Abstract:In this paper the electronic structures and optical properties of Cu:Fe:Mg:LiNbO3 crystals and their comparative groups are investigated by first-principles based on the density functional theory to explore the characteristics of charge transfer in crystals and analyse the parameters of the two-colour holographic storage technology based on optical properties of crystals. The basic crystal model is built as a supercell structure 2 × 2 × 1 of near-stoichiometric pure LiNbO3 crystal with 120 atoms, including 24 Li atoms, 24 Nb atoms and 72 O atoms. Above that the five doped crystal models are established as follows: the copper doped LiNbO3 crystal (Cu:LiNbO3), the ferri doped LiNbO3 crystal (Fe:LiNbO3), the copper and ferri co-doped LiNbO3 crystal (Cu:Fe:LiNbO3), the copper, ferri and magnesium tri-doped LiNbO3 crystal (Cu:Fe:Mg:LiNbO3) with doping ions at Li sites, and the copper, ferri and magnesium tri-doped LiNbO3 crystal (Cu:Fe:Mg(E):LiNbO3) with ferri ions at Nb sites and magnesium ions at both Li sites and Nb sites. The last two models represent the concentration of Mg ions below the threshold (~6.0 mol%) and over the threshold respectively. The charge compensation forms are taken successively as $\small {{\rm{Cu}}_{\rm{Li}}^+}\text-{\rm{V}}_{\rm{Li}}^-$, $\small {{\rm{Fe}}_{\rm{Li}}^{2+}}\text-{2\rm{V}}_{\rm{Li}}^-$, ${{\rm{Fe}}_{\rm{Li}}^{2+}}\text-{\rm{Cu}}_{\rm{Li}}^+ \text-{3\rm{V}}_{\rm{Li}}^- $, ${{\rm{Mg}}_{\rm{Li}}^{+} \text-{\rm{Fe}}_{\rm{Li}}^{2+}}\text- $${\rm{Cu}}_{\rm{Li}}^+\text -{4\rm{V}}_{\rm{Li}}^-$ and ${{\rm{3Mg}}_{\rm{Li}}^{+}}\text-{\rm{Mg}}_{\rm{Nb}}^{3-}\text-{\rm{Fe}}_{\rm{Nb}}^{2-} \text-{2\rm{Cu}}_{\rm{Li}}^+$in doped models. The results show that the extrinsic defect levels within the forbidden band of Cu:LiNbO3 crystal and Fe:LiNbO3 crystal are mainly contributed by the 3d orbits of Cu ions and the 3d orbits of Fe ions respectively. The forbidden band widths are 3.45 eV and 3.42 eV respetively in these two samples. In Cu:Fe:LiNbO3 crystal, the impurity levels are contributed by the 3d orbits of Cu and Fe ions; the forbidden band width is 3.24 eV; the absorption peaks are formed at 1.36, 2.53, and 3.01 eV. The Cu:Fe:Mg:LiNbO3 and Cu:Fe:Mg(E):LiNbO3 crystal presentthe forbidden band width of 2.89 eV and 3.30 eV respectively; the absorption peaks are formed at 2.45, 1.89 eV and 2.89, 2.59 eV, 2.24 eV, respectively. In Cu:Fe:Mg:LiNbO3 crystal, the weak absorption peak at 3.01 eV disappears, beacause of the superposition of the red-shifted absorption edge and the next bigger peak. The peak locations move slightly, which can be explained by the crystal field changing under the different doping concentrations and the different occupying positions of doping ions. In Cu:Fe:Mg(E):LiNbO3 crystal, the absorption peak near 2.5 eV is stronger than that of the other tri-doped crystal, which may be caused by the deference in occupancy among Fe ions. The peak at 2.9 eV can be chosen as erasing light, and the peak at 2.5 eV as write and read light in the two-center nonvolatile holography. The tri-doped crystal with Mg2+ concentration over the threshold shows obvious absorption peak at 2.9 eV and stronger absorption at 2.5 eV, which is beneficial for this application. The strong absorption of write light can shorten the time to reach the saturation of diffraction efficiency, then increase the dynamic range (M/#) and the sensitivity (S). Meanwhile, in this Mg doping condition, write time can be shortened, so optical damage can be weakened, and finally the image quality can be optimized. Keywords:tri-doped lithium niobate crystals/ first-principles/ electronic structure/ absorption spectrum
表3为LN晶体晶格常量的几何优化值与实验值[24], 两者的差值仅在2%左右, 优化后体系结构与实验值基本相同. 优化计算中采用GGA-PW91泛函, 交换关联泛函在局域电荷密度基础上增加电荷密度梯度, 即$V_{{\rm{XC}}}^{{\rm{GGA}}} = {V_{{\rm{XC}}}}[n\left( r \right), $$\nabla n\left( r \right)] $, 但函数中未包括电荷密度高阶微分函数如${\nabla ^2}n\left( r \right)$等, 表中晶格参量的平均绝对差为0.02139 nm, 后面能带计算中与实验值的差0.24 eV, 均与泛函取值相关. 这些误差值较小, 符合模拟计算的一般要求[25], 表明采用的理论模型和计算方法是合理的. 其他一些泛函如局域密度近似LDA只含有均匀电荷密度不含电荷密度的变化函数, 理论上精确度较差; 又如, meta-GGA泛函其中含${\nabla ^2}n\left( r \right)$信息, 理论上在增加了精确度的同时也增加了计算量; 另外, hyper-GGA泛函采用精确非局域性物理量, 但不满足均匀密度极限条件, 对体相材料不适用[25].
Lattice parameter
a/ nm
b/ nm
c/ nm
V/ nm3
Experimental value
1.02966
1.02966
1.38630
1.27284
Optimization result
1.04829
1.04829
1.41321
1.33821
表3LN晶体常数的几何优化值与实验值 Table3.Geometry optimization result and experiment values of LN crystal.
几何优化中LN总能量变化如图2所示, 横坐标为迭代次数. 图2中晶体总能量不断减少, 最终趋于恒定值, 保证晶体结构处于稳定状态, 表明本文所建模型接近晶体的真实结构. 图 2 LN总能量的迭代变化 Figure2. Total energy of geometry optimization in LN crystal.






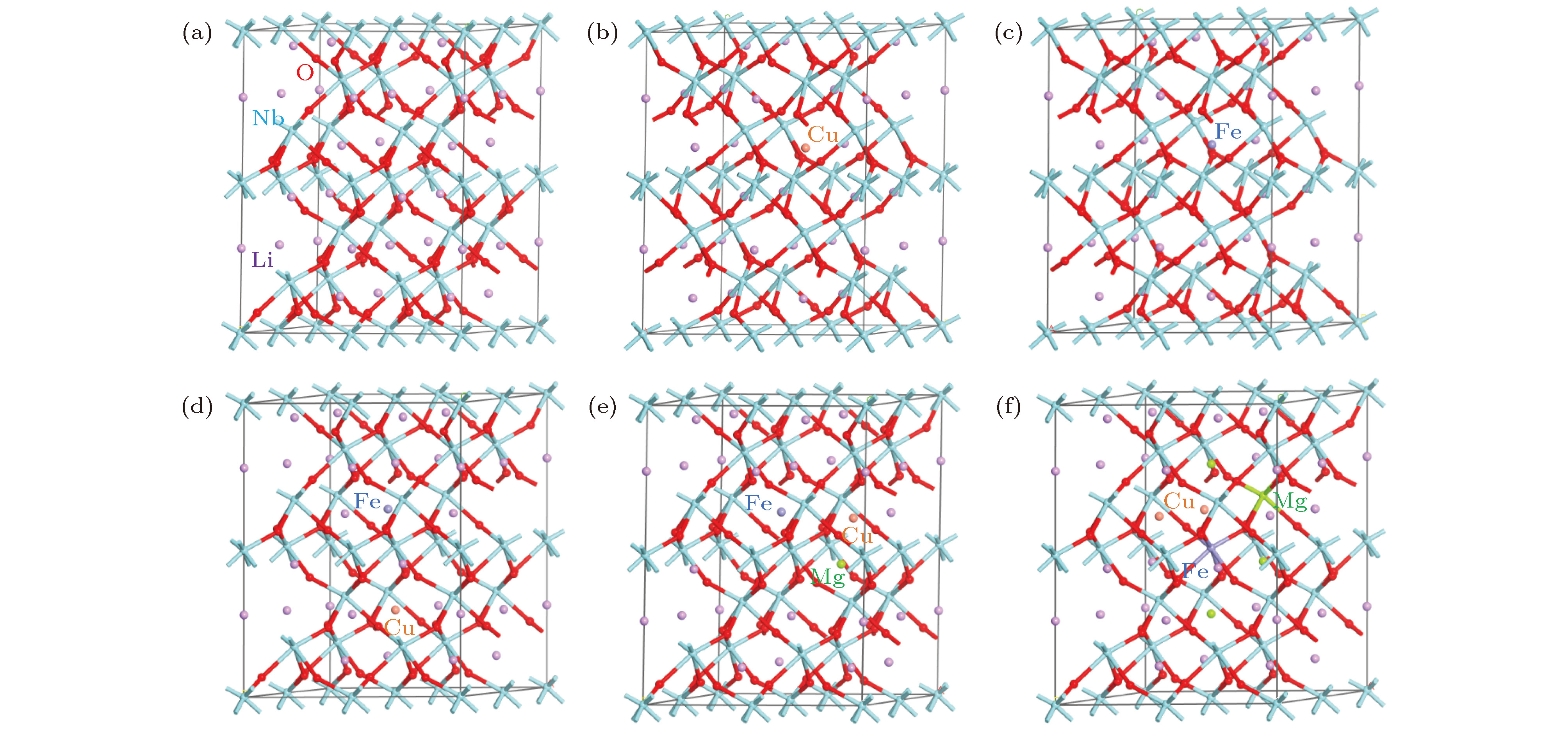 图 1 晶体结构模型 (a) LN; (b) LN1; (c) LN2; (d) LN3; (e) LN4; (f) LN5
图 1 晶体结构模型 (a) LN; (b) LN1; (c) LN2; (d) LN3; (e) LN4; (f) LN5



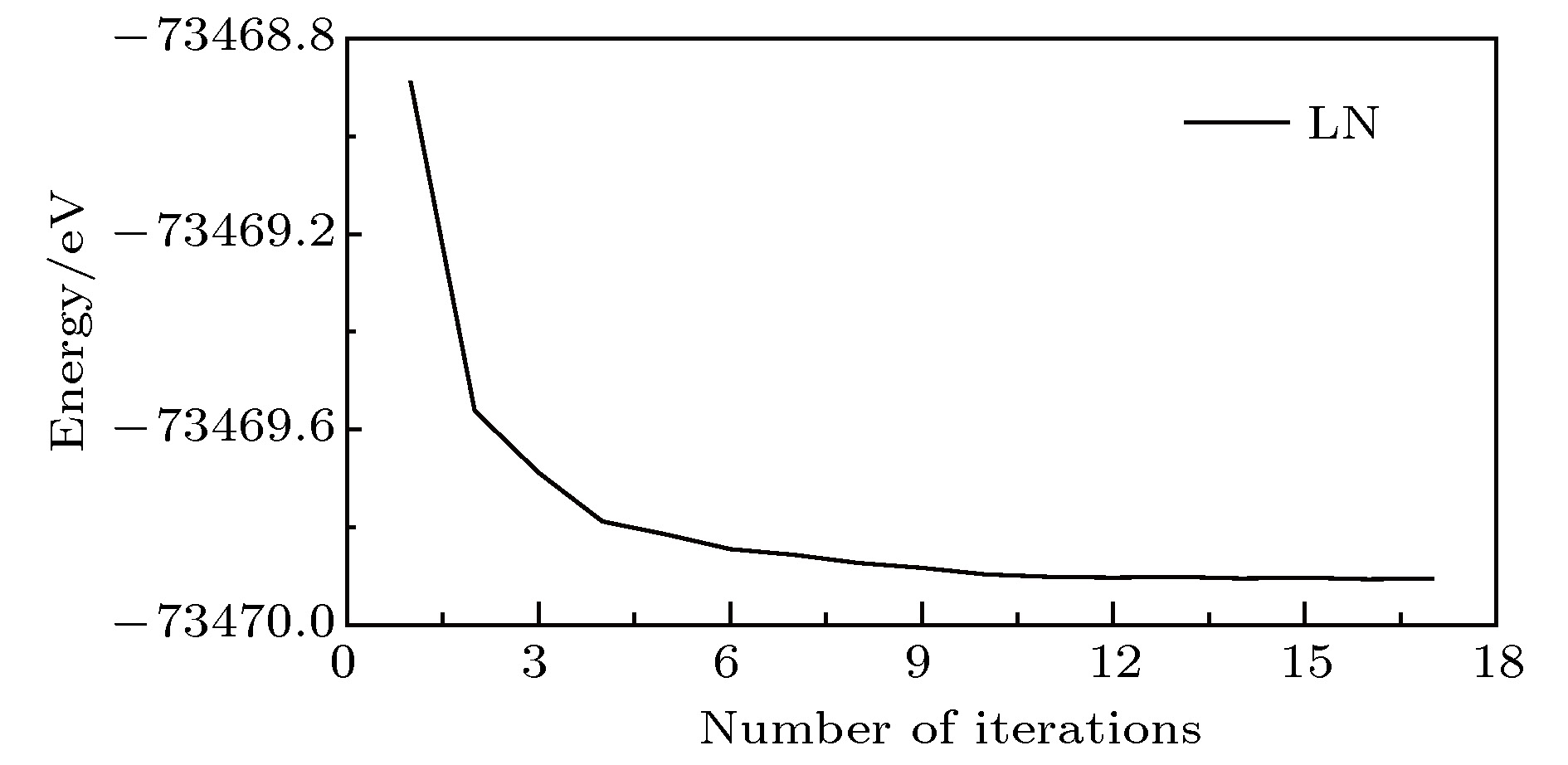 图 2 LN总能量的迭代变化
图 2 LN总能量的迭代变化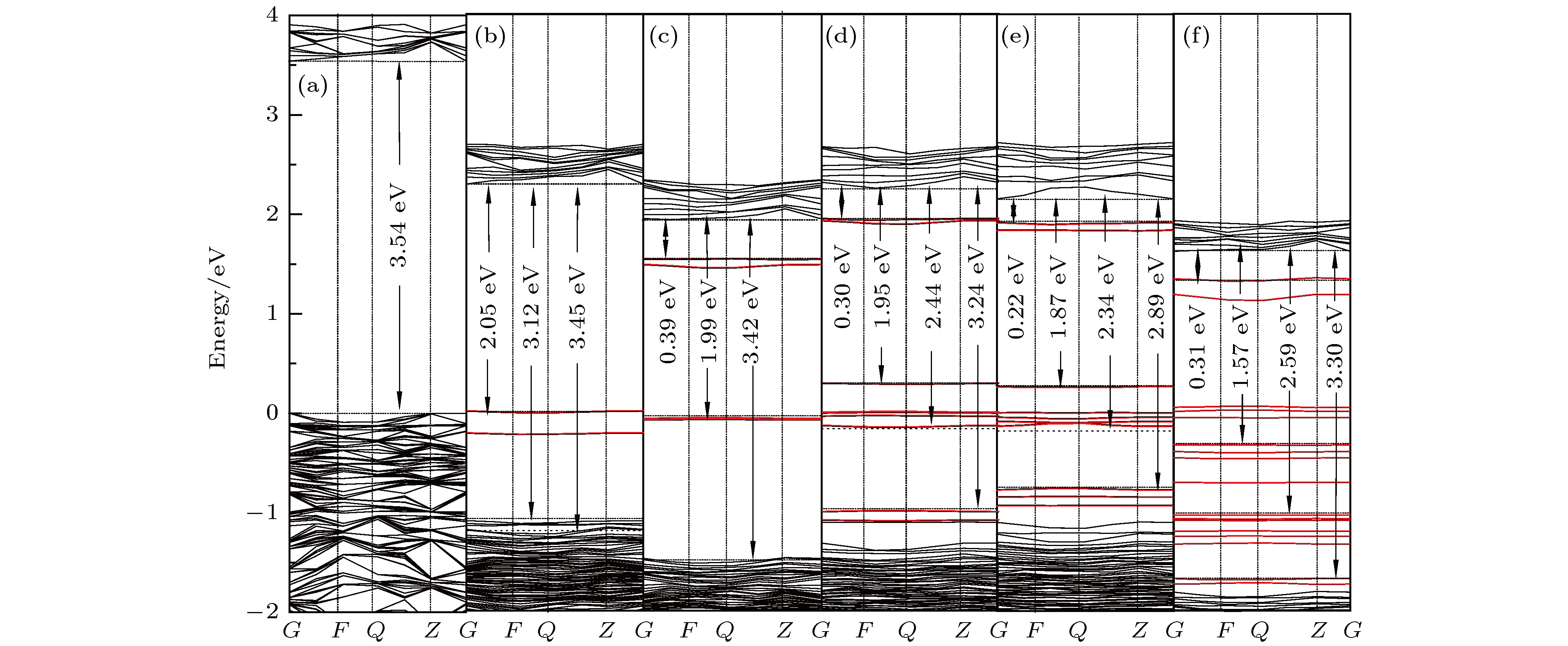 图 3 LN及不同掺杂LN晶体的能带结构图 (a) LN; (b) LN1; (c) LN2; (d) LN3; (e) LN4; (f) LN5
图 3 LN及不同掺杂LN晶体的能带结构图 (a) LN; (b) LN1; (c) LN2; (d) LN3; (e) LN4; (f) LN5

 图 4 晶体禁带附近分态密度图 (a) LN; (b) LN1; (c) LN2; (d) LN3; (e) LN4; (f) LN5
图 4 晶体禁带附近分态密度图 (a) LN; (b) LN1; (c) LN2; (d) LN3; (e) LN4; (f) LN5
 图 5 LN及各掺杂体系光吸收光谱
图 5 LN及各掺杂体系光吸收光谱













