全文HTML
--> --> -->
基于统计物理学的微观振动信息人们也能得到与双原子分子振动相关的宏观热力学量[14]. 对于NO分子微观振动信息的理论研究主要基于1950年Huber和Herzberg[15]建立的计算双原子分子振动能级表达式. 早在1975年, Billingsley[16]便采用优化价态构型多构型自洽场方法得到了基态NO分子的电子偶极矩函数和势能曲线. 2006年, Reddy等[17]依靠实验技术用改进的RKR (Rydberg-Klein-Rees)方法构建了基态NO和NO+的势能曲线, 并采用五参数(Hulburt-Hirschfelder, H-H)函数拟合获得了这些电子态的离解能. 2018年, Qin等[18]使用RKR方法获得了NO分子低阶电子态的势能曲线, 利用该势能曲线通过求解薛定谔方程确定了振动能级, 计算了NO分子平衡和非平衡状态的热力学性质.
以上研究为了解NO分子电子态的势能曲线和振动信息做出了巨大贡献. 然而, 实验上现阶段的光谱测量技术一般只能获得体系低激发振动量子态的物理信息, 对于大多数NO分子电子激发态而言, 高激发振动量子态尤其是处于离解区的数据却很难得到. 由于这一客观事实的存在, 人们通常采用简单的谐振子模型(simple harmonic oscillatormodel, SHO)来计算体系的微观振动信息, 再借助量子统计系综理论来获得系统的宏观热力学量. 如此以来, 用SHO模型计算得到的一部分实际上不存在的虚高能级也参与了统计计算, 从而导致最终的热力学计算结果往往出现较大误差. 为了获得NO分子电子态可靠的完全振动能级从而进一步研究其微观振动对宏观热力学性质的影响, 本文基于课题组前期建立的变分代数法(variational algebraic method, VAM)和能精确计算分子离解能的新解析表达式[19], 研究获得了基态NO分子的完全振动能级, 随后采用量子统计系综理论[14]计算了NO分子的热力学性质, 并将计算获得的理论结果与实验数据进行了分析和比较.











由于NO具有两个电子的双重分裂影响, 其转动配分函数是




对于常见的双原子分子组成的宏观系统, 内能是系统能量在所有微观状态上的平均值, 摩尔内能可表示为






为了获得某分子电子态的完全振动能级, 孙卫国课题组在前期的研究工作中提出了变分代数方法(VAM), 该方法的建立是根据Born-Oppenheimer近似, 利用二阶微扰理论求解薛定谔方程, 获得如下双原子分子体系振动能级的表达式[24]:













综上所述, 对于基态NO分子, 加入微小的变分项抵消实验振动能级误差并利用代数方法求解其完全振动能级










如此一来, 用VAM方法求得的热力学量的具体表现形式就不同于谐振子模型无穷项求和的形式, 减小了对实际上不存在的虚高能级进行统计求和带来的误差.



| $\upsilon $ | $E_{\rm{\upsilon }}^{{\rm{exp}}}$[17] | $E_{\rm{\upsilon }}^{{\rm{MCSCF}}}$[16] | $E_{\rm{\upsilon }}^{{\rm{VAM}}}$ | $E_{\rm{\upsilon }}^{{\rm{exp}}} - E_{\rm{\upsilon }}^{{\rm{MCSCF}}}$ | $E_{\rm{\upsilon }}^{{\rm{exp}}} - E_{\rm{\upsilon }}^{{\rm{VAM}}}$ | υ | $E_{\rm{\upsilon }}^{{\rm{VAM}}}$ |
| 0 | 948.50 | 948.60 | 948.50 | –0.10 | 0 | 26 | 40240.70 |
| 1 | 2824.50 | 2824.60 | 2824.50 | –0.10 | 0 | 27 | 41310.84 |
| 2 | 4627.30 | 4672.40 | 4672.27 | –45.10 | –44.97 | 28 | 42340.94 |
| 3 | 6491.90 | 6492.10 | 6491.90 | –0.20 | 0 | 29 | 43329.70 |
| 4 | 8283.50 | 8283.90 | 8283.43 | –0.40 | 0.07 | 30 | 44275.74 |
| 5 | 10046.90 | 10047.80 | 10046.90 | –0.90 | 0 | 31 | 45177.59 |
| 6 | 11782.30 | 11783.70 | 11782.30 | –1.40 | 0 | 32 | 46033.69 |
| 7 | 13489.40 | 13491.70 | 13489.60 | –2.30 | –0.20 | 33 | 46842.41 |
| 8 | 15171.80 | 15168.73 | 34 | 47602.00 | |||
| 9 | 16823.90 | 16819.60 | 35 | 48310.62 | |||
| 10 | 18448.00 | 18442.06 | 36 | 48966.35 | |||
| 11 | 20044.00 | 20035.93 | 37 | 49567.15 | |||
| 12 | 21611.80 | 21600.99 | 38 | 50110.90 | |||
| 13 | 23151.30 | 23136.97 | 39 | 50595.35 | |||
| 14 | 24662.40 | 24643.56 | 40 | 51018.15 | |||
| 15 | 26144.80 | 26120.38 | 41 | 51376.86 | |||
| 16 | 27598.50 | 27567.04 | 42 | 51668.92 | |||
| 17 | 29023.20 | 28983.04 | 43 | 51891.64 | |||
| 18 | 30367.88 | 44 | 52042.23 | ||||
| 19 | 31720.95 | ||||||
| 20 | 33041.62 | ||||||
| 21 | 34329.18 | ||||||
| 22 | 35582.84 | ||||||
| 23 | 36801.78 | ||||||
| 24 | 37985.07 | ||||||
| 25 | 39131.73 | ||||||
| $D_{\rm{e}}^{{\rm{exp}}}$ | 52155.68 | $D_{\rm{e}}^{{\rm{cal}}}$ | 52155.68 | ||||
| 注: $E_{\rm{\upsilon }}^{{\rm{VAM}}}$中表示VAM计算所需的已知实验振动能级用黑体标出. | |||||||
表1基态NO分子不同振动能级间的比较 (单位: cm–1)
Table1.Comparison of different vibrational levels of NO in the ground state (in cm–1).
将VAM方法和MCSCF方法计算的振动能级与实验能级进行比较, VAM的误差明显小于MCSCF的误差. 在实验上获得的有限精确振动能级区域, 除振动量子数为2的能级外, VAM振动能级与实验值的误差均在[-0.2, 0.067] cm-1范围内. 振动量子数为2的能级误差很大, 实验上获得的数据为4627.300 cm–1, VAM计算值为4672.274 cm–1, MCSCF的计算值为4672.000 cm–1, 两种方法误差均达到45 cm–1左右. 王建坤等[25]用多组态相互作用理论(multireference configuration interaction, MRCI)以cc-pVDZ为基组得到的理论计算结果也在4672 cm–1附近. 更直观地, 从图1可以看出, 使用(18)式和(19)式来判断VAM振动能级的数值结果在其离解区的收敛性是可靠的.
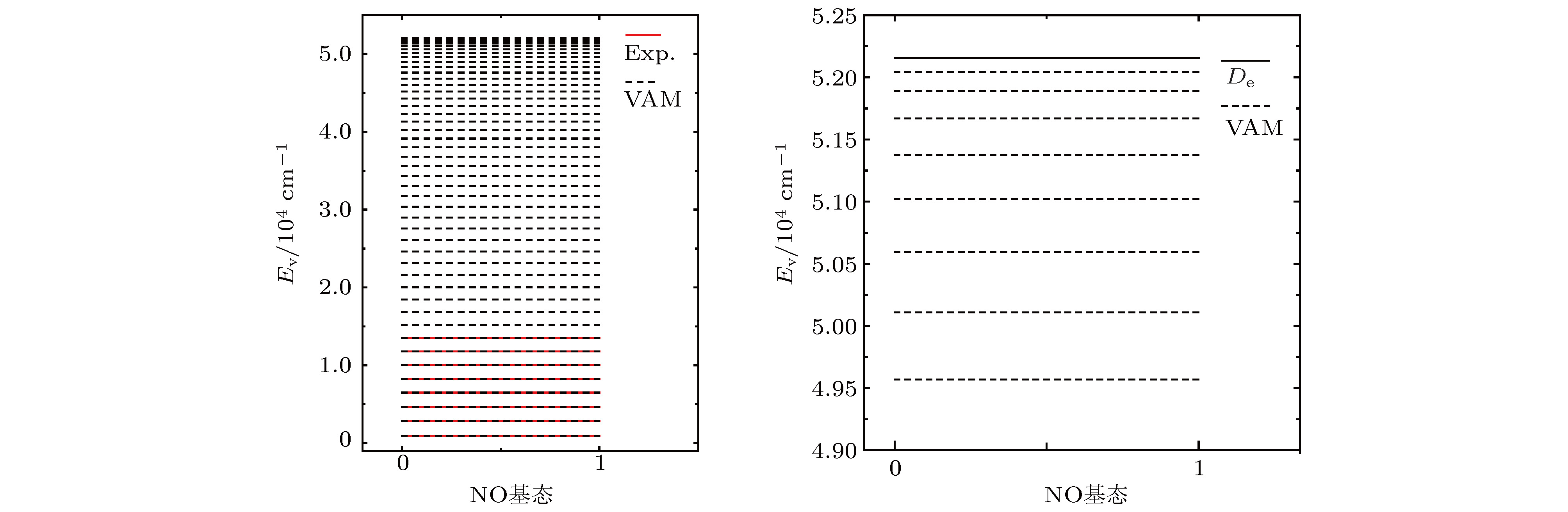 图 1 VAM完全振动能级和实验能级的对比图
图 1 VAM完全振动能级和实验能级的对比图Figure1. Comparison between the VAM full vibrational energies and experimental ones
获得基态NO分子精确的完全振动能级后, 根据量子统计系综理论计算了1000—5000 K温度范围内NO分子气体系统摩尔内能、摩尔熵和摩尔热容. 在高温低压气体条件下, 系统处于理想气体状态, 去掉分子热运动的热容量, 剩余分子振动和转动对应热容量





| T/K | $U_{{\rm{RKR}}}^{{\rm{cal}}}$ | $U_{{\rm{MCSCF}}}^{{\rm{cal}}}$ | $U_{{\rm{SHO}}}^{{\rm{cal}}}$ | $U_{{\rm{VAM}}}^{{\rm{cal}}}$ |
| 1000 | 21.28 | 21.28 | 21.27 | 21.28 |
| 1100 | 22.61 | 22.61 | 22.59 | 22.61 |
| 1200 | 23.98 | 23.99 | 23.95 | 23.98 |
| 1300 | 25.39 | 25.40 | 25.34 | 25.39 |
| 1400 | 26.84 | 26.84 | 26.77 | 26.84 |
| 1500 | 28.31 | 28.31 | 28.23 | 28.31 |
| 1600 | 29.80 | 29.80 | 29.70 | 29.80 |
| 1700 | 31.31 | 31.31 | 31.20 | 31.31 |
| 1800 | 32.84 | 32.84 | 32.71 | 32.84 |
| 1900 | 34.38 | 34.39 | 34.24 | 34.38 |
| 2000 | 35.94 | 35.94 | 35.77 | 35.94 |
| 2100 | 37.51 | 37.51 | 37.32 | 37.51 |
| 2200 | 39.09 | 39.09 | 38.88 | 39.09 |
| 2300 | 40.68 | 40.68 | 40.45 | 40.68 |
| 2400 | 42.28 | 42.28 | 42.03 | 42.28 |
| 2500 | 43.89 | 43.89 | 43.61 | 43.89 |
| 2600 | 45.50 | 45.50 | 45.19 | 45.50 |
| 2700 | 47.12 | 47.12 | 46.79 | 47.12 |
| 2800 | 48.75 | 48.75 | 48.38 | 48.75 |
| 2900 | 50.38 | 50.38 | 49.99 | 50.38 |
| 3000 | 52.01 | 52.01 | 51.59 | 52.01 |
| 3100 | 53.66 | 53.65 | 53.20 | 53.66 |
| 3200 | 55.30 | 55.30 | 54.81 | 55.30 |
| 3300 | 56.95 | 56.95 | 56.43 | 56.95 |
| 3400 | 58.60 | 58.60 | 58.05 | 58.60 |
| 3500 | 60.26 | 60.26 | 59.67 | 60.26 |
| 3600 | 61.92 | 61.92 | 61.29 | 61.92 |
| 3700 | 63.58 | 63.58 | 62.91 | 63.59 |
| 3800 | 65.25 | 65.24 | 64.54 | 65.25 |
| 3900 | 66.91 | 66.91 | 66.17 | 66.92 |
| 4000 | 68.58 | 68.58 | 67.80 | 68.60 |
| 4100 | 70.26 | 70.25 | 69.43 | 70.27 |
| 4200 | 71.93 | 71.93 | 71.07 | 71.95 |
| 4300 | 73.61 | 73.61 | 72.70 | 73.63 |
| 4400 | 75.29 | 75.28 | 74.34 | 75.32 |
| 4500 | 76.97 | 76.96 | 75.97 | 77.01 |
| 4600 | 78.65 | 78.64 | 77.61 | 78.69 |
| 4700 | 80.33 | 80.32 | 79.25 | 80.39 |
| 4800 | 82.01 | 82.01 | 80.89 | 82.08 |
| 4900 | 83.69 | 83.69 | 82.53 | 83.78 |
| 5000 | 85.38 | 85.37 | 84.17 | 85.48 |
表2使用RKR, MCSCF, SHO和VAM振动能级计算的摩尔内能 (单位: J·K–1·mol–1)
Table2.Calculated molar internal energy using RKR, MCSCF, SHO, and VAM vibrational energies as the vibrational contributions, respectively (in J·K–1·mol–1).
| T/K | $S_{{\rm{RKR}}}^{{\rm{cal}}}$ | $S_{{\rm{MCSCF}}}^{{\rm{cal}}}$ | $S_{{\rm{SHO}}}^{{\rm{cal}}}$ | $S_{{\rm{VAM}}}^{{\rm{cal}}}$ |
| 1000 | 60.50 | 60.50 | 60.42 | 60.50 |
| 1100 | 61.76 | 61.76 | 61.67 | 61.76 |
| 1200 | 62.96 | 62.96 | 62.86 | 62.96 |
| 1300 | 64.09 | 64.09 | 63.97 | 64.09 |
| 1400 | 65.16 | 65.16 | 65.03 | 65.16 |
| 1500 | 66.17 | 66.17 | 66.03 | 66.17 |
| 1600 | 67.13 | 67.13 | 66.99 | 67.13 |
| 1700 | 68.05 | 68.05 | 67.89 | 68.05 |
| 1800 | 68.92 | 68.92 | 68.76 | 68.92 |
| 1900 | 69.76 | 69.76 | 69.58 | 69.76 |
| 2000 | 70.56 | 70.56 | 70.37 | 70.56 |
| 2100 | 71.32 | 71.32 | 71.13 | 71.32 |
| 2200 | 72.06 | 72.06 | 71.85 | 72.06 |
| 2300 | 72.77 | 72.77 | 72.55 | 72.77 |
| 2400 | 73.45 | 73.45 | 73.22 | 73.45 |
| 2500 | 74.10 | 74.10 | 73.87 | 74.10 |
| 2600 | 74.73 | 74.73 | 74.49 | 74.73 |
| 2700 | 75.35 | 75.35 | 75.09 | 75.35 |
| 2800 | 75.94 | 75.94 | 75.67 | 75.94 |
| 2900 | 76.51 | 76.51 | 76.23 | 76.51 |
| 3000 | 77.06 | 77.06 | 76.78 | 77.06 |
| 3100 | 77.60 | 77.60 | 77.30 | 77.60 |
| 3200 | 78.12 | 78.12 | 77.82 | 78.12 |
| 3300 | 78.63 | 78.63 | 78.31 | 78.63 |
| 3400 | 79.12 | 79.12 | 78.80 | 79.13 |
| 3500 | 79.60 | 79.60 | 79.27 | 79.61 |
| 3600 | 80.07 | 80.07 | 79.72 | 80.07 |
| 3700 | 80.53 | 80.53 | 80.17 | 80.53 |
| 3800 | 80.97 | 80.97 | 80.60 | 80.97 |
| 3900 | 81.41 | 81.40 | 81.03 | 81.41 |
| 4000 | 81.83 | 81.83 | 81.44 | 81.83 |
| 4100 | 82.24 | 82.24 | 81.84 | 82.25 |
| 4200 | 82.65 | 82.64 | 82.24 | 82.65 |
| 4300 | 83.04 | 83.04 | 82.62 | 83.05 |
| 4400 | 83.43 | 83.42 | 83.00 | 83.43 |
| 4500 | 83.80 | 83.80 | 83.36 | 83.81 |
| 4600 | 84.17 | 84.17 | 83.72 | 84.18 |
| 4700 | 84.53 | 84.53 | 84.08 | 84.55 |
| 4800 | 84.89 | 84.89 | 84.42 | 84.90 |
| 4900 | 85.24 | 85.23 | 84.76 | 85.25 |
| 5000 | 85.58 | 85.57 | 85.09 | 85.60 |
表3使用RKR, MCSCF、SHO和VAM振动能级计算的摩尔熵(单位: J·K–1·mol–1)
Table3.Calculated molar entropy using RKR, MCSCF, SHO, and VAM vibrational energies as the vibrational contributions, respectively (in J·K–1·mol–1).
内能、熵、热容均是重要的热力学参数, 但在实验中常常测定的是热容的值



| T/K | ${C_{{\rm{exp}}}}$ | $C_{{\rm{RKR}}}^{{\rm{cal}}}$ | $C_{{\rm{MCSCF}}}^{{\rm{cal}}}$ | $C_{{\rm{SHO}}}^{{\rm{cal}}}$ | $C_{{\rm{VAM}}}^{{\rm{cal}}}$ | ${\delta _{{\rm{RKR}}}}$a | ${\delta _{{\rm{MCSCF}}}}$b | ${\delta _{{\rm{SHO}}}}$c | ${\delta _{{\rm{VAM}}}}$d |
| 1000 | 13.20 | 13.04 | 13.04 | 10.72 | 13.04 | 1.22% | 1.22% | 18.82% | 1.22% |
| 1100 | 13.68 | 13.52 | 13.52 | 11.24 | 13.52 | 1.18% | 1.18% | 17.85% | 1.18% |
| 1200 | 14.09 | 13.93 | 13.93 | 11.73 | 13.93 | 1.16% | 1.16% | 16.77% | 1.16% |
| 1300 | 14.44 | 14.27 | 14.27 | 12.17 | 14.27 | 1.17% | 1.17% | 15.69% | 1.17% |
| 1400 | 14.74 | 14.56 | 14.56 | 12.58 | 14.56 | 1.19% | 1.19% | 14.66% | 1.19% |
| 1500 | 14.99 | 14.81 | 14.81 | 12.94 | 14.81 | 1.21% | 1.21% | 13.70% | 1.21% |
| 1600 | 15.22 | 15.03 | 15.03 | 13.26 | 15.03 | 1.23% | 1.23% | 12.83% | 1.23% |
| 1700 | 15.41 | 15.22 | 15.21 | 13.55 | 15.22 | 1.26% | 1.26% | 12.04% | 1.26% |
| 1800 | 15.58 | 15.38 | 15.38 | 13.81 | 15.38 | 1.29% | 1.29% | 11.35% | 1.29% |
| 1900 | 15.73 | 15.52 | 15.52 | 14.04 | 15.52 | 1.33% | 1.33% | 10.73% | 1.33% |
| 2000 | 15.86 | 15.65 | 15.64 | 14.25 | 15.65 | 1.36% | 1.37% | 10.18% | 1.36% |
| 2100 | 15.98 | 15.76 | 15.76 | 14.43 | 15.76 | 1.41% | 1.41% | 9.71% | 1.41% |
| 2200 | 16.09 | 15.86 | 15.86 | 14.59 | 15.86 | 1.45% | 1.45% | 9.29% | 1.45% |
| 2300 | 16.18 | 15.95 | 15.94 | 14.74 | 15.95 | 1.48% | 1.49% | 8.92% | 1.48% |
| 2400 | 16.27 | 16.03 | 16.03 | 14.88 | 16.03 | 1.52% | 1.53% | 8.59% | 1.52% |
| 2500 | 16.35 | 16.10 | 16.10 | 15.00 | 16.10 | 1.56% | 1.57% | 8.31% | 1.56% |
| 2600 | 16.43 | 16.17 | 16.17 | 15.10 | 16.17 | 1.61% | 1.61% | 8.07% | 1.61% |
| 2700 | 16.50 | 16.23 | 16.23 | 15.20 | 16.23 | 1.65% | 1.65% | 7.85% | 1.65% |
| 2800 | 16.56 | 16.28 | 16.28 | 15.29 | 16.28 | 1.69% | 1.70% | 7.67% | 1.69% |
| 2900 | 16.62 | 16.34 | 16.33 | 15.38 | 16.34 | 1.74% | 1.74% | 7.51% | 1.73% |
| 3000 | 16.68 | 16.38 | 16.38 | 15.45 | 16.39 | 1.77% | 1.78% | 7.37% | 1.77% |
| 3100 | 16.73 | 16.43 | 16.43 | 15.52 | 16.43 | 1.82% | 1.83% | 7.25% | 1.80% |
| 3200 | 16.78 | 16.47 | 16.47 | 15.58 | 16.47 | 1.87% | 1.88% | 7.16% | 1.85% |
| 3300 | 16.83 | 16.51 | 16.51 | 15.64 | 16.51 | 1.91% | 1.92% | 7.08% | 1.88% |
| 3400 | 16.88 | 16.55 | 16.54 | 15.69 | 16.55 | 1.96% | 1.97% | 7.01% | 1.92% |
| 3500 | 16.92 | 16.58 | 16.58 | 15.74 | 16.59 | 2.01% | 2.02% | 6.96% | 1.95% |
| 3600 | 16.96 | 16.61 | 16.61 | 15.79 | 16.62 | 2.06% | 2.07% | 6.91% | 1.99% |
| 3700 | 17.00 | 16.64 | 16.64 | 15.83 | 16.66 | 2.12% | 2.13% | 6.89% | 2.03% |
| 3800 | 17.04 | 16.67 | 16.67 | 15.87 | 16.69 | 2.18% | 2.19% | 6.86% | 2.06% |
| 3900 | 17.08 | 16.69 | 16.69 | 15.91 | 16.72 | 2.24% | 2.25% | 6.85% | 2.09% |
| 4000 | 17.11 | 16.72 | 16.71 | 15.94 | 16.75 | 2.31% | 2.32% | 6.85% | 2.13% |
| 4100 | 17.15 | 16.74 | 16.74 | 15.97 | 16.78 | 2.38% | 2.40% | 6.86% | 2.16% |
| 4200 | 17.18 | 16.76 | 16.75 | 16.00 | 16.81 | 2.46% | 2.48% | 6.86% | 2.18% |
| 4300 | 17.21 | 16.77 | 16.77 | 16.03 | 16.83 | 2.55% | 2.57% | 6.88% | 2.21% |
| 4400 | 17.24 | 16.79 | 16.79 | 16.05 | 16.86 | 2.64% | 2.66% | 6.90% | 2.24% |
| 4500 | 17.28 | 16.80 | 16.80 | 16.08 | 16.89 | 2.74% | 2.76% | 6.93% | 2.26% |
| 4600 | 17.31 | 16.81 | 16.81 | 16.10 | 16.91 | 2.85% | 2.86% | 6.96% | 2.29% |
| 4700 | 17.34 | 16.82 | 16.82 | 16.12 | 16.94 | 2.96% | 2.99% | 7.00% | 2.31% |
| 4800 | 17.36 | 16.83 | 16.83 | 16.14 | 16.96 | 3.09% | 3.11% | 7.04% | 2.32% |
| 4900 | 17.39 | 16.83 | 16.83 | 16.16 | 16.99 | 3.22% | 3.25% | 7.08% | 2.35% |
| 5000 | 17.42 | 16.84 | 16.83 | 16.18 | 17.01 | 3.37% | 3.38% | 7.13% | 2.36% |
| 注: a, ${\delta _{{\rm{RKR}}}} = \left| {C_{{\rm{exp}}}^{} - C_{{\rm{RKR}}}^{{\rm{cal}}}} \right|/C_{{\rm{exp}}}^{} \times 100\% $; b, ${\delta _{{\rm{MCSCF}}}} = \left| {C_{{\rm{exp}}}^{} - C_{{\rm{MCSCF}}}^{{\rm{cal}}}} \right|/C_{{\rm{exp}}}^{} \times 100\% $; c, $ {\delta _{{\rm{SHO}}}} = \left| {C_{{\rm{exp}}} - C_{{\rm{SHO}}}^{{\rm{cal}}}} \right|/C_{{\rm{exp}}} \times 100\% $; d,$ {\delta _{{\rm{VAM}}}} = \left| {C_{{\rm{exp}}} - C_{{\rm{VAM}}}^{{\rm{cal}}}} \right|/C_{{\rm{exp}}} \times 100\% .$ | |||||||||
表4不同摩尔热容及其与实验值的相对误差 (单位: J·K–1·mol–1)
Table4.Comparisons of different molar capacities with observed experimental

表4还列出了








图2可以更直观地体现不同方法获得的热容计算值与热容实验值的相对误差. 由图2可知VAM热容











 图 2 不同热容相对误差的比较
图 2 不同热容相对误差的比较Figure2. Comparison of the relative errors of different heat capacities
