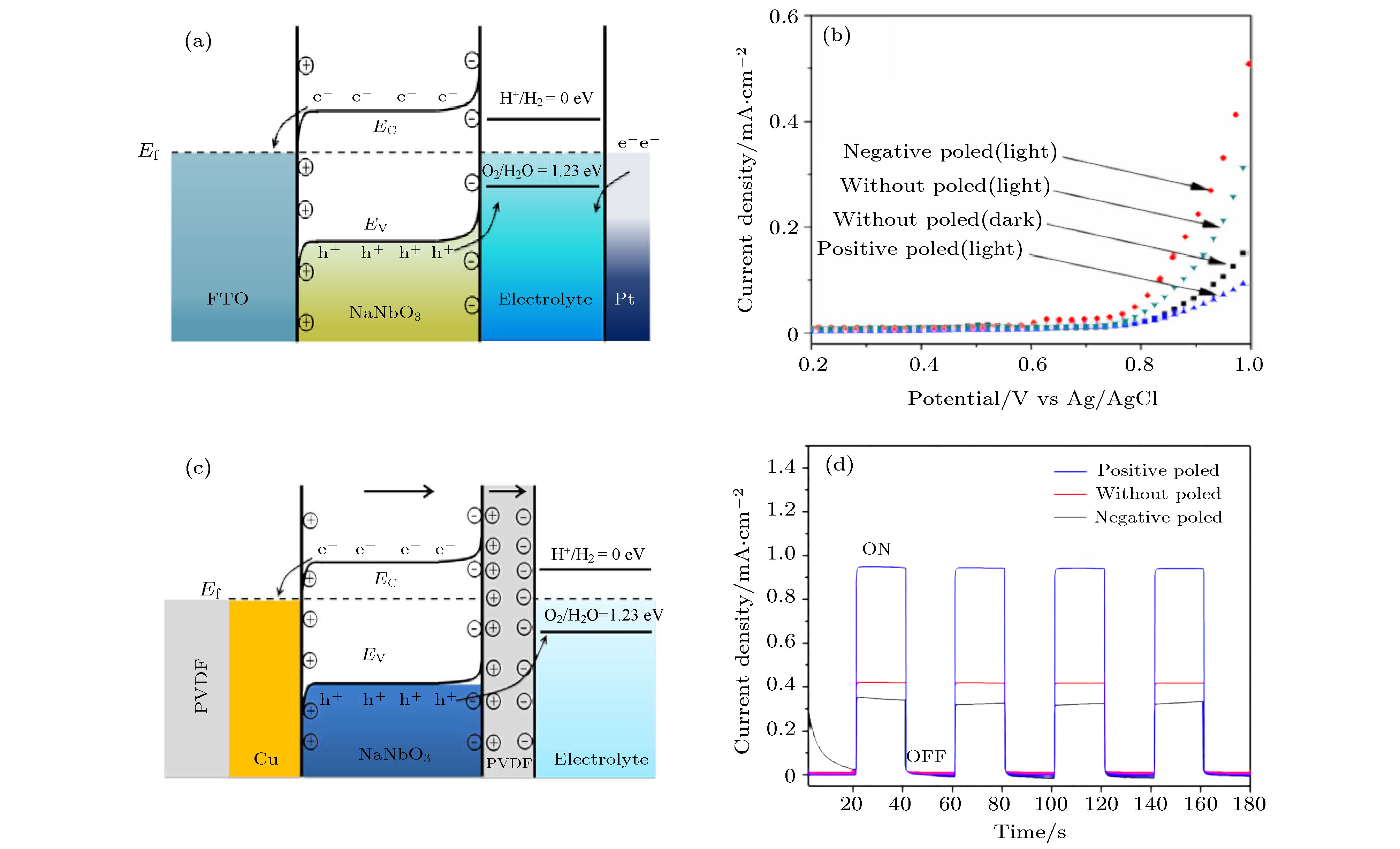 图 12 (a) 铁电极化指向底电极时FTO/NaNbO3光阳极的能带结构示意图; (b) 在0.5 mol/L Na2SO4电解质中以100 mW/cm2的紫外-可见光线照射下, 不同极化条件下光阳极的电流-电位曲线[64]; (c) 铁电极化指向底电极时PVDF/Cu/NaNbO3/PVDF的能带结构示意图; (d) 不同极化条件下NaNbO3/PVDF薄膜的光电流密度-时间曲线[126]
图 12 (a) 铁电极化指向底电极时FTO/NaNbO3光阳极的能带结构示意图; (b) 在0.5 mol/L Na2SO4电解质中以100 mW/cm2的紫外-可见光线照射下, 不同极化条件下光阳极的电流-电位曲线[64]; (c) 铁电极化指向底电极时PVDF/Cu/NaNbO3/PVDF的能带结构示意图; (d) 不同极化条件下NaNbO3/PVDF薄膜的光电流密度-时间曲线[126]Figure12. (a) Band bending of FTO/NaNbO3 for negative polarized; (b) current-potential curves of photoanodes with different polarization conditions[64]; (c) band bending of PVDF/Cu/PVDF-NaNbO3 for negative polarized; (d) current density versus time curves of NaNbO3/PVDF films with different polarization conditions[126]
在各种类型的纳米材料中, 半导体纳米线的垂直排列阵列特别受关注, 因为它们具有较大的半导体/电解质界面接触面积, 可以增强光的散射以及俘获更多光能量的功能, 沿着纳米丝可以进行有效的电荷传输[127,128]. 铌酸钾(KNbO3)是一种经典的钙钛矿氧化物, 具有优秀的铁电性能, 其具有合适的带隙, 出色的化学稳定性以及绿色环保的特性, 已成功用于各种光催化工艺中, 包括氢气生产[129-132], 有机污染物的降解[133,134]和二氧化碳转化[135]. 特别是, Park等[136]报道: 与中性样品相比, 具有宏观铁电极化的K0.5Na0.5NbO3粉末可以增强其光催化效率7倍. 2017年Li 等[137]在掺铌的SrTiO3衬底上外延生长了KNbO3纳米线. 通过控制纳米线的宏观铁电极化状态, 可以有效地调整工作电极的电荷转移特性, 从而优化其光催化性能. 分别用+15 V与–15 V电压极化纳米线后, 在0 V电势下KNbO3的光电流密度从0.7 μA/cm–2增加到11.5 μA/cm–2. 此外, 2018年Fu等[65]通过调节自发极化来改变LiNbO3的光催化电解水的氧化还原活性, 他们发现LiNbO3在经过不同方向的极化之后, 表现出不同的光催化能力.
2018年, Yin等[48]通过调控温度等生长条件可以分别制备出{001}晶面族或{110}晶面族为表面的Bi3TiNbO9纳米片. Bi3TiNbO9具有[Bi2O2][Am–1BmO3m+1]层状钙钛矿结构、带隙为3.3 eV的纳米片, 其铁电极化可以分离光生电子-空穴对, 它们的光催化产氢效率分别为342.6 μmol·h–1·g–1和275.2 μmol·h–1·g–1.
2
3.6.钙钛矿铁电材料中光-力-热协同催化
压电性是指某些介质的单晶体在受到定向压力或张力作用时, 在垂直于应力的晶体两侧表面上分别出现相反的等量电荷的性质, 当外应力方向反转时, 晶体两侧表面的电荷也随之反转[138,139]. 热释电效应是温度变化引起晶体的极化强度改变和电荷释放现象, 宏观上是温度变化使晶体两端出现电压或产生电流. 如图1(c)所示, 铁电材料同时具有铁电、热释电和压电性能, 因此铁电材料都也可以同时具备铁电光催化、热释电冷热催化和压电振动催化的功能. 如图13(a)所示, 光照在铁电体内部产生光生电子-空穴对, 在铁电材料的退极化场EP的作用下电子-空穴对分离和迁移, 引起光催化效应; 具有宏观极化的铁电材料可以在外力或者温度变化的时候在其表面产生动态变化的电荷, 从而进行力催化或者热催化, 将机械能或热能转换为电能、化学能或者分解有机物[4,140]. 压电催化作为新兴的催化反应过程之一, 由于其将机械能转化为化学能的能力而迅速发展[141,142]. ZnO等很多压电材料能够在黑暗中的超声波振动或低频振动下降解有机污染物[143-147], 具有高压电性能的钛酸钡基无铅压电材料已被广泛研究用于压电催化.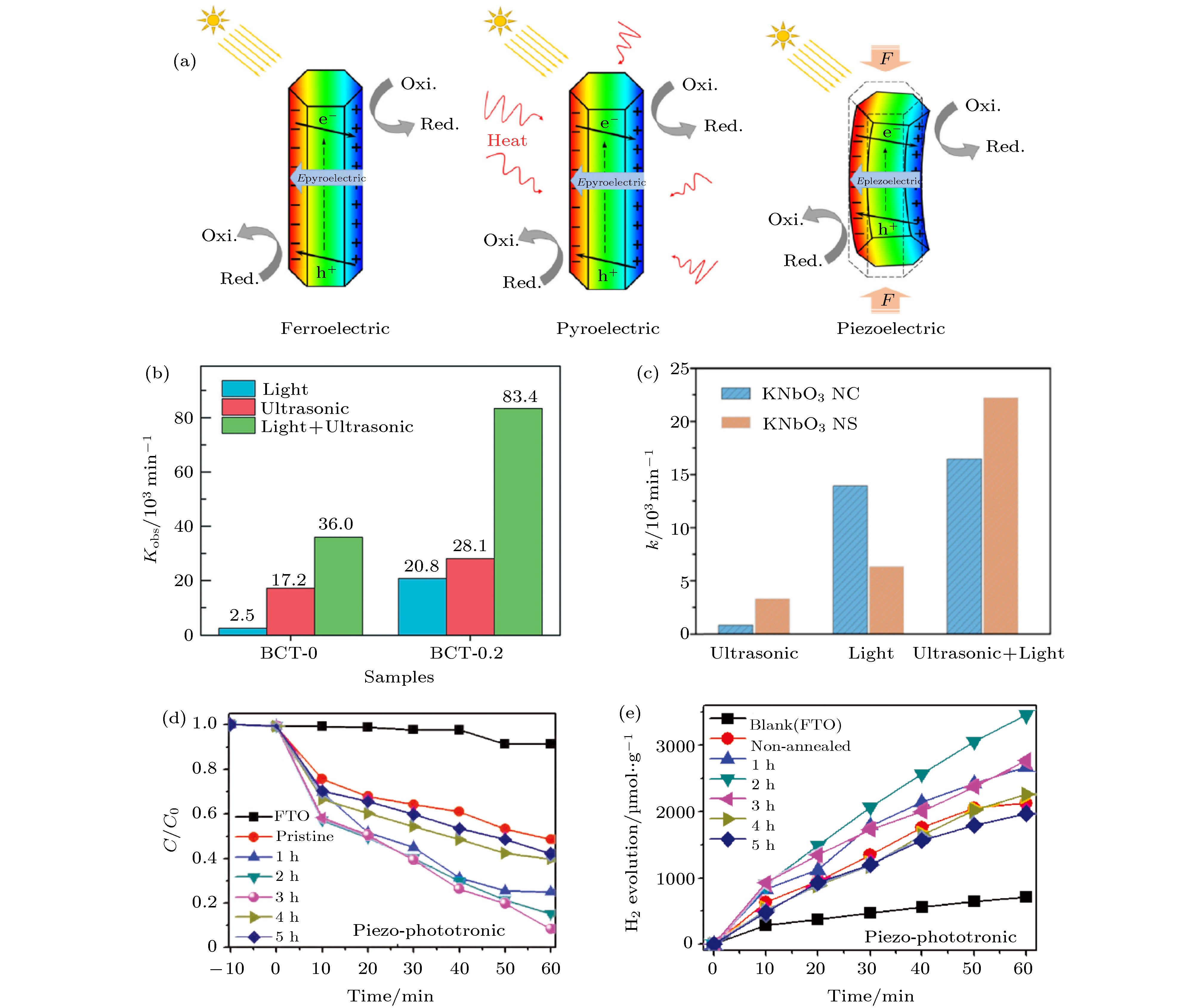 图 13 (a) 铁电、热电和压电材料中自由载流子重组和光激发载流子分离的示意图[140]; (b) 在紫外光照射、超声振动分别和同时存在时, BCT-0和BCT-0.2降解甲基橙染料的Kobs对比[150]; (c) 在氙灯可见光、超时振动分别和同时存在时, KNbO3纳米管(NC)和纳米片(NS)的降解甲基橙染料的Kobs(即k)对比[50]; (d) ZnSnO3–x 纳米线在超声振动和氙灯光照情况下降解罗丹明B染料和(e)分解水产氢的对比图[52]
图 13 (a) 铁电、热电和压电材料中自由载流子重组和光激发载流子分离的示意图[140]; (b) 在紫外光照射、超声振动分别和同时存在时, BCT-0和BCT-0.2降解甲基橙染料的Kobs对比[150]; (c) 在氙灯可见光、超时振动分别和同时存在时, KNbO3纳米管(NC)和纳米片(NS)的降解甲基橙染料的Kobs(即k)对比[50]; (d) ZnSnO3–x 纳米线在超声振动和氙灯光照情况下降解罗丹明B染料和(e)分解水产氢的对比图[52]Figure13. (a) Schematic understanding of free carrier reorganization and photo-excited carrier separation in ferroelectric, pyroelectric and piezoelectric materials under the influence of ferroelectric, pyroelectric and piezoelectric effects respectively[140]; (b) degradation reaction kinetic rate constants (Kobs) of methyl orange over BCT-0 and BCT-0.2 under UV light, ultrasonic vibration and the simultaneous assistance of ultrasonic vibration and UV light[150]; (c) Kobs (i.e. k) of the RhB solution over the KNbO3 nanosheet (NS) and nanotube (NC) under different conditions[50]; (d) the RhB dye solution degradation activity and (e) the amount of hydrogen evolution of ZnSnO3–x nanowires as a function of time under applying light and ultrasonic vibration simultaneously[52]
机械振动可以在材料中通过压电效应产生交变的内电场并增强光生电子-空穴对的分离, 光-力可以协同增强压电材料的催化效应. 在应力或应变作用下压电材料的一端产生正极化电荷, 而在另一端产生负极化电荷; 在压电极化电场的驱动下, 光诱导的电子和空穴可以被有效地分离并向相反的方向迁移, 有利于大量电荷向表面活性位的迁移, 从而参与各种光催化反应[142]. 压电材料在光催化中的应用引起了越来越多的关注, Xue等[57]报道了一种通过将ZnO纳米线垂直编织到碳纤维上而制成的压电光催化系统. 在紫外线照射下由ZnO纳米线产生电子-空穴对, 电子和空穴迅速迁移到相反的表面并产生周期性的压电极化电场, 有效地降解了甲基橙. 通过增加外力的频率, 可以获得更大的压电极化电场, 从而提高了甲基橙分解性能.
部分两相复合的压电材料具有更好的压电性能, 远远超过了其相应的单相材料[148-150], 这表明双相压电材料可能具有更好的压电催化性能. 2019年Lin等[150]发现由正交顺电相Ba0.07Ca0.93TiO3和四方铁电相组成的Ba0.07Ca0.93TiO3固溶体中, 由于顺电正交相的离子极化与铁电相非180°铁电畴之间产生相互耦合, 在两相固溶体的溶解度极限附近(x = 0.23时)获得了更大的电致伸缩性和压电性. 使用H2Ti3O7纳米线模板通过两步水热法合成了一系列Ba1–xCaxTiO3纳米线. 对比Ba0.8Ca0.2TiO3 (BCT-0.2)和BaTiO3 (BCT-0)两份样品, 甲基橙染料分别在40和100 min内完全降解, 这说明同时进行紫光照射和超声振动可以显著地提高甲基橙染料的降解效率. 由于热激发和光诱导的自由电荷载流子浓度的增加和压电极化电场分离电子-空穴对, Ba0.8Ca0.2TiO3纳米线表现出最好的压电催化和光催化性能. 如图13(b)所示, 在超声和光的同时作用下, BCT-0.2系统催化降解过程的Kobs值是83.4 × 10–3 min–1, 比压电催化和光催化降解过程的简单总和Kobs值大71%, 此结果表明在压电催化降解和光催化降解之间存在耦合作用, 两者的耦合作用带来了更高的光催化降解速率.
2013年Zhang等[131]以KNbO3纳米结构为代表材料研究了压电催化和光催化的联合作用, 发现KNbO3纳米片表现出良好的光催化性能. 2019年Yu等[50]制备了KNbO3纳米管和纳米片, 发现超声振动激发的压电效应能够显著增强它们的光催化效应, 并阐明了该压电光催化效应的铁电-光-电-化学过程. 如图13(c)所示, 在可见光和超声振动的激励下, 纳米管降解甲基橙染料的Kobs达到0.016 min–1; 纳米片具有更大的比表面积, 相同测量条件下其压电光催化效果更好, Kobs达到0.022 min–1, 在2 h内降解92.6%的甲基橙染料. 铁电极化可以调控ITO玻璃基片上生长的KNbO3纳米片的光电催化性能, 在AM1.5的模拟太阳光照射和0 V电势下(vs. Ag/AgCl)条件下, 铁电极化指向和背向基片的KNbO3纳米片时的光电流密度分别为0.82和0.53 μC/cm2.
ZnSnO3是一种钙钛矿结构压电材料, 它具有与LiNbO3铁电材料一致的R3c空间群, 其铁电极化Ps约为0.2 μC/cm2[52]. 其机械弯曲产生的应变会产生压电势、极大地增加局部电导并有效地降低势垒高度. 2015年Lo等[51]在FTO玻璃上制备了垂直排列的ZnSnO3纳米线, 在应力作用下产生压电极化电场和能带畸变, 载流子的迁移率增加, 光生电子和空穴的复合减少, 甲基橙的降解表现出更高的光催化活性. 超声振动产生了周期性变化的压电极化电场和相应的能带弯曲, 在超声振动和紫外光照射下Zn1–xSnO3的降解染料的速率达到0.015 min–1, 这是单纯紫外光催化降解染料速率的四倍[53]. 2019年清华大学Wang和Wu[52]制备了具ZnSnO3–x纳米线, 通过在氢氦气体环境中对ZnSO3纳米线进行300 ℃热处理3 h, 制备了氧空位含量适中的ZnSnO3–x纳米线, 其载流子浓度约5 × 1014 /cm3, 载流子迁移率约300 cm2·V–1·s–1, 少数载流子寿命约8.3 ns. 如图13(d)所示, 在40 kHz超声振动和氙灯光照同时进行时, ZnSnO3–x纳米线60 min内降解了92%的罗丹明B染料, 比单纯氙灯光照或者超声振动的效果好1.5和2.3倍; 如图13(e)所示, 超声振动、氙灯光照、超声振动和氙灯光照三种情况下的产氢速率分别达到3562, 3453, 3882 μmol·h–1·g–1. 这说明压电和光协同催化在进行污水净化和制氢方面具有很大的潜力.
此外, 2017年Simrjit和Khare[42] 在机械应变下利用NaNbO3产生的压电电势将甲基橙染料的光降解提高了约115%. Huang等[55]利用非中心对称的压电和光敏性BiOIO3半导体在超声振动和光辐照下有效降解罗丹明B(RhB)染料. 2018年Mushtaq 等[151]采用单晶BiFeO3纳米片和纳米线做催化剂, 通过可见光和机械振动降解有机污染物. 在可见光下, 两种BiFeO3纳米结构均显示出相对较慢的反应速率. 然而, 在压电催化下, 与光催化降解相比, 纳米片和纳米线均显示出更高的反应速率. 当同时使用太阳光和机械振动时, 反应速度甚至进一步提高, BiFeO3纳米线在1 h内降解了97%的RhB染料. BiFeO3纳米线在降解四种不同的有机污染物方面也表现出良好的可重用性和多功能性.
一些光催化材料可以帮助合成有机物[152-155], 然而铁电半导体光催化材料用于有机物合成的工作还很少. 2019年, 日本北海道大学Kubota 等[156]通过球磨振动让BaTiO3粉体产生正压电效应和机械催化效应, 其激发的自由电子还原芳基重氮盐并帮助合成多种有机小分子材料. 该研究说明通过光-振动-热协同催化有望用于合成新材料.
2
3.7.光催化还原CO2生产燃料
CO2分子是热力学最稳定的碳化合物之一. 将CO2光催化还原为烃类燃料需要输入能量才能打破C=O键并形成C—H键, 这涉及多个电子和相应数量的质子的参与[157]. CO2中C原子的化学状态最高(C4+), 它只能在还原剂的支持下发生还原反应. 与H2, S2–, SO32–和胺类相比, H2O由于其丰富以及无毒无污染的特点, 是还原剂的首选. 用H2O将CO2光催化还原为烃类燃料(例如CH4和CH3OH)是一种吸能反应, 其吉布斯自由能增加(ΔG), 如下式所示: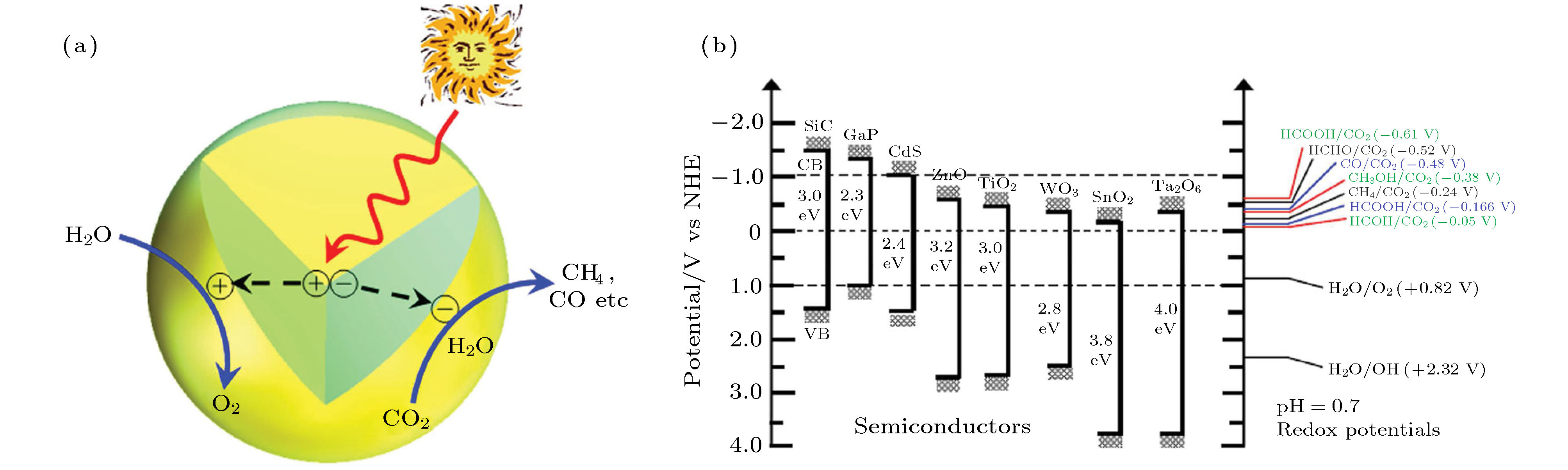 图 14 (a) 半导体催化剂中光照产生电子-空穴对, 电子空穴对分离, 空穴氧化H2O并产生氧气, 电子还原CO2和H2O并产生燃料; (b) 几种半导体光催化剂的导带、价带电势和带隙, 参与CO2还原的化合物在pH=7时的氧化还原电势[158]
图 14 (a) 半导体催化剂中光照产生电子-空穴对, 电子空穴对分离, 空穴氧化H2O并产生氧气, 电子还原CO2和H2O并产生燃料; (b) 几种半导体光催化剂的导带、价带电势和带隙, 参与CO2还原的化合物在pH=7时的氧化还原电势[158]Figure14. (a) Schematic illustration of photoinduced generation of an electron-hole pair in semiconductor that transfers to the surface for CO2 photoredox; (b) conduction band, valence band potentials, and band gap energies of various semiconductor photocatalysts relative to the redox potentials at pH 7 of compounds involved in CO2 reduction[158].
太阳光驱动的CO2转化为可再生燃料的潜力巨大, 但是由于CO2分子在环境大气中的惰性以及光催化剂反应的效率较低, 该方向的研究仍然具有很高的挑战性. 2019年Tu等[164]利用铁电材料SrBi4Ti4O15的EP增强了光催化分解CO2并产生燃料的效率. SrBi4Ti4O15属于层状铋基铁电半导体材料, 具有(Bi2O2)2+和交错的带负电的离子/多面体层组成的层状结构, 形成了静电场以促进电子-空穴对的分离, 从而受到研究人员的重视. 通过水热法制备SrBi4Ti4O15纳米片, 再将SrBi4Ti4O15纳米片旋涂至ITO玻璃上(SBTO-1). 为了增强铁电性并排除附着在催化剂表面的有机杂质的影响, 将制备好的SrBi4Ti4O15纳米片在350 °C (SBTO)和650 °C (SBTO-2)下退火, 其中SBTO展示出了最强的铁电性能. 如图15(a)和图15(b)所示, 受光照激发产生的电子-空穴对分离, 电子进入导带进行还原反应生产CH4与CO, 空穴进入价带并参与氧化反应产生氧气. SrBi4Ti4O15在365 nm光照下的外量子效率为1.33%. Bi4Ti3O12和BiOBr纳米片, 被用作参比样品. 由于这是气固反应, 并且催化剂直接与CO2反应, 因此只有气态产物, 而没有液态产物. 如图15(c)所示, SrBi4Ti4O15催化剂具有还原CO2的超强能力, 产生CH4和CO的远胜于其他三种催化剂. SrBi4Ti4O15催化剂生产CH4的概率高达93%, 速率为19.8 μmol·h–1·g–1, 是Bi4Ti3O12和BiOBr的283倍. 重复进行3次循环测试, 连续4 h进行光催化CO2还原, 确认了SrBi4Ti4O15的耐久性和可重复性. 如图15(d)所示, 光催化测试表明, 通过350 °C退火可以改变SrBi4Ti4O15的铁电性能并提高SrBi4Ti4O15纳米片的还原CO2的活性. 三种SrBi4Ti4O15光催化样品还原CO2并生产CH4和CO的活性顺序依次为SBTO > SBTO-2 > SBTO-1, 与铁电性能的强弱相一致. SrBi4Ti4O15纳米片的CO2还原活性超过了先前报道的基于铋的光催化剂和大多数其他光催化剂.
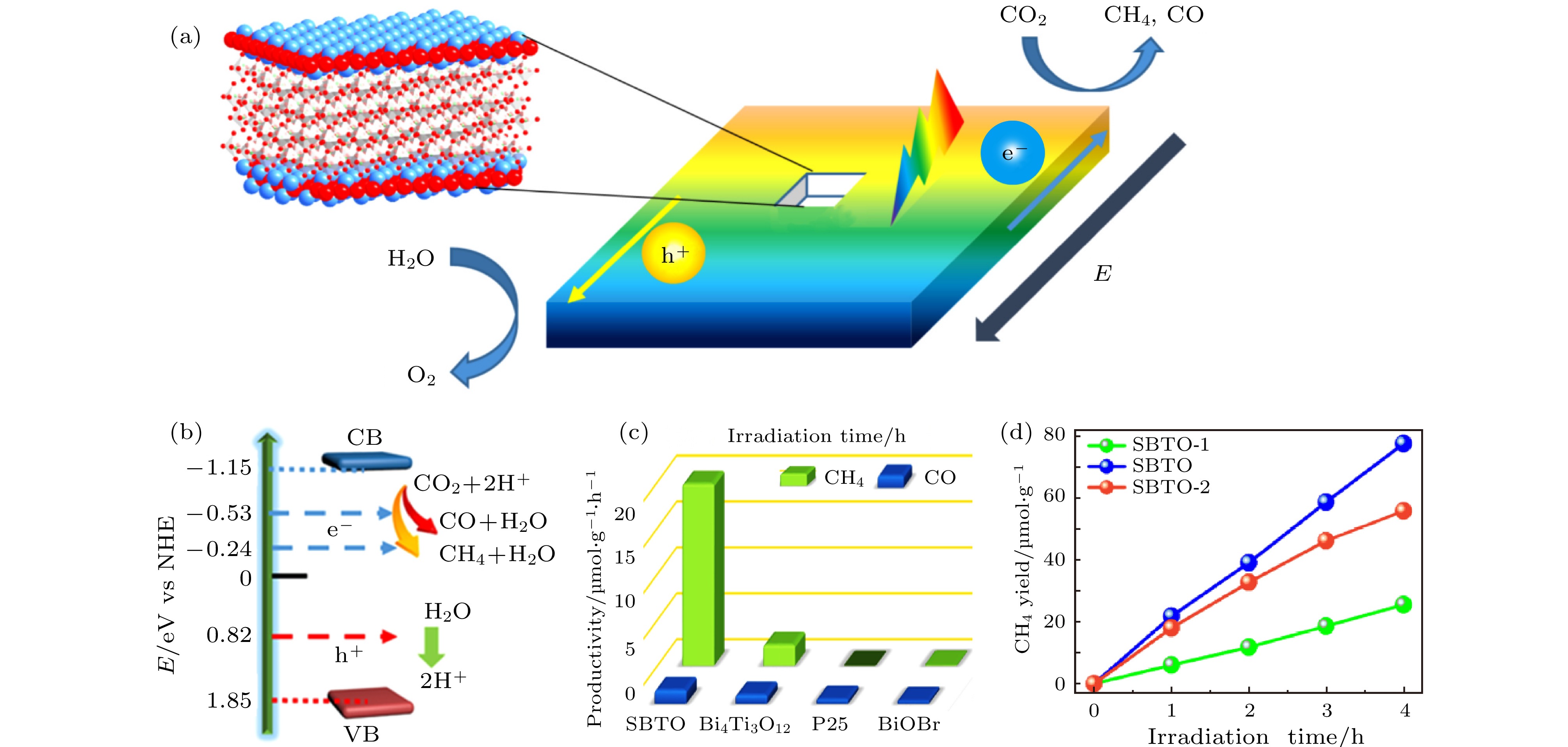 图 15 (a) SrBi4Ti4O15的铁电极化增强光生电子-空穴对分离效率的示意图; (b) SrBi4Ti4O15的能级图; (c) SrBi4Ti4O15, Bi4Ti3O12, P25和BiOBr通过光催化产生CH4和CO的速率; (d) 不同退火温度的SrBi4Ti4O15通过光催化生产CH4的数量-时间曲线[164]
图 15 (a) SrBi4Ti4O15的铁电极化增强光生电子-空穴对分离效率的示意图; (b) SrBi4Ti4O15的能级图; (c) SrBi4Ti4O15, Bi4Ti3O12, P25和BiOBr通过光催化产生CH4和CO的速率; (d) 不同退火温度的SrBi4Ti4O15通过光催化生产CH4的数量-时间曲线[164]Figure15. (a) Schematic diagram of polarization-field enhanced separation of photogenerated charge carriers; (b) diagram for the band energy levels of SrBi4Ti4O15; (c) the corresponding rates over SrBi4Ti4O15, Bi4Ti3O12, P25 and BiOBr; (d) CH4 yield curves of SrBi4Ti4O15 with different annealing temperatures[164]
尽管CH3NH3PbI3在光电领域的研究已经取得了很大的进展, 但是CH3NH3PbI3是否真正的属于铁电材料还存在一定争议, 理论研究说明CH3NH3PbI3的铁电性有助于帮助载流子获得更长的平均扩散长度和有利于电子-空穴对的分离和迁移. 有趣的是, CH3NH3PbI3材料会随着温度、压力以及电场的改变而发生相变, 在立方相(α)、四方相(β)与正交相之间经历可逆转变[170]. 第一性原理计算发现, 四方相(β) CH3NH3PbI3材料存在铁电性. 如果CH3NH3PbI3是铁电材料, 应该满足一些铁电材料特有的特征, 例如: 非中心对称、电场可控的极性以及自发极化. 目前大部分研究更倾向于将CH3NH3PbI3铁电极化的起源归咎于材料中CH3NH3+的旋转. 同时, 一些研究者也在CH3NH3PbI3材料中观察到了铁电畴, 2014年Kutes等[171]通过压电力显微镜在四方相的CH3NH3PbI3材料中发现了铁电畴, 并且铁电极化可以在外电场下发生翻转. 同时, 四方相CH3NH3PbI3也存在电滞回线, 但是在室温下, 其铁电极化保持性比较差[172]. Kim等[173]在没有外电场的情况下, 观察到了CH3NH3PbI3材料的自发铁电极化, 且铁电极化保持时间可以长达60 min, 保持时间的延长是因于材料内部铁电畴尺寸的增加, 这与钙钛矿晶体的大小有关. 2017年Rakita等[174]研究介电响应, 间接测量了四方相CH3NH3PbI3材料的P-E回线, 这是证明铁电材料的直接证据. 图16(a)显示了MAPbI3晶体的介电响应[174], 在铁电半导体中, 介电响应主要由虚部决定, 通过积分介电常数在外电场下的介电常数虚部εim, 可得到图16(b)所示的P-E电滞回线. 以上实验证实了四方相的CH3NH3PbI3材料是铁电材料, 但是目前还没有在CH3NH3PbI3材料中观察到可靠的铁电剩余极化. 尽管最近取得了许多进展, 但是还需要更多的工作来阐明CH3NH3PbI3半导体材料的铁电性.
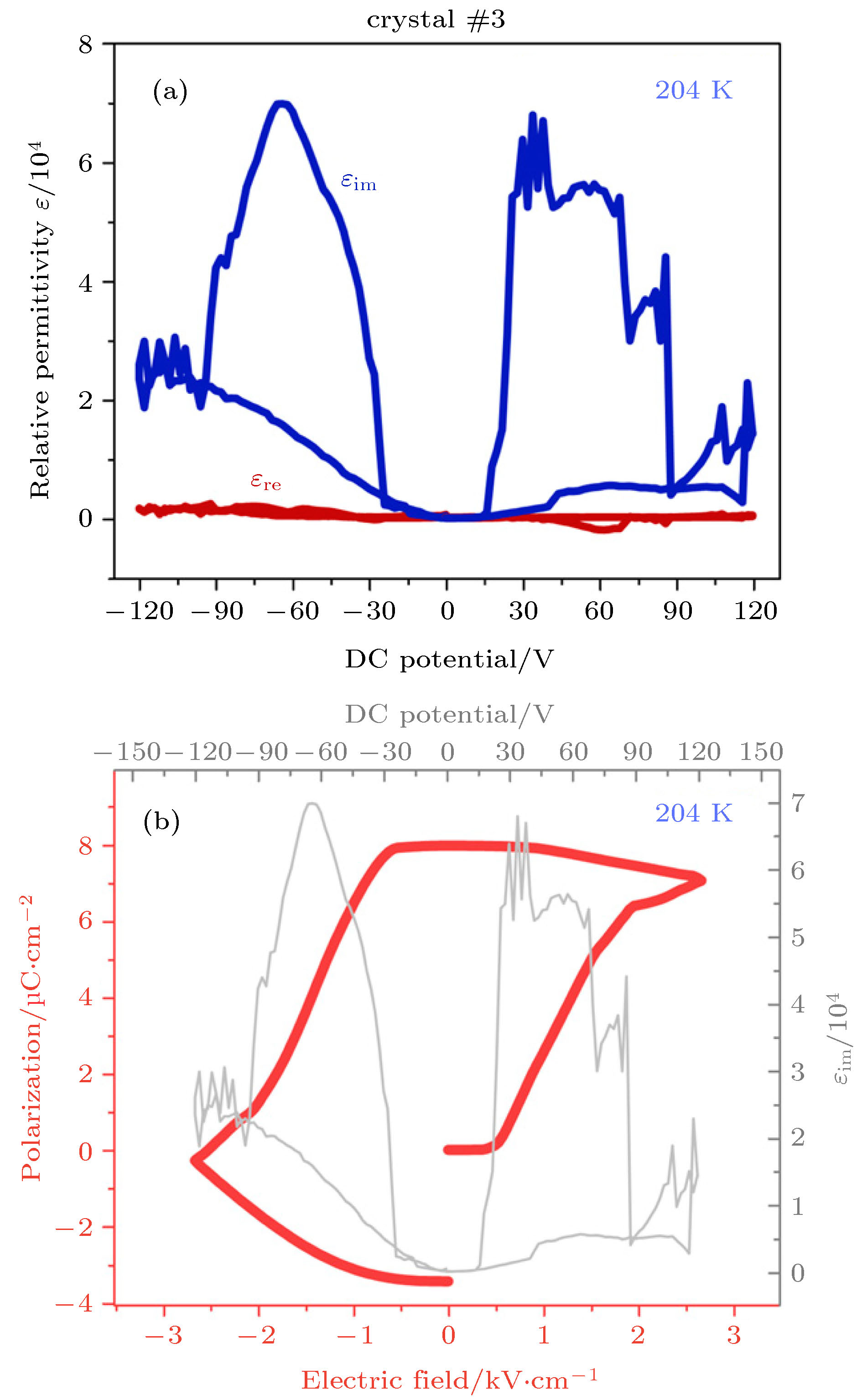 图 16 (a) CH3NH3PbI3晶体在204 K时的介电常数实部εre和虚部εim与外电压的曲线; (b) 通过对εim进行积分得到的P-E电滞回线[174]
图 16 (a) CH3NH3PbI3晶体在204 K时的介电常数实部εre和虚部εim与外电压的曲线; (b) 通过对εim进行积分得到的P-E电滞回线[174]Figure16. (a) Dielectric response at 204 K of a CH3NH3PbI3 crystal, showing that εre dominates the dielectric response; (b) P-E hysteresis loop obtained from integration of εim over applied electric field[174]
CH3NH3PbI3作为光催化材料具有很光明的前景, 但是铅卤钙钛矿材料可以在多种溶液中分解, 本身并不稳定性, 因此其在光电催化领域的实际应用面临很大的难题[175]. 影响光催化器件稳定性的因素包括温度、湿度、光强以及pH值等, 目前研究者主要通过不同的封装技术来解决这些问题[176,177]. 接下来将一一介绍目前铅卤钙钛矿材料光催化器件方面的重要进展.
2014年Luo等[71]采用CH3NH3PbI3制备了高效的太阳能光伏电池, 并将其与光催化电池串联, 从而显著提高了系统的光电催化和能量转换效率. 他们用旋涂法制备了CH3NH3PbI3薄膜, 测试其带隙为1.5 eV, 吸收的光谱范围可以达到800 nm, 在AM1.5G的模拟太阳光下, 其太阳能电池的光电流密度达到了21.3 mA/cm–2, 开路电压为1.06 V. 相比于传统光催化材料的光电流密度以及可见光的吸收范围, 这是一次很大的提升. 与此同时, 串联的CH3NH3PbI3薄膜光伏电池给光催化电极提供电子和空穴, 从而进行光催化析氢与产氧. 为了用高性价比的普通电极取代昂贵的贵金属电极Pt, IrO2或RuO2, 研究人员已经进行了大量的研究: MoS2, NiMo可用于光催化电极材料并在酸性电解液中加快H2析出反应[178-180], 金属氧化物和氢氧化物可以在碱性电解液中用于光催化电极材料并加快O2析出反应[179-181], 但是大多数催化剂不可以同时在酸性或者碱性中保持稳定的析氢与产氧的能力. 如图17(a)所示, 通过将铁(Fe)掺Ni(OH)2, 在Ni泡沫上制备了NiFe层状双氢氧化物(即NiFe-LDH)电极. 光伏电池和光催化装置串联后分别向碱性电解液中的两个NiFe-LDH电极传输电子、空穴进行并进行还原反应产氢、氧化反应产氧, 其能量及载流子传输路径如图17(b)所示. 利用NiFe-LDH/Ni泡沫电极层制备的功能性析氢产氧器件可以稳定在碱性或者酸性条件下析氢产氧, 在实验误差范围内, 通过气相色谱法证实了氢氧比例为2∶1. 在黑暗和模拟光照下(AM1.5G; 100 mW/cm2), 串联光催化器件表现出如图17(c)所示的光电流密度-电压曲线, 并产生2.00 V的开路电压, 同时保持15.7%的光电能量转换效率. 在该光伏电池-串联催化结构中, 在光电流为10 mA/cm2时, 相应的太阳能-氢能的能量转换效应达到12.3%. 同时, 该串联光催化装置表现出了良好的稳定性, 可以稳定工作两个小时以上. 图17(d)为该装置在开与关的模拟光AM1.5G, 100 mW/cm2照明下, 无外电压条件下稳定工作8 min的电流-时间曲线. 此实验为开创高活性、低成本的铅卤钙钛矿材料光催化器件提供了良好的基础与思路.
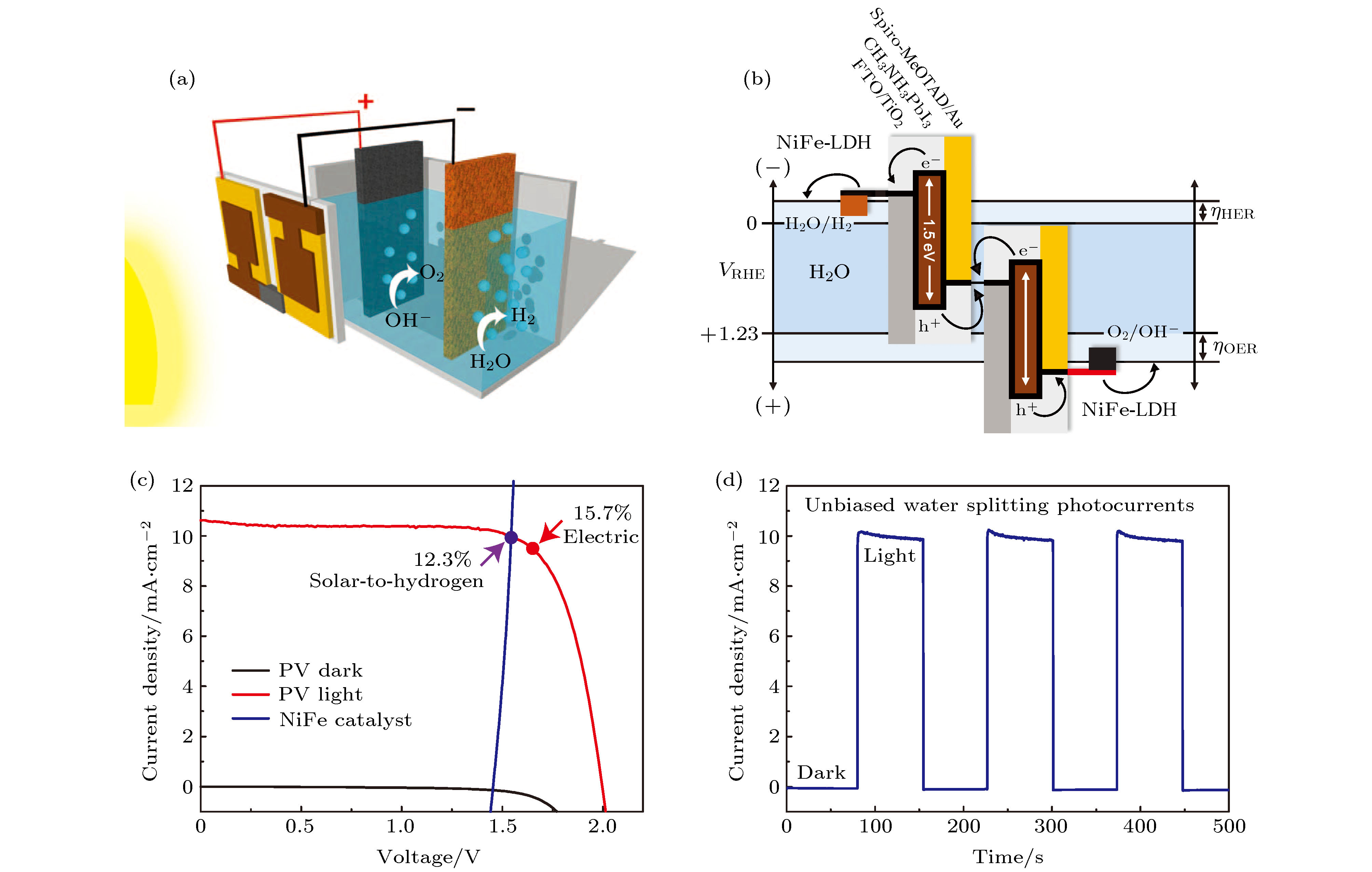 图 17 (a) CH3NH3PbI3薄膜光伏电池和串联催化装置的宏观结构示意图; (b) 材料能带-电荷输运示意图; (c)钙钛矿串联电池两个NiFe-LDH电极之间的光电流密度-电压曲线; (d) 光伏电池串联催化分解水装置的光电流密度-时间曲线[71]
图 17 (a) CH3NH3PbI3薄膜光伏电池和串联催化装置的宏观结构示意图; (b) 材料能带-电荷输运示意图; (c)钙钛矿串联电池两个NiFe-LDH电极之间的光电流密度-电压曲线; (d) 光伏电池串联催化分解水装置的光电流密度-时间曲线[71]Figure17. (a) Schematic diagram of the water-splitting device based on CH3NH3PbI3 film; (b) generalized energy schematic of the perovskite tandem cell for water splitting; (c) J-V curves of the perovskite tandem cell, and the NiFe/Ni foam electrodes in a two-electrode configuration; (d) current density-time curve of the integrated water-splitting device[71]
普通的铅卤钙钛矿材料和器件结构极易受到水的影响, 因此怎样封装铅卤钙钛矿器件是提高其稳定性的重要因素. 水通过顶层空穴传输层的“针孔”进入CH3NH3PbI3是其分解的重要原因, 同时CH3NH3PbI3对外电压、光照和pH值极其敏感[182]. 如图18(a)所示, 为了进一步提高了CH3NH3PbI3光催化器件的稳定性, 2016年Hoang等[68]构建FTO/m-TiO2/CH3NH3PbI3/Spiro-MeOTAD/Au/Ni的结构, 利用介孔TiO2 (m-TiO2)为电子传输层, 无孔Spiro-MeOTAD作为空穴传输层, Au薄膜做电极和防水保护层, Ni薄膜做光阳极催化剂, 有效地增加了器件的产氧能力, 将CH3NH3PbI3器件的光催化分解水的稳定时间提升至数十分钟. 如图18(b)所示, 在1.23 V (vs. SHE)电势下, FTO/m-TiO2/CH3NH3PbI3/Spiro-MeOTAD/Au光阳极的光电流密度为9.2 mA/cm2, Au表面增加Ni催化剂之后该光阳极的光电流密度可以达到17.4 mA/cm2. 2015年Da Peimei等[183]也同样利用Ni层与电极层增大了CH3NH3PbI3光催化器件的光电流密度, 开路电压为0.95 V, 最大光电流密度可以达到19.0 mA/cm2. 2016年Crespo-Quesada等[69]将铅卤钙钛矿光催化器件与金属保护层复合, 在连续照明下, 光催化器件显示出很好的稳定性. 如图18(c)所示, 可以利用一种简单的易溶合金In0.51Bi0.325Sn0.165 (FM)对CH3NH3PbI3器件进行封装, 构建了FTO/PEDOT:PSS/CH3NH3PbI3/PCBM/PEIE/Ag/FM结构, 其中PEDOT:PSS (聚苯乙烯磺酸盐(3, 4-乙撑二氧噻吩))是空穴传输层, PCBM ([6, 6]-苯基-C61-丁酸甲酯)是电子传输层, PEIE (乙氧基化聚乙烯亚胺)可以调节功函数并提高电子从PCBM到Ag的效率, FM不仅阻止电解液渗透而且利于电子迁移到电解液中参与还原反应. 如图18(d)所示, 在AM1.5G的模拟太阳光从FTO玻璃一侧照射条件下, 钙钛矿太阳能电池的短路电流为(15.0 ± 1.4) mA/cm2, 开路电压为(1.0 ± 0.09) V, 光电转换效率分别为7.7% ± 1.5%. 最为突出的是, 器件的光电流密度可以在1.8 h内维持最大值的80%以上.
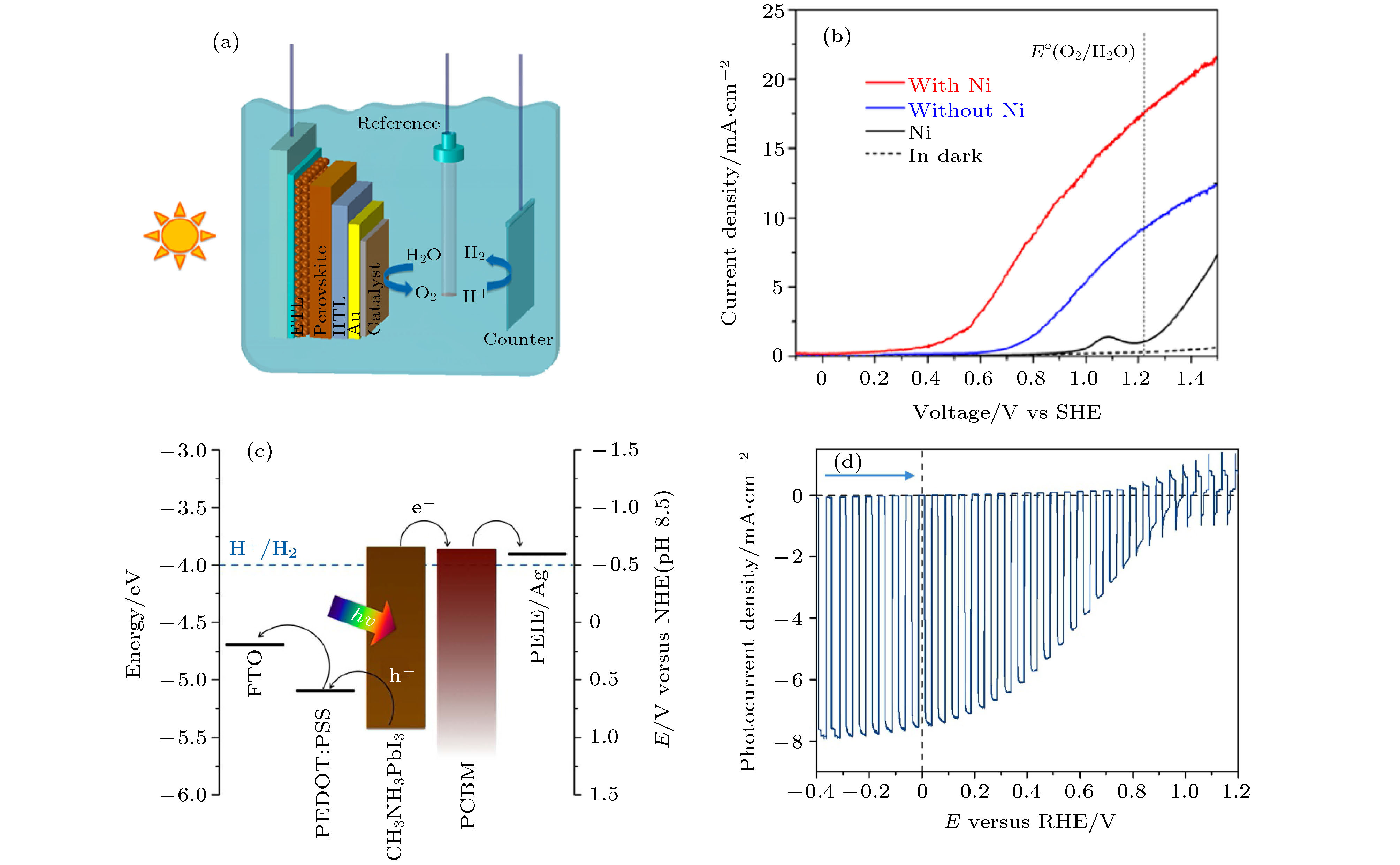 图 18 (a) FTO/m-TiO2/CH3NH3PbI3/Spiro-MeOTAD/Au/Catalyst光催化结构的集成光电解装置示意图; (b) 在模拟光照下Au表面含Ni催化剂(红色曲线)和不含Ni催化剂(蓝色曲线)时钙钛矿光阳极的光电流-外电势曲线[68]; (c) FTO/PEDOT:PSS/CH3NH3PbI3/PCBM/PEIE/Ag的材料能带和功函数匹配图; (d) 光催化器件在开关光循环条件下的I-V图[69]
图 18 (a) FTO/m-TiO2/CH3NH3PbI3/Spiro-MeOTAD/Au/Catalyst光催化结构的集成光电解装置示意图; (b) 在模拟光照下Au表面含Ni催化剂(红色曲线)和不含Ni催化剂(蓝色曲线)时钙钛矿光阳极的光电流-外电势曲线[68]; (c) FTO/PEDOT:PSS/CH3NH3PbI3/PCBM/PEIE/Ag的材料能带和功函数匹配图; (d) 光催化器件在开关光循环条件下的I-V图[69]Figure18. (a) Schematic diagram of FTO/m-TiO2/CH3NH3PbI3/Spiro-MeOTAD/Au/Catalyst integrated photoelectrolysis device with perovskite photoelectrode; (b) photocurrent verus potential comparison diagram of perovskite photoanode with Ni catalyst and Ni catalyst under simulated light[68]; (c) energy and work function matching of FTO/PEDOT:PSS/CH3NH3PbI3/PCBM/PEIE/Ag; (d) photocurrent verus potential diagram of photocatalytic device switching[69]
2018年Zhang等[70]构建了特殊CH3NH3PbI3光催化器件结构, 在不影响铅卤钙钛矿材料本身光催化效率的前提下, 有效提高了光催化器件的使用寿命. 他们构建了Glass/ITO/NiO/CH3NH3PbI3/PCBM/Ag/Ti/Pt结构, NiO层作为空穴传输层, PCBM作为电子传输层. 为了对器件进行保护, 他们在最顶层涂刷一层银浆, 并覆盖一层钛箔, 但是钛箔层的存在会影响器件产氢的效率, 因此, 他们在钛层上通过磁控溅射的方法生长一层Pt颗粒, 有效的避免了钛层对光催化分解水效率的影响. 这种结构在保证铅卤钙钛矿材料本身催化效率的同时, 有效地解决了其在水中易分解和催化效率不稳定的问题. 经过测试, 在AM1.5G的模拟太阳光, 0.5 mol/L H2SO4溶液中, 此结构器件的开路电压为0.95 V, 光电流密度可以达到18 mA/cm2, 并且可以在溶液中稳定工作12 h.
2016年Fu等[184]将CH3NH3PbI3与经典光催化材料TiO2复合来增强光催化效率. 众所周知, TiO2材料3.2 eV的带隙导致其只能吸收紫外光, 但是紫外光只占太阳光的4%, 大大限制了TiO2在光催化方面的应用, 如果可以将光催化器件的吸收光谱扩展到可见光范围内, 那么光催化器件的转换效率将得到巨大的提升[185,186]. 带隙1.5 eV的CH3NH3PbI3可以高效地吸收可见光并产生电子-空穴对, 可以满足以上条件. 因此, 在碳纤维上利用“水热法”生长棒状TiO2, 再通过“两步旋涂法”将CH3NH3PbI3镶嵌到TiO2中, 最后利用金属氧化物Cu2O材料进行封装. 可见光在CH3NH3PbI3中激发大量的电子-空穴对, 电子通过TiO2空穴传输层到达Pt电极参与还原反应并生产氢气, 而空穴通过Cu2O空穴传输层到达电解液(1 mol/L Na2SO4)参与氧化反应并产生氧气. 同时, Cu2O可以有效地隔离钙钛矿材料与水溶液的接触, 保证了该复合器件在光催化过程中的稳定性.
TiO2载流子迁移率较高, 可见光吸收系数比较高, 稳定性好, 无毒无害, 是当前光催化和光电催化材料中的杰出代表, FTO/H:TiO2在AM1.5G模拟太阳光照射下的光电流密度为1.97 mA/cm2. 随着铁电材料研究的不断发展, 铁电材料在光催化领域将会得到足够的重视. 根据目前的研究表明, 对于铁电材料用于光电极, 通过极化, 光电流密度大部分可以提升50%左右. 目前更为流行的研究是将经典的氧化物光催化材料与钙钛矿铁电材料复合, 例如: TiO2与BaTiO3, BiVO4与BiFeO3等, 这样的异质结构结合了二者的优点, 同时铁电层对氧化物起到保护作用, 延长了TiO2等材料的使用寿命. 对于将铁电材料与传统氧化物材料复合作为光电极, 退极化场EP可以将大部分材料的光电流提升40%左右. 此外, CH3NH3PbI3材料具有合适的带隙(1.5 eV), 可见光吸收系数很高, 少数载流子不容易被缺陷俘获, 其少数载流子寿命τm长、扩散长度Lm可以达到毫米量级, 因此其能源转换效应很高. 比如ITO/NiO/CH3NH3PbI3/PCBM/Ag/Ti/Pt样品在AM1.5G模拟太阳光照射下的光电流密度为18 mA/cm2, 这远大于氧化物铁电材料的光电流密度. 但是, CH3NH3PbI3的Pb元素有毒, 它在光照、潮湿环境以及加热等条件下很不稳定, 难以得到大规模的实际应用.
当前, 对氧化物钙钛矿铁电材料以及卤素钙钛矿材料在光催化方面的研究不断深入, 更多的问题也有待解决, 将光催化材料进行商业化应用, 还有较远的路程. 钙钛矿铁电材料在光催化领域的应用存在若干挑战, 包括: 提高光催化材料的电荷分离效率、能量转换效率、结构和性能稳定性, 确保光催化材料对环境友好无毒无害, 提高光催化材料的性价比以便能够被市场接受等. 为了实现预期的光催化水分解和能源合成, 在未来钙钛矿铁电材料光电极的研究中可以优先考虑以下四方面.
1)铁电光催化材料的设计和制备. BiFeO3, PbTiO3等铁电材料虽然具有外电场容易调控的较大铁电极化, 但是其带隙较大, 载流子迁移率较低, 不适合做光催化材料吸光层; SrTiO3的带隙较大; Bi2FeCrO6也存在载流子迁移率较低的问题; MAPbI3具有优异的半导体性能, 然而其在光照和液体环境中稳定性仍然面临巨大挑战. 因此, 需要设计和制备具有较大铁电极化和优异半导体性能的新型铁电半导体材料. 其次, 需要制备比表面积较大的铁电材料纳米片或者其他纳米结构, 并让铁电极化背向或者面向其表面或界面, 从而确保退极化场EP能够高效地分离光生电子-空穴对.
2)光电化学过程的系统研究. 应综合考虑包含铁电材料的光催化系统的光收集、电荷分离和表面反应的特性, 实现更有效的水分解、能源合成和分解污染物. 应仔细研究特定钙钛矿铁电材料光电极的性能决定因素, 阐明铁电、压电和热释电效应对分离电子-空穴对的贡献, 以便进一步提高光生电子-空穴对的分离和迁移效率. 另外, 钙钛矿铁电材料光电极上的光催化能源合成比光催化水分解具有更复杂的动力学机理, 为了确定相关的科学问题, 有必要密切关注有机物的电化学合成研究.
3)催化性能稳定性研究. 对于实际的光催化水分解、能源合成和分解污染物, 对钙钛矿铁电材料光电极的要求不仅包括优异的初始光催化性能, 而且还具有可接受的稳定性. 在设计光催化结构时, 应谨慎地避免光电极与氧化还原产物之间可能发生的物理和化学相互作用. 迄今为止, 大多数研究都集中在提高三元金属氧化物光电极的光催化水分解方面, 但对其光催化水分解稳定性的研究较少. 更详细地研究钙钛矿铁电材料光电极/电解质的界面非常重要, 这将增加我们对三元金属氧化物光电极的光催化水分解稳定性的认识. 特别是, 应分析光催化水分解和有机合成的动力学过程对三元金属氧化物光电极稳定性的影响.
4)高效筛选和数据库. 为了能够快速地、准确地从大量钙钛矿铁电材料中找到有效的钙钛矿铁电材料光电电极, 高识别的筛选方法特别是实验研究和计算预测相结合的方法具有重要意义. 有必要建立一个基于鉴定和筛选结果的标准数据库, 从而提供钙钛矿铁电材料的带隙、能带位置和光电性能, 这将促进钙钛矿铁电材料光电极的发现和进一步研究.
总而言之, 尽管本文对钙钛矿铁电半导体材料光催化和光电催化的讨论并不详尽, 我们希望它能够为研究钙钛矿铁电光催化材料降解染料、分解水制氢和其他能源材料提供有价值的信息和启发.
