全文HTML
--> --> -->随着现代科学技术的进步和发展, 压电陶瓷从最初的BaTiO3 (BT)体系发展到以Pb(Zr1–xTix)O3 (PZT)为代表的铅基压电陶瓷体系. 由于在准同型相界(MPB)附近的PZT基陶瓷具有非常优异的综合电学性能, 因此目前应用最为广泛. 但是, PZT基陶瓷中Pb的含量约为原料总量的60%, 而Pb的毒性以及易挥发性使铅基压电陶瓷在生产、使用及废弃后处理过程中都会给生态环境和人类自身健康造成严重损害[3,4]. 近年来, 世界各国都立法禁止使用含铅的电子材料, 如欧盟颁布执行的《关于限制在电子电器设备中使用某些有害成分的指令》(Restriction on the Use of Hazardous Substancces), 日本通过的《家用电子产品回收法案》, 我国信息产业部出台的《电子信息产品污染防治管理办理》等. 因此, 发展环境友好的无铅压电陶瓷成为当前国际压电材料研究的热点之一[3,5-8].
目前, 主要的无铅压电陶瓷可分为以下几种结构: 钙钛矿结构、钨青铜结构、铋层状结构. 因为钙钛矿结构压电陶瓷的压电性能优异, 制备工艺与传统铅基陶瓷工艺兼容, 是目前研究最广泛的一大类无铅压电陶瓷. 常见的钙钛矿结构无铅压电陶瓷主要包括BT基、钛酸铋钠(Bi0.5Na0.5TiO3, BNT)基和铌酸钾钠(K0.5Na0.5NbO3, KNN)基等.
作为最早发现的BT压电陶瓷, 具有介电常数高、机电耦合系数大、介电损耗较小等特点, 但是因为该类陶瓷的居里温度较低(TC ≈ 120 ℃), 其物理性能的温度稳定性差, 并且烧结温度高(大于1350 ℃), 因此, 需要大幅度提升其压电性能来满足实际应用的需求. 目前, 更多地是利用BT陶瓷具有高介电性能的特点而作为介质材料进行应用.
BNT陶瓷具有较高居里温度(TC ≈ 320 ℃)、高剩余极化强度(Pr ≈ 38 μC/cm2)、较好的压电性能以及较低的烧结温度[9], 但BNT陶瓷室温下矫顽场很大(Ec ≈ 73 kV/cm)、去极化温度较低(约为100 ℃)等, 从而限制了该类陶瓷的进一步应用.
KNN基陶瓷具有高居里温度(TC ≈ 420 ℃)和较好的压电性能, 但是纯的KNN陶瓷存在高温下K, Na等元素易挥发、工艺敏感性强、难以通过传统陶瓷工艺实现致密化等问题, 长期以来研究进展缓慢. 2004年, Saito等[10]通过化学掺杂改性, 利用反应模板晶粒生长(RTGG)法制备的KNN基织构陶瓷性能达到d33 ≈ 416 pC/N, 机电耦合系数kp ≈ 0.61, TC ≈ 253 ℃. 2018年, Li等[11]采用织构化工艺, 加入3 mol%的NaNbO3作为模板制备的0.96(K0.5Na0.5)(Nb0.965Sb0.035)O3-0.01CaZrO3-0.03(Bi0.5K0.5)HfO3织构陶瓷体系, 其d33高达700 pC/N, kp ≈ 0.76, TC ≈ 242 ℃. 2019年, Tao等[12]采用固相合成法, 制备的(0.96–x)K0.48Na0.52Nb0.95Sb0.05O3-0.04Bi0.5(Na0.82K0.18)0.5HfO3-0.4%Fe2O3-xAgSbO3非织构陶瓷, 其d33可达到(650 ± 20) pC/N, TC ≈ 180 ℃. 这些陶瓷的性能接近甚至超过了PZT4的性能, 表明KNN基陶瓷是最有可能取代铅基压电陶瓷的无铅压电材料体系之一.
本文基于国际最新研究结果和研究工作, 综合分析并简要介绍了KNN基无铅压电陶瓷高压电活性的国内外的研究, 特别是高性能KNN基无铅压电陶瓷、制备工艺优化与相关理论基础的研究进展, 并对今后的研究方向进行了展望.
2
2.1.高压电性能
32.1.1.相界构建
与PZT基压电陶瓷类似, KxNa1–xNbO3陶瓷是由铁电体KNbO3和反铁电体NaNbO3 形成的二元固溶体. 随着温度的变化, KNN陶瓷会在–123 ℃时经历三方相(R)-正交相(O)转变, 在210 ℃时发生正交相-四方相(T)转变, 在410 ℃下发生四方相-立方相(C)转变[8]. 利用离子掺杂或取代、添加第二组元或多组元化合物等方法, 可将KNN基无铅压电陶瓷的相转变温度移动到室温附近, 构建室温下的新型相界. 在新型相界区域, 因为多相共存, 有更多可能的极化状态, 不同相之间的各向异性减小, 电畴更易偏转, 从而使KNN基无铅压电陶瓷的性能得到提高[8,14].在KNN基中添加一定的离子(Li+, Ag+, Sb5+, Ta5+)或Bi0.5(K/Na/Li)0.5TiO3, (Ba/Ca/Sr)TiO3等钙钛矿结构化合物, 可以提升KNN基陶瓷中四方相的比例, 把TO-T由200 ℃左右调节至室温附近, 使KNN基陶瓷在室温下从单一的正交相结构转变为O-T相共存, 构建出室温下的O-T相界, 提升KNN基陶瓷的压电性能. 表1 归纳了一些构建室温下O-T相界的KNN基陶瓷体系的研究进展[15-23].
| Material system | d33/pC·N–1 | kp | TC/℃ |
| KNLANT[15] | 252 | 0.454 | 438 |
| KNLN-BCZT[16] | 180 | 0.34 | 425 |
| KNN-KLN[17] | 121 | 0.39 | — |
| KNLN-AS[18] | 230 | 0.39 | 430 |
| KNLN-BNCT[19] | 262 | 0.36 | ~400 |
| KNN-BC[20] | 165 | 0.40 | ~390 |
| KNN-LS[21] | 280 | 0.494 | 364 |
| KNN-BLZ[22] | 265 | 0.365 | 364 |
| KNN-BC-BNH[23] | 272 | 0.47 | 333 |
表1室温下具有O-T相界的KNN压电陶瓷性能
Table1.Properties of KNN ceramics with O-T phase boundary at room temperature.
从表1可以看出, 可以通过一定的离子掺杂或者多组元复合, 提升KNN基陶瓷的压电性能, 尤其是添加了Li+的陶瓷体系, 能够在获得较高压电性能的同时保持较高的居里温度.
通过构建室温下的O-T相界, 可以提升KNN基陶瓷的压电活性, 但是相对于铅基陶瓷仍有一定的差距. 在KNN基陶瓷中降低TO-T的同时, 添加一定的离子(Sb5+, Ta5+)或Bi0.5(K/Na/Li/Ag)0.5(Zr/Hf)O3, (Ba/Ca/Mg)ZrO3, Bi(Sc/Co/Fe)O3等化合物, 可以提升KNN基陶瓷中三方相的比例, 把TR-O由–123 ℃左右调节至室温附近, 在室温下构建出三方-正交-四方(R-O-T)或三方-四方(R-T)新型相界. 其中R-O-T新型相界是同时调控TO-T和TR-O至室温附近, 使其室温下同时存在R, O, T三相. R-T新型相界是进一步压缩陶瓷体系的TO-T和TR-O之间的温区, 使其在室温附近重叠, 形成新的相界, 室温下存在R相和T相. 多相共存时, 相与相之间存在较低的能量势垒, 促进极化翻转, 优化KNN基陶瓷的电学性能. 表2和表3分别列出了室温下构建R-O-T[12,24-34]和R-T[35-47]相界的部分研究成果.
| Material system | d33/pC·N–1 | kp | TC /℃ |
| KNN-BNZ-BG[24] | 312 | 0.44 | 341 |
| KNN-BZ-BNZ[25] | 345 | 0.50 | ~260 |
| KNN-NS-BNKZH[26] | 452 | 0.63 | ~270 |
| KNNS-BNCZ[27] | 415 | 0.46 | 245 |
| KNNTS-BNKZ[28] | 400 | 0.46 | 240 |
| KNN-BNZN[29] | 318 ± 10 | — | 360 |
| KNNS-BKZH[30] | 451 | 0.52 | 258 |
| KNNS-BLKZ[31] | 385 | — | 245 |
| KNNS-SZ-BNH[32] | 470 ± 5 | 0.51 ± 0.02 | 244 |
| KNNS-BS-BNZ[33] | ~480 | — | ~225 |
| KNNS-BNZ-BZ[34] | 610 | 0.58 | 241 |
| KNNS-BNKZ-Fe-AS[12] | 650 | — | ~180 |
表2室温下具有R-O-T相界的KNN压电陶瓷的性能
Table2.Properties of KNN ceramics with R-O-T phase boundary at room temperature.
| Material system | d33 /pC·N–1 | kp | TC/℃ |
| KNNS-BNZSn[35] | 465 | 0.51 | 240 |
| KNNS-BZH[36] | 410 | — | 255 |
| KNNS-BNKZ[37] | 490 | 0.46 | 227 |
| KNNTS-BNKZ[38] | 460 | 0.40 | ~220 |
| KNNS-BNH[39] | 419 | 0.45 | 242 |
| KNNS-BKZS[40] | 430 | — | 243 |
| KNNS-BNLCZ[41] | 485 | 0.48 | 227 |
| KNNS-BNKH[42] | 525 | — | ~210 |
| KNNS-BF-BNZ[43] | 550 | — | 237 |
| KNNS-CZ-BKHT-MnO2[44] | 425 | 0.49 | 215 |
| KNNS-BZ-BKH[45] | 570 ± 10 | — | ~190 |
| KNNS-BNZ-BF[46] | 511 | 0.515 | 269 |
| KNANS-BNZ[47] | 440 | 0.50 | 250 |
表3室温下具有R-T相界的KNN压电陶瓷的性能
Table3.Properties of KNN ceramics with R-T phase boundary at room temperature.
从表1—3可以看出, 相较于O-T相界, 具有R-O-T/R-T相界的KNN基无铅压电陶瓷的电学性能提升程度更大, 表明相界类型对其压电活性影响很大. 相较于只降低TO-T构建室温下O-T相界, 在KNN陶瓷中构建R-O-T/R-T相界则更加复杂, 需要引入多种添加离子或者化合物才能实现. 值得注意的是, 由于KNN陶瓷对组分和工艺的敏感性以及掺杂离子的相互影响, 在KNN基陶瓷中构建新型相界需要进行组分和工艺的精细调控.
图1(a)给出了KNN基无铅压电陶瓷的压电常数d33的历史演变图. 2014年Wang等[37]利用固相烧结法合成的 (1–x)(K1–yNay)(Nb1–zSbz)O3-xBi0.5(Na1–wKw)0.5ZrO3非织构陶瓷, 室温下具有R-T相界, 陶瓷的d33 ≈ 490 pC/N, TC ≈ 190 ℃. 随后该团队合成的KNNS-BZ-BKH非织构陶瓷, 其d33高达(570 ± 10) pC/N, TC ≈ 190 ℃; KNNS-BF-BNZ非织构陶瓷[43], 其d33≈550 pC/N, TC≈ 237 ℃. 2018年, Li等[11]采用织构化工艺制备的KNNS-CZ-BKH织构陶瓷, 其d33 ≈ 700 pC/N, kp≈0.76. Liu等[48]报道了具有高压电性能兼具优异的温度稳定性的KNN-BZ-BNH-MnO2非织构陶瓷, 其

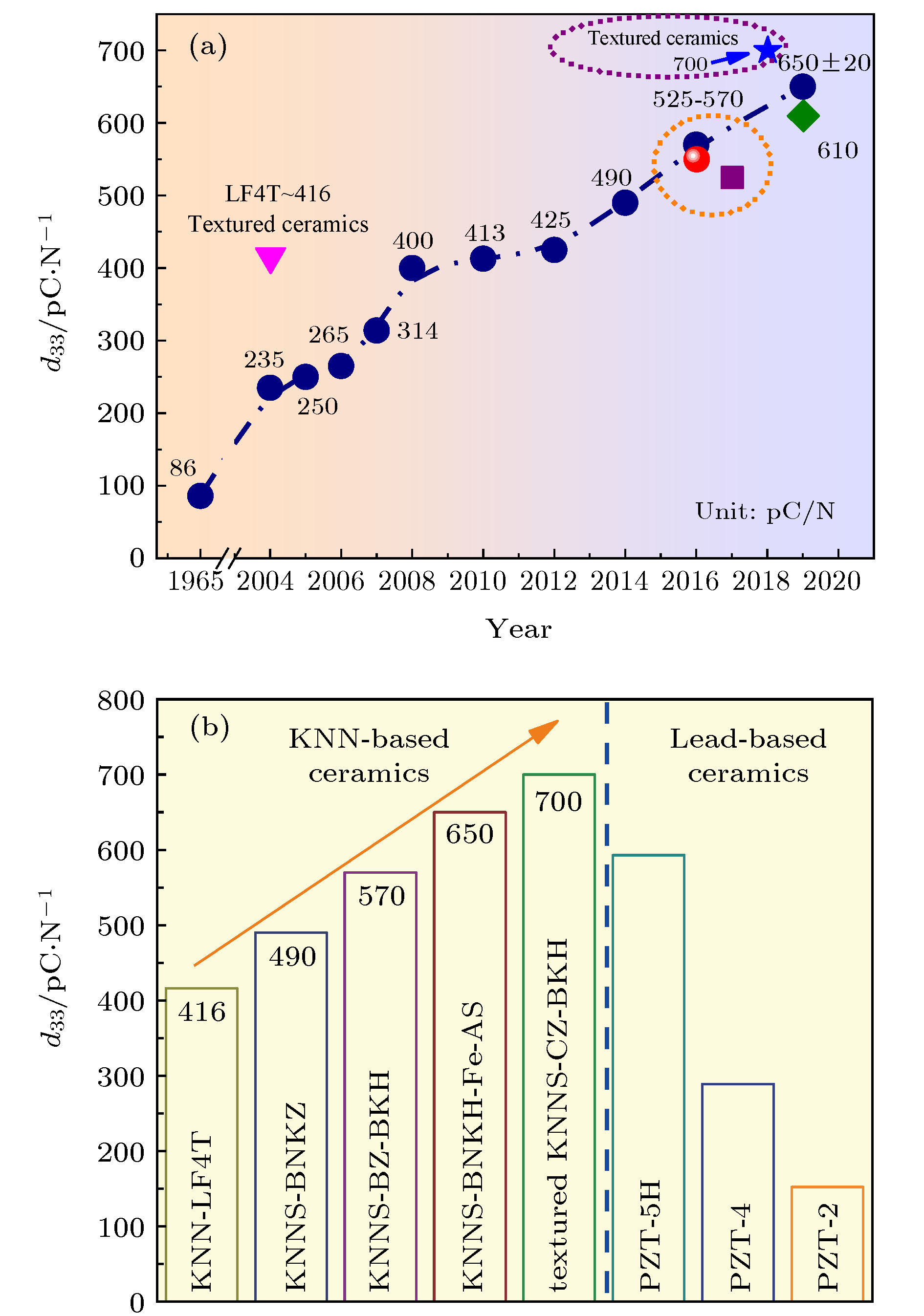 图 1 (a) KNN基无铅压电陶瓷d33的历史演变图; (b) KNN基无铅压电陶瓷与铅基陶瓷d33对比图[11,12,34,37,43,45]
图 1 (a) KNN基无铅压电陶瓷d33的历史演变图; (b) KNN基无铅压电陶瓷与铅基陶瓷d33对比图[11,12,34,37,43,45]Figure1. (a) Historical evolution in d33 values of KNN-based ceramics as a function of time; (b) comparison of d33 values among KNN-based ceramics and PZT materials[11,12,34,37,43,45].
3
2.1.2.畴结构调控
铁电材料在降温过程中, 在低于居里温度时, 铁电体从顺电相转变为铁电相, 其晶体结构从高对称性转变为低对称性. 晶体的正负中心不再重合而形成自发极化, 并且在同一个铁电相中, 自发极化存在多个等效的自发极化方向. 对于铁电晶体来说, 由于自发极化使其正负两端产生束缚电荷, 束缚电荷产生的电场与极化方向相反, 会在铁电晶体中产生退极化场. 为了降低退极化场形成的静电能以及晶格畸变产生的应变能, 在铁电单晶或者陶瓷中会出现许多小的区域, 每个区域内的晶胞自发极化方向相同, 而相邻区域内自发极化方向取向不同. 自发极化方向一致的小区域称为电畴. T相中的自发极化方向为


压电陶瓷的压电响应分为本征与非本征贡献两部分, 其中本征贡献主要取决于晶格畸变, 而非本征贡献与电畴运动有关, 包括畴壁振动、电畴翻转、畴壁运动. 因此, 除了构建相界外, 畴结构调控对于提升KNN基无铅压电陶瓷的压电性能至关重要.
电子显微技术是常用的电畴观测方法, 包括透射扫描电子显微镜(TEM)、压电力显微镜(PFM)以及扫描电子显微镜(SEM)等. 将合成后的KNN基无铅压电陶瓷样品进行减薄处理后可以通过TEM进行电畴结构的观测[49-51], 在测量畴结构尺寸的同时还可以利用选区电子衍射进行特定区域的结构分析[49]. 使用原位透射电镜观测, 则能够观察外电场作用下电畴结构的变化[52]. 因为不同极性电畴被酸腐蚀的程度不同, 通过该特点可以采用SEM直接观察腐蚀后的KNN基陶瓷电畴结构. 文献[53-55]利用混合酸溶液(浓度为37%的盐酸与40%氢氟酸溶液体系比1∶1)对KNN基陶瓷进行腐蚀, 并通过SEM观测了其相应的畴结构. López-Juárez等[56]使用48 vol.%的氢氟酸腐蚀(K0.5Na0.5)NbO3陶瓷体系后, 观测到正交相的陶瓷中90°畴和180°畴结构. 压电力显微镜(PFM)因为分辨率高、对样品要求低, 是一种研究畴结构的重要检测方法, 被广泛应用在KNN基无铅压电陶瓷中[57,58]. 其中, 利用原位PFM中电场、温度场的变化可以判断KNN基无铅压电陶瓷中畴的翻转难易程度以及温度稳定性[59,60].
通过电子显微技术可以在高压电性能、多相共存的KNN基陶瓷中同时观察到条纹畴以及亚微米级不规则的畴结构. 图2(a)—(c)给出了在室温下不含相界, O-T, R-O-T, R-T相界的KNN基陶瓷的畴结构. 未掺杂的K0.5Na0.5NbO3陶瓷, 其畴结构多为鱼骨状、水痕状, 其中鱼骨状畴结构对应90°畴, 水痕状畴结构对应180°畴[61]. K0.5Na0.5 NbO3陶瓷在室温下为正交相, 其90°畴结构尺寸为0.5—1 μm宽以及3—7 μm长, 180°畴的尺寸为3—10 μm宽, 20—40 μm长[56]. 通过构建室温下的新型相界, KNN陶瓷体系中的畴结构形状或者尺寸发生改变. 如图2(b)和图2(c)所示, 对于KNN基陶瓷中构建了室温下的O-T/R-O-T/R-T相界时[34,43,62,63], 可以利用TEM或者SEM观察到条纹畴或者不规则形状的复杂畴结构, 并且畴结构的尺寸下降到亚微米级或者纳米级. 构建室温下新型相界可以使KNN陶瓷具有更多极化方向. 相对于室温下为O相的KNN陶瓷, 位于多相共存区域的KNN陶瓷含有更多的极化状态和晶体取向度, 不同相之间的各向异性减小, 因此畴的翻转和移动比单相陶瓷更容易[8,14]. 有研究表明, 新型相界施加一定的外加电场后, 除了发生电畴翻转外还会产生电诱导相变, 诱导相为单斜相(M), 极化矢量通过中间诱导相更易翻转, 从而增强陶瓷的压电性能[49,64].
 图 2 (a)正交相(K0.5Na0.5NbO3陶瓷[56,61]); (b)室温下O-T相界(KNNL-BZ-BNT陶瓷体系[62], KNNSL-BNZ-BZ-MnO2陶瓷体系[63]); (c)室温下R-T/R-O-T相界KNN基陶瓷的畴结构(KNNS-BF-BNZ陶瓷体系[43], KNNS-BNZ-BZ陶瓷体系[34])
图 2 (a)正交相(K0.5Na0.5NbO3陶瓷[56,61]); (b)室温下O-T相界(KNNL-BZ-BNT陶瓷体系[62], KNNSL-BNZ-BZ-MnO2陶瓷体系[63]); (c)室温下R-T/R-O-T相界KNN基陶瓷的畴结构(KNNS-BF-BNZ陶瓷体系[43], KNNS-BNZ-BZ陶瓷体系[34])Figure2. Domain structures of KNN-based ceramcis with different phase boundaries at room temperature: (a) Orthorhombic (K0.5Na0.5NbO3 ceramics[56,61]); (b) O-T phase boundaries (KNNL-BZ-BNT ceramics[62], KNNSL-BNZ-BZ-MnO2 ceramics[63]); (c) R-T/R-O-T phase boundaries (KNNS-BF-BNZ ceramics[43], KNNS-BNZ-BZ ceramics[34]).
在KNN基无铅压电陶瓷中进行多组元掺杂, 可以显著地降低各元素的扩散速率, 破坏晶体的长程有序, 从而容易形成纳米级尺寸的电畴. 例如, Sun等[52]发现 (K0.48Na0.52)(Nb0.955Sb0.045)O3-0.01SrZrO3陶瓷体系未掺入(Bi0.5Ag0.5)ZrO3前的畴结构尺寸为0.2—1 μm, 在掺入(Bi0.5Ag0.5)ZrO3后的陶瓷体系中同时观察到了30—65 nm, 65— 160 nm的条状畴以及30—45 nm尺寸的纳米畴. 该陶瓷体系掺杂后的d33 ≈ 487 pC/N. Liu等[48]构建的KNNS-BZ-BNH-MnO2 体系具有10—100 nm尺寸的畴结构, 其压电系数为600 pm/V. Zheng等[42]通过TEM研究发现在KNN陶瓷中掺入Sb和Bi(Na, K)HfO3可以使陶瓷具有宽度为10—30 nm, 长度为100—300 nm尺寸的纳米畴, 从而提高该陶瓷的压电性能(d33 ≈ 525 pC/N). Tao等[12]制成的高压电性能(d33 ≈ (650 ± 20) pC/N)的KNN基陶瓷体系的畴结构约为2 nm的纳米畴. Zhou等[34]获得的KNN基陶瓷压电性能d33 ≈ 610 pC/N, 畴结构尺寸为50—70 nm. 表4列举了一些高性能KNN基无铅压电陶瓷压电常数与畴结构尺寸[12,34,42,46,48,51,52,54,57,65-70]. 从表4可以看出, 畴结构尺寸与陶瓷压电性能的优异密切相关, 尤其是纳米尺寸的畴结构对陶瓷的高压电性能起着重要的作用. 这是因为畴壁能与畴尺寸的平方根成正比[56], 纳米尺度的畴结构在外电场作用下更容易翻转, 从而对KNN陶瓷的电学性能起到增强作用. 因此, 对畴结构进行调控可以提升KNN基体系的压电性能.
| Material system | d33 or $ {d}_{33}^{*} $ | Domain size |
| KNNS-SZ-BAZ[52] | 487 pC/N | 30—65 nm, 65—160 nm, 30—45 nm |
| KNNS-BZ-BNH[48] | 600 pm/V | 10—100 nm |
| KNNS-BNKH[42] | 525 pC/N | 10—30 nm |
| KNNS-BNKZ-Fe-AS[12] | (650 ± 20) pC/N | 2 nm |
| KNNS-BNZ-BZ[34] | 610 pC/N | 50—70 nm |
| KNNT-BNKZ-CZ[51] | 482 pm/V | 60 nm |
| KNNS-BZ-BNZ[65] | 300 pC/N | 150 nm—1.0 μm |
| KNNS-CZ-BKH[66] | 550 pC/N | 30—230 nm |
| KNNS-BNH[67] | 512 pC/N | 100 nm |
| KNNS-SZ-BNZ[68] | 450 pC/N | 50—200 nm |
| KNLNTS[54] | 455 pC/N | 110—310 nm |
| KNNS-BNZ-BF[46] | 510 pC/N | < 1 μm |
| KNN-BNZ-MnO2-Sb2O3[69] | 318 pC/N | < 1 μm |
| KNN-BI-BNZ[57] | 317 pC/N | ~200 nm |
| KNNdNS-BNZ[70] | 400 pC/N | ~ 1 μm |
表4KNN基无铅压电陶瓷压电常数与畴结构尺寸
Table4.Piezoelectric constant of KNN ceramics with domain size.
2
2.2.温度稳定性
在KNN基无铅压电陶瓷中添加三方或四方诱导物构建室温下新型相界, 其本质是将KNN基陶瓷的相变点移动至室温附近, 获得高的压电性能. 目前, KNN基无铅压电陶瓷的压电性能已经可以满足一部分压电器件及应用的性能要求. 但是, 与PZT铅基陶瓷材料中的准同型相界不同, KNN基陶瓷中构建的新型相界为多晶型相界(PPB), 不仅有组分依赖性, 还有温度依赖性. 当温度上升或者下降时, KNN基无铅压电陶瓷会发生相变恢复至单相结构, 从而导致该陶瓷体系压电性能下降, 因此, 在小于TC的温度范围内存在另一个多晶型相变是KNN压电陶瓷温度稳定性差的本质原因. 因此, 为了能够解决无铅压电陶瓷的应用问题, 还需要考虑KNN基无铅压电陶瓷的高居里温度以及良好的温度稳定性.3
2.2.1.居里温度
压电陶瓷中压电性能的提升往往伴随着该体系居里温度的下降, 如图3所示[12,15-47]. 压电材料可以在大约1/2TC—2/3TC的温度范围内安全使用. 因此, 在构建室温下新型相界的同时, 应该考虑其实际应用的温度范围, 调控压电性能与居里温度之间的关系, 发展同时具有高压电性能以及高居里温度的KNN基无铅压电陶瓷体系, 这将对无铅压电陶瓷的发展具有极大的推动作用. 图 3 KNN基无铅压电陶瓷d33与TC对比图[12,15-47]
图 3 KNN基无铅压电陶瓷d33与TC对比图[12,15-47]Figure3. Comparison of d33 and TC values of KNN-based ceramics[12,15-47].
表5列举了部分兼具高压电性能高居里温度的KNN陶瓷体系[24,57,71-81]. 其中Jiang等[74]通过在KNN基陶瓷中添加BiAlO3组元, 可以有效提升陶瓷的电学性能, 保持高居里温度, 该陶瓷体系 d33 ≈ 355 pC/N, TC ≈ 335 ℃. 另外, 在KNN基陶瓷中添加BiGaO3[24], BiInO3[57]等组元也可以使陶瓷同时具有高d33和高TC.
| Material system | d33 /pC·N–1 | TC/℃ |
| KNN-BNH[71] | 385 | 315 |
| KNN-BNZ-LF[72] | 345 | 314 |
| KNN-BNZ-MnO2[73] | 300 | 345 |
| KNN-BNZ-BG[24] | 312 | 341 |
| KNN-BNZ-BA[74] | 355 | 335 |
| KNN-BAZ[75] | 347 | 318 |
| KNN-BNZ[76] | 360 | 329 |
| KNN-BKZ-BZ[77] | 305 | ~300 |
| KNLNS-BS[78] | 325 | 358 |
| KNN-BNZS[79] | 350 | 315 |
| KNN-BS-BNKLZ[80] | 366 | 335 |
| KNN-BNT-BNZ[81] | 318 | 326 |
| KNN-BNZ-BI[57] | 317 | 336 |
表5同时具有高压电性能和高居里温度的KNN陶瓷体系
Table5.The KNN-based ceramics with high piezoelectric constant and high Curie temperature.
在KNN中如果设计陶瓷体系的 A位碱金属为非化学计量比, 可以通过弥补高温下的碱金属挥发提高陶瓷体系的致密度, 从而提升KNN基压电陶瓷的压电性能[82-84], 而非化学计量比的Nb5+对KNN基压电陶瓷的影响研究报道较少. 本课题组通过调节B位Nb5+非化学计量比的量来对KNNS-BNZ陶瓷相界进行调控[70]. 通过该陶瓷体系的微观形貌表征以及致密度的测试发现, 适量的过量Nb5+能够形成A位空位, 加速原子的扩散, 从而使陶瓷致密度增加, 最终提高陶瓷的TC. 并且在不降低TC的前提下, 构建室温下的R-T相界提升KNN陶瓷的压电性能, 从而获得既具有压电性能又具有高TC的KNN基压电陶瓷, 该陶瓷体系获得的综合性能如下: d33 = 421 pC/N, kp = 0.48, TC = 256 ℃.
3
2.2.2.温度稳定性
陶瓷的温度稳定性对KNN基陶瓷能否规模化应用至关重要. 研究者们通过多种方法增加KNN基无铅压电陶瓷的温度稳定性. 将相变温度移动至室温以下能够有效地改善KNN基陶瓷的温度稳定性. Zhang等[85]在KNN-LS陶瓷体系中添加CaTiO3, 将TO-T移动到室温以下, 使其温度稳定性增强, 但压电性能大幅度下降.构建弥散的新型相界, 在室温附近形成一个温度变化范围比较大的新型相界, 能够在增加陶瓷体系压电性能的同时增加陶瓷体系的温度稳定性. Yao等[86]通过在KNN基陶瓷中添加CaZrO3组元, 使KNN基陶瓷的O-T相变变得弥散, 延长相变的温度范围, 从而提升陶瓷的温度稳定性, 使得该陶瓷体系的


与非织构化的KNN基陶瓷相比, KNN基陶瓷的织构化也可以有效地改善陶瓷的温度稳定性. 相对于非织构化的LF4陶瓷在160 ℃的应变变化量为30%, 织构化的 LF4T陶瓷在160 ℃时的应变变化量为6.5%[10]. Li等[66]制备的织构化KNNS-CZ-BKH陶瓷体系, 通过加入NaNbO3模板, 获得了纳米尺寸的电畴结构, 因此在温度为200 ℃时, d33值仍有室温d33的70%, 同时, x = 0.03和0.04组分在室温到150 ℃温度范围内应变的变化值小于20%.
KNN基无铅压电陶瓷中构建的新型相界类型是与陶瓷的温度稳定性有关. Zhao等[60]通过对KNN基陶瓷构建不同相界, 并对比各种相界的温度稳定性. 对比R-O, O-T, R-O-T相界, 发现具有R-T相界的KNN陶瓷体系具有优异的电学性能以及最好的温度稳定性.
研究者们还发现调控晶粒尺寸或者畴结构也可以增加KNN基陶瓷体系的温度稳定性. Cen等[51]认为KNN陶瓷体系的晶粒尺寸可以影响陶瓷的压电性能以及温度稳定性, 通过探讨不同烧结温度下制备的KNN基陶瓷体系的相结构以及晶粒尺寸, 发现在1180 ℃烧结温度下的陶瓷具有适宜的晶粒尺寸. 晶粒尺寸的适当增长不仅能够改变该陶瓷体系的相结构, 并增加了畴结构的尺寸, 从而降低了铁电相到顺电相相转变温度的弛豫性, 以此增加了该陶瓷体系的温度稳定性. 1180 ℃烧结温度下制备的陶瓷具有优异的压电性能, 其


| d33/pC·N–1 | d33 variation/% | $ {d}_{33}^{*} $/pm·V–1 | $ {d}_{33}^{*} $ variation/% | |
| KNLNT-CZ[86] | — | — | — | almost unchanged @140 ℃ |
| KNN-BNZ-LF[72] | 345 | — | 420 | 8%@100 ℃ |
| KNNT-BNKZ-SZ-MnO2[49] | — | — | 400 | 10%@180 ℃ |
| KNNT-BNKZ-CZ-MnO2[51] | — | — | 482 | 10%@120 ℃ |
| KNNS-BNZ-SZ[87] | 390 | — | — | 13%@180 ℃ |
| KNN-BLT-BZ-MnO2[88] | — | — | 470 | 8.5%@100 ℃, 21.2%@170 ℃ |
| KNNS-BZ-BNZ[65] | 300 | 10@100 ℃ | — | — |
| KNNS-(BHo)NHf[89] | — | — | ~386 | almost unchanged @140 ℃ |
| KNNT-BNZ-CZ[90] | — | — | 502 | 10%@135 ℃ |
| KNNS-BNKH[42] | 525 | — | 460 | 10%@80 ℃ |
| KNN-BZ-BNH-MnO2[91] | 300 | 15@120 ℃ | 540 ± 10 | 5%@100 ℃ |
| KNN-BNH-BF-MnO2[92] | 450 | — | — | 28%@160 ℃ |
| KNN-BNZ-MnO2-Sb2O3[69] | 318 | — | — | 9%@170 ℃ |
| KNNS-BZH-BNZ[36] | 410 | — | 441 | 2.5%@100 ℃, 16.1%@180 ℃ |
| 注: 16.1%@180 ℃表示到180 ℃性能下降16.1%. | ||||
表6温度稳定性高的KNN陶瓷体系的压电常数以及变化量
Table6.Comparison of piezoelectric constant and variation among KNN-based ceramics.
虽然通过掺杂改性或者结构优化可以在一定程度上提升KNN基无铅压电陶瓷的温度稳定性, 然而与PZT基铅基压电陶瓷相比还有一定的差距, 并且如何从结构上解决KNN基无铅压电陶瓷的温度敏感性还需要进一步的研究.
2
3.1.粉体制备
尽管传统固相法合成陶瓷粉体具有成本低、制备工艺简单的优点, 但是该方法制备的粉体容易出现成分不均匀, 颗粒尺寸大, 不利于后续烧结工艺. 除了固相法之外, 合成KNN粉体的主要方法有水热合成法[93-96]、熔盐法[97,98]、共沉淀法[99]、喷雾热解法[100]等.水热合成法可以在处于低温及高压的水热釜中一次性合成相应的超细粉体, 该方法对环境友好, 能够精确控制粉体形貌及化学组成, 因此可以通过优化粉体微观结构而改善KNN基陶瓷的性能[101]. Takeshi等[102]通过水热法合成了尺寸小于1 μm的KNbO3, NaNbO3粉体, 制备了相应的KNN陶瓷体系. Liu等[94]通过水热法分别合成KNbO3, NaNbO3以及LiSbO3粉体, 通过优化粉体配比, 制备了(1–x)K0.5Na0.5NbO3-xLiSbO3陶瓷体系, 获得了d33 = 183 pC/N, 机械品质因数 Qm = 99.83以及kp = 0.33的陶瓷体系.
熔盐法制备工艺简单, 能够通过调控原料与盐的比例, 精确控制合成纳米粉体的形貌, 改善陶瓷的性能[97]. Li等[98]采用熔盐法制备了KxNa1–xNbO3粉体, 通过调控原料与盐的比例为1∶3以及合成温度, 获得了平均尺寸为1.5 μm的立方晶粒KNN粉体, 并在获得的粉体中添加1 mol% ZnO为烧结助剂, 制备了性能为d33 = 120 pC/N, TC = 406 ℃, Qm = 126以及kp = 0.302的KNN陶瓷体系.
共沉淀法可以直接通过化学反应获得所需粉体, 并且制备的粉体颗粒尺寸均匀. Kumar等[99]采用共沉淀方法制备了K0.5Na0.5NbO3陶瓷, 该陶瓷具有高致密度以及增强的电学性能.
喷雾热解法制备的粉体纯度高, 分布均匀, 同时工艺简单, 能够连续化生产, 并且该方法所使用的水溶液前驱体成本低, 对环境友好, 有广阔的工业应用前景[100]. Haugen等[100]通过喷雾热解法制备了平均粒径为130 nm的KNN粉体, 将该粉体进行常规烧结后制备出95%密度的KNN陶瓷, 该陶瓷体系具有

2
3.2.烧结工艺
烧结是陶瓷坯体致密化的重要工艺, 传统固相烧结具有工艺简单、成本低等优点, 但是因为高温烧结时K, Na元素易挥发, 该烧结工艺制备的陶瓷样品难以致密. 优化烧结工艺对获得高性能KNN陶瓷体系是十分重要的. 目前, KNN陶瓷的烧结工艺除了传统固相烧结外, 还有二步烧结法[34,67,103]、热压烧结[104]、放电等离子体烧结[105]、微波辅助烧结[106]、冷烧结辅助烧结法[107,108]等.两步烧结法是先升温至一个较高的温度, 保温一定时间后, 降低到一个较低的烧结温度长时间进行保温后随炉冷却, 这种方法能够充分有效地提升KNN陶瓷的致密度, 从而增强陶瓷的电学性能[8]. Liu等[67]通过两步烧结法(快速升温至1180 ℃, 保温1 min后, 快速降温至1075 ℃, 在该温度下保温7 h, 并随炉冷却), 制备的0.963(K0.48Na0.52)(Nb0.955Sb0.045)O3-0.037(Bi0.50Na0.50)HfO3陶瓷的d33 ≈ 512 pC/N, kp ≈ 0.54. 该课题组还通过两步烧结法制备了(0.96–x)(K0.48Na0.52)(Nb0.96Sb0.04)O3-0.04(Bi0.50Na0.50)ZrO3-xBaZrO3陶瓷体系, 获得了d33 ≈ 610 pC/N, kp ≈ 0.58, k33 ≈ 0.69的高压电性能[34].
热压烧结法是在高温烧结的同时对样品进行加压, 从而提高陶瓷致密度. Yu等[109]通过热压法制备的K0.5Na0.5NbO3-0.5 mol%Al2O3陶瓷体系, 其d33为137 pC/N,

放电等离子体烧结法是一种低温快速烧结方法. 该方法因为烧结温度低, 能够快速完成烧结过程, 抑制碱金属元素的挥发, 从而提高陶瓷致密度, 提高陶瓷压电性能. Li等[110]采用该方法制备了K0.5Na0.5NbO3陶瓷, 该陶瓷的密度为4.47 g/cm3, 为理论密度的99%, 压电性能为148 pC/N. 但是该方法同样存在烧结设备昂贵、不利于工业化生产的缺点.
微波烧结法是利用微波加热进行陶瓷烧结的方法. 该方法具有加热时间短、能量利用率高(80%—90%)、安全清洁不污染等优点[106]. Feizpour等[106]利用微波烧结在短时间内制备的KNN陶瓷与传统烧结法制备的陶瓷性能相匹配. 但是该方法存在升温速率难以精确控制的弱点.
冷烧结辅助烧结法是一种将陶瓷粉体与去离子水、酒精或者溶液充分混合, 将混合后粉体在一定压力下进行热处理, 之后再进行烧结或者热处理的一种方法. 该方法有助于提高陶瓷致密度, 降低陶瓷烧结温度. Ma等[107]采用该方法制备了K0.5Na0.5NbO3 陶瓷, 将K0.5Na0.5NbO3陶瓷粉体与不同质量百分比的去离子水进行混合, 在350 MPa, 120 ℃下保温30 min压制成型, 在120 ℃下继续保温6 h去除残余水后在1070—1145 ℃烧结4 h, 最终获得d33 ≈ 131 pC/N的KNN陶瓷. Chi等[108]将K0.5Na0.5NbO3陶瓷粉体与NaCl水溶液进行混合, 在450 MPa下压制成型, 在120 ℃下烘干后8 h后, 900—1100 ℃下烧结3 h. 该陶瓷的压电常数为d33 ≈ 115 pC/N, kp ≈ 0.32, TC ≈ 448 ℃. 但是该方法在提升压电性能方面还需要进一步研究.
烧结气氛对陶瓷性能的影响也十分重要. Cen等[111]在还原气氛下(PO2 = 1 × 10–11 MPa)烧结制备的CaZrO3掺杂KNN基无铅压电陶瓷, 具有d33 ≈ 270 pC/N,


另外, 在KNN无铅压电陶瓷中进行不同金属氧化物的掺杂在一定程度上都能够有效地改善陶瓷的烧结特性, 提高陶瓷的致密度, 改善KNN基压电陶瓷的相关性能, 而金属氟化物掺杂的研究报道甚少. 2012年, Li等[112]通过在K0.5Na0.5Nb0.95Ta0.05O3陶瓷中添加1.0 wt% NaF作为烧结助剂, 降低烧结温度, 促进晶粒生长, 提高陶瓷致密度, 相对于未添加NaF时陶瓷体系的d33 ≈ 110 pC/N, 添加NaF后陶瓷体系的d33提升了40%左右, 达到153 pC/N. CuF2也是一种能够有效降低KNN陶瓷烧结温度, 提升陶瓷压电性能的烧结助剂[113]. Wu等[114]通过在KNN基陶瓷中掺杂ZnF2, 探讨ZnF2对陶瓷微观结构、电学性能以及温度稳定性的影响. Liao等[115]通过合成K0.5Na0.5Nb0.996Cu0.01O3-xFx陶瓷体系, 探讨了F-O取代对氧空位以及缺陷复合体的抑制作用, 通过进行F取代O, 陶瓷的d33 ≈103 pC/N, kp ≈0.42、机械品质因子Qm≈ 1031.
2
3.3.制备工艺
陶瓷的织构化可以使陶瓷内部晶粒沿一定方向排列取向生长, 陶瓷在该方向上具有类似单晶的性能. 模板晶粒生长(TGG)法是在KNN粉料中按照一定比例加入片状模板晶粒、黏结剂、增塑剂, 配制成流延浆料后, 通过流延成型制备织构化的KNN基陶瓷. Li等[11]采用该方法制备了一系列KNN基陶瓷, 利用传统固相法制备出KNN基陶瓷粉体, 并采用熔盐法制备了NaNbO3模板, 坯体在600 ℃下预热处理5 h后, 再采用两步烧结法1190 ℃以及1090 ℃下烧结10 h制备, 制备出d33 ≈ 700 pC/N, kp ≈ 0.76, TC ≈ 242 ℃的KNN基织构陶瓷[11]. 研究发现, KNN基织构化压电陶瓷的优异压电性主要来源于三方面: 1)对于

相较于传统固相烧结法, 其他烧结方法或者工艺制备的KNN陶瓷能够有效降低相应的粉体烧结温度, 并且晶粒生长更为均匀, 从而较好地提高陶瓷的致密度. 但是这些方法工艺过程复杂, 生产成本较高, 难以实现大规模生产. 因此, 通过离子掺杂或组元取代方法提升KNN基陶瓷压电性能仍然是目前研究的重点和主要方向.
2
4.1.铁电相变
在相界处压电陶瓷的介电和压电系数等都会出现异常的增大, 目前绝大多数的高压电材料都是在材料的相界处获得的, 比如目前最常见的PZT陶瓷, 其性能最优的配方就处于三方-四方的准同型相界(MPB).根据钙钛矿铁电体的位移型相变理论, 钙钛矿铁电体中的电极化形成主要来自于离子位移, 当温度高于TC时, 材料处于顺电相, 此时ABO3结构中A位离子位于晶胞顶角, B位离子位于体心, 而O离子在面心, 正负离子中心相互重合, 由此宏观上不存在电极化. 而当温度低于TC时, 离子相对于中心位置发生偏移, 正负电荷中心分离, 在晶胞内形成电偶极子, 由此造成了晶格结构的改变. 电极化的形成引起晶格结构发生畸变, 并改变晶格的对称性, 晶格会沿极化方向明显拉伸, 其余方向则相对压缩, 铁电相变因而形成. 位移型相变显示出铁电相中, 正负离子总是沿极化方向进行拉伸, 整体的晶体结构显示出长程有序性.
铁电相变的成因主要归因于B位离子与O离子之间的强共价作用, 例如BaTiO3中, 按照位移型相变理论, 立方相的BaTiO3结构中Ti原子应该位于TiO6八面体的中心位置, 而当温度低于TC时, Γ点的横光学支振动模软化, Ti因此偏离中心位移产生位移, 并形成电极化. 而通过X射线吸收精细结构谱(XAFS)在10–15 s时间测得的结果显示[116], Ti总是倾向于与最之靠近的3个O原子成键, 从而偏离中心位置, 形成TiO3的配合体, 这种情况即使在高于TC的情况下仍旧存在. 但是由于Ti的这种偏离较小, 因而其势垒较低, 在高温下可以通过较强热振动在附近8个靠近O3构型的位置频繁移动. 这种行为在长程过程下即显示出Ti原子的平均位置位于晶胞的体心. 第一性原理计算也证实, Ti—O键的强共价成分来源于Ti 3d轨道和O 2p轨道的杂化耦合, 是诱发BaTiO3和PbTiO3之间铁电相的成因[117]. 因此, Ti总是靠近同一平面的3个O原子, 形成TiO3配合体, 在高温立方相时, Ti在统计上平均地与周围邻近的6个O原子形成配位; 而四方相时, Ti则只与邻近的5个O原子配位; 正交相Ti与4个O原子配位; 直到三方相结构, Ti只稳定与其中3个O原子配位, 不再移动.
Atern和Yacoby[118]对KTaO3:9%Nb使用XAFS直接测得的O原子相对于近邻Nb分布概率显示更直接的证据. 由于纯的KTaO3为先兆铁电体[119], 高于0 K下显示为立方顺电结构, 而引入Nb可以提高其铁电相转变温度TC. 9%的Nb掺杂的KTaO3显示的TC为86 K, 但实验测得结果显示当温度达到300 K时, Nb原子在立方相结构下仍然有很大的离子位移, 这说明晶体宏观畸变的变化并不是通过降低离子位移来实现, 而是通过打乱位移方向来降低畸变程度. 这个结果显示钙钛矿晶格的相变存在很强的有序-无序型机制.
Shuvaeva等[120]通过XAFS研究了KNbO3的相变, 发现该晶体的相变同时受位移型和有序-无序型机制的作用. 低温下的三方相为完全的位移型结构, 而在高温下其有序-无序型机制则明显增强. 图4给出了KNbO3中不同相结构下Nb原子的实际位置, 该示意图所示模型来自于XAFS测量所得结果[120]. 其中八位模型(eight-site model)对应为理论的有序-无序机制下Nb所在位置, Nb总是沿

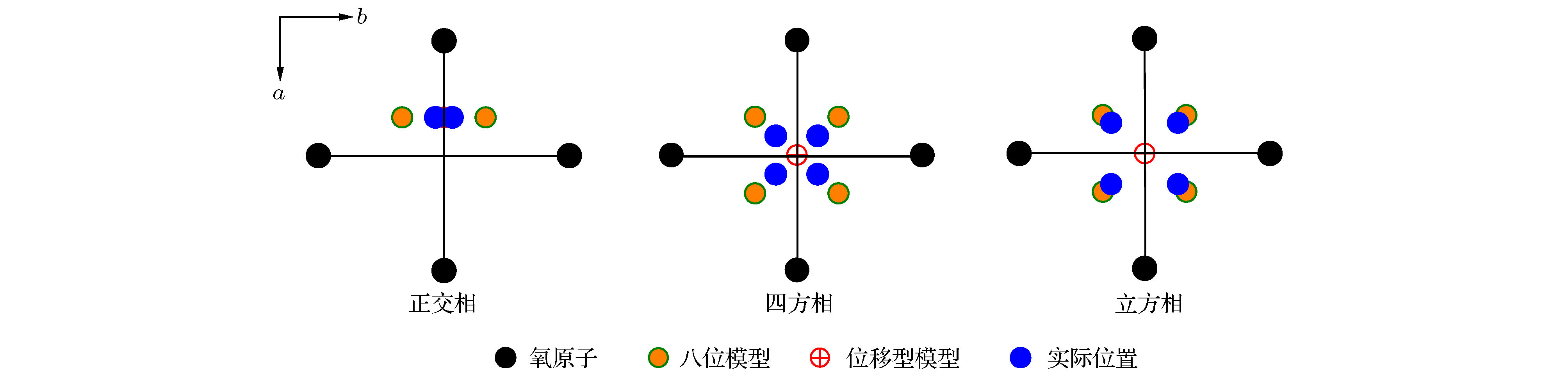 图 4 根据文献[120]重画的KNbO3中Nb原子位置在(001)平面的投影示意图
图 4 根据文献[120]重画的KNbO3中Nb原子位置在(001)平面的投影示意图Figure4. Projections of real Nb off-center displacements on the (001) plane redrawn from the Ref. [120].
根据Landau-Ginsburg-Devonshire热力学唯象理论[121,122], 铁电相变实际上是中心对称顺电相的自发对称破缺行为, 产生的铁电相可以电极化P为序参量去描述系统状态. Devonshire[121,122]将立方相作为原型, 认为铁电相仅仅是顺电相下的微扰, 因此可以将立方相附近的自由能F以电极化P的形式进行展开, 并用以描述铁电相的畸变程度. 而因为立方相的中心对称性,


由于




而对于g4为负值的情况, 自由能必须重新包含g6项, 且g6为正值. 因此, 对于E = 0的平衡条件, 可以写为
Cochran[123]等通过对Landau-Ginsburg-Devonshire自由能进行八阶展开, 仅考虑二阶系数为温度相关系数, 其余高阶系数为常数, 利用KNbO3和K0.5Na0.5NbO3实验数据计算出总共十个高阶项的Landau系数, 成功构建出了K1–xNaxNbO3的自由能函数, 然后对其相图进行了预测, 其预测值与实验结果十分符合.
2
4.2.铁电材料中的压电行为
压电材料的压电系数的表达式为铁电材料的压电性最显著的特点是, 在铁电-铁电相界处具有极高的压电系数. 这一现象发生的原因在于, 对于非本征压电效应来说, 在铁电-铁电相界处, 两个或几个铁电相的自由能相等, 比如KNN中的三方-正交-四方相界, 电畴可能沿



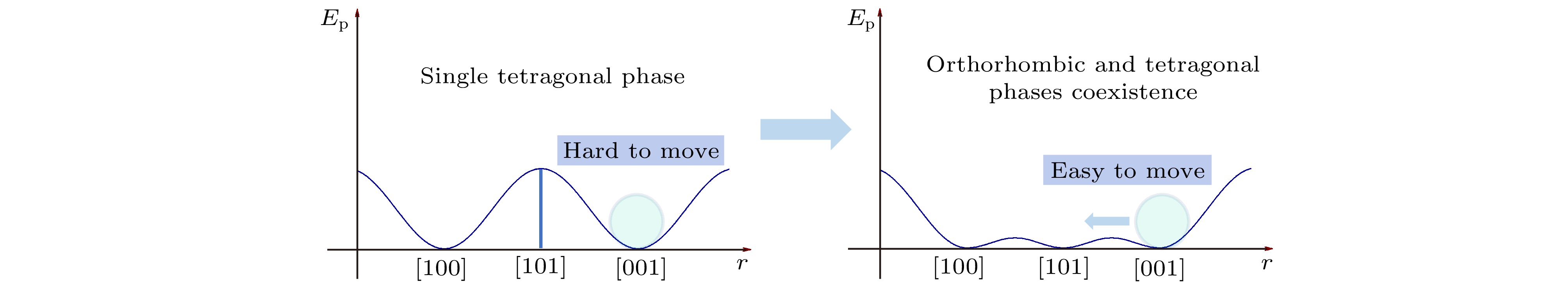 图 5 B位原子在单四方相与两相共存时沿[
图 5 B位原子在单四方相与两相共存时沿[
Figure5. Energy distribution for B-site atom in single tetragonal phase and two-phase coexistence along [

2
4.3.相界构建机制
传统的Pb基陶瓷中, PZT由三方相的反铁电体PbZrO3和四方相铁电体PbTiO3按照一定比例混合构建出准同型相界MPB, 并由此获得极高的压电性能. 而在无铅压电陶瓷, 比如KNN中, KNbO3和NaNbO3的等量混合并没有造成大的结构变化, 仅仅是其β角出现一点改变, K含量增加而β角下降[126]. 进一步的研究揭示, 室温下的KNN在Na含量偏高时是空间群为Pm的单斜结构, 而在K偏高的部分一侧为Amm2的正交相结构, 但总体来说两者在极化方向上差别不大, 用Glazer符号[127]表示两相分别为


目前, 提高KNN基陶瓷压电性最普遍的做法是通过离子掺杂取代, 提升三方-正交相变温度或降低正交-四方相变温度, 诱导在室温下形成多个铁电相的共存态. 例如, Li取代A位离子, 诱导正交-四方相变温度降低至室温, 形成室温下的铁电相界, 该方法可以将KNN陶瓷的d33提升至235 pC/N, 并将TC提升约470 ℃[128]. 第一性原理研究表明, Li掺杂可能大幅提高了A位离子p轨道与O 2p的轨道杂化[129]. 这与PbTiO3的形成原因类似, Pb与O的轨道杂化诱导了四方相的形成, 并提高极化强度, 增加TC. 但这仍有疑问, 由于以上研究均是结合虚晶近似(virtual crystal approximation, VCA)进行的, 难以区分到底是Li, Na和K中的具体哪一种元素造成的A位离子与O的强杂化, 且Li的电子轨道中不包含p轨道. 更关键的是无论Li, Na还是K的电负性都明显低于Pb, 最高的Li也仅仅与Ca的电负性类似, 但CaTiO3中A位不存在强共价作用, 且其是典型的立方相结构, 因此关于Li掺杂KNN诱导四方相的起源尚有待进一步研究.
从B位离子取代的角度来看, 通常来说, 认为大尺寸的B位可以给O6八面体内部更高的化学压力, 从而驱动晶体结构向三方相转变. 这在PbTiO3的计算中也有所证实, 四方相的PbTiO3可以在高静水压下转变为三方相结构[130]. 因此, Zr和Hf等容易驱动KNN向三方相转变, 并且由于掺入的B位离子半径较大, 其在氧八面体内的离子位移受限降低, 因而正交-四方转变温度和居里温度随之降低. 半径与Nb相似的Ta则对KNN的相变温度影响十分微弱, 平均每0.01 mol降低正交-四方转变温度TO-T约4 ℃, 提升三方-正交相变温度TR-O约2.3 ℃[8]. 值得注意的是, Sb的取代对提升KNN材料的压电性质十分明显, Sb可以迅速地提高TR-O, 同时降低TO-T, 由于这个特性, Sb已经成为KNN基材料掺杂过程中最重要的取代元素, 绝大部分高压电的KNN材料都含有Sb. 实际上Sb的取代行为与Zr和Hf十分类似, 但是由于其名义上的化合价为+5价, 与Nb相同, 因而其不容易破坏晶格的长程有序性, 相对容易进入晶格. 而Zr和Hf的化合价都是+4价, 研究显示过量的Zr很容易聚集在晶界处, 影响晶粒生长, 因此Zr和Hf的取代通常都伴随着Ba2+, Sr2+, Ca2+或者(Bi0.5Na0.5)2+等+2价离子. 但是, 根据Shanon[131]给出的离子半径(见表7), Sb5+的离子半径比Nb5+要更小, 在KNN中取代时不会对O6八面体提供内生的化学压力, 导致TR-O上升和TO-T下降. 本课题组研究Ga2O3掺杂对KNN的影响时发现[132], 小半径的Ga3+进入KNN晶格B位反而会提升TO-T, 与大尺寸离子对相界的作用基本相反. 在考虑到Sb在高温下容易被还原为三价离子, 怀疑KNN陶瓷中可能是+3价态的Sb在推动KNN相变温度的改变, 这需要进一步的研究去证实. 总之, 根据以上提及的掺杂机制, 通过调整不同掺杂物的比例, 控制各个铁电相之间的相变温度, 就可以在KNN基材料中设计出室温下三方-正交共存、正交-四方共存甚至三方-四方共存的铁电相界, 从而获取高压电性的无铅材料.
| Nb5+ | Ta5+ | Zr4+ | Hf4+ | Sn4+ | Ti4+ | Sb5+ | Sb3+ | Ga3+ | |
| 离子半径/? | 0.64 | 0.64 | 0.72 | 0.71 | 0.69 | 0.605 | 0.60 | 0.76 | 0.62 |
表7处于6配位时的离子半径表[131]
Table7.The ionic radii in six-fold coordination[131].
另外, 需要注意的是, 不同价态的离子在掺杂过程中容易造成晶格点缺陷, 常见的如CuO掺杂, 六配位的Cu2+离子半径为0.73 ?, 容易进入晶格B位, 此时的化学反应为



2
4.4.第一性原理计算
第一性原理计算基于密度泛函理论(DFT), DFT理论将晶格内部的多电子薛定谔方程转化成为一个求解电子气密度函数, 在形式上近似于单电子薛定谔方程的Kohn-Sham方程来求解体系基态能量. 然后通过静态晶格来实现对材料力学、声子频率、有效电荷、介电常数、压电常数、弹性常数和极化强度等宏观性质的计算. 在这个过程中, 由于第一性原理计算本质上是直接求解材料本身的多电子薛定谔方程, 因此不需要提供任何实验收集的经验参数. 目前许多对钙钛矿压电材料的重要机理解释都来自于第一性原理计算, 比如Ti—O键的轨道杂化, 这直接证明了铁电性起源于长程库仑力之间的竞争, 并导致了离子位移; 相反, 短程排斥力则容易促使形成高对称的顺电相.第一性原理计算在Pb基材料之中研究相对广泛, 比如证明了弛豫铁电体中的高压电应变来自于极化方向的偏转[134], 预测了高压下纯PbTiO3存在一个MPB相界, 并存在高压电活性等[130]. 而在无铅压电材料中, 相关研究仍然相对较少, 尤其是近些年热门的KNN材料. 在此, 本文对第一性原理计算在KNN中的研究进行简要介绍.
长久以来, Ta通常被用作KNN压电材料的掺杂取代元素之一, 并通过和Li, Sb等元素共同掺杂诱导室温下形成铁电相界, 获得了具有高压电活性的KNN基陶瓷, 但Ta取代Nb占据B位是如何对晶格产生影响的却缺少研究. Suewattana和Singh[135]使用第一性原理计算研究了纯的KNN合金与Ta掺杂的KNN (KNNT)合金的局域结构和动力学. 研究发现Ta取代后的局域结构具有比Nb更小的离子位移. 尽管Nb与O的距离相对Ta更近, 但Nb的力常数反而比Ta更小. 另外, 通过获得的谐性径向分布函数(PDF)发现, 纯的KNN和KNNT在高温下的第3个分布峰即Nb-Nb或Ta-Ta的分布峰仍然保持尖锐, 说明Nb和Ta的分布仍然保持相对有序的结构, 但KNNT中Ta全部的峰形都相对Nb更加尖锐, 显示出Ta具有更大的力常数, 这被认为是导致实验中出现Ta掺杂后居里温度降低和介电常数增加的主要原因.
Voas等[136]使用第一性原理结合准随机结构(SQS)研究了K0.5Na0.5NbO3在R3c和Pm结构下的A位离子分布. 通过与中子衍射测试的数据进行对比, SQS能够准确给出钙钛矿结构中A位离子的局域分布形式. 通过结构计算显示, Na更倾向于向靠近K远离Na的方向产生位移, 而又同时尽可能与整体极化方向保持一致. 而在基态相R3c和室温相Pm之间, Na-O的交互作用变化最为明显, 这显示Na-O的交互作用是导致从R3c到Pm相变的驱动力.
室温下的KNN陶瓷具有比KNbO3 (KN)更高的压电系数[137], 但两者都为正交相结构, 铁电相变在室温下未对两者的压电性能产生贡献. 本课题组通过第一性原理计算结合取向平均的方法[138,139], 直接比较计算了正交相KN和KNN的单晶与陶瓷的压电性能, 研究发现KNN陶瓷明显具有比KN更高的压电性能, 其d33相较KN增加了约70%, 这是与实验观察所得一致, 计算结果显示两者正交相陶瓷的d33都主要来源于单晶中d15和d33的贡献. 进一步研究发现, KNN与KN的Born有效电荷相差不大, 说明电子部分的贡献两者几乎没有差别. 在该研究中, KNN中的K和Na按照沿钙钛矿

| K | Nb | OⅡ | OⅠ | ||||
| KN | ?u3/?η3 | 0.108 | 0.166 | –0.092 | –0.091 | ||
| ?u1/?η5 | 0.115 | 0.210 | –0.151 | –0.024 | |||
| K | Na | Nb | OⅡ | OI,1 | OI,2 | ||
| KNN | ?u3/?η3 | 0.103 | 0.542 | 0.125 | –0.158 | –0.125 | –0.135 |
| ?u1/?η5 | 0.094 | 0.828 | 0.194 | –0.235 | –0.061 | –0.309 | |
表8不同结构下原子内坐标随应变的梯度, 注意OI位于Bmm2不包含Nb原子的(010)平面, KNN中OI,1和OI,2沿a方向分别靠近K和Na原子[138]
Table8.Internal atomic coordinate gradients as a function of strains in different structure, noted that OI is located at the (010) plane without Nb atoms in Bmm2, OI,1 and OI,2 are close to K and Na along a axis, respectively[139].
Yang等[129]则利用DFT加上虚拟晶胞近似(VCA)方法调查了Li掺杂的KNN材料, 对比了其电子态密度分布及压电应力常数e33, 验证了Li掺杂后增强的压电性能. 其结果显示Li掺杂增强了O 2p轨道和Nb 4d轨道的杂化, 减小了Nb-O键距离并增强其畸变程度. 这些结果被认为是Li掺杂后压电性能增强的主要原因.
Li等[140]对CaZrO3掺杂的KNN的电子结构进行了计算. 研究发现Ca在A位相对更容易取代Na, 并且Ca掺杂会导致KNN的费米面向高能量的导带移动, 从而降低带隙; 而Zr的掺杂则会让费米面向价带移动, 提高带隙宽度. 此外, Ca取代A位具有更大的电负性, 从而诱导KNN沿c方向拉伸, 因此KNN逐渐从O相转变为T相, 这是导致CaZrO3取代让KNN具有更高性能的主要原因. 关于K/Na = 1附近KNN的相变问题一直争论已久, Liu等[141]研究K1–xNaxNbO3体系的相变, 发现随Na组分的增加, 该体系可能经历O-M-O的相变过程, 显示出该类材料在0.3 < x < 0.8所具有的并不是严格意义上的正交相O, 反而是一个低对称性的单斜相M, 并推测该单斜相可在常规R, O和T三相中扮演中间相的角色, 让各个极化态之间相互转向更容易, 从而提高材料的压电活性. 哈尔滨工业大学的Li等[142]利用第一性原理计算结合VCA的方法详细研究了K1–xNaxNbO3体系的压电系数, 并预测其MPB出现在x = 0.52附近. 该研究同时计算了体系的结构参数、体积模量和禁带宽度, 并指出极化强度在x = 0.5时从[011]到[001]转向具有比纯KNbO3更低的能量壁垒, 这是导致其具有增强压电的主要原因. Yang等[143]使用第一性原理计算了不同Na浓度下的KNN结构与体系总能, 发现当Na浓度逐渐增加时, 出现一个O-T相转变, 并导致了增强的压电特性, 该结果证明了高d33来源于相变. 计算结果还表明NbO6八面体的形变与材料整体的相变起源于氧八面体内Nb—O键长度的变化. 此外, 在计算中, KNN在Na含量为0.55时出现一个最高的压电常数e33 = 6.77 C/m2, 而通过与实验进行对比, 实验中陶瓷的d33也在该组分展现了最高的压电性d33 = 203 pC/N, 与计算结果的趋势一致, Yang等[143]认为KNN陶瓷中压电性能的这些变化可能来源于组分波动与相结构的转变.
对于KNN基无铅压电陶瓷, 压电性能的提升会伴随着陶瓷体系TC的下降; 当KNN陶瓷在小于居里温度的温度范围内存在另一个多晶型相变时会导致陶瓷热稳定性下降. 这是KNN基陶瓷与PZT陶瓷相比最大的不足之处, 这将影响KNN陶瓷的实际应用. 如何获得同时具有高压电性与高温度稳定性的KNN基无铅压电陶瓷仍然是未来研究的难点与重点之一.
另外, KNN基无铅压电陶瓷因为性能对成分的敏感性, 烧结温区较窄以及工艺重复性差, 不利于KNN基无铅压电陶瓷的大规模生产. 以实际应用为前提, 如何解决KNN基无铅压电陶瓷的组分、温度敏感性是关系到该体系规模化应用的关键.
而在理论方面, 一方面KNN中K/Na的分布结构仍值得进一步研究, 因为尽管K和Na的价电荷较低, 离子位移对极化强度贡献相对较Nb和O更小, 但K/Na的分布结构对压电性也具有一定影响[138,139], 尤其是对邻近的O影响最为明显. 另一方面, 有限温度下的压电活性在KNN中仍然值得探讨, 关于该方面的论述可能可以从理论上对PPB相界的低热稳定性进行解释. 此外, 离子掺杂、多组元复合对晶格的作用需要深入研究, 尤其是Li和Sb的掺杂, CaZrO3, Bi(Zr, Hf)O3与KNN的复合, 以便更好地理解相变的驱动力来源和高压电活性的起源.
