全文HTML
--> --> -->对于有机-无机杂化钙钛矿, 构建二维(2D)-三维(3D)异质结构是提高钙钛矿稳定性的有效手段. 由于全无机钙钛矿铯离子极其稳定, 当用有机盐进行表面处理时, 有机阳离子不会和铯离子发生离子交换形成2D钙钛矿. Wang等[27]通过引入有机阳离子对CsPbI3全无机钙钛矿进行表面处理, 实现了在CsPbI3全无机钙钛矿表面构筑单分子层的有机阳离子. 该表面单分子层在提高全无机钙钛矿的热、湿稳定性的同时, 也有效钝化了钙钛矿的表面缺陷, 显著提升了器件效率. Yoo和Park[28]较为系统地评估了碘化苯烷基铵的碳链长度对于有机-无机杂化钙钛矿电流电压滞回效应的影响, 发现随着烷基碳链长度增加, 对滞回效应的改善更有效. 本文选取CsPbI2Br为研究对象, 采用乙胺碘、丙胺碘、丁胺碘三种有机铵碘盐对钙钛矿薄膜进行表面处理, 探究不同碳链长度铵盐对钙钛矿薄膜结构和湿稳定性的影响. 实验结果表明长碳链铵盐—碘化丁铵(butylamine iodine, BAI)可以显著提高钙钛矿的湿稳定性. 在此基础上组装电池, 探究了表面处理对于器件光电性能的影响, 得出处理的最佳浓度为2—4 mg/mL.
2.1.HPbI3+x的合成
将PbI2与HI (57%wt)按1 ∶ 1.4的摩尔比混合, 溶于N,N-二甲基甲酰胺(dimethylformamide, DMF)中, 冰浴搅拌2 h. 冰浴反应得到的产物在70 ℃下旋蒸. 溶剂蒸发完全后将所得的固体通过4次乙醚离心清洗, 得到无色固体粉末. 将该固体粉末于50 ℃恒温箱中干燥.2
2.2.溶液配制
按化学计量比2 ∶ 1 ∶ 1将CsI, HPbI3+x, PbBr2混合, 溶于DMF中, 形成0.5 mol/L CsPbI2Br前驱体溶液.为确定不同长度碳链对钙钛矿层稳定性的影响, 调控表面处理剂的组分和浓度, 配制以下溶液: 1) 2 mg/mL碘化乙铵(ethylammonium iodide, EAI)乙酸乙酯(EtOAc)溶液; 2) 2 mg/mL碘化丙铵(propylammonium iodide, PAI)乙酸乙酯溶液; 3) 2 mg/mL BAI乙酸乙酯溶液; 4) 4 mg/mL BAI乙酸乙酯溶液; 5) 8 mg/mL BAI乙酸乙酯溶液.
2
2.3.器件组装
将TiO2/FTO基底于70 ℃加热台上预热, 将CsPbI2Br前驱体溶液在3000 r/min转速下旋转30 s, 旋涂在基底上, 得到前驱体薄膜, 在150 ℃下退火4 min得到深棕色的钙钛矿薄膜. 将不同碳链的碘盐溶液旋涂与全无机钙钛矿薄膜表面, 其中转速为3000 r/min, 旋涂时间为30 s, 所得薄膜在100 ℃下退火4 min. 再将空穴传输材料(0.1 mol/L Spiro-MeOTAD氯苯溶液, 0.035 mol/L叔丁基锂(Li-TISF)乙腈溶液和0.12 mol/L 4-叔丁基吡啶(tBP))旋涂于钙钛矿薄膜上层, 其中旋涂条件为: 首先转速为500 r/min, 旋涂时间为2 s; 然后转速为4500 r/min, 旋涂时间为25 s, 形成空穴传输层. 最后通过热蒸发沉积的方式在空穴传输层上沉积Ag金属电极, 其中Ag层厚度为100 nm.2
2.4.性能测试
用紫外可见分光光度计测定薄膜吸光性. 用X射线衍射(X-ray diffraction, XRD)测试所制备的CsPbI2Br薄膜的晶体结构. 用扫描电子显微镜(scanning electronic microscopy, SEM)测量CsPbI2Br薄膜的表面形貌. 器件光电性能在AM 1.5G, 100 mW/cm2标准太阳光测试仪上测定, 测量条件为3 A光源. 电池光电转化效率(incident photon-to-electron conversion efficiency, IPCE)由IPCE测试仪测量.3.1.铵盐表面处理对薄膜的影响
对新制CsPbI2Br薄膜进行不同碳链的铵盐处理, 通过紫外-可见吸收和XRD来考察研究各不同碳链铵盐对钙钛矿薄膜结构的影响. 图1 (a)显示, 铵盐处理前后薄膜紫外可见吸收光谱没有发生改变, 均在670 nm左右有一吸收边, 是典型CsPbI2Br钙钛矿吸收峰, 说明在该过程中没有发生明显的I-Br离子交换. 图1(b)表明 EAI, PAI, BAI处理后的薄膜XRD图谱与未处理薄膜一致, 不同碳链的有机阳离子对CsPbI2Br钙钛矿处理时, 并未发生离子交换形成2D钙钛矿薄膜, 只是在钙钛矿表面形成了阳离子端基化. 这一现象也与先前的研究结果相符合, 即全无机钙钛矿中Cs+阳离子极其稳定, 不易发生离子交换.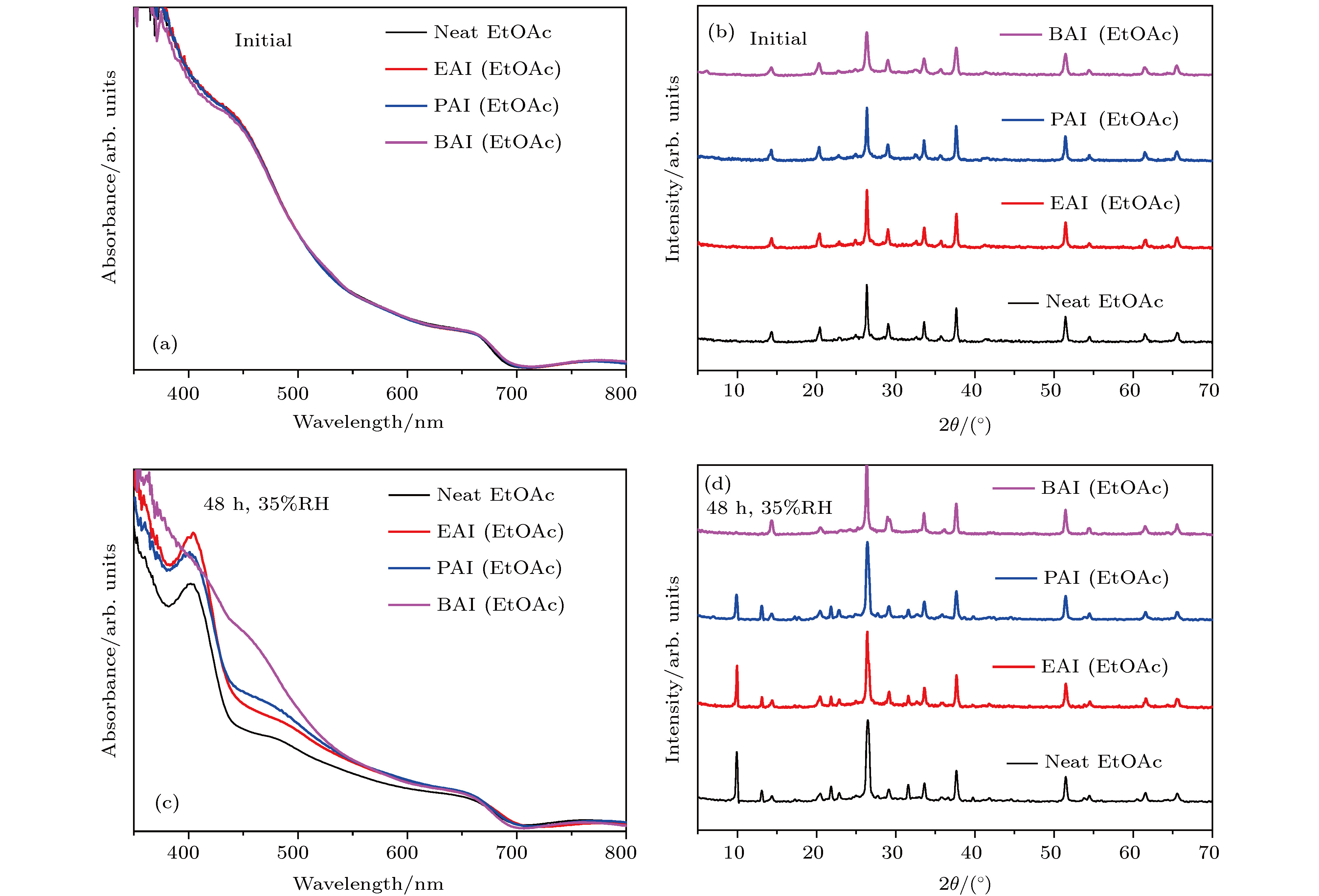 图 1 未处理、EAI、PAI和BAI处理后的CsPbI2Br薄膜 (a) 紫外可见吸收光谱(新制); (b) XRD图谱; (c) 紫外可见吸收光谱(35% RH, 48 h); (d) XRD图谱(35% RH, 48 h)
图 1 未处理、EAI、PAI和BAI处理后的CsPbI2Br薄膜 (a) 紫外可见吸收光谱(新制); (b) XRD图谱; (c) 紫外可见吸收光谱(35% RH, 48 h); (d) XRD图谱(35% RH, 48 h)Figure1. (a) UV-vis spectra and (b) XRD patterns of CsPbI2Br films under EtOAc, EAI, PAI, BAI treatments, respectively. After placed in 35% RH for 48 h, (c) UV-vis spectra and (d) XRD patterns of CsPbI2Br films under EtOAc, EAI, PAI, BAI treatments, respectively.
为了探究不同铵盐对钙钛矿薄膜湿稳定性的影响, 我们将不同铵盐处理过的全无机钙钛矿薄膜置于温度为25 ℃、相对湿度(relative humidity, RH)为35%的环境48 h. 通过图1(c)紫外可见吸收光谱发现, 未处理、EAI和PAI处理后的CsPbI2Br薄膜均在450 nm左右出现δ相吸收, 表明薄膜已经发生相变[25]. 而BAI处理的CsPbI2Br薄膜吸收与新制CsPbI2Br薄膜相比无明显变化. 图1(d) XRD图谱中显示除BAI处理外, 其余各组均在10°出现δ相衍射峰, 与紫外可见吸收光谱结果一致[27]. 实验结果表明, 与EAI, PAI相比, BAI处理能够提高全无机钙钛矿薄膜的湿稳定性.
基于以上讨论, 进一步探究BAI的浓度对处理后的CsPbI2Br全无机钙钛矿湿稳定性的影响. 因此, 设置BAI乙酸乙酯溶液的浓度分别为0, 2, 4, 8 mg/mL, 进行对比实验, 结果如图2所示. 图2(a)表明不同浓度BAI处理后的CsPbI2Br薄膜的吸收曲线相似, 证明BAI处理浓度对CsPbI2Br的带隙无影响. 图2(b)显示当薄膜在35% RH空气环境下放置48 h后, 未处理的CsPbI2Br薄膜在450 nm处出现明显的δ相吸收边, 经过BAI处理的CsPbI2Br薄膜均无明显变化. 图2 (c)和图2(d)显示2 mg/mL和4 mg/mL BAI处理的钙钛矿薄膜在分别暴露56 h和64 h后, 在450 nm处出现明显的δ相吸收, 而8 mg/mL BAI的钙钛矿薄膜则未出现明显的δ相特征. 这表明随着BAI处理浓度的提高, 钙钛矿表面有机阳离子层增厚使得CsPbI2Br钙钛矿薄膜的湿稳定性逐渐增强. 图3(b)—(d)的XRD图谱表明, 无BAI处理的钙钛矿薄膜在48 h时出现δ相特征峰(10°), 而4 mg/mL和8 mg/mL BAI处理分别在56 h和64 h出现, XRD结果与紫外可见吸收光谱图结果一致. 上述实验结果证明有机铵盐BAI处理CsPbI2Br钙钛矿时, 钙钛矿表层有机阳离子端基化的量能够显著影响其湿稳定性[29]. 但是在稳定性实验中, BA表面端基化的CsPbI2Br在降解过程中δ相特征峰强度比未经BAI处理的CsPbI2Br降解后的δ相峰更强, 表明这些端基化的BA对于CsPbI2Br钙钛矿的降解过程有显著影响.
 图 2 在35% RH空气环境下, 不同浓度BAI处理后CsPbI2Br薄膜在不同暴露时间时的紫外可见吸收光谱 (a) 0 h; (b) 48 h; (c) 56 h; (d) 64 h
图 2 在35% RH空气环境下, 不同浓度BAI处理后CsPbI2Br薄膜在不同暴露时间时的紫外可见吸收光谱 (a) 0 h; (b) 48 h; (c) 56 h; (d) 64 hFigure2. UV-visible absorption spectra of CsPbI2Br films under different BAI treatments exposed to 35% RH environment in air after various exposure time: (a) 0 h; (b) 48 h; (c) 56 h; (d) 64 h.
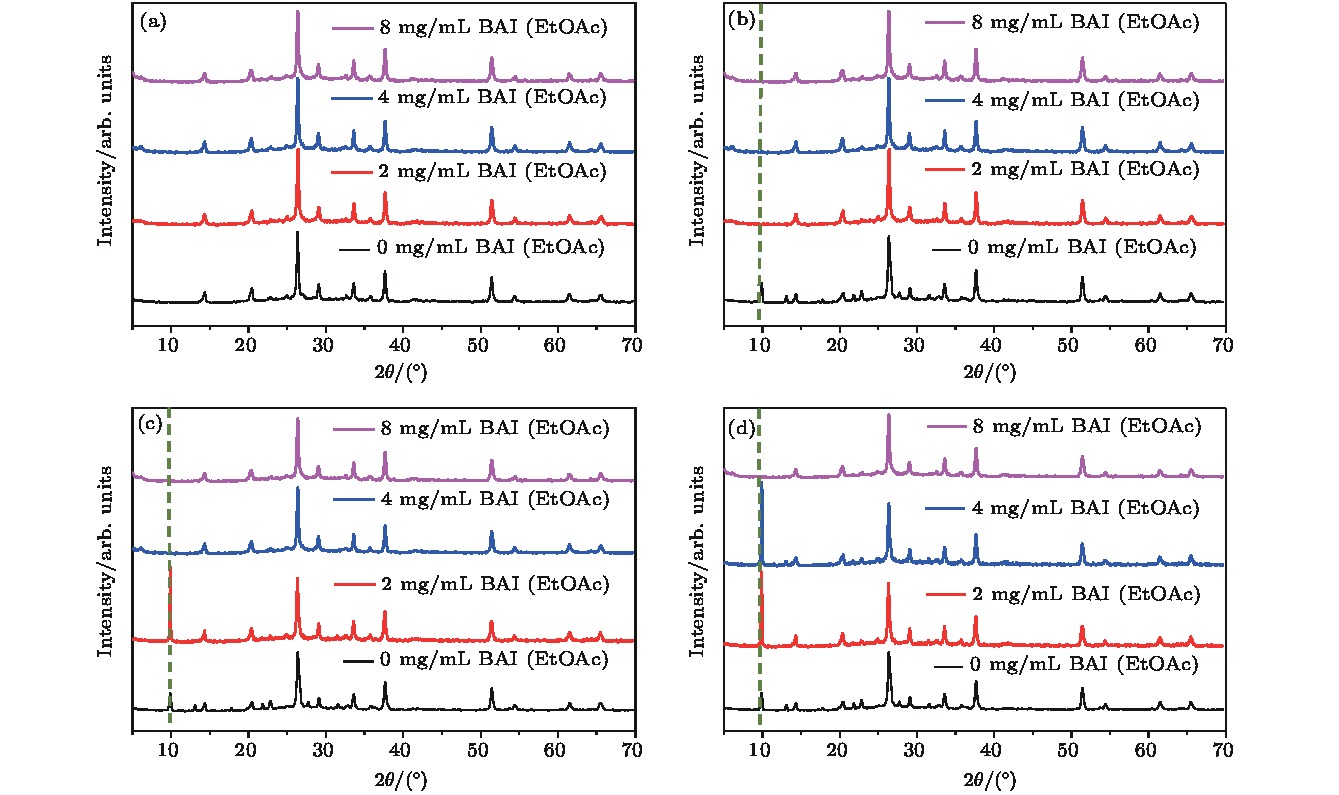 图 3 在35% RH空气环境下, 不同浓度BAI处理后CsPbI2Br薄膜在不同暴露时间时的XRD图谱 (a) 0 h; (b) 48 h; (c) 56 h; (d) 64 h
图 3 在35% RH空气环境下, 不同浓度BAI处理后CsPbI2Br薄膜在不同暴露时间时的XRD图谱 (a) 0 h; (b) 48 h; (c) 56 h; (d) 64 hFigure3. XRD patterns of CsPbI2Br films under different BAI treatments exposed to 35% RH environment in air after various exposure time: (a) 0 h; (b) 48 h; (c) 56 h; (d) 64 h.
使用SEM观察不同浓度表面处理后CsPbI2Br薄膜的形貌特征. 如图4(a)和图4(b)所示, CsPbI2Br薄膜致密没有明显孔洞, 但是钙钛矿晶粒大小不均匀, 平均尺寸约为50 nm, 呈紧密堆积. 不同浓度的BAI 处理后, CsPbI2Br钙钛矿晶粒大小没有明显增加, 但是颗粒大小变得更加均匀, 特别是晶界不再明显. 这显示BAI的处理引发了晶体的再生长, 但是晶粒的进一步增长和BAI的浓度没有显著相关. 并且BAI处理后的CsPbI2Br薄膜的电子显微镜图像出现由于导电性变差引起的变白, 从图4(c)和图4(d)可以看出, 随着BAI浓度增加到4 mg/mL和8 mg/mL, 钙钛矿晶粒大小并没有进一步增加和明显变化. BAI浓度越高表面泛白现象越严重, 这显示有机阳离子的表面端基化的增加显著改变了钙钛矿表面的电学性质.
 图 4 不同浓度BAI处理后CsPbI2Br 薄膜的SEM图谱 (a) 0 mg/mL; (b) 2 mg/mL; (c) 4 mg/mL; (d) 8 mg/mL
图 4 不同浓度BAI处理后CsPbI2Br 薄膜的SEM图谱 (a) 0 mg/mL; (b) 2 mg/mL; (c) 4 mg/mL; (d) 8 mg/mLFigure4. SEM images of CsPbI2Br films under the various BAI (EtOAc) treatments: (a) 0 mg/mL; (b) 2 mg/mL; (c) 4 mg/mL; (d) 8 mg/mL
2
3.2.器件光电性能测试
按照 FTO/TiO2/CsPbI2Br/Spiro-MeOTAD/Ag典型器件结构组装电池, 对比不同浓度BAI处理后的CsPbI2Br钙钛矿太阳能电池性能. 如表1和图5所示, 当以较低浓度BAI (2 mg/mL)处理CsPbI2Br薄膜时, 各光伏参数变化不大, 说明该条件处理能保持电池的效率. 当BAI处理浓度提高到4 mg/mL和8 mg/mL时, 器件效率明显下降. 这主要是因为BAI处理浓度增加后, 钙钛矿表面有机阳离子层增厚会阻碍电荷的传输, 从而使得短路电流显著降低(表1, 其中Jsc为短路电流密度, Uoc为开路电压, FF为填充因子, PCE为光电转化效率), 最终降低电池性能.| 处理方式 | Jsc/mA·cm–2 | Uoc/V | FF/% | PCE/% |
| 0 mg/mL BAI | 15.7 ± 0.13 | 1.05 ± 0.015 | 69 ± 3 | 11.4 ± 0.6 |
| 2 mg/mL BAI | 15.8 ± 0.1 | 1.07 ± 0.01 | 68 ± 1.8 | 11.6 ± 0.4 |
| 4 mg/mL BAI | 14.3 ± 0.1 | 1.05 ± 0.01 | 68.5 ± 2.2 | 10.3 ± 0.37 |
| 8 mg/mL BAI | 10.7 ± 0.14 | 1.04 ± 0.013 | 66 ± 2.7 | 7.5 ± 0.5 |
表1不同浓度BAI处理后CsPbI2Br钙钛矿太阳能电池的光伏参数(取32个样品均值)
Table1.Photovoltaic parameters of CsPbI2Br perovskite solar cells under different BAI treatments (average of 32 devices)
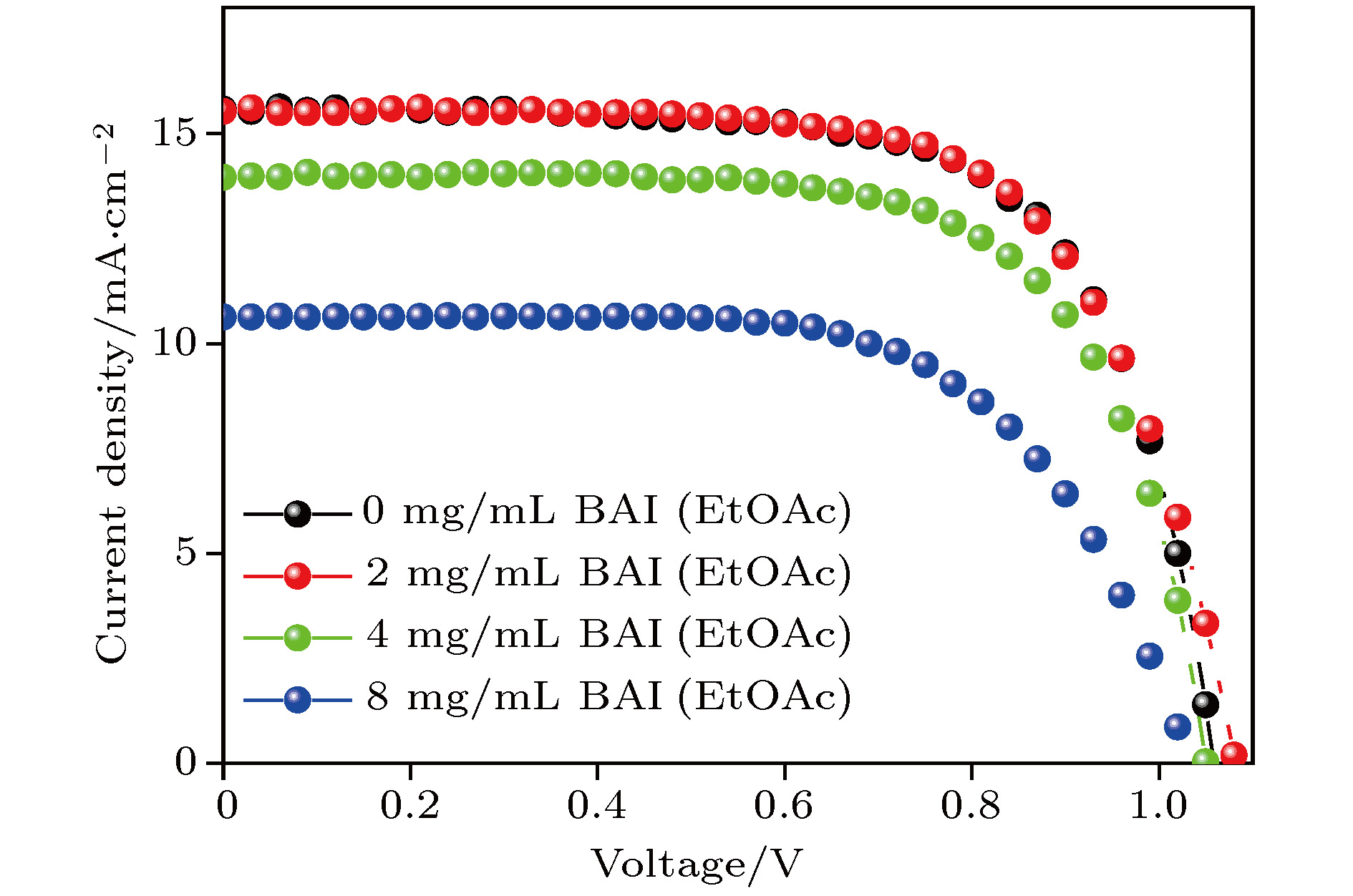 图 5 不同浓度BAI处理CsPbI2Br钙钛矿太阳能电池伏安特性曲线
图 5 不同浓度BAI处理CsPbI2Br钙钛矿太阳能电池伏安特性曲线Figure5. Voltage-current characteristics of CsPbI2Br perovskite solar cells under different BAI treatments.
