全文HTML
--> --> -->本文通过理论计算对含空位α-Fe中H的存在状态及分布位置进行系统分析, 基于此对不同条件下H在含α-Fe平衡溶解度进行了计算, 并开展实验验证.
3.1.α-Fe晶体与间隙H原子的相互作用计算及结构预测
33.1.1.α-Fe晶体与间隙H原子的晶体结构和能量分析
分别计算了H原子溶于α-Fe的四面体间隙位置(tetrahedral interstice site, T-site)和八面体间隙位置(octahedral interstice site, O-site)中所得的α-Fe+H晶体弛豫后稳定结构, 计算结果如图1和表1所列. 由图1和表1可见, H原子溶入α-Fe晶格间隙后, 晶格发生一定程度的畸变, 晶格常数发生变化, 晶体发生不同程度的膨胀, 其对称元素减少, 由立方晶系转变为四方晶系. H原子处于八面体间隙位置时α-Fe-H的体积膨胀量更大, 这与α-Fe中八面体间隙半径小于四面体间隙半径有关. 图 1 α-Fe+H的晶体结构 (a) T-site; (b) O-site
图 1 α-Fe+H的晶体结构 (a) T-site; (b) O-siteFigure1. Crystalline structure of α-Fe+H: (a) T-site; (b) O-site.
| 晶体类型 | a/? | b/? | c/? | α/(°) | β/(°) | γ/(°) | V/? | 空间群 |
| α-Fe+H(T-site) | 5.6843 | 5.6612 | 5.6804 | 89.999 | 89.999 | 90.000 | 5.6843 | 115(P-4m2) |
| α-Fe+H(O-site) | 5.8019 | 5.6112 | 5.6112 | 90.000 | 90.000 | 90.000 | 5.8019 | 123(P4/mmm) |
表1α-Fe+H的晶格常数和晶体结构
Table1.Lattice parameters and crystalline structure of α-Fe+H.
从能量的角度分析间隙H在α-Fe中的占位倾向, 定义晶体结合能




定义含缺陷的α-Fe晶体形成能






H原子的溶解热










由于计入H原子的零点能非常重要[20], 需要根据H原子在晶格内的声子谱计算其零点能, 零点能






间隙H原子晶体的能量计算结果如表2所列. 由于实际计算过程中八面体间隙中的H原子会向四面体间隙弛豫, 这从侧面说明四面体间隙是H原子的优先占位. 因此表2中仅给出四面体间隙的α-Fe+H能量计算结果.
| 晶体类型 | ${E_{{\rm{crystal}}}}$/eV | ${E_{{\rm{ZP}}}}$/eV | ${E_{{\rm{bind}}}}$/eV | $E_{{\rm{form}}}$/eV | $\Delta H_{{ {\rm{sol} } } }^{\rm{H} }$/eV |
| α-Fe+H(T-site) | –13861.050 | 0.246 | 5.182 | 0.390 | 0.390 |
| α-Fe | –13845.344 | — | 5.530 | — |
表2α-Fe和α-Fe+H结合能、形成能和溶解热
Table2.Binding energy, formation energy, and heat of solution of α-Fe and α-Fe+H.
由表2可见, H在α-Fe的间隙溶解热



3
3.1.2.α-Fe晶体与间隙H原子电子结构分析
弛豫后间隙H原子与最近邻Fe原子的距离提示二者可能会成键. 为研究四面体间隙中的H原子与α-Fe晶体的相互作用, 对图1(a)的构型进行了原子布居及键布居、态密度及差分电荷密度分析.计算自由H原子, α-Fe晶体和α-Fe+H(T-site) 晶体中的原子轨道布居如表3所列. 根据H和Fe原子的核外电子表达式, H原子的价电子位于1s, Fe原子的价电子位于3d和4s, 其中3d轨道能量较高. 对比表3数据可知H在四面体间隙固溶时, H原子的1s轨道得电子, 而Fe原子的4s轨道明显失电子, 3d轨道获得了部分电子. 据此推断α-Fe-H中H近邻的Fe原子中的4s轨道受激与3d轨道杂化, 并与H原子的1s电子成键, H的电负性较强, 故得电子. 在距离间隙H原子较远处, 由于受到了间隙H原子及其近邻Fe2 (Fe4), Fe3 (Fe11)的共同作用, 晶体中的Fe1, Fe8, Fe12等原子的净电子得失只有微弱起伏, 在间隙H原子含量较多时, 这种作用可能会导致晶体内部电子浓度的不均匀, 使晶体中广泛存在相互排斥的区域, 从而降低了晶体中原子的平均结合强度[26].
| 晶体类型 | 原子 | 轨道电荷占据数 | 总布居 | 净布居 | ||
| s | p | d | ||||
| α-Fe-H(T-site) | H | 1.34 | 0 | 0 | 1.34 | –0.34 |
| Fe2, Fe4 | 0.62 | 0.66 | 6.65 | 7.93 | 0.07 | |
| Fe3, Fe11 | 0.62 | 0.67 | 6.65 | 7.94 | 0.06 | |
| Fe12, Fe10 | 0.65 | 0.72 | 6.62 | 7.99 | 0.01 | |
| Fe1, Fe9, Fe13, Fe14, Fe16 | 0.65 | 0.72 | 6.62 | 7.99 | 0.01 | |
| Fe8, Fe6 | 0.65 | 0.74 | 6.61 | 8.01 | –0.01 | |
| α-Fe | Fe | 0.68 | 0.70 | 6.62 | 8.00 | 0 |
| 自由态 | H | 1.00 | 0 | 0 | 1.00 | 0 |
表3α-Fe+H (T-site)晶体原子轨道布居
Table3.Atomic orbital population of α-Fe+H (T-site).
对原子间的键布居进行分析, 结果列于表4. 除间隙H原子的四个近邻Fe原子之间存在反键外, 还有部分Fe原子之间结合减弱,且晶体内原子之间的距离发生变化, 这说明间隙H原子固溶后弱化了晶体的整体结合强度, 且在局部形成薄弱区域. 比较α-Fe+H (T-site)和α-Fe+H (O-site)晶体的键布居数, 八面体间隙H原子的近邻Fe原子之间存在更强的反键, 这可能是α-Fe+H (O-site)晶体具有更高的内能和更低的结合强度的原因之一.
| 晶体类型 | 原子对 | 距离/? | 键布居 |
| α-Fe-H (T-site) | Fe2-H | 1.6494 | 0.16 |
| Fe3-H | 1.6507 | 0.16 | |
| Fe2-Fe3 | 2.5558 | –0.09 | |
| Fe2-Fe4 | 2.7286 | –0.14 | |
| Fe8-Fe11 | 2.4783 | 0.17 | |
| Fe3-Fe12 | 2.4471 | 0.18 | |
| Fe8-Fe16 | 2.8401 | 0.05 | |
| Fe7-Fe8 | 2.4601 | 0.15 | |
| α-Fe-H (O-site) | Fe2-Fe4 | 2.6287 | –0.28 |
| α-Fe | Fe-Fe | 2.4400 | 0.14 |
| Fe-Fe | 2.8174 | 0.06 |
表4α-Fe-H (T-site)晶体键布居
Table4.Bond population of α-Fe+H (T-site).
分析间隙H原子及其近邻Fe原子的电子态密度, 结果如图2所示. 可见间隙H原子固溶后1s电子态密度波峰向低能级迁移, 且能级发生分裂, 存在多个波峰, 其–10 — –7.5 eV的波峰与近邻Fe原子4s态电子波峰发生重叠, 说明H和Fe存在微弱的共轭效应. Fe原子的3p电子态密度在固溶前后无明显变化, 说明H与Fe的作用主要来源于H的1s电子和4s的共轭作用; 另外Fe的3d电子在该位置的波峰也有一定的增强, 说明较内层的价电子也部分参与了成键, 这和原子轨道布居分析结果较一致.
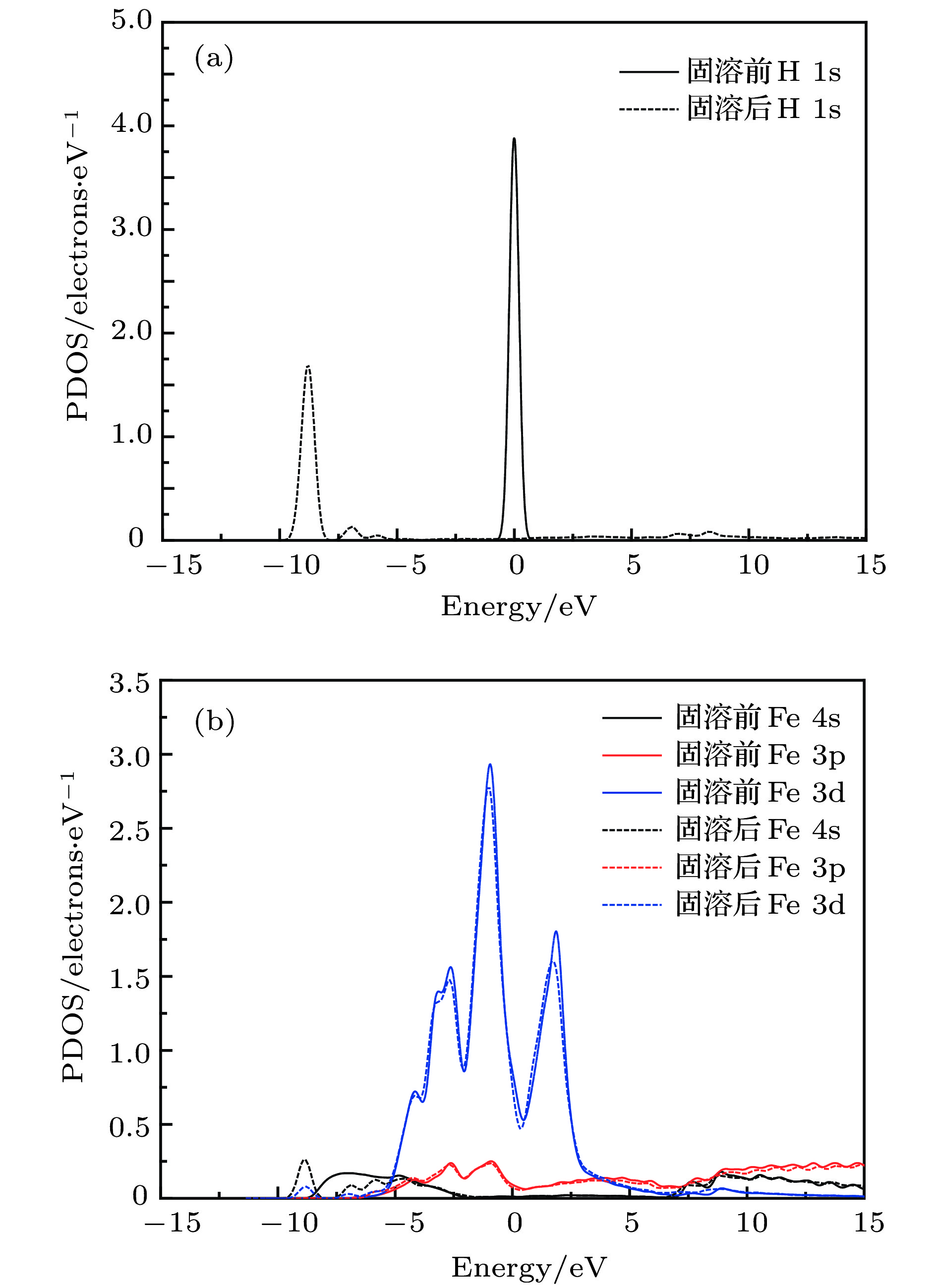 图 2 α-Fe+H (T-site)的分波电子态密度 (a) 固溶前后 H 原子; (b) 固溶前后最近邻Fe原子; (c) 四面体间隙H原子; (d) 间隙H原子最近邻Fe原子
图 2 α-Fe+H (T-site)的分波电子态密度 (a) 固溶前后 H 原子; (b) 固溶前后最近邻Fe原子; (c) 四面体间隙H原子; (d) 间隙H原子最近邻Fe原子Figure2. Partial electronic density of state of α-Fe+H (T-site): (a) Free H atom and interstitial H atom; (b) Fe atom in perfect α-Fe crystal and the nearest neighbour of interstitial H atom; (c) H atom in tetrahedral interstice; (d) the nearest neighbour Fe atom of interstitial H atom.
计算图1(a)中α-Fe+H (T-site)的差分电荷密度, 结果如图3所示. 图3中蓝色为得电子, 红色为失电子. 可见H得电子, Fe2, Fe4, Fe11为H近邻原子, 与图3中出现的其他Fe相比, 其周围空间有较明显的失电子. 由于不同位置的Fe价电子受H的影响不同, 各Fe净电荷在长程范围内稍有起伏. 这说明虽然间隙H原子在α-Fe中的直接作用范围较小, 但其间接带来的晶格电荷密度变化的范围较大. 电荷密度的这种变化可以一定程度解释α-Fe+H(T-site)结合能降低的原因.
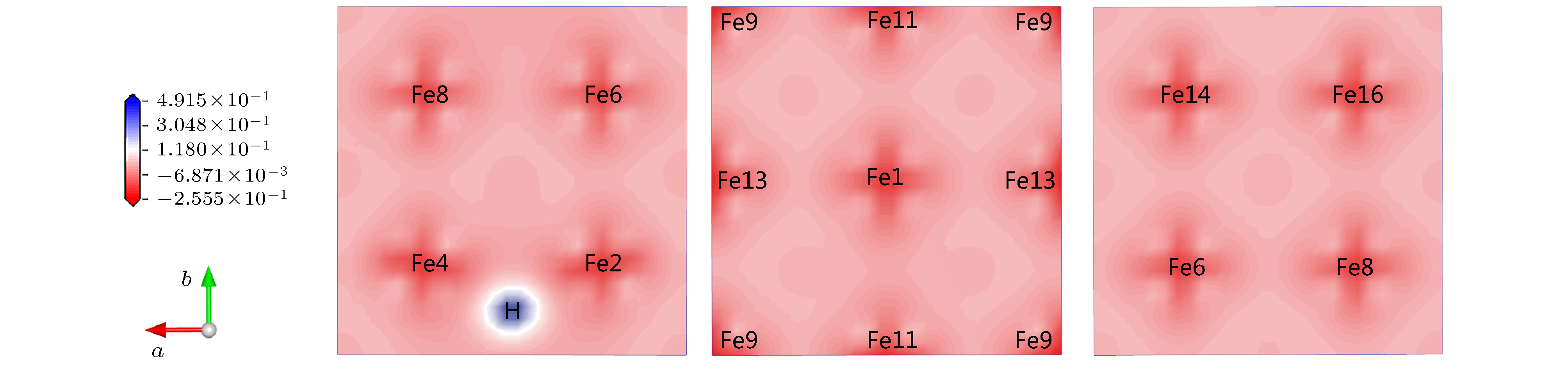 图 3 α-Fe+H(T-site)的差分电荷密度
图 3 α-Fe+H(T-site)的差分电荷密度Figure3. Electron density difference of α-Fe+H(T-site).
2
3.2.含空位的α-Fe+Vac晶体与H原子相互作用计算及结构预测
空位(vacancy, 以下用Vac代替)是高强钢中广泛存在的一种点缺陷, 由于空位尺寸比间隙更大, 因此可能会容纳较多的H, 在晶格中形成Vac-H复合体. 分别计算了α-Fe+Vac及含有Vac-H复合体的α-Fe+(1H-Vac)—α-Fe+(6H-Vac)晶体稳定结构, 并进行能量分析, 结果如表5, 图4和表6所列.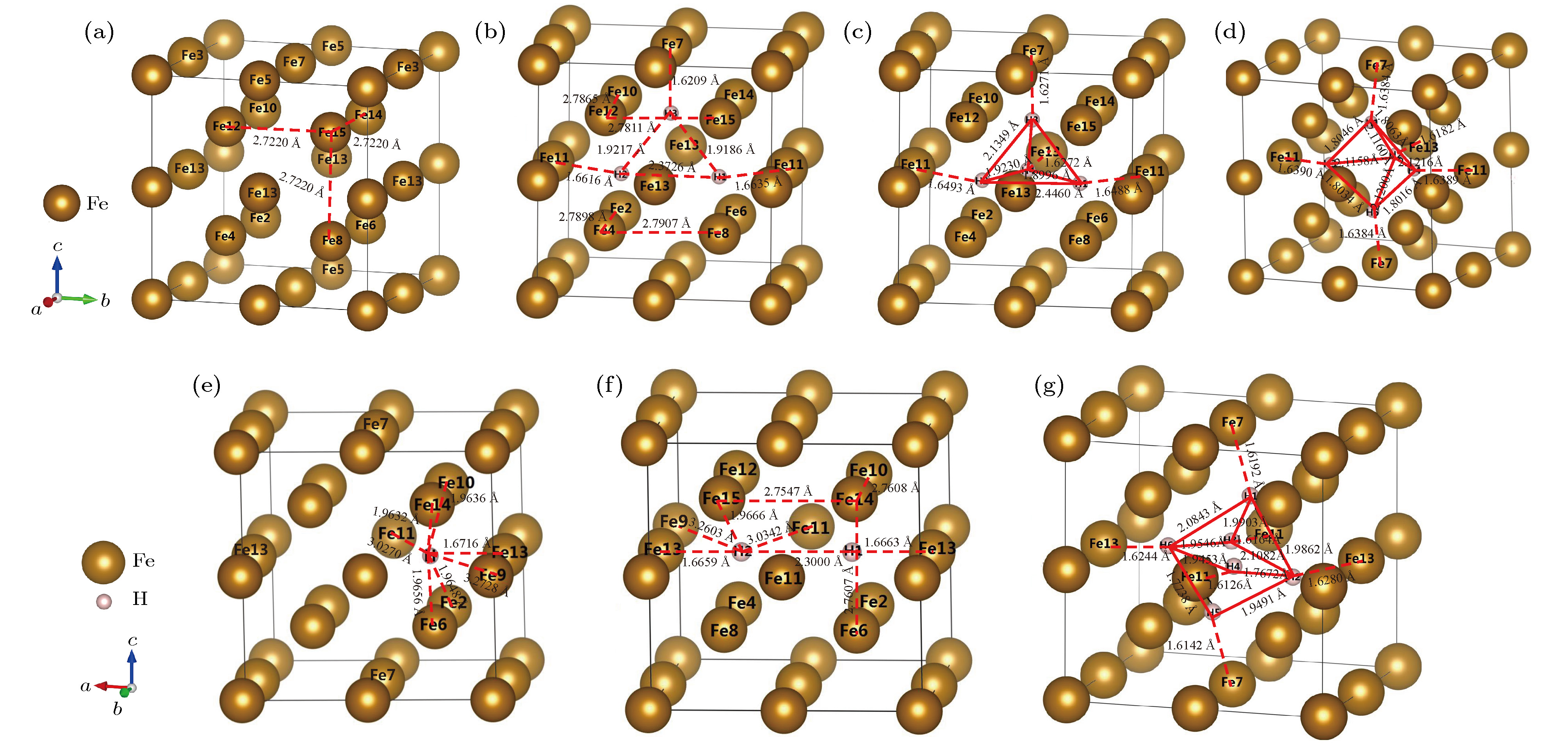 图 4 α-Fe+(nH-Vac)的晶体结构 (a) α-Fe+Vac; (b) α-Fe+(3H-Vac); (c) α-Fe+(4H-Vac); (d) α-Fe+(5H-Vac); (e) α-Fe+(1H-Vac); (f) α-Fe+(2H-Vac); (g) α-Fe+(6H-Vac)
图 4 α-Fe+(nH-Vac)的晶体结构 (a) α-Fe+Vac; (b) α-Fe+(3H-Vac); (c) α-Fe+(4H-Vac); (d) α-Fe+(5H-Vac); (e) α-Fe+(1H-Vac); (f) α-Fe+(2H-Vac); (g) α-Fe+(6H-Vac)Figure4. Crystalline structure of α-Fe+(nH-Vac): (a) α-Fe+Vac; (b)α-Fe+(3H-Vac); (c) α-Fe+(4H-Vac); (d) α-Fe+(5H-Vac); (e) α-Fe+(1H-Vac); (f) α-Fe+(2H-Vac); (g) α-Fe+(6H-Vac).
| 晶体类型 | a/? | b/? | c/? | α/(°) | β/(°) | γ/(°) | V/? | 空间群 |
| α-Fe+Vac | 5.6033 | 5.6033 | 5.6033 | 90.000 | 90.000 | 90.000 | 175.923 | 221${\rm{(}}Pm\overline {{\rm{3}}m} )$ |
| α-Fe+(Vac-1H) | 5.6321 | 5.6103 | 5.6103 | 90.000 | 90.001 | 89.999 | 177.270 | 99${\rm{(}}P{\rm{4}}MM)$ |
| α-Fe+(Vac-2H) | 5.6285 | 5.6285 | 5.6484 | 90.000 | 90.000 | 90.000 | 178.940 | 123(P4/MMM) |
| α-Fe+(Vac-3H) | 5.6297 | 5.6598 | 5.6853 | 90.004 | 90.011 | 90.002 | 181.154 | 25(PMM2) |
| α-Fe+(Vac-4H) | 5.6727 | 5.6943 | 5.6723 | 89.966 | 90.540 | 89.973 | 183.221 | 38(AMM2) |
| α-Fe+(Vac-5H) | 5.6905 | 5.7086 | 5.7093 | 90.000 | 90.004 | 90.002 | 185.467 | 99${\rm{(}}P{\rm{4}}MM)$ |
| α-Fe+(Vac-6H) | 5.7407 | 5.7270 | 5.7208 | 89.433 | 89.691 | 89.692 | 188.064 | 5(C2) |
表5α-Fe+(nH-Vac)的晶格常数和晶体结构
Table5.Lattice parameters and crystalline structure of α-Fe+(nH-Vac).
| 晶体类型 | ${E_{{\rm{crystal}}}}$/eV | ${E_{{\rm{ZP}}}}$/eV | ${E_{{\rm{bind}}}}$/eV | $E_{{\rm{form}}}$/eV | $\Delta H_{_{ {\rm{sol} } } }^{\rm{H} }$/eV |
| α–Fe+Vac | –12977.593 | — | 5.369 | 2.416 | — |
| α–Fe+(Vac-1H) | –12993.933 | 0.141 | 5.055 | 1.928 | –0.347 |
| α–Fe+(Vac-2H) | –13010.262 | 0.295 | 4.777 | 1.450 | –0.324 |
| α–Fe+(Vac-3H) | –13026.330 | 0.478 | 4.513 | 1.234 | –0.034 |
| α–Fe+(Vac-4H) | –13042.359 | 0.670 | 4.275 | 1.056 | 0.014 |
| α–Fe+(Vac-5H) | –13058.299 | 0.889 | 4.055 | 0.968 | 0.131 |
| α–Fe+(Vac-6H) | –13073.995 | 1.149 | 3.842 | 1.123 | 0.438 |
表6α-Fe+(nH-Vac)的结合能、形成能和溶解热
Table6.Binding energy, formation energy, and heat of solution of α-Fe+(nH-Vac).
由表5及表6可见, 随着H原子向空位中溶入, 晶体的体积不断增大, 而α-Fe+(nH-Vac)结合能不断降低, 因此可以认为空位中H原子的溶入会降低晶体中原子的结合能力. 随着空位所结合的H原子数量增多, 总零点能加速升高, 其原因就在于空位中H原子数量增加后其自由体积减小, 振动频率增加. 通过表6中的





| 晶体类型 | $E_{{\rm{trap}}}^{\rm{H}}$/eV | |
| 不考虑${E_{{\rm{ZP}}}}$ | 考虑${E_{{\rm{ZP}}}}$ | |
| α-Fe+(Vac-1H) | 0.633 | 0.778 |
| α-Fe+(Vac-2H) | 0.623 | 0.627 |
| α-Fe+(Vac-3H) | 0.361 | 0.211 |
| α-Fe+(Vac-4H) | 0.322 | –0.011 |
| α-Fe+(Vac-5H) | 0.227 | –0.297 |
| α-Fe+(Vac-6H) | –0.028 | –0.772 |
表7α-Fe+(nH-Vac)对H原子的陷阱能
Table7.Hydrogen trapping energy of α-Fe+(nH-Vac)
结合晶体内部电子结构对图4中各α-Fe + (nH-Vac)晶体稳定构型做进一步解释和说明. 图4(a)表明含有单一空位的α-Fe+Vac晶体结构仍然属于对称性最高的立方晶系, 空位中心最近邻的8个原子Fe2, Fe4, Fe6, Fe8, Fe10, Fe12, Fe14, Fe15净电荷均为0.07, 相对失电子, 而Fe7, Fe11, Fe13相对得电子.
空位中溶入1个H原子时, 超胞中H原子并未处于弛豫后结构的空位中心, 而是占据了空位八面体间隙附近的内壁位置, 这与其他金属中的情况类似[27,28]. 如图4(e)所示, H原子最近邻原子是Fe13, 且H-Fe13距离为1.6716 ?, 两原子连线平行于X轴, 从图4(e)可知单空位中H原子可能占据的位置共有6个.
当α-Fe+(Vac-1H)继续溶入第二个H原子, 两个H原子依然占据等电荷密度面, 且第二个H原子与第一个H原子关于空位中心对称, 二者的Y轴和Z轴坐标与Fe13一致, 此时系统能量最低, 两个H原子距离明显超过成键距离, 相比于空位中存在1个H原子的情况, 该晶体具有更高的对称性. 由表8、图5(d)和图5(e)可知, 两个H原子净电荷均为–0.20并形成反键, H原子之间区域差分电荷密度为负并接近于0, 因此空位内的H原子趋于相互排斥而不是形成H2分子. 由图4(f)可知H在空位内的可能构型有3个. 对比图5(c)和图5(e)的等电荷密度面(电荷密度为0.24 ?–3)和电荷密度等高线, 当空位中溶入H原子时, 空位腔中的电荷总量整体增加, 且空位远处存在电荷密度降低的区域. 可推断空位中的H能够影响较大范围的晶格电荷密度分布.
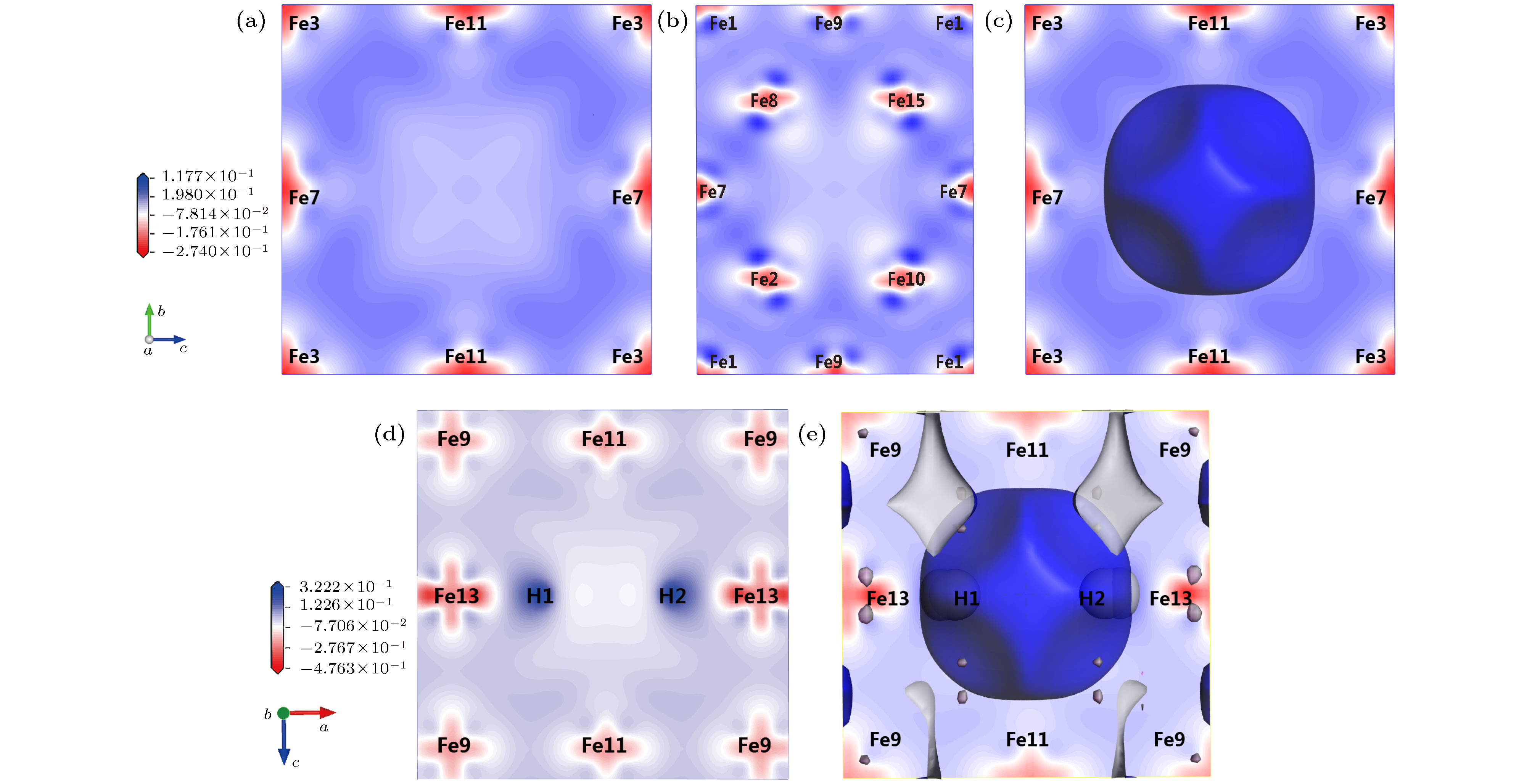 图 5 α-Fe+(nH-Vac)的等电荷面和差分电荷密度 (a) α-Fe+Vac ((100)面); (b) α-Fe+Vac ((110)面); (c) α-Fe+Vac等电荷面; (d) α-Fe+(2H-Vac) ((010)面); (e) α-Fe+(2H-Vac)等电荷面
图 5 α-Fe+(nH-Vac)的等电荷面和差分电荷密度 (a) α-Fe+Vac ((100)面); (b) α-Fe+Vac ((110)面); (c) α-Fe+Vac等电荷面; (d) α-Fe+(2H-Vac) ((010)面); (e) α-Fe+(2H-Vac)等电荷面Figure5. Isoelectronic density surface and electron density difference of α-Fe+(nH-Vac): (a) Electron density difference of α-Fe+Vac in surface (100); (b) electron density difference of α-Fe+Vac in surface (110); (c) isoelectric density surface of α-Fe+Vac; (d) electron density difference of α-Fe+(2H-Vac) in surface (010); (e) isoelectric density surface of α-Fe+(2H-Vac).
| 晶体类型 | 原子 | 轨道电荷占据数 | 总布居 | 净布居 | ||
| s | p | d | ||||
| α-Fe+Vac | Fe7, Fe11, Fe13 | 0.74 | 0.72 | 6.68 | 8.13 | –0.13 |
| Fe3, Fe5, Fe9 | 0.68 | 0.73 | 6.63 | 8.04 | –0.04 | |
| 其余Fe原子 | 0.66 | 0.67 | 6.60 | 7.93 | 0.07 | |
| α-Fe+(Vac-2H) | Fe13 | 0.71 | 0.71 | 6.73 | 8.15 | –0.15 |
| Fe9 | 0.66 | 0.70 | 6.62 | 7.98 | 0.02 | |
| Fe11 | 0.71 | 0.71 | 6.65 | 8.07 | –0.07 | |
| α-Fe+Vac—α-Fe+(Vac-6H) | H1 | 1.20—1.22 | 0 | 0 | 1.20—1.22 | –0.20— –0.22 |
| 自由态 | H | 1.00 | 0 | 0 | 1.00 | 0 |
表8α-Fe+(nH-Vac)晶体原子轨道布居
Table8.Atomic orbital population of α-Fe+(nH-Vac).
当空位溶入第3个H原子时, 第3个H原子处于靠近Fe7原子的空位内壁, 二者X和Y坐标基本一致, 三个H原子的净电荷均为负值, 因此H1和H2受H3的排斥, 沿着Z轴负方向移动, 由图4(b)可知空位内的可能构型有12个.
空位溶入第4个H原子时, 弛豫构型如图4(c), 4个H原子形成四面体结构, 由于净电荷均为负值, 因此仍然有相互排斥的倾向, H3和H4的存在使得H1和H2的X和Z坐标相比于Fe11均发生较大改变, H1和H2的存在又使H3和H4与最近邻Fe原子距离更近, H3和H4的相互作用导致其X轴和Z轴方向坐标分别发生了较大改变, 这种结构能使晶格内应力和总能达到最低, 共有12个可能构型.
空位溶入第5个H原子时, 弛豫构型如图4(d), 晶体对称性相对于α-Fe+(Vac-4H)有所提高. H原子仍形成四面体结构, 且第5个H原子位于四面体底面, 底面近似正方形, 实际上该晶体与α-Fe+(Vac-1H)具有相同的对称性, 其可能构型有6个.
空位溶入第6个H原子时, 弛豫构型如图4(g), α-Fe+(Vac-6H)晶体为三斜晶, 空位内的6个H原子形成倾斜的扁八面体, 其可能构型有6个, 根据表6可知, 该晶体的形成能和溶解热突然升高, 其稳定存在所需的氢分压远远高于本文研究对象的使用条件, 故在后续研究中不再考虑.
2
3.3.空位对α-Fe四面体间隙H原子固溶的影响
由3.2节计算结果可知, α-Fe中的空位

 图 6 α-Fe+Vac+H(T-site)的晶体结构
图 6 α-Fe+Vac+H(T-site)的晶体结构Figure6. Crystalline structure of α-Fe+Vac+H(T-site).
| 晶体类型 | ${E_{{\rm{crystal}}}}$/eV | ${E_{{\rm{ZP}}}}$/eV | ${E_{{\rm{bind}}}}$/eV | $E_{{\rm{form}}}$/eV | $\Delta H_{{ {\rm{sol} } } }^{\rm{H} }$/eV |
| α–Fe+Vac+H(T-site) | –12993.354 — –12993.353 | 0.248—0.250 | 5.012 | 2.755—2.756 | 0.339—0.340 |
表9α-Fe+Vac+H(T-site)的结合能、形成能和溶解热
Table9.Binding energy, formation energy, and heat of solution of α-Fe+Vac+H(T-site).
4.1.α-Fe中H原子溶解问题的物理模型
假设待研究晶体内共有2 × 2 × 2超胞M个, 其中只含有单一空位的超胞数量为















2
4.2.0 K下H原子溶解倾向随化学势的变化情况
如前所述,













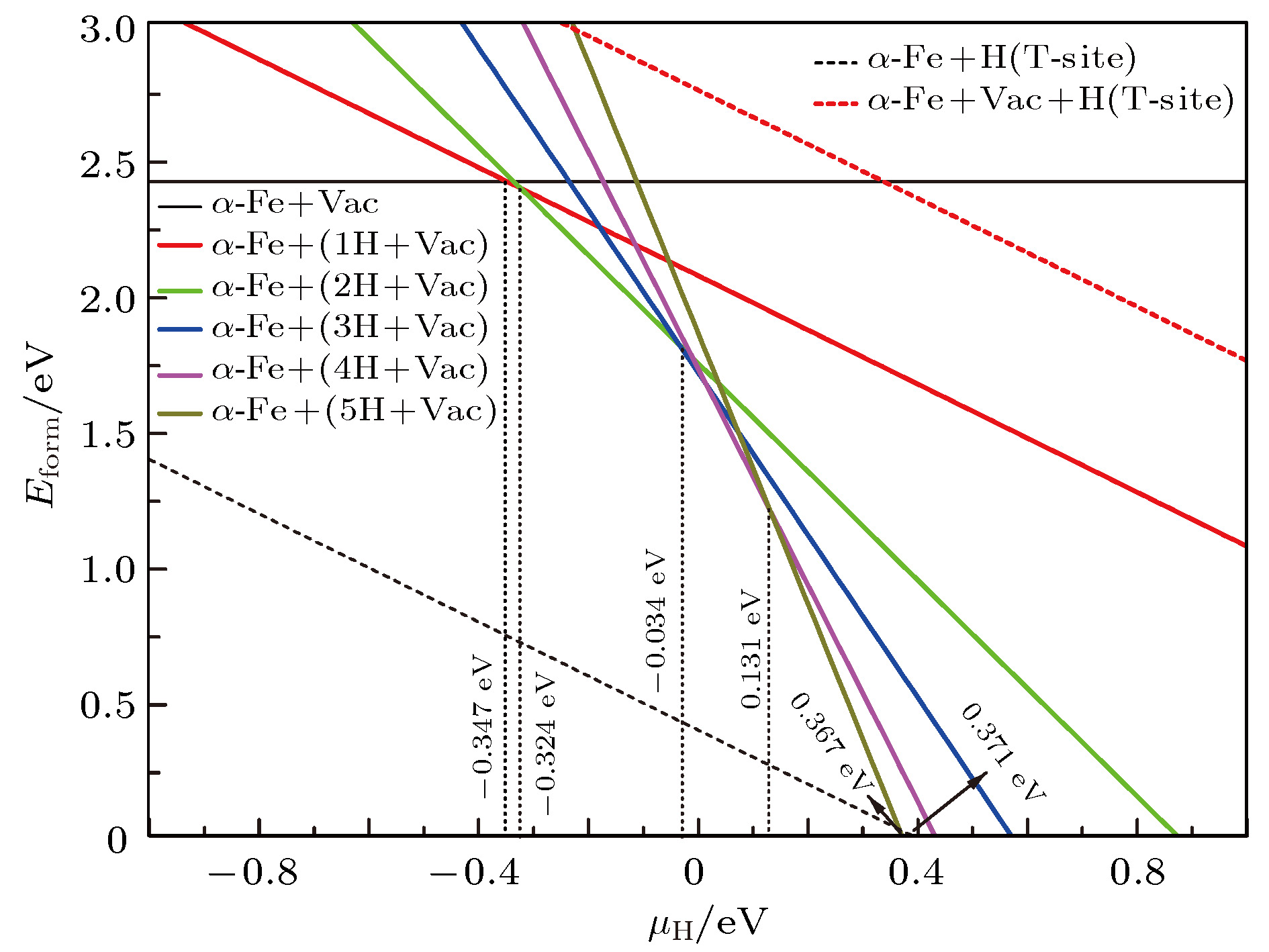 图 7 0 K下缺陷形成能随μH变化情况
图 7 0 K下缺陷形成能随μH变化情况Figure7. Formation energy of defects at 0 K for different H chemical potentials.
2
4.3.变温条件下空位和H原子溶解倾向随化学势的变化情况
根据4.1节建立的物理模型计算单空位、空位溶H、间隙溶H的含量如图8(a),(b),(c)所示, 可见只有富氢条件及高温条件下, 在热力学平衡条件下α-Fe才能溶有较多的H, 其中空位容纳H原子的比例较低. 基于上述结果得到含空位缺陷α-Fe中的H平衡溶解度如图9所示.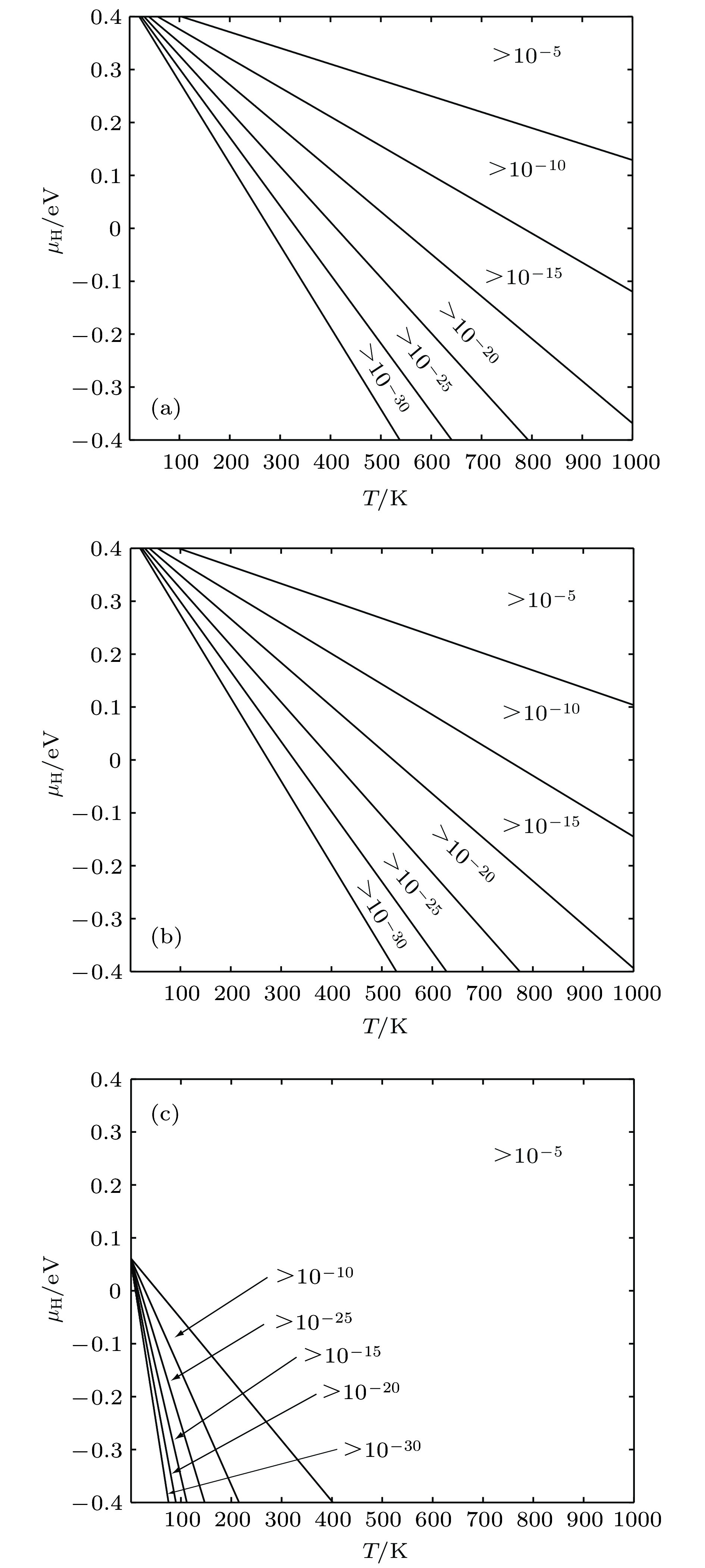 图 8 缺陷随T和μH变化情况 (a) 空位平衡浓度; (b) 空位内H平衡浓度; (c) 间隙H平衡浓度
图 8 缺陷随T和μH变化情况 (a) 空位平衡浓度; (b) 空位内H平衡浓度; (c) 间隙H平衡浓度Figure8. Equilibrium concentration of defects for different temperature and H chemical potentials: (a) Equilibrium concentration of vacancies; (b) equilibrium concentration of H occupying in vacancies; (c) equilibrium concentration of H occupying in interstitial positions.
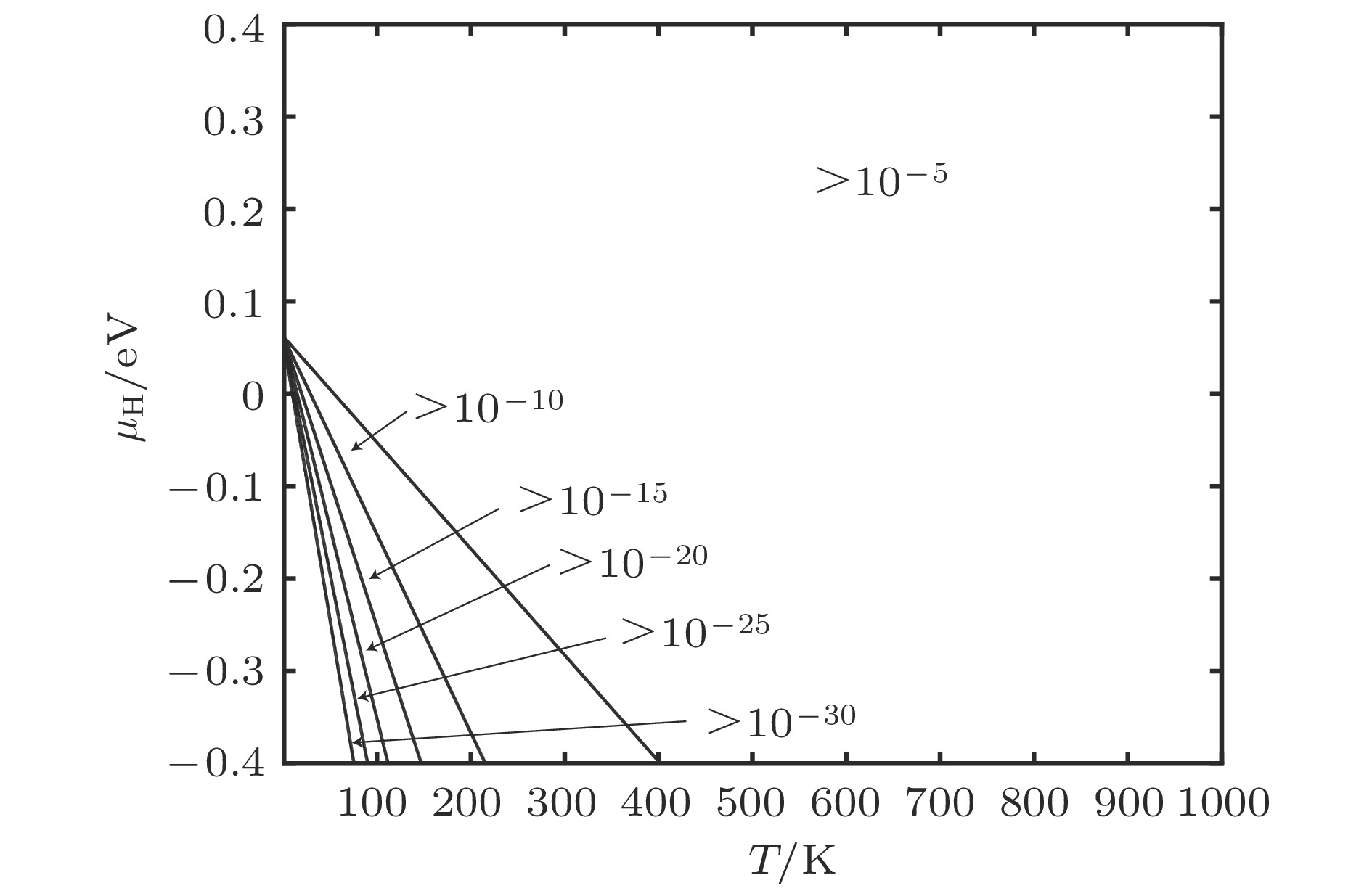 图 9 含空位α-Fe晶体H总平衡浓度
图 9 含空位α-Fe晶体H总平衡浓度Figure9. Equilibrium total concentration of H of α-Fe containing vacancies.
2
4.4.实验验证
采用Devnathan-Stachurski双电解池技术对计算结果进行验证. 双电解池实验装置如图10所示, 电解池分为阴极室和阳极室, 待侧纯Fe试样夹在阴极和阳极室中间. 主单元工作电极为阳极并连接金属薄片, 辅助电极和参比电极置于NaOH溶液中. 从单元为恒电流充氢单元, 工作电极作为阴极连接于纯Fe试样上, 辅助电极和阳极置于0.1 mol/L的HCl溶液中, 阴极充氢时电解液中加入少量Na2S. 实验开始前将试样阳极表面镀镍, 之后在阳极室施加电位, 使金属薄板相对于参比电极有一个恒正电位, 当残余电流小于1 μA时, 阴极室立即施加恒定充氢电流, 这样就在试样的阴极侧表面形成扩散H浓度为




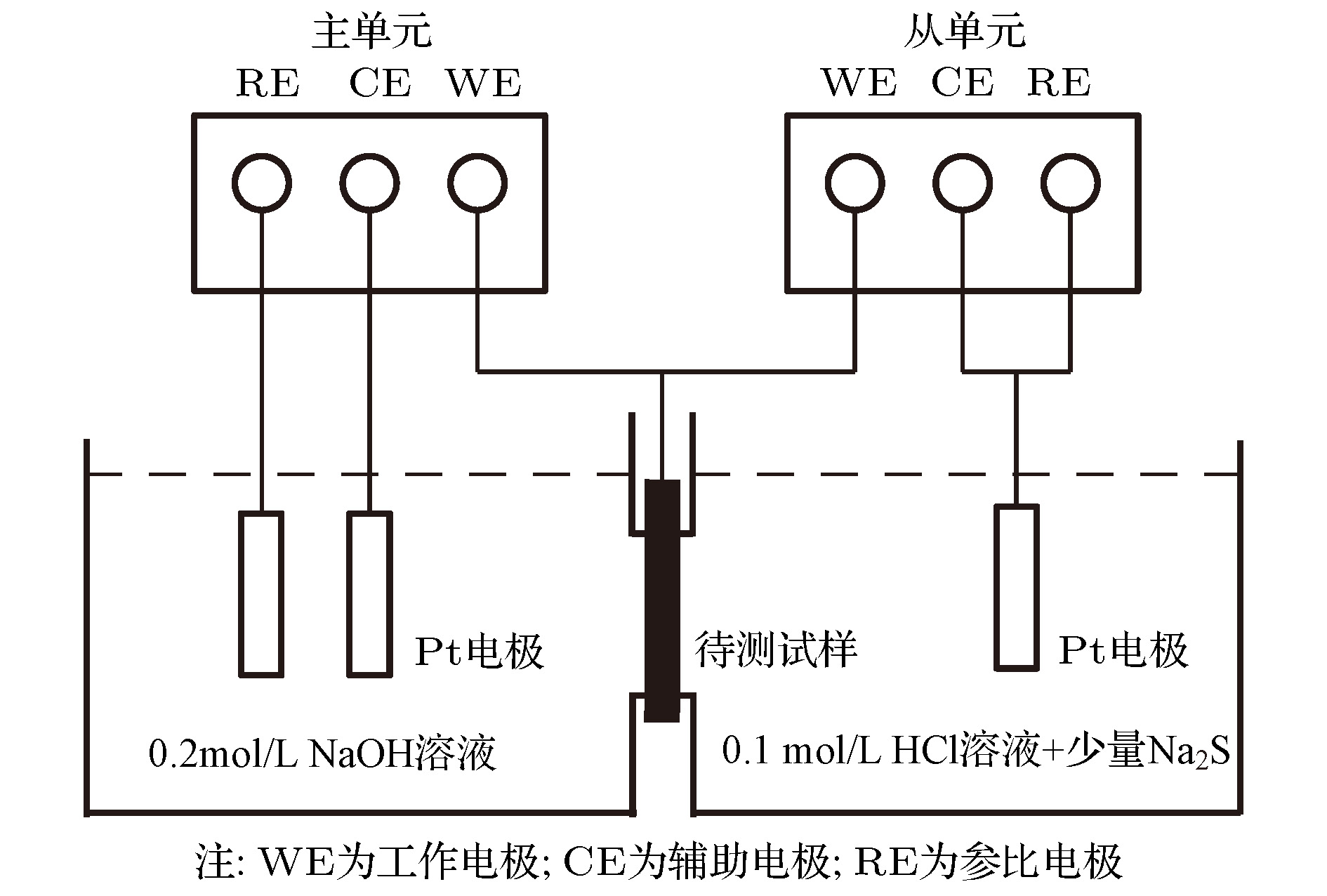 图 10 Devnathan-Stachurski双电解池示意图
图 10 Devnathan-Stachurski双电解池示意图Figure10. Schematic diagram of Devnathan-Stachurski double electrolytic cell.



| 温度/K | μH/eV | cH/% | |
| 计算值 | 实验值 | ||
| 298.15 | –0.239 | 2.08 × 10–2 | 4.41 × 10–2 2.88 × 10–2, 其中晶格溶H占总扩散H含量的43%[2] |
表10H平衡溶解度计算值和实验值
Table10.Calculated and test value of equilibrium concentration of H atom.
1) H原子与α-Fe内最近邻Fe原子主要依靠H的1s轨道电子和Fe的4s和3p轨道杂化而微弱成键, 与以往研究结果一致.
2) 空位内的H原子优先占据八面体间隙附近的空位内壁位置, 在考虑零点能情况下, 真空0 K下单空位最多只能稳定容纳3个H原子, 且H原子之间没有自发形成H2分子的倾向.
3) 结合能计算结果表明H原子的溶入会降低α-Fe晶体的稳定性, 并在α-Fe晶体中产生了较大范围的电荷密度起伏, 弱化晶体中原子之间的结合.
4) 通过热力学分析得出四面体间隙是α-Fe中H原子固溶的主要位置, 且随着

5)计算结果与扩散H平衡浓度实验值吻合良好, 说明计算和分析过程合理. 但本文的研究对象限于α-Fe晶格间隙和空位缺陷, 后续将对α-Fe晶体中的晶界、位错及合金原子与H的相互作用开展研究.
