1.State Grid Heilongjiang Electric Power Company Limited Electric Power Research Institute, Harbin 150040, China 2.Key Laboratory of Engineering Dielectrics and Its Application, Ministry of Education, Heilongjiang Provincial Key Laboratory of Dielectric Engineering, School of Electrical and Electronic Engineering, Harbin University of Science and Technology, Harbin 150080, China
Fund Project:Project supported by China Postdoctoral Science Foundation (Grant No. 2013M531058) and the 2018 Comprehensive Plan of Heilongjiang Electric Power Co., Ltd., China (Grant No. 52243717000V).
Received Date:04 November 2018
Accepted Date:18 December 2018
Available Online:01 March 2019
Published Online:05 March 2019
Abstract:According to the pseudopotential plane-wave method of first-principles calculation based on the spin density functional theory, the electronic structure, magnetic and optical properties of laminated molybdenum oxides (orthonormal and monoclinic MoO3) are studied theoretically. The interlaminar dissociation energy, band-structure, spin polarization, dielectric function, and the optical absorption/reflectivity in a charged state are systematically calculated to explore the potential technology applications of laminated MoO3 as electrochromic or electromagnetic materials in optoelectronic devices. The semilocal GGA-PW91 and nonlocal HSE06 exchange-correlation functional are employed to obtain the more accurate crystal structure and band gap respectively. The cleavage energy results indicate that the single layers can easily flake off from the bulk material of these molybdenum oxides. The band structure and atomic-projected density of states prove that the conduction band minimum and valence band maximum are mainly derived from the atom-orbitals bonding oriented in layer-plane, representing characteristic two-dimensional electronic structure. The spin polarized calculations imply that the evident magnetic-moment will engender in MoO6 octahedron layers of the perfect MoO3 due to the substantial spin polarization of Mo and vertex O atoms which are ferromagnetic-coupling to produce significant net magnetic moments, essentially accounting for the magnetic source of bulk MoO3. The Mo vacancy reduces the electronic density of states derived from the spin polarized d-orbitals, leading the net magnetic moment to decrease, while the OI vacancy can reduce the density of spin-down states in the MoO3, resulting in the significant improvement of net magnetic moment. The existence of OII vacancy leads to the energetic spin-splitting of O-2p and Mo-4d orbital states, and thus increasing net magnetic moment by raising the electronic density of polarized spin-up states. The electron spin polarization of Mo-4d orbital component dominantly contributes to the bulk magnetism. The laminated MoO3 presents a significant optical response in the visible region with obvious anisotropy of optical absorption spectra, which will represent a considerable blue shift or new low-frequency absorption peaks for visible light when loading charges. The calculation results demonstrate that the laminated molybdenum oxides have evident electrochromic property with controllable magnetic moment, which provides theoretical basis and technical data for developing novel functional materials with high performance to be used in electromagnetic or optoelectronic devices. Keywords:laminated molybdenum oxide/ first-principles calculation/ electronic structure/ electrochromic material
a = 3.954 ?, b = 3.687 ?, c = 7.095 ?, $\beta $ = 103.745°
表1$\alpha $-MoO3和MoO3-Ⅱ的晶格参数 Table1.Crystal parameters of $\alpha $-MoO3 and MoO3-Ⅱ.
图 1 (a) $\alpha $-MoO3和(b) MoO3-Ⅱ晶体结构, 顶点、共角和共棱氧原子分别标识为OⅠ, OⅡ和OⅢ Figure1. The crystal structures of (a) $\alpha $-MoO3 and (b) MoO3-Ⅱ. The apical, corner-sharing and edge-sharing oxygen atoms are denoted by OⅠ, OⅡ and OⅢ respectively.
23.2.片层解离能 -->
3.2.片层解离能
由于MoO3中的原子层受较弱的vdW力束缚, 因此有可能被剥离成具有原子厚度的2D纳米片. 为了成为一个独立的单层结构, 在剥落过程中需要克服层间解离能(Ed–E0)/S0 (Ed–E0和E0分别是层间距离为d和vdW力结合平衡时的氧化钼单胞能量, S0为单胞中层平面的面积), 当d→∞时即为独立单片层的解离能. 用PW91泛函结合OBS方案的DFT-D方法计算$\alpha $-MoO3和MoO3-Ⅱ体材料的解理能, 剥落过程中的能量差与层间距d的变化关系如图2所示. 能量随分离距离d逐渐收敛, 两种材料的计算结果非常相似. 由于缺少对vdW作用的完整描述, 用DFT-D校正方法能够精确计算vdW相互作用, 计算的解离能为0.428 J/m2 ($\alpha $-MoO3)和0.419 J/m2 (MoO3-Ⅱ), 与石墨烯(0.36 J/m2)相当, 表明从层状氧化钼中剥离出单层是可行的. 最近的实验研究报道了采用多种方法从$\alpha $-MoO3和MoO3-Ⅱ体材料剥离出二维纳米片[43], 与本文理论计算的结果相一致. 图 2$\alpha $-MoO3和MoO3-Ⅱ在解离过程中的解离能随层间距离d的变化 Figure2. The cleavage energies of $\alpha $-MoO3 and MoO3-Ⅱ, varying with inter-layer distance d.
23.3.电子结构 -->
3.3.电子结构
用PW91泛函第一原理能量最小化的几何优化得到的比较准确的氧化钼晶体结构, 再分别由PW91泛函结合LDA + U方法和HSE06泛函计算电子结构, 可以比较两种方法的可靠性. 在布里渊区中选择高对称的能量色散路径G-Z-T-Y-S-X-U-G (正交相$\alpha $-MoO3)和C-B-A0-E0-C-Z-T (单斜相MoO3-Ⅱ), 计算的能带结构能量色散关系E(k)和电子能态密度(DOS)和原子投影态密度(PDOS)如图3和图4所示(以费米能级作为能量参考零点). 电子自旋极化导致$\alpha $-MoO3的能带在费米能级附近产生明显的自旋分裂, 导带底的上旋电子态能量降低至费米能级以下, 与价带顶的下旋电子态发生交叠, 使能带呈现金属特征. 但是由于层堆叠方向相邻MoO6八面体层之间的电子不发生明显交叠, 如图3能带结构中Z -T和S -X路径上的能量不发生色散, 因此在层堆叠方向上电子不能形成定向运动, 这说明即使是费米能级横跨导带底上旋带, 在$\alpha $-MoO3的层堆叠方向上也不能导电, 而在MoO6八面体层平行方向电子输运具有金属电导特征. 由于DFT计算是一种基态方法, 系统在激发过程中从(N )电子到(N1)电子的交换-不连续导致激发态的计算存在误差, 但是$\alpha $-MoO3能带计算结果和实验结果的差异并不主要是由计算误差引起的, 而是因为实验测量的带隙实际为光激发电子跃迁的光学带隙(吸收或发射光子的能量), 必须满足能量、动量和自旋态守恒. 虽然自旋极化计算表明$\alpha $-MoO3能带在带边附近发生明显的自旋分裂导致$\alpha $-CB1自旋带穿过费米能级, 但是带边光电子跃迁只能是由$\beta $-VB1能带向$\beta $-CB1能带或由$\alpha $-CB1能带向$\alpha $-CB2能带垂直跃迁, 因此会出现明显的光吸收边(光学带隙), 如图3右图所示. 表2列出了光学带隙计算值, 结果表明PW91泛函计算的光学带隙偏小, 而用HSE06泛函计算的光学带隙与其他文献报道的实验结果非常接近, 能够计算出精确的电子结构, 证明了本文采用的理论计算方法的合理性和计算结果的精确度. 用PW91泛函计算的$\alpha $-MoO3光学带隙宽度为1.38—1.64 eV, 明显小于文献报道的$\alpha $-MoO3带隙宽度3.0 eV, 而HSE06泛函的计算值分别为2.14—2.85 eV, 与实验值非常接近[29]. 此外, PW91和HSE06计算得到的能带结构色散曲线变化特征类似, 除了光学带隙的大小有所不同, HSE06泛函计算的色散曲线更加准确, 即$\alpha $-MoO3费米能级附近的$\beta $-VB1,$\beta $-CB1,$\alpha $-CB1和$\alpha $-CB2能带沿C -X和R -T路径的能量本征值发生简并, 这与文献报道的理论计算结果相一致[41,44]. 因此计算光学性质时采用HSE06泛函会获得较为精确的结果. 与$\alpha $-MoO3相比, 单斜相MoO3-II的光学带隙与$\alpha $-MoO3非常接近, 但自旋极化导致的自旋分裂更为明显, 如图3(b)所示. 这样的结果表明MoO6八面体层之间通过vdW作用形成堆叠, 堆叠方式不影响能带结构的基本特征, 但层间可能存在由自旋极化导致的自旋磁耦合. 值得注意的是, 由PW91泛函结合LDA + U方法预测MoO3 -Ⅱ能带结构与采用HSE06泛函计算结果非常一致, 说明不同交换相关泛函适用于不同的材料体系, 选择合适的交换相关泛函对提高计算电子结构的准确度具有重要意义. 图 3 用PW91泛函(左图)和HSE06泛函(右图)计算的(a) $\alpha $-MoO3和(b) MoO3-Ⅱ能带结构, 费米能级作为能量参考零点(水平虚线) Figure3. Band structures of (a) $\alpha $-MoO3 and (b) MoO3-Ⅱ calculated by PW91 functional (left panels) and HSE06 functional (right panels), with the Fermi energy level being set as reference energy zero (horizontal dot line).
表2$\alpha $-MoO3和MoO3 -Ⅱ光学能带带隙的计算值和文献报道实验数据 Table2.The calculated optical band-gaps of $\alpha $-MoO3 and MoO3 -Ⅱ in comparison with reported experimental data.
图 4 (a) $\alpha $-MoO3和(b) MoO3-Ⅱ的DOS,Mo和O的PDOS Figure4. The total density of states, Mo and O atomic projected density of states for (a) $\alpha $-MoO3 and (b) MoO3-Ⅱ.
用HSE06泛函计算了两种层合钼氧化物的DOS和PDOS, 如图4和5所示. 价带顶(VBM)的电子态主要由O原子的2p轨道(O-2p)组成, 而O-2p态和Mo-4d态(Mo原子的4d轨道)组成的杂化态的能量本征值分布在6—2 eV范围. 导带(CB)由主要的Mo-4d轨道与少部分O-2p电子态杂化而成. 另外, 不同位置氧原子的PDOS存在明显的差异, 如图4所示. 两种化合物的扭转共棱MoO6八面体上都有三种不同的氧原子(不同的物理化学环境), 分别由不同的颜色标识: 顶点原子OI单独与Mo原子配位成键, 共角氧原子OⅡ为与两个Mo原子形成不对称配位, 而共棱氧原子OIII则与三个Mo原子三重配位成键. 两氧化钼晶体结构模型中分别定义a和c轴(晶格矢量a和c)为弱vdW力结合方向(与层平面方向垂直), 所以OI─Mo─OI和OⅡ─Mo─OⅡ分别沿着c轴和a轴方向成键, 而OIII─Mo─OIII成键沿着b轴(晶格矢量b)方向. VBM电子态主要由OⅡ和OIII的2p轨道组成(如图4和图5所示), 这些氧原子轨道位于层平面上, 并垂直于相应的O─Mo─O键. 导带底(CBM)电子态主要由层平面内的Mo-4d轨道组成, 与少部分OⅡ原子2p轨道形成杂化. 这些结果表明, 层状氧化钼的VBM主要由平面内OⅢ和OⅡ原子2p轨道杂化而成, 而CBM则来自于OⅡ和Mo原子平面内2p和4d轨道的贡献. 此外, 用HSE06泛函计算的MoO3 EA和IP与文献报道的UPS和IPES测量实验数据比较接近, 进一步证明了本文所采用的理论计算方法的可靠性和计算结果的精确度. 表3中列出了两种MoO3的EA和IP计算值以及用于比较的实验数据[29]. 图 5 (a) $\alpha $-MoO3和(b) MoO3 -Ⅱ PDOS的轨道成分: Mo原子的4d轨道成分(上图)和三种等价氧原子OⅠ, OⅡ和OⅢ的2p轨道成分(下图) Figure5. Partial orbital components of atomic projected density of states for (a) $\alpha $-MoO3 and (b) MoO3 -Ⅱ: Mo-4d orbital (above panels) and O-2p orbital (below panels) of three equivalent oxygens OⅠ, OⅡ and OⅢ.
$\alpha $-MoO3
MoO3 -Ⅱ
EA/eV
IP/eV
EA/eV
IP/eV
计算
PW91
6.555
7.94
6.11
8.12
HSE06
6.615
8.76
6.17
8.29
实验
6.700
9.68
—
—
表3用PW91梯度校正泛函和HSE06杂化泛函计算的两种层堆叠钼氧化物的EA和电IP, 最后一行中列出文献[29]中$\alpha $-MoO3的实验结果 Table3.The calculated electron affinity (EA) and ionization potential (IP) of two laminated molybdenum oxides employing PW91 and HSE06 functionals respectively. The experimental results of $\alpha $-MoO3 from Ref. [29] are also listed for comparison.
23.4.磁特性 -->
3.4.磁特性
虽然能带结构和DOS表现出的$\alpha $-MoO3电子自旋能级分裂并不明显, 但上旋和下旋电子态的完整自旋对称性遭到破坏. 图6(a)中的$\alpha $-MoO3自旋密度的等值面空间分布表明, 自旋极化导致不同电子自旋态在空间非对称分布, 使MoO6八面体构成的二维原子层的内部和表面氧原子OⅠ周围分别分布上旋和下旋态电子, 并且上旋态密度高于下旋态电子密度, 因而MoO6层内部和表面呈现亚铁自旋磁耦合, $\alpha $-MoO3总的自旋或净自旋(磁矩)较小. MoO3-Ⅱ的自旋密度分布与$\alpha $-MoO3类似, 因自旋极化使自旋态失去了空间对称性(如图6(b)所示), 下旋电子分布在MoO6八面体内的Mo和OⅠ附近, 而其他氧原子周围分布着较高密度的上旋电子, 因此呈现亚铁自旋磁有序, 净自旋密度较低. 由此可以推测OⅠ空位将会对$\alpha $-MoO3的磁矩产生重要影响, 并且由于实验报道表明氧化钼的磁性来源于原子空位缺陷, 因此为了研究原子空位对磁性的影响, 通过构建$\alpha $-MoO3的1 × 2 × 2和MoO3-Ⅱ的2 × 2 × 2的超胞并删除其中的某一原子来模拟含有不同点空位缺陷的晶体结构(空位的浓度为6%), 并进行几何优化和自旋极化电子结构计算, 从而分析磁性产生的原因和机理. 含点空位MoO3体材料的部分DOS (区分上旋和下旋态)计算结果如图7所示, 表明费米能级附近的电子本征态出现了明显的自旋分裂, 主要来源于Mo-4d轨道的自旋极化, 形成的磁矩随空位的种类和含量发生变化, 其中Mo空位和三种不等价氧原子空位分别表示为VM, VO-Ⅰ, VO-Ⅱ和VO-Ⅲ. 引入氧空位引起的磁矩明显大于钼空位, 产生的磁矩来源于相邻MoO6层之间的自旋铁磁耦合. 图 6$\alpha $-MoO3(左图)和MoO3-Ⅱ(右图)的(a)自旋分布的等密度面空间分布, 自旋密度等值面密度为0.05 electrons/?3; (b) Mo-4d和O-2p的部分DOS(上旋态$\alpha $和下旋态$\beta $), 费米能量为能量参考零点(垂直虚线) Figure6. (a) The spin distribution representing as spin densityisosurface contoured at 0.05 electrons/?3 and (b) the spin-resolved partial DOS of Mo-4d and O-2p with Fermi energy being referenced as energy zero indicated by vertical dashed line, for $\alpha $-MoO3 (left panels) and MoO3-Ⅱ (right panels).
图 7 含有6% Mo空位、OⅠ空位、OⅡ空位和OⅢ空位的 (a) $\alpha $-MoO3和 (b) MoO3 -II缺陷晶体的Mo-4d和O-2p的部分DOS (上旋态$\alpha $和下旋态$\beta $), 费米能量为能量参考零点(垂直虚线) Figure7. The spin-resolved partial DOS of Mo-4d and O-2p for (a) $\alpha $-MoO3 and (b) MoO3 -Ⅱ crystals with 6% Mo, OⅠ, OⅡ and OⅢ vacancies respectively. The Fermi energy is referenced as energy zero indicated by vertical dashed line.
图8为含OⅠ和OⅡ空位氧化钼的等自旋密度面的空间分布图. OⅠ空位周围的自旋密度呈现平面等腰三角形状, 表明空位与近邻的三个Mo原子发生较强的自旋极化, 在远离Mo和O原子的位置只出现少量自旋磁矩. 一般情况下, 一个氧空位作为施主可以在系统中留下两个电子, 而Muliken电荷布居分析表明, 三个相邻Mo原子中每个原子上的电荷为1.29 (无空位的计算值为1.46), 相当于具有3.87 (4.38)个价电子. 这表明0.51个电子已经从OⅠ空位转移到了三个相邻的Mo原子上, 导致钼离子的价态降低. Mop+离子具有部分占据的d轨道子壳层, 在Mop+离子的上旋和下旋d轨道上占据的电子数不同会产生净磁矩. 表4列出了不同空位的氧化钼总自旋值(净磁矩), 可以推测引入OⅠ和OⅡ空位所产生的净磁矩将会随空位浓度增加逐渐增大. 此外, 采用不同非局域交换相关泛函计算结果都表明, Mo空位的存在使可提供的单电子占据d轨道数减少而使总自旋磁矩降低, 但OⅢ空位的存在对MoO3净磁矩影响不大. 对于$\alpha $-MoO3缺陷晶体, OⅠ和OⅡ空位的存在导致费米能级附近的Mo-4d轨道电子自旋分裂增大, 使自旋极化产生的净自旋升高, 虽然Mo和OⅢ空位并没有明显造成各轨道电子态的自旋能级分裂发生变化, 但Mo空位使Mo-4d轨道成分的电子态密度降低, 导致总自旋略微减小. 对于MoO3 -Ⅱ晶体, OⅠ和OⅡ空位使较高能级的O-2p和Mo-4d轨道电子产生自旋分裂, 上旋态能级降低导致费米能级附近提高了自旋极化的上旋电子态密度, 从而导致净磁矩增加, 而Mo和OⅢ空位对电子自旋极化状态的影响与$\alpha $-MoO3类似. MoO3的磁性主要来源于Mo-4d轨道成分的电子自旋极化. 图 8$\alpha $-MoO3(左图)和MoO3-Ⅱ(右图)含有6% OⅠ(上图)和OⅡ(下图)空位的自旋密度等值面, 等值面的密度为0.05 electrons /?3 Figure8. The isosurface of spin density of $\alpha $-MoO3 (left images) and MoO3-Ⅱ (right images) with 6% OⅠ (above images) and OⅡ (below images) vacancies respectively. The isosurfaces are contoured at 0.05 electrons/?3.
总自旋h·bar/2/单胞
完整晶体
VMo
VO-Ⅰ
VO-Ⅱ
VO-Ⅲ
$\alpha $-MoO3
8.206
7.7855
8.235
8.244
8.214
MoO3 -Ⅱ
4.395
3.995
4.770
4.800
4.378
表4不同点空位类型MoO3晶体的总自旋值(每个单胞), 点空位浓度为6% Table4.The total spin values (per unit cell) of MoO3 configurations with different vacancies of 6% concentration.
23.5.光学特性 -->
3.5.光学特性
层状氧化钼是一种有效的光电子和光催化材料, 因此有必要对层状氧化钼块体材料$\alpha $-MoO3和MoO3 -Ⅱ进行详细的光学性能研究. 由于采用PW91泛函, 带隙2.0 eV偏低, 吸收边(截至频率)比用HSE06泛函计算的结果低得多, 因此用HSE 06泛函计算$\alpha $-MoO3和MoO3 -Ⅱ的光学性质, 包括介电函数的虚部随频率的变化以及光吸收谱, 结果如图9和图10所示. 在本研究中只考虑了垂直光激发过程, 而更准确地描述光吸收需要将声子辅助激发包括在内, 并且入射光采用如实际应用相似的非偏振光分别沿着MoO6八面体层堆叠的层平面垂直和平行方向入射. 层状MoO3介电函数谱线在0—12 eV频段出现多个谱峰, 谱线波动幅度随着频率的增加逐渐减小, 在超过12 eV以后趋近于稳定值. 非偏振光的入射方向对$\alpha $-MoO3介电函数的影响较小, 但MoO3 -Ⅱ的光各向异性非常明显. 介电函数虚部的峰值来自于价带到导带的电子激发, 根据虚部介电谱峰可以分析对应的电子跃迁. 两种MoO3复介电函数虚部的谱峰主要源自从价带O离子2p态向导带Mo离子4d态的电子跃迁. 由于$\alpha $-MoO3结构中的扭曲共棱MoO6八面体片层以ABA模式按照层间错位排列方式堆叠, 所以在相邻MoO6八面体层之间形成电偶极矩, 而按照AAA模式层堆叠的MoO3-Ⅱ不存在层间偶极矩. 这种层间电偶极矩阵在外电场作用下产生转向极化所需的时间(弛豫时间)相当长, 因此只有在长时间的静电场作用下才能完成极化过程, 而在交变电场作用下则不能参与介电响应, 导致$\alpha $-MoO3介电函数虚部在频率为零时出现一个不为零的峰值(如图9(a)所示). $\alpha $-MoO3能带结构中的上旋态能带$\alpha $-CB1在G-Z-T-Y区域出现了微小的简并分裂并且费米能级穿过该能带(如图3(a)右图所示), 所以在频率(能量)极小的光激发下产生光电子跃迁, 导致在频率f → 0时$\alpha $-MoO3介电函数虚部不为零. 而MoO3-Ⅱ能带在费米能级附近不存在这种微小的简并分裂, 如图3(b)右图所示, 在频率f → 0时介电函数虚部为零. 图 9 (a) $\alpha $-MoO3和(b) MoO3-Ⅱ介电函数谱线, 入射非偏振光分别沿着原子层平面的垂直(上图)和平行(下图)方向入射 Figure9. The calculateddielectric functions of (a) $\alpha $-MoO3 and (b) MoO3-Ⅱ with the light incident along the directions perpendicular (above panels) and parallel (below panels) to atomic-layer plane respectively.
图 10 (a) $\alpha $-MoO3和(b) MoO3-Ⅱ在不同带电(–2e—+2e/单胞)状态下的光吸收谱, 入射非偏振光分别沿着原子层平面的垂直(左图)和平行(右图)方向入射 Figure10. The calculatedabsorption spectra of (a) $\alpha $-MoO3 and (b) MoO3-Ⅱ with the light incident along the directions perpendicular (left panels) and parallel (right panels) to atomic-layer plane respectively, under different charge loading (–2e—+2e per unit cell).
















 图 1 (a)
图 1 (a) 




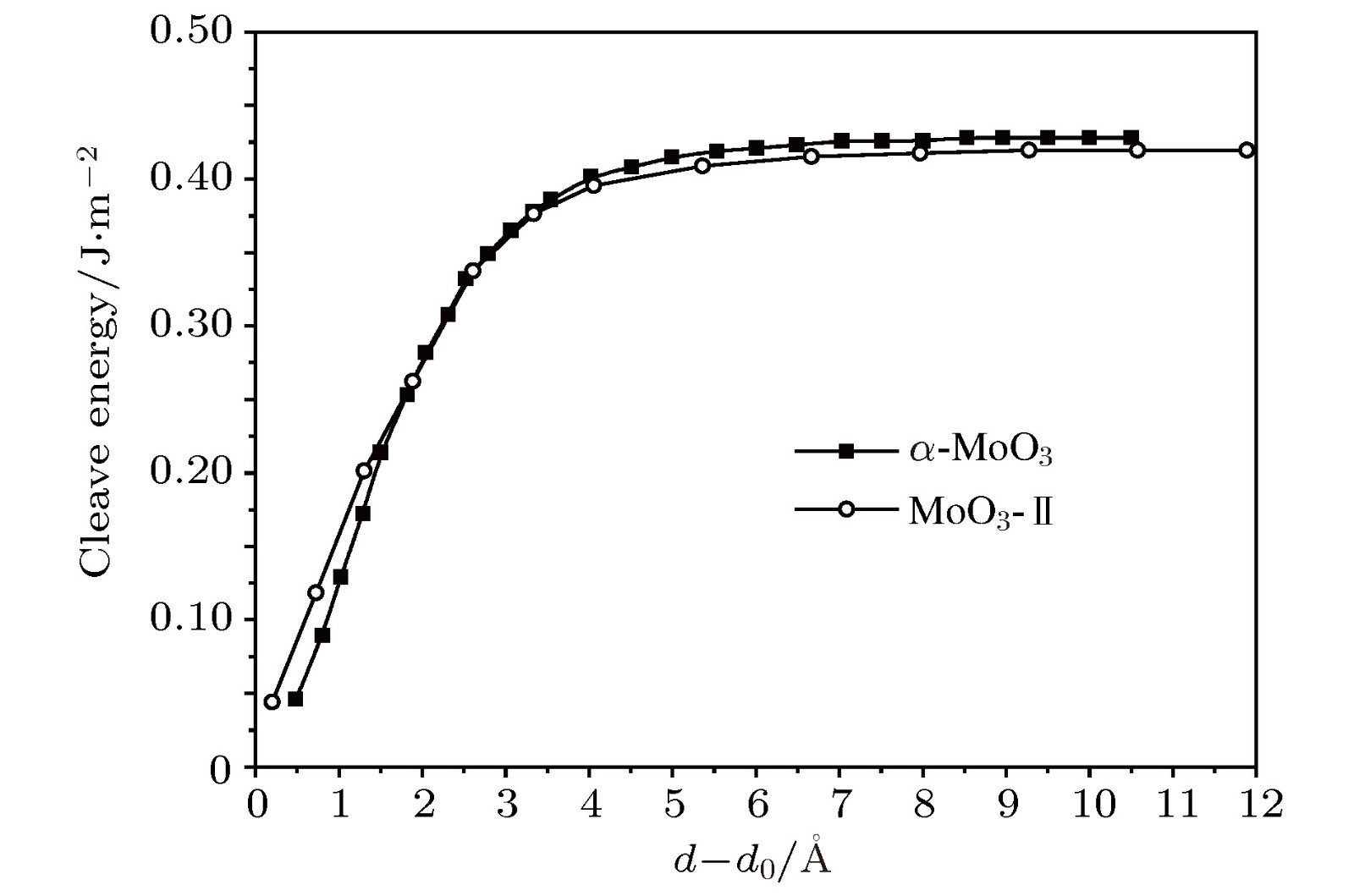 图 2
图 2 




















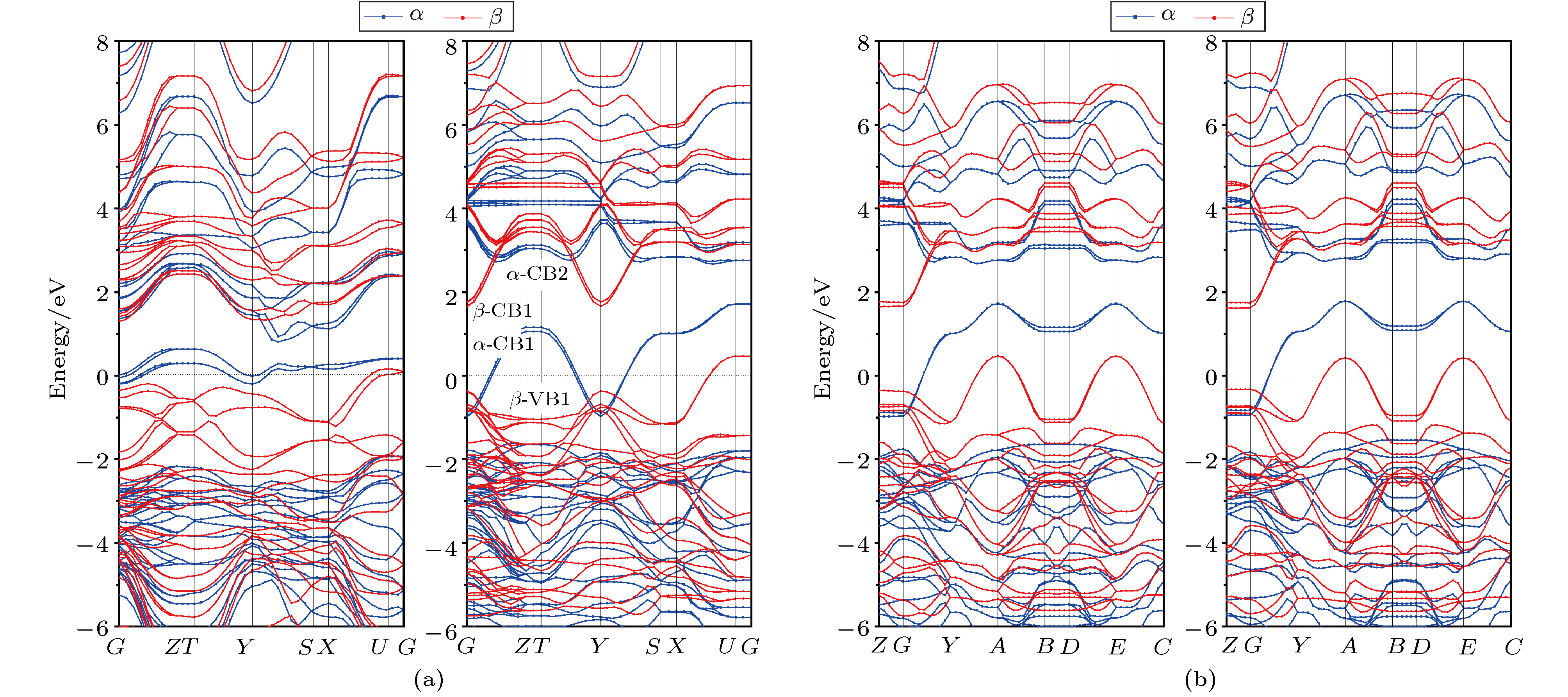 图 3 用PW91泛函(左图)和HSE06泛函(右图)计算的(a)
图 3 用PW91泛函(左图)和HSE06泛函(右图)计算的(a) 



 图 4 (a)
图 4 (a) 

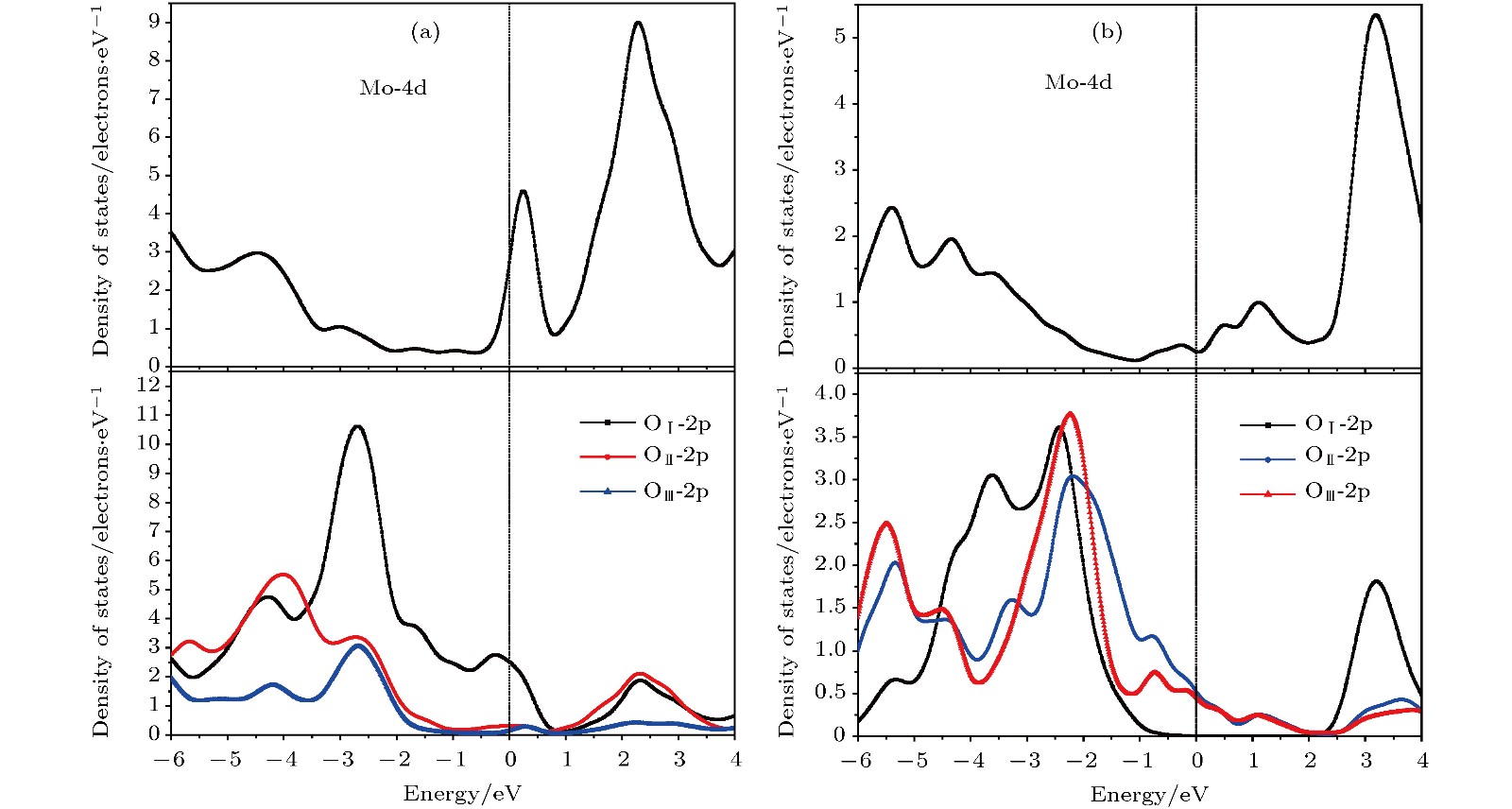 图 5 (a)
图 5 (a) 









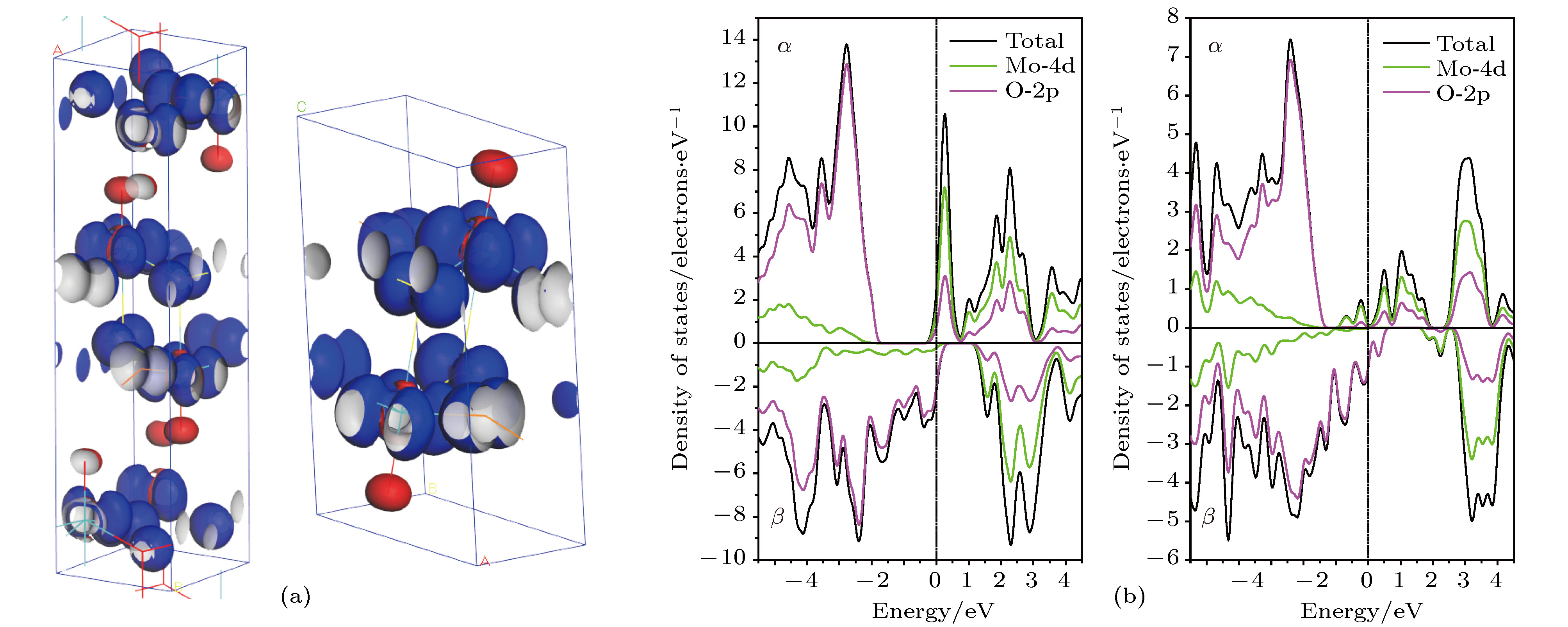 图 6
图 6 



 图 7 含有6% Mo空位、OⅠ空位、OⅡ空位和OⅢ空位的 (a)
图 7 含有6% Mo空位、OⅠ空位、OⅡ空位和OⅢ空位的 (a) 





 图 8
图 8 









 图 9 (a)
图 9 (a) 

 图 10 (a)
图 10 (a) 









