 ,1,2, 胡鹏飞1,2, 李春义,1,2
,1,2, 胡鹏飞1,2, 李春义,1,2Progress on deer genome research
Hengxing Ba,1,2, Pengfei Hu1,2, Chunyi Li,1,2通讯作者: 李春义,博士,研究员,研究方向:鹿茸生物学。E-mail:lichunyi1959@163.com;巴恒星,博士,研究员,研究方向:特种动物基因组学。E-mail:bahengxing@caas.cn
编委: 姜雨
收稿日期:2020-10-28修回日期:2020-12-29网络出版日期:2021-04-16
| 基金资助: |
Received:2020-10-28Revised:2020-12-29Online:2021-04-16
| Fund supported: |
作者简介 About authors
巴恒星,博士,研究员,研究方向:特种动物基因组学。E-mail:

摘要
鹿科动物是世界上最丰富的大型哺乳动物之一,在极北地区、热带地区和高海拔地区都有分布。中国占世界鹿科动物40%以上,是鹿科动物进化的主战场。鹿科动物除了具有反刍动物常见的独特表型特征外,更是进化出周期性再生新器官——鹿茸角。鹿科动物是研究生态学、行为学和进化生物学非常有价值的动物模型,特别是在研究哺乳动物器官再生方面具有重要科学价值。鹿基因组是系统阐述鹿的进化和演变,解析鹿科动物独特生物学性状的依据,对遗传资源保护和利用具有重要意义。目前,随着鹿科动物参考基因组的不断完善,在鹿基础科学研究上取得了诸多重要成果。本文详细综述了鹿科动物基因组学研究进展,主要包括鹿遗传变异数据、适应性进化分子基础、独特性状鹿茸角的起源进化关键基因和功能基因组学,以期为鹿科动物的深入研究奠定理论基础。
关键词:
Abstract
Deer family is one of the most abundant mammalian families in the world. Deer species are distributed in wide geographic ranges including the North Pole, tropical regions and high-altitude mountains. Of these deer species, China accounts for more than 40% of them and is the main site for deer evolution. Besides the common phenotypical attributes for ruminants, deer family is evolved to possess the unique head gears with periodic regeneration, i.e. antlers. It is currently well accepted that deer is a very valuable model for the studies of ecology, behavior, evolution and biology, especially for the study of mammalian organ regeneration. Reference deer genome is the basis for systematically illustrating deer evolution, deciphering unique biological attributes of deer species, and is significant in protection and utilization of deer genetic resources. In this review, we summarize the recent progress in the field of deer genome research, including data of deer genetic variation, molecular basis of adaptive evolution, and key genes and functional genomics involved in deer antler origin and evolution. The overall aim of the paper is to provide the reference neccessary for in depth investigation of deer species.
Keywords:
PDF (850KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
巴恒星, 胡鹏飞, 李春义. 鹿科动物基因组学研究进展. 遗传[J], 2021, 43(4): 308-322 doi:10.16288/j.yczz.20-362
Hengxing Ba.
鹿科(Cervidae)包括4个亚科,分别是鹿亚科(Cervinae)、獐亚科(Hydropotinae)、麂亚科(Muntiacinae)和空齿鹿亚科(Odocoileinae),共16属约52种,在反刍亚目里,种类上仅次于牛科(Bovidae),在极北地区、热带地区和高海拔地区都有分布。中国是鹿类动物最丰富的国家之一,占世界鹿科动物40%以上,是鹿科动物进化的主战场[1]。鹿科动物除了具有反刍动物常见的独特表型特征外,更是进化出周期性再生新器官——鹿茸角。科学家们普遍认为,鹿科动物是非常有价值的生态学、行为学和进化生物学的研究模型[1],特别是哺乳动物器官再生的研究模型[2],具有重要科学价值。
近年来高通量测序技术日新月异,推动了动物基因组学的飞速发展,基因组学研究方法也在不断地创新和改进,这些都为鹿科动物基因组学研究创造了前所未有的机遇,迎来鹿科学研究的新时代。目前,相关领域的研究人员已产生了大量鹿科动物的基因组学数据,在此基础上使解析鹿科动物的适应性进化分子基础、探究鹿茸角起源进化及鹿茸角再生发育相关通路和基因成为可能。不仅如此,基因组学研究还有利于加速家养鹿类动物品种培育和遗传改良进程。本文对近年来鹿科动物基因组学研究领域所取得的重要进展进行了系统地分析和总结,也对今后该领域研究方向进行了展望,以期为解析鹿科动物独特的生物学性状和遗传资源保护利用提供参考依据。
1 鹿科动物基因组资源
1.1 基因组参考序列
鹿科动物基因组测序起步较晚,2011年首次由美国贝勒医学院在NCBI GenBank上公开了白尾鹿(Odocoileus virginianus)基因组,但未见文章发表。2014年也发布了西方狍(Capreolus capreolus)基因组Contig序列,仍未见文章报道。2017年,Li等[3]在GigaScience和中国国家基因库(China National GeneBank, CNGB)上发表了驯鹿(Rangifer tarandus)基因组。通过构建9个不同长度插入片段测序文库,基于Illumina HiSeq 4000平台测序,de novo组装2.64 Gb的驯鹿基因组草图,Contig和Scaffold N50大小分别为89.7 kb和0.94 Mb,注释了21,555个蛋白编码基因。随着基因组测序成本快速降低,最近3年,鹿科动物基因组序列发表数量迅速增加,已达16个鹿种(表1),涵盖了鹿科动物所有4个亚科,它们基因组组装大小在2.5~2.8 Gb之间,但这些鹿基因组仍处于草图水平,质量还有待进一步提升。为了更好地利用基因组数据解析鹿科动物的特殊表型性状,本文归纳了这16个鹿种的系统进化关系,并进一步总结了它们的染色体核型、形态学数据(图1)及自然地理分布(图2)。Table 1
表1
表1鹿科动物16个参考基因组基本信息
Table 1
| 物种 | 发表时间 (年) | 测序/组装技术 | 基因组大小 (去除Gap后) (Gb) | 测序 深度(×) | Contig/ Scaffold N50 (kb/Mb) | Scaffolds 数量 |
|---|---|---|---|---|---|---|
| 白尾鹿 (Odocoileus virginianus)a | 2011 | Illumina HiSeq/Allpath-lg | 2.38 (2.36) | 150 | 122.0/0.9 | 17,025 |
| 西方狍 (Capreolus capreolus)b | 2014 | Illumina HiSeq/SOAPdenovo | 2.78 (2.74) | 24 | 4.1/0.01 | 3,088,511 |
| 驯鹿 (Rangifer tarandus)[3] | 2017 | Illumina HiSeq/SOAPdenovo | 2.64 (2.54) | 220 | 89.7/0.94 | 58,765 |
| 赤鹿 (Cervus elaphus)[8] | 2017 | Illumina HiSeq/AllPaths | 3.40 (1.95) | 74 | 7.9/0.27 | 34,724 |
| 麋鹿 (Elaphurus davidianus)[9] | 2017 | Illumina HiSeq/SOAPdenovo | 2.52 (2.46) | 82 | 32.7/3.0 | 46,381 |
| 豚鹿 (Axis porcinus)[10] | 2018 | Illumina HiSeq/SOAPdenovo | 2.68 (2.64) | 197 | 172.8/20.6 | 136,093 |
| 黑尾鹿 (Odocoileus hemionus)[11] | 2018 | Illumina HiSeq/BWA; SAMtools | 2.34 (2.34) | 25 | 113.3/0.8 | 838,758 |
| 小麂 (Muntiacus reevesi)[6] | 2019 | Illumina HiSeq/Supernova; Hi-C | 2.58 (2.51) | 34 | 225.1/9.4 | 29,705 |
| 赤麂 (Muntiacus muntjak)[6] | 2019 | Illumina HiSeq/SOAPdenovo | 2.57 (2.52) | 68 | 215.5/- | 25,651 |
| 白唇鹿 (Przewalskium albirostris)[12] | 2019 | Illumina HiSeq/Platanus | 2.69 (2.64) | 214 | 39.6/3.8 | 171,874 |
| 獐 (Hydropotes inermis)[12] | 2019 | Illumina HiSeq/Supernova | 2.53 (2.48) | 76 | 131.4/13.8 | 22,246 |
| 黑麂 (Muntiacus crinifrons)[12] | 2019 | PacBio/FALCON | 2.68 (2.67) | 116 | 8.2/1.3 | 21,052 |
| 驼鹿 (Alces alces)c | 2019 | Illumina HiSeq/Supernova | 2,74 (2,54) | 35 | 131,8/4.1 | 48,219 |
| 东方狍 (Capreolus pygargu)[13] | 2020 | Illumina HiSeq/Supernova | 2.61 (2,55) | 100 | -/6.6 | 92,100 |
| 塔里木马鹿 (Cervus elaphus yarkandensis)[4] | 2020 | Illumina NextSeq/Supernova; Genetic maps | 2.60 (2.56) | 63 | 275.5/31.7 | 19,010 |
| 梅花鹿 (Cervus nippon)d | 2020 | PacBio/wtdbg2; Hi-C; Optical maps | 2.50 (2.50) | 58 | 23,600/78.8 | 588 |
新窗口打开|下载CSV
目前,仅有5个鹿科动物的基因组组装到染色体水平,分别是塔里木马鹿(Cervus elaphus yarkandensis)、赤鹿(Cervus elaphus)、小麂(Muntiacus reevesi)、赤麂(Muntiacus muntjak)和梅花鹿(Cervus Nippon)。Ba等[4]利用由38,083个单核苷酸多态性位点(single nucleotide polymorphism, SNP)组成的高密度遗传连锁图谱[5]将塔里木马鹿基因组95.9%序列组装到34对染色体。其中,3号、8号和31号染色体仅由1个Scaffold组成。这34对染色体组装大小与遗传图谱评估大小相关系数高达0.987。由于构建大型野生动物遗传图谱十分困难,利用Hi-C技术de novo组装基因组染色体也是目前最有力技术手段。Mudd等[6]利用Hi-C技术完成了小麂(2n?=?46)和赤麂(2n?=6♀/7♂)基因组的染色体组装。类似地,梅花鹿基因组也是通过Hi-C技术挂载到33对染色体上,但未见文章报道。
图1
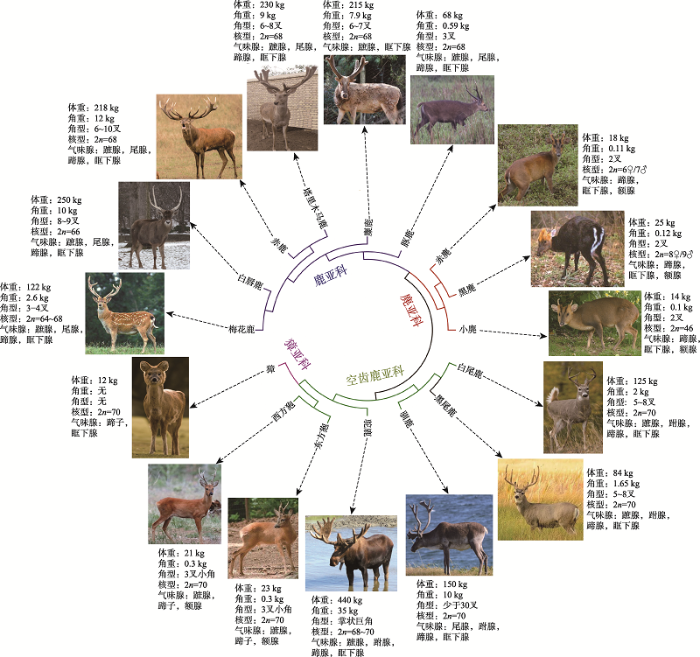 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图116个基因组测序鹿种系统进化关系及表型数据
系统进化拓扑关系参考文献[14,15,16],表型数据参考图书《中国鹿科动物》[1]和《The Biology of Deer》[17],核型数据参考王宗仁等[18],鹿科动物照片来源于Wikimedia共享(
Fig. 1Phylogenetic relationships and phenotypic data of 16 genome-sequenced deer species
马鹿/赤鹿(Cervus elaphus)是鹿科动物中最大的类群,由22个亚种组成,分布在欧洲南部和中部、北美洲、非洲北部和亚洲。在生态学、生物多样性和种间杂交等研究领域中,该类群研究最为广泛而备受关注。塔里木马鹿是中亚仅有的亚种,被认为是最原始亚种[7],有特殊的进化地位,其染色体水平基因组图谱将为马鹿/赤鹿类群,乃至整个鹿类动物的科学研究提供最有价值的参考基因组数据资源。
图2
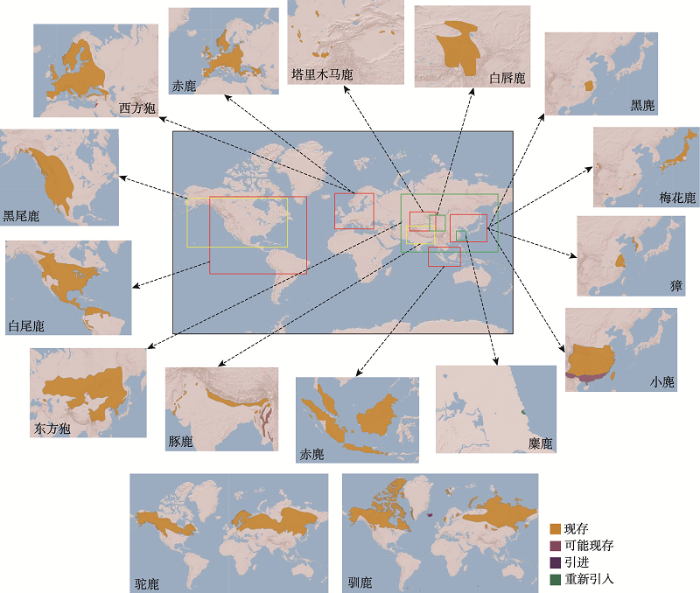 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图216个基因组测序鹿种的自然地理分布示意图
数据来源于IUCM Red List网站(
Fig. 2Natural geographical distribution of 16 genome-sequenced deer species
1.2 基因组变异资源
由于鹿科动物参考基因组缺乏,而且对野生动物进行大规模基因组测序显然不太现实。目前,鹿科动物的遗传变异数据主要是基因组SNP位点,其主要来源于简化基因组测序[11,19~23]、牛和羊SNP芯片跨科物种分型[24,25]以及保守牛外显子目标测序[26]和低覆盖率重测序[27,28](表2),这严重制约了鹿科动物基因组变异数据的产出效率和质量。比如,通常简化基因组测序仅产生相当于1%~2%的基因组的SNP数据[22],另外简化基因组测序短序列聚类时,多拷贝重复序列容易导致SNP位点的假阳性[29]。Table 2
表2
表2鹿科动物基因组变异资源
Table 2
| 物种 | 发表时间 (年) | 测序平台/方法 | 个体数量 | 标记类型 | 标记数量 |
|---|---|---|---|---|---|
| 白尾鹿 (Odocoileus virginianus)[22] | 2011 | Roche 454/简化基因组 | 16 | SNP | 13,734c |
| 白尾鹿 (Odocoileus virginianus)[24] | 2012 | Illumina BovineSNP50 | 10 | SNP | 469a |
| 骡鹿 (Odocoileus hemionus)[24] | 2012 | Illumina BovineSNP50 | 4 | SNP | 429a |
| 黑尾鹿 (Odocoileus hemionus)[24] | 2012 | Illumina BovineSNP50 | 14 | SNP | 434a |
| 驯鹿 (Rangifer tarandus)[25] | 2015 | Illumina BovineSNP50+OvineSNP50 | 4 | SNP | 4572+1471a |
| 赤鹿/马鹿 (Cervus elaphus)[28] | 2015 | Illumina GAII-X/重测序 | 7 | SNP | 1.8×105c |
| 骡鹿 (Odocoileus hemionus)[26] | 2016 | Illumina HiSeq2000/外显子捕获 | 7 | SNP | 23,204c |
| 梅花鹿 (Cervus nippon)[19] | 2017 | Illumina HiSeq2500/双酶切简化基因组 | 42 | SNP | 96,188c |
| 梅花鹿 (Cervus nippon)[27] | 2017 | Illumina Hiseq Xten/重测序 | 100 | SNP | 38,319,467c |
| 豚鹿 (Axis porcinus)[23] | 2017 | Illumina HiSeq2500/酶切简化基因组 | 11 | SNP | 11,155c |
| 梅花鹿(Cervus nippon)/ 马鹿(Cervus elaphus)/F1[20] | 2018 | Illumina HiSeq2500/双酶切简化基因组 | 30 | SNP | 2015b |
| 驼鹿 (Alces alces)[21] | 2018 | Illumina HiSeq2500/酶切简化基因组 | 34 | SNP | 336c |
| 梅花鹿 (Cervus nippon)[42] | 2018 | Applied Biosystems 3730/基因组 | 96 | SSR | 29a |
| 骡鹿(Odocoileus hemionus)/ 白尾鹿(Odocoileus virginianus)/F1/F2[11] | 2019 | Illumina CervusSNP50 | 30 | SNP | 40b |
| 狍 (Capreolous capreolus)[13] | 2019 | Illumina HiSeq2500/酶切简化基因组 | 250 | SNP | 83,893c |
| 梅花鹿 (Cervus nippon)[43] | 2020 | PCR扩增 | 478 | Y-SNP | 9c |
| 梅花鹿 (Cervus nippon)[40] | 2020 | Applied Biosystems 3730/转录组 | 140 | SSR | 29a |
| 赤鹿 (Cervus nippon)[44] | 2020 | PCR扩增 | 123 | X-/Y-SSR | 10/5a |
新窗口打开|下载CSV
在跨科物种SNP分型上,Haynes等[24]利用牛BovineSNP50芯片对北美白尾鹿和黑尾鹿(Odocoileus hemionus)进行分型,分型成功率仅为38.7%。Kharzinova等[25]利用BovineSNP50和OvineSNP50芯片对驯鹿进行分型,分型成功率也仅为43.0%和47.0%。Shafer等[30]阐述在牛科和鹿科(分化时间大约2~3千万年)之间跨芯片分型是不合适的,不仅分型成功率低,而且多态位点仅为1%。
Brauning等[28]对马鹿/赤鹿类群8只个体基因组进行了低覆盖率重测序,利用牛基因组做参考比对并结合严格的SNP筛选策略,共获得了1.8×105个SNP位点,产生了Illumina CervusSNP50芯片。Rowe等[31]利用CervusSNP50芯片对9个鹿种共396只个体进行SNP分型检测,分型成功率为82.3%,远远高于牛羊芯片的分型成功率。Johnston等[5]利用CervusSNP50芯片构建了马鹿/赤鹿类群的遗传连锁图谱,其标记密度和图谱的完整性远远好于Slate等[32]在2002年发表的仅含有600个标记的鹿遗传图谱。
我国鹿类动物遗传资源十分丰富,同时也是重要的家养经济动物。Hu等[33]对我国5个主要鹿种(包含21个亚种/群体)进行了简化基因组测序,平均测序深度16×,获得197,543个SNP位点,并进行了遗传多样性和系统进化分析。Ba等[19]对我国东北7个地区饲养的42只梅花鹿纯种个体进行双酶切简化基因组测序,平均测序深度23×,共筛选98,166个SNP位点,平均个体SNP密度为0.74 SNPs/kb,略高于经历过严重群体瓶颈的麋鹿(0.51 SNPs/kb)[34],但低于大熊猫(1.32 SNPs/kb)[35]和荷斯坦奶牛(1.35 SNPs/kb)[36]约2倍,而且出现杂合子缺陷。这些结果表明人们长期以茸重性状为单一育种目标,强烈的人工选择导致梅花鹿饲养群体经历了严重的净化和遗传漂变。Hu等[27]通过基因组重测序对梅花鹿鹿茸高产和低产性状进行关联分析,共获得94个与鹿茸重量相关的候选SNP位点。Hu等[37]进一步利用这94个SNP位点对341只个体进行分型,并结合高、低产鹿茸转录组学基因表达数据[38],筛选出16个SNP位点与茸重性状显著相关。
由于不同鹿种之间能够杂交且后代可育,人们为了繁育高产性状的茸鹿,经常在饲养梅花鹿和马鹿之间进行杂交。Ba等[20]对纯种梅花鹿和马鹿及杂交F1共30只个体进行双酶切简化基因组测序,平均测序深度15×,获得2015个梅花鹿和马鹿种特异SNP标签,这些标签为纯种和杂交种鉴定奠定了基础。在鹿种鉴定上,Xie等[39]也通过挖掘鹿科动物基因组中保守且与牛羊比较存在突变位点的编码基因,最终在CEP295NL基因序列上开发出1对鹿科动物PCR通用引物,与鹿种特异引物相比,通用引物节省了更多的人力和物力。
在鹿科动物基因组测序之前,科学家通常利用几个牛微卫星多态(short tandem repeats, STRs)标记对鹿科动物进行分型,由于标记数量少,多态信息严重不足。随着高通量测序技术发展,鹿科动物STRs也被快速批量开发。Jia等[40]在梅花鹿多种组织转录组数据[41]中开发了29个STRs,对140只个体进行茸重性状关联分析,找到8个显著关联的标记。Wang等[42]在梅花鹿基因组数据中开发了29个STRs,其中10个高度多态的STRs被用作梅花鹿亲子鉴定标记,排除准确率达99.99%。总之,鹿科动物遗传变异数据的获得不仅对鹿科学研究极具价值,也能为开发和利用经济价值极高的家养鹿类动物提供强有力的遗传分析和改良工具。
2 比较基因组学
2.1 染色体核型进化
染色体重排在驱动进化中的作用一直是进化生物学的长期问题。Farre等[45]重构了反刍动物祖先染色体核型,发现反刍动物祖先与鲸偶蹄目(Cetartiodactyla)分离后染色体重排主要发生在染色体内部,而在反刍亚目(Ruminantia)的有角下目(Pecora)谱系中,染色体重排也发生在染色体之间。反刍动物染色体断点区域附近的基因在物种之间,尤其是在牛中表现出更多的差异表达,并且在具有增强子的基因中差异更大,与这些断点区域的系统发生起源相一致。这些结果表明与染色体重排共定位的谱系特异性调控元件可能提供了有助于塑造反刍动物进化有价值的功能修饰。在鹿科动物中,麂属动物的核型在较短的分化时间内(仅数百万年)发生了巨大变化。除小麂(2n= 46)外,其他麂属动物的染色体数目通常只有6~9条,其中赤麂(2n=6♀/7♂)是哺乳动物中染色体数目最少的物种。因此,麂属动物成为研究哺乳动物染色体核型生物学研究的理想材料。通过细菌人工染色体文库的荧光原位杂交建立了麂属动物之间的高分辨比较染色体图谱,确定了麂属动物染色体的串联融合进化方式[46,47,48,49,50,51]。最近,Mudd 等[6]通过基因组测序和染色体组装,进一步发现小麂和赤麂这两个近缘麂显著的核型差异主要是在基因组上发生了大范围的染色体串联融合事件,而核型急剧变化对局部染色体改变影响甚小,甚至在融合位点附近参与染色体维护的基因很少显示出快速进化的证据。对于不同鹿种的染色体进化,Huang等[52]证实梅花鹿(2n= 33)和马鹿(2n=34)的染色体核型进化过程中存在唯一的罗伯逊易位。
另外,性染色体起源及演化机制一直是进化生物学家最感兴趣的科学问题之一。Huang等[53]研究发现雄性黑麂基因组中1号常染色体短臂(1p)和4号常染色体发生罗伯逊易位,之后又经历了复杂的内部重排形成年轻新性染色体1p+4。Zhou等[54]研究发现黑麂与哺乳动物Y染色体的演化历程相似,可以成为哺乳动物性染色体演化研究的珍贵模型。关于麂属动物的染色体核型进化,Huang等[55]在2012年进行了详细综述,限于篇幅限制,本文不再综述。
2.2 适应性进化
鹿科动物在北极和热带均有分布,一些鹿种在长期进化中已经适应了极端环境。最近,Lin等[56]揭示了驯鹿适应北极环境分子机制。通过比较基因组发现驯鹿维生素D代谢通路中的两个关键基因(POR和CYP27B1)受到了强烈的自然选择,基因编码酶的活性比山羊和狍子(Capreolous capreolus)高,这可能使驯鹿对钙的吸收能力大大增强。脂蛋白转运(APOB)和脂质合成(FASN)两个重要基因在驯鹿中也发生了选择性突变,这两个基因也参与了企鹅(Pygoscelis adeliae)和北极熊(Ursus maritimus)脂肪代谢进化,表明极地动物能量代谢经历了趋同进化。驯鹿节律通路中的核心调控基因(PER2)发生了驯鹿特异性突变,导致PER2基因与另一个节律核心基因(CRY)无法结合,使得驯鹿丧失了昼夜节律分子钟而适应北极极昼和极夜的环境。这些结果使人们对极地动物的适应性在分子水平上有了更深入和全面的了解,也为解决人类一些健康问题提供了重要线索,如维生素D对钙沉积的影响、生物钟调控与人类睡眠障碍的关系。另外,Weldenegodguad等[57]发现西伯利亚人极地寒冷适应性基因在驯鹿基因组上发生了基因家族扩张和正选择,特别是离子通道受体和离子交换调控基因,如TRPV5 和TRPV6等,这可能是对温度敏感适应性的主要原因。在类似的研究中,Yang等[58]对不同栖息环境下的脊椎动物重要的冷感受器蛋白TRPM8进行了氨基酸成像与蛋白质三维结构计算建模的研究,发现TRPM8通道的功能结构在南极洲的帝企鹅(Aptenodytes forsteri)与非洲大陆的非洲象(Loxodonta)之间存在重大差异,帝企鹅的TRPM8冷敏感性显著低于非洲象。在鹿科动物中,塔里木马鹿生存于极端干燥、炎热和强烈太阳辐射的沙漠环境。Ababaikeri等[59]通过对塔里木马鹿比较基因组学研究发现该物种的沙漠环境适应性候选基因,主要参与了氧化应激、水分再吸收、能量代谢、热应激、呼吸系统适应、DNA损伤和修复等生物过程。同时,这些候选基因与其他沙漠动物适应性基因均有重叠,表明沙漠物种对沙漠环境适应经历了趋同进化。
麋鹿被认为是急剧恢复和拯救高度濒危动物的经典例子。我国现存麋鹿群体是来自于英国动物园的18只个体繁育后代,这预示着严重的瓶颈效应和近交衰退。Zhu等[34]利用比较基因组学对麋鹿种群的适应性进行了评估,尽管麋鹿群体存在极端的种群瓶颈,但其仍具有较高的遗传多样性,延长的近亲繁殖历史可能有助于清除有害的隐性等位基因,同时,与生殖,胚胎发育和免疫反应相关的17个基因忍受着高选择压力。另外,麋鹿对高盐食物的潜在适应可能是由于SCNN1A基因受到强烈正选择,参与控制体内钠重吸收。另外的29个正选择基因涉及血压调节、心血管发育、胆固醇调节、血糖控制和甲状腺激素合成。这些正选择的基因可能也反应了麋鹿对高盐食物的适应与响应。
鹿科动物进化不仅仅体现在极端环境适应性方面,Ba等[60]发现鹿嗅觉系统适应性进化的分子证据。有51个快速进化基因与感受器官细胞纤毛(cilia)组装有关,同时,实现细胞纤毛波形功能的轴突纤毛丝动力蛋白(dynein)家族重链基因在基因组上发生了扩张,也发现了14个嗅觉受体基因中89个位点在鹿上发生了正选择。由于鹿存在多处皮肤腺体,散发的化学小分子是彼此之间通讯的重要信息素,推测嗅觉系统在接受信息素的长期进化过程中一定也受到了适应性进化的选择压力。
2.3 再生器官(鹿茸角)起源进化关键基因
鹿茸角是哺乳动物中唯一的胚胎组织在成体后发育的器官[61],是鹿科动物防御天敌和争夺配偶的武器。更重要的是,鹿茸角能周期性再生而保持最佳战斗状态,被认为是鹿科动物长期进化过程中一个器官创新。最近,Wang等[14]对反刍动物角起源进化的比较基因组研究发现,鹿茸角新器官的起源依赖招募骨组织、皮肤组织、脑组织和睾丸组织的基因。鹿科动物正选择基因、加速进化元件以及生茸组织高表达基因都参与了神经脊细胞迁移通路,揭示神经脊干细胞在鹿角起源发育中扮演重要角色[62]。Price等[63]也报道了生茸组织特异性高表达神经脊细胞的关键标志物,如SNAI1等。鹿科动物中仅有獐在自然条件下是不长茸角的,被认为是在进化过程中发生了茸角丢失[64],因此,利用比较基因组学研究鹿角关键再生基因,獐是唯一的选择。Wang等[14]发现绵羊角的关键调控基因RXFP2在麝科和獐两个无角反刍动物支系中发生趋同的假基因化,认为RXFP2也是决定鹿科动物茸角再生的关键基因。Lin等[56]发现驯鹿基因组上促进细胞增殖的细胞周期因子CCND1基因上游增加了一个雄性激素受体结合区域,这可能使驯鹿在更低的雄激素水平下能促成雌性驯鹿长角。结果表明,鹿科动物进化过程中,细胞周期相关基因与鹿茸角的发生与再生关联紧密。
总之,比较基因组学是从进化的视角去解析复杂的生物学问题和探寻调控基因。随着多个鹿基因组序列完成,基因组质量也越来越高,将进一步加快鹿科动物比较基因组学研究进程,探索适应性进化和新器官起源的奥秘。
3 功能基因组学
目前,鹿类动物功能基因组学研究主要集中在鹿茸干细胞、鹿茸再生以及鹿茸生长中心的快速生长发育等研究,本文主要针对这几方面进行了总结。3.1 鹿茸干细胞
鹿茸发生和再生过程是由干细胞驱动的,被称为鹿茸干细胞(antler stem cell, ASC)[2,65,66]。国内外多个实验室已对ASC进行了分离鉴定,其不仅表达多种成体干细胞标记因子,如CD73、CD90、CD29、CD44、CD146、CD105、CD166、STRO-1、Vimentin和Nestin,还表达部分胚胎干细胞标记因子,如C-Myc和OCT4[67,68,69,70]。最近,Wang等[69]通过膜蛋白组学发现RXFP2在ASC中高表达,而在对照的面部骨膜细胞中几乎不表达,该蛋白与反刍动物长角有关,可能作为ASC鉴定的一个新标记。在microRNA水平上,Ba等[71]通过测序显示ASC表达小鼠胚胎期特异表达的miR-296。Ba等[72]也对培养的ASC进行单细胞RNA-seq测序,表明ASC在培养过程中仍然保持着高度的干性和同质性。Wang等[70]构建胚胎嵌合实验模型证实了ASC不同于胚胎干细胞嵌合到整个宿主的组织中,也不同于间充质干细胞完全消失,而是有限地嵌合到一些组织中,特别是嵌合到了生殖系统中。这些结果证实ASC不仅是成体干细胞,还具有胚胎干细胞部分属性。干细胞的自我更新、分化与成熟均与其所处的微环境有关。研究发现鹿皮可能为ASC提供了类似的微环境[73]。在鹿茸发生时,雄性激素刺激ASC缓慢增殖和分化,而当分化的组织与头部皮肤变得接触并紧密相贴时,皮肤给ASC提供了特殊的微环境,这个微环境触发了ASC快速增殖(分裂速度比接触前至少快10倍)和定向分化,最终导致鹿茸的发生。类似地,当骨化的鹿角脱落后,伤口开始愈合,ASC再次与皮肤紧密接触并被致敏,ASC再由致敏状态转变成激活状态,启动鹿茸再生。Dong等[74,75,76]通过蛋白质组学研究表明鹿茸再生的起始阶段是一系列蛋白调控的,可能与上皮-间质转化过程有关,包括无疤痕愈合的第一阶段,源于ASC分化细胞的移动性也受到高度调节,休眠期的ASC和面部骨膜细胞可能使用相似机制来维持干细胞的休眠状态。Sun等[77]通过建立ASC与毛乳头细胞共培养体系,鉴定出128个小分子,60%以上与外泌体相关,信号通路富集分析表明这些分子参与PI3K/AKT信号,可能影响ASC成骨细胞分化和血管生成。Li等[78]利用双向电泳方法对ASC与面部骨膜细胞进行蛋白质组学比较研究,也发现了PI3K/AKT信号在ASC细胞差异表达蛋白中富集。Liu等[79]利用小分子抑制剂影响PI3K/AKT信号的细胞生物学实验进一步证实PI3K/AKT信号在ASC生物学功能中扮演重要角色。
钙网蛋白CALR是一种多功能蛋白,在内质网管腔内主要起钙离子结合蛋白的作用,与多种信号系统有关,例如蛋白质折叠和钙的调节体内平衡。最新证据表明,CALR可在细胞核中与雄激素受体结合,抑制雄激素受体下游转录活动[80]。Dong等[76]通过蛋白质组学研究发现CALR在开始分化的ASC中高表达,并在蛋白互作网络中扮演核心(hub)基因的角色。Akhtar等[81]通过对鹿施以外源雄性激素,也发现雄性激素在低水平时ASC开始分化,高表达CALR,进一步证实了该因子可能是雄激素依赖性鹿茸角再生的主要调节因子。
3.2 鹿茸生长中心
2004年和2012年,研究人员分别利用蛋白质组学和转录组学研究鹿茸生长中心的基因表达。Park等[82]鉴定到了130个蛋白,其中MAPK1、COL1A1、SMUBP2和ZFP28等仅在鹿茸生长中心高表达,推测这些蛋白对鹿茸的快速生长极其重要。Yao等[83,84]建立了鹿茸生长中心快速生长和骨化的转录组表达谱,涉及转录因子、信号分子和细胞外基质等。鹿茸快速生长发育是基于鹿茸顶端生长中心的ASC增值和分化的。鹿茸发育60天进入快速生长期,90天时开始快速骨化。为了研究不同时间点鹿茸生长发育相关基因表达动态,很多****都对不同发育时间的鹿茸生长中心进行了转录组和蛋白组分析[83,85~87]。然而,由于鹿茸生长中心组织层结构发育关系复杂,这些研究的样品采集标准不尽相同,难以在不同的研究之间进行有效地比较。Li等[88]根据发育生物学理论对鹿茸生长中心进行了组织层空间划分,即从远端到近端划分5个连续发育状态的组织层,包括间充质层、前软骨层、过渡层、软骨层和矿化层。基于生长中心组织层发育关系,Ba等[89]对这5个组织层进行了转录组测序,发现不同组织层之间有明显的表达谱特征,而且共表达网络分析鉴定了9个基因表达模块,其中370个核心(hub)基因参与调控了软骨、骨和血管生成,为鹿茸生长中心生长发育研究奠定了分子基础。Ker等[90]建立了鹿茸顶端ASC和人间充质细胞的比较模型,通过转录组测序及体外实验验证阐明UHRF1对鹿茸细胞快速增殖起主要作用,而S100A10是鹿茸矿化的关键基因。Chen等[91]分析lncRNA在鹿茸生长中心间充质组织和软骨组织中的表达动态。与软骨组织相比,间充质组织中有1212个lncRNA和518个mRNA的转录水平发生了显着改变,表明lncRNA通过调节细胞增殖、迁移和成骨相关基因,促进了鹿茸间充质组织向软骨组织的分化。
Hu等[38]对具有快速和慢速两种表型的鹿茸生长中心进行microRNA表达谱比较,发现与鹿茸快速生长的18个microRNA分子,构建microRNA- mRNA调控网络,确定14个基因与鹿茸快速生长相关。Yao等[92]也对鹿茸主干和眉支两个生长中心进行转录组测序,比较发现主干生长中心的软骨发育基因显著上调,如SOX9、COL2A1和ACAN等,而骨化基因显著下调,如COL10A1、SPP1和IBSP等。
4 结语与展望
“人类基因组计划”的完成标志着人类对于生命现象和过程有了本质的认识。鹿科动物在长期自然进化过程中,产生了诸多优异性状,如鹿茸角的再生和快速生长[65,93~95]、低癌症发生率[96,97]、无伤疤伤口愈合能力[98,99]、骨质疏松快速逆转[100,101,102]和大型哺乳动物滞育模型(狍子)[103,104,105,106,107]等。这些都触及了重大基础生物学和医学问题,其背后所蕴藏的遗传机理非常值得人们去认知和掌握。随着测序技术的发展,以单分子荧光和纳米孔为代表的三代测序技术将进一步提升鹿科动物基因组质量,完整的基因组学数据将为这些优异独特的生物学性状相关基因发现奠定了基础。最近,家养梅花鹿和马鹿也已被纳入我国畜禽遗传资源目录。鹿遗传变异数据大规模产出和利用能有效加快家养鹿类动物的遗传改良和品种保护,比如,利用基因组学数据全面评估我国茸鹿品种遗传结构多样性,利用连锁分析和关联分析等基因组学方法,高效挖掘茸鹿品种中有利等位基因,提出其利用途径和具体方案,并在品种资源创新过程中充分利用基因组学研究成果提高创新效率。
重要的是,针对鹿茸角这一进化过程中独特进化的再生器官,尚有不少重要问题有待解决,如多种细胞类型参与和调控下的鹿茸再生和发育,其它再生模式生物中扮演“总开关”调控角色的免疫细胞是否也扮演了类似的角色。常规组学技术无法详细系统地研究鹿茸角再生和发育过程中细胞类型、发育状态和分化路径。然而,近年来快速发展的单细胞测序技术给发育生物学带来了革命性的变化,能够对单个细胞内基因表达和调控进行无差别的分析,通过更多维、更精准的研究视角,去系统地、全面地揭示和解析细胞的功能。目前,本研究团队正在利用单细胞测序技术绘制鹿茸角再生和发育过程中单细胞图谱,解析不同细胞类型之间互作和通讯以及细胞谱系演化路径等,在单细胞水平上诠释鹿茸干细胞如何再生和发育成完整器官的关键科学问题。未来,可以通过建立以鹿茸为再生医学研究模型开展人类再生医学研究。在实现这些研究目标过程中,鹿基因组学研究将起到不可替代的作用,由此可见,鹿科动物基因组学研究才刚刚开始。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 3]
DOI:10.2174/157488809789057446URLPMID:19492976 [本文引用: 2]

Mammalian organ regeneration is the
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 4]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:30620341 [本文引用: 1]
[本文引用: 3]
[本文引用: 3]
DOI:10.1111/mec.15450URLPMID:32306438 [本文引用: 2]
Species that evolved in temperate regions during the Pleistocene experienced periods of extreme climatic transitions. Consequent population fragmentation and dynamics had the potential to generate small, isolated populations where the influence of genetic drift would be expected to be strong. We use comparative genomics to assess the evolutionary influence of historical demographics and natural selection through a series of transitions associated with the formation of the genus Capreolus, speciation within this genus during the Quaternary and during divergence among European roe deer (C. capreolus) populations. Our analyses were facilitated by the generation of a new high-coverage reference genome for the Siberian roe deer (C. pygargus). We find progressive reductions in effective population size (Ne ), despite very large census sizes in modern C. capreolus populations and show that low Ne has impacted the C. capreolus genome, reducing diversity and increasing linkage disequilibrium. Even so, we find evidence for natural selection shared among C. capreolus populations, including a historically documented founder population that has been through a severe bottleneck. During each phylogenetic transition there is evidence for selection (from dN/dS and nucleotide diversity tests), including at loci associated with diapause (delayed embryonic development), a phenotype restricted to this genus among the even-toed ungulates. Together these data allow us to assess expectations for the origin and diversification of a mammalian genus during a period of extreme environmental change.
[本文引用: 3]
[本文引用: 1]
URLPMID:15522810 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 5]
DOI:10.1093/jhered/esv081URLPMID:26447215 [本文引用: 3]
Two sets of commercially available single nucleotide polymorphisms (SNPs) developed for cattle (BovineSNP50 BeadChip) and sheep (OvineSNP50 BeadChip) have been trialed for whole-genome analysis of 4 female samples of Rangifer tarandus inhabiting Russia. We found out that 43.0% of bovine and 47.0% of Ovine SNPs could be genotyped, while only 5.3% and 2.03% of them were respectively polymorphic. The scored and the polymorphic SNPs were identified on each bovine and each ovine chromosome, but their distribution was not unique. The maximal value of runs of homozygosity (ROH) was 30.93Mb (for SNPs corresponding to bovine chromosome 8) and 80.32Mb (for SNPs corresponding to ovine chromosome 7). Thus, the SNP chips developed for bovine and ovine species can be used as a powerful tool for genome analysis in reindeer R. tarandus.
[本文引用: 2]
[本文引用: 3]
[本文引用: 3]
[本文引用: 3]
URLPMID:23110438 [本文引用: 1]
DOI:10.1093/jhered/esv137URLPMID:26774056 [本文引用: 1]
Cross-species application of single-nucleotide polymorphism (SNP) chips is a valid, relatively cost-effective alternative to the high-throughput sequencing methods generally required to obtain a genome-wide sampling of polymorphisms. Kharzinova et al. (2015) examined the applicability of SNP chips developed in domestic bovids (cattle and sheep) to a semi-wild cervid (reindeer). The ancestors of bovids and cervids diverged between 20 and 30 million years ago (Hassanin and Douzery 2003; Bibi et al. 2013). Empirical work has shown that for a SNP chip developed in a bovid and applied to a cervid species, approximately 50% genotype success with 1% of the loci being polymorphic is expected (Miller et al. 2012). The genotyping of Kharzinova et al. (2015) follows this pattern; however, these data are not appropriate for identifying runs of homozygosity (ROH) and can be problematic for estimating linkage disequilibrium (LD) and we caution readers in this regard.
[本文引用: 1]
[本文引用: 1]
URLPMID:31101010 [本文引用: 1]
DOI:10.1111/eva.12705URLPMID:30459847 [本文引用: 2]
The Milu (Pere David's deer, Elaphurus davidianus) were once widely distributed in the swamps (coastal areas to inland areas) of East Asia. The dramatic recovery of the Milu population is now deemed a classic example of how highly endangered animal species can be rescued. However, the molecular mechanisms that underpinned this population recovery remain largely unknown. Here, different approaches (genome sequencing, resequencing, and salinity analysis) were utilized to elucidate the aforementioned molecular mechanisms. The comparative genomic analyses revealed that the largest recovered Milu population carries extensive genetic diversity despite an extreme population bottleneck. And the protracted inbreeding history might have facilitated the purging of deleterious recessive alleles. Seventeen genes that are putatively related to reproduction, embryonic (fatal) development, and immune response were under high selective pressure. Besides, SCNN1A, a gene involved in controlling reabsorption of sodium in the body, was positively selected. An additional 29 genes were also observed to be positively selected, which are involved in blood pressure regulation, cardiovascular development, cholesterol regulation, glycemic control, and thyroid hormone synthesis. It is possible that these genetic adaptations were required to buffer the negative effects commonly associated with a high-salt diet. The associated genetic adaptions are likely to have enabled increased breeding success and fetal survival. The future success of Milu population management might depend on the successful reintroduction of the animal to historically important distribution regions.
URLPMID:23242367 [本文引用: 1]
DOI:10.1007/s00335-015-9606-7URLPMID:26475143 [本文引用: 1]
Despite the growing number of sequenced bovine genomes, the knowledge of the population-wide variation of sequences remains limited. In many studies, statistical methodology was not applied in order to relate findings in the sequenced samples to a population-wide level. Our goal was to assess the population-wide variation in DNA sequence based on whole-genome sequences of 32 Holstein-Friesian cows. The number of SNPs significantly varied across individuals. The number of identified SNPs increased with coverage, following a logarithmic curve. A total of 15,272,427 SNPs were identified, 99.16 % of them being bi-allelic. Missense SNPs were classified into three categories based on their genomic location: housekeeping genes, genes undergoing strong selection, and genes neutral to selection. The number of missense SNPs was significantly higher within genes neutral to selection than in the other two categories. The number of variants located within 3'UTR and 5'UTR regions was also significantly different across gene families. Moreover, the number of insertions and deletions differed significantly among cows varying between 261,712 and 330,103 insertions and from 271,398 to 343,649 deletions. Results not only demonstrate inter-individual variation in the number of SNPs and indels but also show that the number of missense SNPs differs across genes representing different functional backgrounds.
[本文引用: 1]
DOI:10.1007/s00438-018-1520-8URLPMID:30539301 [本文引用: 2]
Velvet antler displays the fastest and most robust tissue proliferation in the animal world, it is a model for a complete organ development/regeneration, and alternative medicine, tonic made from velvet antler, was beneficial for human. The weight of velvet antler had high biomedical and economic value, but the related regulation mechanisms controlling velvet antler weight remain unclear. In this study, extremely heavy and light velvet antler groups were selected from a sika deer population of 100 individuals with extreme velvet antler weight. A combination of full-length transcriptome sequencing and microRNA sequencing to the proliferation zone in the tip of velvet antler was applied. A total of 55306 transcripts and 1082 microRNAs were identified. Some highly expressed genes (COL1A1, COL1A2, COL3A1, FN1, and ATP6) and microRNAs (miR-21, let-7i, and miR-27b) were highly correlated with the physiological and growth characteristics of velvet antlers. Among the 334 differentially expressed genes, we found that most of the genes were located in the developmental process, especially animal organ development process. It is exciting to see that more blood vessels were found in the growing tip of heavy velvet antler through histological observation, and GO term of blood vessel development was also significant different between two groups. The combination analysis with mRNA and microRNA data in velvet antler showed a specific regulation network involved in the development of bone, mesenchyme, cartilage, and blood vessel, and helped us clearly find out the candidate 14 genes and 6 microRNAs, which could be used for selecting significant DNA markers of velvet antler weight.
[本文引用: 1]
DOI:10.1186/s41065-020-00137-xURLPMID:32591015 [本文引用: 2]
BACKGROUND: Sika deer is one of the most popular and valued animals in China. However, few studies have been conducted on the microsatellite of Sika deer, which has hampered the progress of genetic selection breeding. To develop and characterize a set of microsatellites for Sika deer which provide helpful information for protection of Sika deer natural resources and effectively increase the yield and quantity of velvet antler. RESULTS: We conducted a transcriptome survey of Sika deer using next-generation sequencing technology. One hundred eighty-two thousand two hundred ninety-five microsatellite markers were identified in the transcriptome, 170 of 200 loci were successfully amplified across panels of 140 individuals from Shuangyang Sika deer population. And 29 loci were found to be obvious polymorphic. Number of alleles is from 3 to 14. The expected heterozygosity ranged from 0.3087 to 0.7644. The observed heterozygosity ranged from 0 to 0.7698. The polymorphism information content values of those microsatellites varied ranged from 0.2602 to 0.7507. The marker-trait association was tested for 6 important and kernel characteristics of two-branched velvet antler in Shuangyang Sika deer through one-way analysis of variance. The results showed that marker-trait associations were identified with 8 different markers, especially M009 and M027. CONCLUSIONS: This study not only provided a large scale of microsatellites which were valuable for future genetic mapping and trait association in Sika deer, but also offers available information for molecular breeding in Sika deer.
DOI:10.1007/s00438-016-1231-yURLPMID:27423230 [本文引用: 1]
Sika deer is of great commercial value because their antlers are used in tonics and alternative medicine and their meat is healthy and delicious. The goal of this study was to generate transcript sequences from sika deer for functional genomic analyses and to identify the transcripts that demonstrate tissue-specific, age-dependent differential expression patterns. These sequences could enhance our understanding of the molecular mechanisms underlying sika deer growth and development. In the present study, we performed de novo transcriptome assembly and profiling analysis across ten tissue types and four developmental stages (juvenile, adolescent, adult, and aged) of sika deer, using Illumina paired-end tag (PET) sequencing technology. A total of 1,752,253 contigs with an average length of 799 bp were generated, from which 1,348,618 unigenes with an average length of 590 bp were defined. Approximately 33.2 % of these (447,931 unigenes) were then annotated in public protein databases. Many sika deer tissue-specific, age-dependent unigenes were identified. The testes have the largest number of tissue-enriched unigenes, and some of them were prone to develop new functions for other tissues. Additionally, our transcriptome revealed that the juvenile-adolescent transition was the most complex and important stage of the sika deer life cycle. The present work represents the first multiple tissue transcriptome analysis of sika deer across four developmental stages. The generated data not only provide a functional genomics resource for future biological research on sika deer but also guide the selection and manipulation of genes controlling growth and development.
URLPMID:29223356 [本文引用: 2]
[本文引用: 1]
DOI:10.1371/journal.pone.0242506URLPMID:33226998 [本文引用: 1]
Microsatellites are widely applied in population and forensic genetics, wildlife studies and parentage testing in animal breeding, among others, and recently, high-throughput sequencing technologies have greatly facilitated the identification of microsatellite markers. In this study the genomic data of Cervus elaphus (CerEla1.0) was exploited, in order to identify microsatellite loci along the red deer genome and for designing the cognate primers. The bioinformatics pipeline identified 982,433 microsatellite motifs genome-wide, assorted along the chromosomes, from which 45,711 loci mapped to the X- and 1096 to the Y-chromosome. Primers were successfully designed for 170,873 loci, and validated with an independently developed autosomal tetranucleotide STR set. Ten X- and five Y-chromosome-linked microsatellites were selected and tested by two multiplex PCR setups on genomic DNA samples of 123 red deer stags. The average number of alleles per locus was 3.3, and the average gene diversity value of the markers was 0.270. The overall observed and expected heterozygosities were 0.755 and 0.832, respectively. Polymorphic Information Content (PIC) ranged between 0.469 and 0.909 per locus with a mean value of 0.813. Using the X- and Y-chromosome linked markers 19 different Y-chromosome and 72 X-chromosome lines were identified. Both the X- and the Y-haplotypes split to two distinct clades each. The Y-chromosome clades correlated strongly with the geographic origin of the haplotypes of the samples. Segregation and admixture of subpopulations were demonstrated by the use of the combination of nine autosomal and 16 sex chromosomal STRs concerning southwestern and northeastern Hungary. In conclusion, the approach demonstrated here is a very efficient method for developing microsatellite markers for species with available genomic sequence data, as well as for their use in individual identifications and in population genetics studies.
DOI:10.1101/gr.239863.118URLPMID:30760546 [本文引用: 1]
The role of chromosome rearrangements in driving evolution has been a long-standing question of evolutionary biology. Here we focused on ruminants as a model to assess how rearrangements may have contributed to the evolution of gene regulation. Using reconstructed ancestral karyotypes of Cetartiodactyls, Ruminants, Pecorans, and Bovids, we traced patterns of gross chromosome changes. We found that the lineage leading to the ruminant ancestor after the split from other cetartiodactyls was characterized by mostly intrachromosomal changes, whereas the lineage leading to the pecoran ancestor (including all livestock ruminants) included multiple interchromosomal changes. We observed that the liver cell putative enhancers in the ruminant evolutionary breakpoint regions are highly enriched for DNA sequences under selective constraint acting on lineage-specific transposable elements (TEs) and a set of 25 specific transcription factor (TF) binding motifs associated with recently active TEs. Coupled with gene expression data, we found that genes near ruminant breakpoint regions exhibit more divergent expression profiles among species, particularly in cattle, which is consistent with the phylogenetic origin of these breakpoint regions. This divergence was significantly greater in genes with enhancers that contain at least one of the 25 specific TF binding motifs and located near bovidae-to-cattle lineage breakpoint regions. Taken together, by combining ancestral karyotype reconstructions with analysis of cis regulatory element and gene expression evolution, our work demonstrated that lineage-specific regulatory elements colocalized with gross chromosome rearrangements may have provided valuable functional modifications that helped to shape ruminant evolution.
[本文引用: 1]
DOI:10.1159/000133525URLPMID:8485991 [本文引用: 1]
The Indian muntjac is believed to have the lowest chromosome number in mammals (2n = 6 in females and 2n = 7 in males). It has been suggested that a series of tandem chromosome fusions from an ancestral Chinese muntjac-like species (2n = 46) may have occurred during the karyotypic evolution of the Indian muntjac. In an earlier study, hybridization signals generated by the Chinese muntjac centromeric heterochromatin DNA probe (C5) were found to be distributed interstitially in the chromosomes of the Indian muntjac, providing supportive evidence for the tandem chromosome fusion theory. In this study, the highly conserved human telomeric DNA sequence (TTAGGG)n was localized by fluorescence in situ hybridization (FISH) on the metaphase chromosomes of three Cervidae species: the Indian muntjac, Chinese muntjac, and woodland caribou. As expected, hybridization signals were observed at the termini of almost every chromosome in all three species. In addition, interstitial hybridization signals were detected in chromosomes 1 and 2 of the Indian muntjac. The observed interstitial telomeric signals appeared to correspond to specific interstitial centromeric heterochromatin sites. These interstitial telomeric signals could represent remnant DNA sequences from the ancestral species telomeres, further supporting the tandem chromosome fusion theory. Furthermore, these observations permit the elucidation of the chromosome sites where breakage and fusion most likely occurred during the restructuring of the ancestral Chinese muntjac-like chromosomes to form the present day Indian muntjac karyotype.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/gb-2008-9-10-r155URLPMID:18957082 [本文引用: 1]
BACKGROUND: Indian muntjac (Muntiacus muntjak vaginalis) has an extreme mammalian karyotype, with only six and seven chromosomes in the female and male, respectively. Chinese muntjac (Muntiacus reevesi) has a more typical mammalian karyotype, with 46 chromosomes in both sexes. Despite this disparity, the two muntjac species are morphologically similar and can even interbreed to produce viable (albeit sterile) offspring. Previous studies have suggested that a series of telocentric chromosome fusion events involving telomeric and/or satellite repeats led to the extant Indian muntjac karyotype. RESULTS: We used a comparative mapping and sequencing approach to characterize the sites of ancestral chromosomal fusions in the Indian muntjac genome. Specifically, we screened an Indian muntjac bacterial artificial-chromosome library with a telomere repeat-specific probe. Isolated clones found by fluorescence in situ hybridization to map to interstitial regions on Indian muntjac chromosomes were further characterized, with a subset then subjected to shotgun sequencing. Subsequently, we isolated and sequenced overlapping clones extending from the ends of some of these initial clones; we also generated orthologous sequence from isolated Chinese muntjac clones. The generated Indian muntjac sequence has been analyzed for the juxtaposition of telomeric and satellite repeats and for synteny relationships relative to other mammalian genomes, including the Chinese muntjac. CONCLUSIONS: The generated sequence data and comparative analyses provide a detailed genomic context for seven ancestral chromosome fusion sites in the Indian muntjac genome, which further supports the telocentric fusion model for the events leading to the unusual karyotypic differences among muntjac species.
DOI:10.1007/s10709-005-2449-5URLPMID:16850210 [本文引用: 1]
A set of Chinese muntjac (Muntiacus reevesi) chromosome-specific paints has been hybridized onto the metaphases of sika deer (Cervus nippon, CNI, 2n = 66), red deer (Cervus elaphus, CEL, 2n = 62) and tufted deer (Elaphodus cephalophus, ECE, 2n = 47). Thirty-three homologous autosomal segments were detected in genomes of sika deer and red deer, while 31 autosomal homologous segments were delineated in genome of tufted deer. The Chinese muntjac chromosome X probe painted to the whole X chromosome, and the chromosome Y probe gave signals on the Y chromosome as well as distal region of the X chromosome of each species. Our results confirmed that exclusive Robertsonian translocations have contributed to the karyotypic evolution of sika deer and red deer. In addition to Robertsonian translocation, tandem fusions have played a more important role in the karyotypic evolution of tufted deer. Different types of chromosomal rearrangements have led to great differences in the genome organization between cervinae and muntiacinae species. Our analysis testified that six chromosomal fissions in the proposed 2n = 58 ancestral pecoran karyotype led to the formation of 2n = 70 ancestral cervid karyotype and the deer karyotypes is more derived compare with those of bovid species. Combining previous cytogenetic and molecular systematic studies, we analyzed the genome phylogeny for 11 cervid species.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
DOI:10.1038/s41598-020-65487-yURLPMID:32488117 [本文引用: 1]
Reindeer are semi-domesticated ruminants that have adapted to the challenging northern Eurasian environment characterized by long winters and marked annual fluctuations in daylight. We explored the genetic makeup behind their unique characteristics by de novo sequencing the genome of a male reindeer and conducted gene family analyses with nine other mammalian species. We performed a population genomics study of 23 additional reindeer representing both domestic and wild populations and several ecotypes from various geographic locations. We assembled 2.66 Gb (N50 scaffold of 5 Mb) of the estimated 2.92 Gb reindeer genome, comprising 27,332 genes. The results from the demographic history analysis suggested marked changes in the effective population size of reindeer during the Pleistocene period. We detected 160 reindeer-specific and expanded genes, of which zinc finger proteins (n = 42) and olfactory receptors (n = 13) were the most abundant. Comparative genome analyses revealed several genes that may have promoted the adaptation of reindeer, such as those involved in recombination and speciation (PRDM9), vitamin D metabolism (TRPV5, TRPV6), retinal development (PRDM1, OPN4B), circadian rhythm (GRIA1), immunity (CXCR1, CXCR2, CXCR4, IFNW1), tolerance to cold-triggered pain (SCN11A) and antler development (SILT2). The majority of these characteristic reindeer genes have been reported for the first time here. Moreover, our population genomics analysis suggested at least two independent reindeer domestication events with genetic lineages originating from different refugial regions after the Last Glacial Maximum. Taken together, our study has provided new insights into the domestication, evolution and adaptation of reindeer and has promoted novel genomic research of reindeer.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s13258-019-00911-wURLPMID:31902105 [本文引用: 1]
BACKGROUND: Cervids have evolved very successful means for survival and thriving to adapt to various climates and environments. One of these successful means might be the effective and efficient way of communication. To support this notion, cervids are well equipped with a variety of skin glands that distribute in different body regions. However, studies relevant to adaptive evolution in cervids, particularly on olfactory reception at the molecular level, have thus far not been reported. OBJECTIVE: To provide valuable insights into molecular evidence for the adaptive evolution of olfactory-related gene in cervids. METHODS: Based on recently sequenced genomes of cervids and closely-related-species, we performed comparative genomic analysis at genome level using bioinformatics tools. RESULTS: Tree topology strongly supported that Bovidae was the sister group of Moschidae and both formed a branch that was then clustered with Cervidae. Expansion of heavy chain genes of the dynein family and 51 rapidly evolving genes could be associated with adaptation of cilia that serve as sensory organelles and act as cellular antennae. Based on the branch-site model test along the deer branch spanning 7-21 mammalian species, 14 deer olfactory receptor genes were found to be undergoing positive selection pressure and 89 positive selection sites (probability > 60%) had amino acid substitutions unique to deer. CONCLUSION: This study, for the first time, provides significant molecular evidence for adaption of olfactory-related genes of cervids according to their olfactory behavior.
[本文引用: 1]
DOI:10.1016/j.semcdb.2008.11.011URLPMID:19084608 [本文引用: 1]
Deer antlers are periodically replaced cranial appendages that develop from permanent outgrowths of the frontal bones known as pedicles. Antler re-growth is a unique regenerative event in mammals which in general are unable to replace bony appendages. Recent evidence suggests that antler regeneration is a stem cell-based process that depends on the activation of stem cells located in the pedicle periosteum which are presumed to be neural crest-derived. It has been demonstrated that several developmental pathways are involved in antler regeneration that are also known to play a role in the control of skeletal development and regeneration in other vertebrates. However, in contrast to most other natural examples of regeneration of complete body structures, antler regeneration apparently neither depends on a functional nerve supply nor involves a direct contact between wound epithelium and mesenchymal tissue. Antlers thus demonstrate that regeneration of a large bony appendage in a mammal can be achieved by a process that differs in certain aspects from epimorphic regeneration in lower vertebrates.
DOI:10.1111/j.1469-7580.2005.00478.xURLPMID:16313394 [本文引用: 1]
Many organisms are able to regenerate lost or damaged body parts that are structural and functional replicates of the original. Eventually these become fully integrated into pre-existing tissues. However, with the exception of deer, mammals have lost this ability. Each spring deer shed antlers that were used for fighting and display during the previous mating season. Their loss is triggered by a fall in circulating testosterone levels, a hormonal change that is linked to an increase in day length. A complex 'blastema-like' structure or 'antler-bud' then forms; however, unlike the regenerative process in the newt, most evidence (albeit indirect) suggests that this does not involve reversal of the differentiated state but is stem cell based. The subsequent re-growth of antlers during the spring and summer months is spectacular and represents one of the fastest rates of organogenesis in the animal kingdom. Longitudinal growth involves endochondral ossification in the tip of each antler branch and bone growth around the antler shaft is by intramembranous ossification. As androgen concentrations rise in late summer, longitudinal growth stops, the skin (velvet) covering the antler is lost and antlers are 'polished' in preparation for the mating season. Although the timing of the antler growth cycle is clearly closely linked to circulating testosterone, oestrogen may be a key cellular regulator, as it is in the skeleton of other male mammals. We still know very little about the molecular machinery required for antler regeneration, although there is evidence that developmental signalling pathways with pleiotropic functions are important and that novel 'antler-specific' molecules may not exist. Identifying these pathways and factors, deciphering their interactions and how they are regulated by environmental cues could have an important impact on human health if this knowledge is applied to the engineering of new human tissues and organs.
DOI:10.1098/rspb.1998.0362URLPMID:9628037 [本文引用: 1]
The entire mitochondrial cytochrome b (cyt b) gene was compared for 11 species of the artiodactyl family Cervidae, representing all living subfamilies, i.e., the antlered Cervinae (Cervus elaphus, C. nippon, Dama dama), Muntiacinae (Muntiacus reevesi), and Odocoileinae (Odocoileus hemionus, Mazama sp., Capreolus capreolus, C. pygargus, Rangifer tarandus, Alces alces); and the antlerless Hydropotinae (Hydropotes inermis). Phylogenetic analyses using Tragulidae, Antilocapridae, Giraffidae and Bovidae as outgroups provide evidence for three multifurcating principal clades within the monophyletic family Cervidae. First, Cervinae and Muntiacus are joined in a moderately-to-strongly supported clade of Eurasian species. Second, Old World Odocoileinae (Capreolus and Hydropotes) associate with the Holarctic Alces. Third, New World Odocoileinae (Mazama and Odocoileus) cluster with the Holarctic Rangifer. The combination of mitochondrial cyt b and nuclear k-casein sequences increases the robustness of these three clades. The Odocoileini + Rangiferini clade is unambiguously supported by a unique derived cranial feature, the expansion of the vomer which divides the choana. Contrasting with current taxonomy, Hydropotes is not the sister group of all the antlered deers, but it is nested within the Odocoileinae. Therefore, Hydropotes lost the antlers secondarily. Thus, the mitochondrial cyt b phylogeny splits Cervidae according to plesiometacarpal (Cervinae + Muntiacinae) versus telemetacarpal (Odocoileinae + Hydropotinae) conditions, and suggests paraphyly of antlered deer.
URLPMID:22457177 [本文引用: 2]
[本文引用: 1]
DOI:10.1371/journal.pone.0002064URLPMID:18446198 [本文引用: 1]
The annual regeneration of deer antlers is a unique developmental event in mammals, which as a rule possess only a very limited capacity to regenerate lost appendages. Studying antler regeneration can therefore provide a deeper insight into the mechanisms that prevent limb regeneration in humans and other mammals, and, with regard to medical treatments, may possibly even show ways how to overcome these limitations. Traditionally, antler regeneration has been characterized as a process involving the formation of a blastema from de-differentiated cells. More recently it has, however, been hypothesized that antler regeneration is a stem cell-based process. Thus far, direct evidence for the presence of stem cells in primary or regenerating antlers was lacking. Here we demonstrate the presence of cells positive for the mesenchymal stem cell marker STRO-1 in the chondrogenic growth zone and the perivascular tissue of the cartilaginous zone in primary and regenerating antlers as well as in the pedicle of fallow deer (Dama dama). In addition, cells positive for the stem cell/progenitor cell markers STRO-1, CD133 and CD271 (LNGFR) were isolated from the growth zones of regenerating fallow deer antlers as well as the pedicle periosteum and cultivated for extended periods of time. We found evidence that STRO-1(+) cells isolated from the different locations are able to differentiate in vitro along the osteogenic and adipogenic lineages. Our results support the view that the annual process of antler regeneration might depend on the periodic activation of mesenchymal progenitor cells located in the pedicle periosteum. The findings of the present study indicate that not only limited tissue regeneration, but also extensive appendage regeneration in a postnatal mammal can occur as a stem cell-based process.
DOI:10.3727/096368912X661391URLPMID:23294672 [本文引用: 1]
Recent studies have reported that stem cells can be isolated from various tissues such as bone marrow, fatty tissue, umbilical cord blood, Wharton's jelly, and placenta. These types of stem cell studies have also arisen in veterinary medicine. Deer antlers show a seasonal regrowth of tissue, an unusual feature in mammals. Antler tissue therefore might offer a source of stem cells. To explore the possibility of stem cell populations within deer antlers, we isolated and successfully cultured antler-derived multipotent stem cells (MSCs). Antler MSCs were maintained in a growth medium, and the proliferation potential was measured via an assay called the cumulative population doubling level. Immunophenotyping and immunostaining revealed the intrinsic characteristic stem cell markers of antler MSCs. To confirm the ability to differentiate, we conducted osteogenic, adipogenic, and chondrogenic induction under the respective differentiation conditions. We discovered that antler MSCs have the ability to differentiate into multiple lineages. In conclusion, our results show that deer antler tissue may contain MSCs and therefore may be a potential source for veterinary regenerative therapeutics.
[本文引用: 2]
DOI:10.1038/s41419-019-1686-yURLPMID:31165741 [本文引用: 2]
Deer antlers are extraordinary mammalian organs that can fully regenerate annually. Antler renewal is a stem cell-based epimorphic process and antler stem (AS) cells can initiate de novo generation of antlers in postnatal mammals. However, although being called stem cells, the AS cells have not been characterized at molecular level based on the stem cell criteria. Comprehensive characterization of the AS cells would undoubtedly help to decipher the mechanism underlying the full regeneration of deer antlers, the only case of stem cell-based epimorphic regeneration in mammals. In the present study, three types of AS cells (antlerogenic periosteal cells APCs, for initial pedicle and first antler formation; pedicle periosteal cells PPC, for annual antler regeneration; and reserve mesenchyme cells RMCs, for rapid antler growth), were isolated for comprehensive molecular characterization. A horn-growth-related gene, RXFP2, was found to be expressed only in AS cells lineages but not in the facial periosteal cells (FPCs, locates geographically in the vicinity of the APCs or PPCs), suggesting the RXFP2 might be a specific marker for the AS cell lineage in deer. Our results demonstrated that AS cells expressed classic MSC markers including surface markers CD73, CD90, CD105 and Stro-1. They also expressed some of the markers including Tert, Nestin, S100A4, nucleostemin and C-Myc, suggesting that they have some attributes of the ESCs. Microinjection of male APC into deer blastocysts resulted in one female foetus (110 days gestation) recovered with obvious pedicle primordia with both male and female genotype detected in the ovary. In conclusion, the AS cells should be defined as MSCs but with partial attributes of ESCs.
DOI:10.1007/s00438-015-1158-8URLPMID:26738876 [本文引用: 1]
MicroRNAs (miRNAs) can effectively regulate gene expression at the post-transcriptional level and play a critical role in tissue growth, development and regeneration. Our previous studies showed that antler regeneration is a stem cell-based process and antler stem cells reside in the periosteum of a pedicle, the permanent bony protuberance, from which antler regeneration takes place. Antlers are the only mammalian organ that can fully regenerate and hence provide a unique opportunity to identify miRNAs that are involved in organ regeneration. In the present study, we used next generation sequencing technology sequenced miRNAs of the stem cells derived from either the potentiated or the dormant pedicle periosteum. A population of both conserved and 20 deer-specific miRNAs was identified. These conserved miRNAs were derived from 453 homologous hairpin precursors across 88 animal species, and were further grouped into 167 miRNA families. Among them, the miR-296 is embryonic stem cell-specific. The potentiation process resulted in the significant regulation (>+/-2 Fold, q value <0.05) of conserved miRNAs; 8 miRNA transcripts were down- and 6 up-regulated. Several GO biology processes and the Wnt, MAPK and TGF-beta signaling pathways were found to be up-regulated as part of antlerogenic stem cell potentiation process. This research has identified miRNAs that are associated either with the dormant or the potentiated antler stem cells and identified some target miRNAs for further research into their role played in mammalian organ regeneration.
DOI:10.1007/s10142-019-00659-2URLPMID:30673893 [本文引用: 1]
Antler regeneration, a stem cell-based epimorphic process, has a potential as a valuable model for regenerative medicine. A pool of antler stem cells (ASCs) for antler development is located in the antlerogenic periosteum (AP). However, whether this ASC pool is homogenous or heterogeneous has not been fully evaluated. In this study, we produced a comprehensive transcriptome dataset at the single-cell level for the ASCs based on the 10x Genomics platform (scRNA-seq). A total of 4565 ASCs were sequenced and classified into a large cell cluster, indicating that the ASC resident in the AP are likely to be a homogeneous population. The scRNA-seq data revealed that tumor-related genes were highly expressed in these homogeneous ASCs, i.e., TIMP1, TMSB10, LGALS1, FTH1, VIM, LOC110126017, and S100A4. Results of screening for stem cell markers suggest that the ASCs may be considered as a special type of stem cell between embryonic (CD9) and adult (CD29, CD90, NPM1, and VIM) stem cells. Our results provide the first comprehensive transcriptome analysis at the single-cell level for the ASCs and identified only one major cell type resident in the AP and some key stem cell genes, which may hold the key to why antlers, the unique mammalian organ, can fully regenerate once lost.
URLPMID:17177282 [本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.jprot.2019.01.004URLPMID:30641233 [本文引用: 1]
The ability to activate and regulate stem cells during wound healing and tissue/organ regeneration is a promising field which could bring innovative approaches to regenerative medicine. The regenerative capacity of invertebrates has been well documented, however in mammals, stem cells that drive organ regeneration are rare. Deer antler is unique in providing a mammalian model of complete organ regeneration based on stem cells. The present study investigated the differentially regulated proteins (DRPs) between different antler stem cell populations (n=3) using 2D-DIGE. Western blotting was used to validate the proteomics results. Comparative proteomics resulted in protein profiles which were similar for the biological replicates but different between the cells derived from two different stem cell niches involved in antler growth/regeneration and cells derived from facial periosteum. Ninety-two up- and down-regulated proteins were identified by MALDI-TOF MS. The work indicates that the epithelial-mesenchymal transition process may participate in the initiation of antler regeneration including the first stage of scar-less wound healing. Cell mobility is also highly regulated during antler regeneration. Energy and nucleotide metabolism may however be less active in antler regeneration as compared to that in antler generation phase. These results provide new insights into the underlying mechanisms of stem cell-based regeneration of mammalian organs.
DOI:10.1021/acs.jproteome.0c00026URLPMID:32155067 [本文引用: 2]
As the only known mammalian organ that can fully and annually regenerate, deer antler has significant advantages over lower-order animal models when investigating the control of stem-cell-based organ regeneration. Antler regeneration is known to be initiated and maintained by neural-crest-derived stem cells in different states of activation. Antler stem cells can therefore be used as a model to study proteins and pathways involved in the maintenance of a stem cell niche and their activation and differentiation during organ formation. In this study, the MSC markers CD73, CD90, and CD105 were examined within the antler tip. Label-free quantification was performed to investigate the protein profiles of antler stem cells under different stages of activation and included dormant pedicle periosteum (DPP), antler growth center (GC), post-active stem cells from mid-beam antler periosteum (MAP), and deer facial periosteum (FP) as a control (n = 3 per group). PEAKS and IPA software were used to analyze the proteomic data. Our research confirmed the central role of stem cell activation in the development of this mammalian organ by localizing the MSC markers within the antler growth center. Label-free quantification revealed that the greatest number of unique proteins (87) was found in the growth center. There were only 12 proteins found with expression levels that significantly differed between DPP and FP. Protein profiles of these two groups indicated that antler stem cells may use similar mechanisms to maintain dormancy within a stem cell niche. The number of significantly regulated proteins among DPP, MAP, and GC was 153. Among them, the majority were upregulated in the growth center. Activation of antler stem cells was associated with many biological processes and signaling pathways, such as Hippo and canonical Wnt signaling. This work identifies the key pathways, molecular/cellular functions, and upstream regulators involved in mammal organ regeneration. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD016824.
DOI:10.1007/s10735-019-09853-9URLPMID:31858326 [本文引用: 1]
Deer antlers are the only mammalian organs capable of complete renewal. Antler renewal is a stem cell-based [antler stem cells (ASCs)] process. Maintenance and activation of the ASCs require them to be located in a specialized microenvironment (niche), and to interact with the cells resident in the niche. Based on previous experiments we found that niche of the ASCs is provided by the closely associated enveloping skin, which currently was known includes dermal papilla cells (DPCs) and epidermal cells. Antler generation/regeneration are triggered by the interactions between ASCs and the niche. In the present study, we established an in vitro co-culture system in which ASCs and DPCs, were cultured together to mimic the in vivo state. A MLEFF strategy was adopted to identify the interactive molecules from the co-culture system. In total, 128 molecules were identified and over 60% belonged to exosomes. Important biological processes that were activated by these molecules included osteoblast differentiation, angiogenesis, and the PI3K-AKT signaling pathway. In so doing, we have significantly simplified the process for identifying interactive molecules, which may be the key signals for triggering antler formation/renewal. Further study of these molecules will help us to gain insights into the mechanism of mammalian organ regeneration.
URLPMID:22279561 [本文引用: 1]
[本文引用: 1]
DOI:10.1038/367480a0URLPMID:8107809 [本文引用: 1]
We have shown that a polypeptide of M(r) 60,000 (60K) that shares N-terminal homology with a calcium-binding protein, calreticulin, can bind to an amino-acid sequence motif, KXGFFKR, found in the cytoplasmic domains of all integrin alpha-subunits. The homologous amino-acid sequence, KXFFKR (where X is either G, A or V), is also present in the DNA-binding domain of all known members of the steroid hormone receptor family; amino acids in this sequence make direct contact with nucleotides in their DNA-responsive elements and are crucial for DNA binding. Here we show that both the 60K protein (p60), purified on a KLGFFKR-Sepharose affinity matrix, and recombinant calreticulin can inhibit the binding of androgen receptor to its hormone-responsive DNA element in a KXFFKR-sequence-specific manner. Calreticulin can also inhibit androgen receptor and retinoic acid receptor transcriptional activities in vivo, as well as retinoic acid-induced neuronal differentiation. Our results indicate that calreticulin can act as an important modulator of the regulation of gene transcription by nuclear hormone receptors.
DOI:10.1016/j.ygcen.2019.113235URLPMID:31369730 [本文引用: 1]
Deer antlers offer a unique model to study organ regeneration in mammals. Antler regeneration relies on the pedicle periosteum (PP) cells and is triggered by a decrease in circulating testosterone (T). The molecular mechanism for antler regeneration is however, unclear. Label-free liquid chromatography-mass spectrometry (LC-MS/MS) was used to identify differentially-expressed proteins (DEPs) in the regeneration-potentiated PP (under low T environment) over the non-regeneration-potentiated PP (under high T environment). Out of total 273 DEPs, 189 were significantly up-regulated and 84 were down-regulated from these comparisons: after castration vs before castration, natural T vs before castration, and exogenous T vs before castration. We focused on the analysis only of those DEPs that were present in fully permissive environment to antler regeneration (low T). Nine transduction pathways were identified through the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, including the estrogen signaling pathway. A total of 639 gene ontology terms were found to be significantly enriched in regeneration-potentiated PP (low T) from the DEPs. Reliability of the label free LC-MS/MS was determined by qRT-PCR to estimate the expression level of selected genes. The results suggest that up-regulated heat shock proteins (HSP90AB1, HSP90B1), peptidyl-prolyl cis-trans isomerase 4 (FKBP4), mitogen-activated protein kinase 3 (MAPK3) and calreticulin (CALR) and down-regulated SHC-transforming protein 1 (SHC1), heat shock protein family A member 1A (HSPA1A) and proto-oncogene tyrosine-protein kinase (SRC) may be associated directly or indirectly with antler regeneration. Further studies are required to investigate the roles of these proteins in regeneration using appropriate in vivo models.
DOI:10.1002/pmic.200401027URLPMID:15529405 [本文引用: 1]
Deer antlers are the only mammalian organs capable of repeated regeneration. Although antlers are known to develop from pedicles, which arise from antlerogenic cells of cranial periosteum, their developmental process is not fully elucidated. For example, while endocrine and environmental factors influence the antler development, it is still unclear which signaling pathways are involved in the transduction of such stimuli. To study the developmental process of antlers and identify proteins functioning in their growth, we have established proteome maps of red deer (Cervus elaphus) antlers. With two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization mass spectrometry, we analyzed more than 800 protein spots and identified approximately 130 individual proteins derived from the growing tip of antlers. The overall profile of the antler proteome was dissimilar to those of other types of tissue. Also comparison of proteomes derived from proximal bony tissue and the growing tip of antlers revealed substantial differences. Moreover several cell growth or signaling-related proteins are expressed exclusively in the growing tip, suggesting that these proteins function in the growth and differentiation of antlers. Currently, using the antler proteome maps, we are actively searching for the regulatory factor(s) that may control the antler development.
DOI:10.1007/s11010-011-1209-3URLPMID:22198337 [本文引用: 2]
Deer antlers are well known for their regeneration and rapid growth. However, little is known about the genes that are involved in their development, especially the molecular mechanisms responsible for rapid growth. In the present study, we produced more than 41 million sequencing reads using the Illumina sequencing platform. These reads were assembled into 89,001 unique sequences (mean size: 450 bp), representing more than 58 times as many Sika deer sequences previously available in the NCBI database (as of Sep 15, 2011). Based on a similarity search with known proteins, we identified 40,088 sequences with a cut-off E value of 10(-5). Assembled sequences were then annotated using Gene ontology terms, Clusters of Orthologous Groups classifications, and Kyoto Encyclopedia of Genes and Genomes pathways. In addition, we found a number of highly expressed genes involved in the regulation of Sika deer antler rapid growth, including transcription factors, signaling molecules, and extracellular matrix proteins. Our data represent the most comprehensive sequence resource available for the deer antler and provide a basis for new research on deer antler molecular genetics and functional genomics.
[本文引用: 1]
DOI:10.1007/s11033-012-2216-5URLPMID:23073784 [本文引用: 1]
Deer antlers serve as useful models of rapid growth and mineralization in mammals. During the period of rapid growth, the antlers of many species of deer will elongate by more than 2 cm per day, after which the antlers gradually ossify. However, little is known about the genes that are involved in their development, particularly the molecular mechanisms responsible for rapid growth and ossification. In our previous studies, we have reported on the transcriptome analysis of deer antlers at rapid growth and ossification stages. With the aim to get a comprehensive understanding of gene expression patterns during antler growth, in the present study, we performed a rigorous algorithm to identify differentially expressed genes between two different stages (60 and 90 days) during antler growth. A total of 16,905 significantly changed transcripts were identified. Those sequences were mapped to 5,573 genes with 2,217 genes up-regulated and 3,356 genes down-regulated (60 days vs. 90 days), including ribosomal proteins, translation initiation and elongation factors, transcription factors, signaling molecules and extracellular matrix proteins. We also performed the gene ontology (GO) functional enrichment and pathway enrichment analysis of gene expression patterns with hypergeometric test and Bonferroni Correction. Both the two stages were enriched with members of GO categories and distinct pathways. Our data represent the most comprehensive sequence resource available for the deer antler and provide a basis for further research on deer antler molecular genetics and functional genomics.
URLPMID:32133026
[本文引用: 1]
[本文引用: 1]
DOI:10.1002/ar.10120URLPMID:12221718 [本文引用: 1]
The utilization of a deer antler model to study gene expression in tissues undergoing rapid growth has been hampered by an inability to sample the different tissue types. We report here a standardized procedure to identify different tissue types in growing antler tips and demonstrate that it can help in the classification of expressed sequence tags (ESTs). The procedure was developed using observable morphological markers within the unstained tissue at collection, and was validated by histological assessments and virtual Northern blotting. Four red deer antlers were collected at 60 days of growth and the tips (top 5 cm) were then removed. The following observable markers were identified distoproximally: the dermis (4.86 mm), the subdermal bulge (2.90 mm), the discrete columns (6.50 mm), the transition zone (a mixture of discrete and continuous columns) (3.22 mm), and the continuous columns (8.00 mm). The histological examination showed that these markers corresponded to the dermis, reserve mesenchyme, precartilage, transitional tissue from precartilage to cartilage, and cartilage, respectively. The gene expression studies revealed that these morphologically identified layers were functionally distinct tissue types and had distinct gene expression profiles. We believe that precisely defining these tissue types in growing antler tips will greatly facilitate new discoveries in this exciting field.
DOI:10.1186/s12864-019-5560-1URLPMID:30836939 [本文引用: 1]
BACKGROUND: With the unprecedented rapid growth rate (up to 2.75 cm/day), velvet antler is an invaluable model for the identification of potent growth factors and signaling networks for extremely fast growing tissues, mainly cartilage. Antler growth center (AGC) locates in its tip and consists of five tissue layers: reserve mesenchyme (RM), precartilage (PC), transition zone (TZ), cartilage (CA) and mineralized cartilage (MC). The aim of this study was to investigate the transcription dynamics in the AGC using RNA-seq technology. RESULTS: Five tissue layers in the AGC were collected from three 3-year-old male sika deer using our previously reported sampling method (morphologically distinguishable). After sequencing (15 samples; triplicates/tissue layer), we assembled a reference transcriptome de novo and used RNA-seq to measure gene expression profiles across these five layers. Nine differentially expressed genes (DEGs) were selected from our data and subsequently verified using qRT-PCR. The results showed a high consistency with the RNA-seq results (R(2) = 0.80). Nine modules were constructed based on co-expression network analysis, and these modules contained 370 hub genes. These genes were found to be mainly involved in mesenchymal progenitor cell proliferation, chondrogenesis, osteogenesis and angiogenesis. Combination of our own results with the previously published reports, we found that Wnt signaling likely plays a key role not only in stimulating the antler stem cells or their immediate progeny, but also in promoting chondrogenesis and osteogenesis during antler development. CONCLUSION: We have successfully assembled a reference transcriptome, generated gene expression profiling across the five tissue layers in the AGC, and identified nine co-expressed modules that contain 370 hub genes and genes predorminantly expressed in and highly relevant to each tissue layer. We believe our findings have laid the foundation for the identification of novel genes for rapid proliferation and chondrogenic differentiation of antler cells.
DOI:10.1186/s13287-018-1027-6URLPMID:30376879 [本文引用: 1]
BACKGROUND: Deer antlers are bony structures that re-grow at very high rates, making them an attractive model for studying rapid bone regeneration. METHODS: To identify the genes that are involved in this fast pace of bone growth, an in vitro RNA-seq model that paralleled the sharp differences in bone growth between deer antlers and humans was established. Subsequently, RNA-seq (> 60 million reads per library) was used to compare transcriptomic profiles. Uniquely expressed deer antler proliferation as well as mineralization genes were identified via a combination of differential gene expression and subtraction analysis. Thereafter, the physiological relevance as well as contributions of these identified genes were determined by immunofluorescence, gene overexpression, and gene knockdown studies. RESULTS: Cell characterization studies showed that in vitro-cultured deer antler-derived reserve mesenchyme (RM) cells exhibited high osteogenic capabilities and cell surface markers similar to in vivo counterparts. Under identical culture conditions, deer antler RM cells proliferated faster (8.6-11.7-fold increase in cell numbers) and exhibited increased osteogenic differentiation (17.4-fold increase in calcium mineralization) compared to human mesenchymal stem cells (hMSCs), paralleling in vivo conditions. Comparative RNA-seq identified 40 and 91 previously unknown and uniquely expressed fallow deer (FD) proliferation and mineralization genes, respectively, including uhrf1 and s100a10. Immunofluorescence studies showed that uhrf1 and s100a10 were expressed in regenerating deer antlers while gene overexpression and gene knockdown studies demonstrated the proliferation contributions of uhrf1 and mineralization capabilities of s100a10. CONCLUSION: Using a simple, in vitro comparative RNA-seq approach, novel genes pertinent to fast bony antler regeneration were identified and their proliferative/osteogenic function was verified via gene overexpression, knockdown, and immunostaining. This combinatorial approach may be applicable to discover unique gene contributions between any two organisms for a given phenomenon-of-interest.
DOI:10.1038/s41598-020-66383-1URLPMID:32528134 [本文引用: 1]
Long non-coding RNA (lncRNA) is a transcription product of the mammalian genome that regulates the development and growth in the body. The present study aimed to analyze the expression dynamics of lncRNA in sika antler mesenchymal and cartilage tissues by high-throughput sequencing. Bioinformatics was applied to predict differentially expressed lncRNAs and target genes and screen lncRNAs and mRNAs related to osteogenic differentiation, cell proliferation, and migration. Finally, the expression of the lncRNAs and target genes were analyzed by qRT-PCR. The results showed that compared to the cartilage tissue, the transcription levels of lncRNA and mRNA, 1212 lncRNAs and 518 mRNAs, in mesenchymal tissue were altered significantly. Thus, a complex interaction network was constructed, and the lncRNA-mRNA interaction network correlation related to osteogenic differentiation, cell proliferation, and migration was analyzed. Among these, the 26 lncRNAs and potential target genes were verified by qRT-PCR, and the results of qRT-PCR were consistent with high-throughput sequencing results. These data indicated that lncRNA promotes the differentiation of deer antler mesenchymal tissue into cartilage tissue by regulating the related osteogenic factors, cell proliferation, and migration-related genes and accelerating the process of deer antler regeneration and development.
DOI:10.1186/s11658-020-00234-9URLPMID:32944020 [本文引用: 1]
Background: Deer antlers have become a valuable model for biomedical research due to the capacities of regeneration and rapid growth. However, the molecular mechanism of rapid antler growth remains to be elucidated. The aim of the present study was to compare and explore the molecular control exerted by the main beam and brow tine during rapid antler growth. Methods: The main beams and brow tines of sika deer antlers were collected from Chinese sika deer (Cervus nippon) at the rapid growth stage. Comparative transcriptome analysis was conducted using RNA-Seq technology. Differential expression was assessed using the DEGseq package. Functional Gene Ontology (GO) enrichment analysis was accomplished using a rigorous algorithm according to the GO Term Finder tool, and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis was accomplished with the R function phyper, followed by the hypergeometric test and Bonferroni correction. Quantitative real-time polymerase chain reaction (qRT-PCR) was carried out to verify the RNA levels for differentially expressed mRNAs. Results: The expression levels of 16 differentially expressed genes (DEGs) involved in chondrogenesis and cartilage development were identified as significantly upregulated in the main beams, including transcription factor SOX-9 (Sox9), collagen alpha-1(II) chain (Col2a1), aggrecan core protein (Acan), etc. However, the expression levels of 17 DEGs involved in endochondral ossification and bone formation were identified as significantly upregulated in the brow tines, including collagen alpha-1(X) chain (Col10a1), osteopontin (Spp1) and bone sialoprotein 2 (Ibsp), etc. Conclusion: These results suggest that the antler main beam has stronger growth capacity involved in chondrogenesis and cartilage development compared to the brow tine during rapid antler growth, which is mainly achieved through regulation of Sox9 and its target genes, whereas the antler brow tine has stronger capacities of endochondral bone formation and resorption compared to the main beam during rapid antler growth, which is mainly achieved through the genes involved in regulating osteoblast and osteoclast activities. Thus, the current research has deeply expanded our understanding of the intrinsic molecular regulation displayed by the main beam and brow tine during rapid antler growth.
URLPMID:25046387 [本文引用: 1]
DOI:10.1016/j.bone.2019.115046URLPMID:31446115
Antlers are bony appendages of deer that undergo periodic regeneration from the top of permanent outgrowths (the pedicles) of the frontal bones. Of the
DOI:10.1080/03036758.1984.10426948URL [本文引用: 1]
URLPMID:13629476 [本文引用: 1]
[本文引用: 1]
DOI:10.1186/s13287-019-1457-9URLPMID:31744537 [本文引用: 1]
BACKGROUND: When the deer antler is cast, it leaves a cutaneous wound that can achieve scarless healing due to the presence of antler stem cells (ASCs). This provides an opportunity to study regenerative wound healing. METHODS: In this study, we investigated the therapeutic effects and mechanism of antler stem cell-conditioned medium (ASC-CM) on cutaneous wound healing in rats. In vitro, we investigated the effects of the ASC-CM on proliferation of HUVEC and NIH-3T3 cell lines. In vivo, we evaluated the effects of ASC-CM on cutaneous wound healing using full-thickness skin punch-cut wounds in rats. RESULTS: The results showed that ASC-CM significantly stimulated proliferation of the HUVEC and NIH-3T3 cells in vitro. In vivo, completion of healing of the rat wounds treated with ASC-CM was on day 16 (+/- 3 days), 9 days (+/- 2 days) earlier than the control group (DMEM); the area of the wounds treated with ASC-CM was significantly smaller (p < 0.05) than the two control groups. Further molecular characterization showed that the ratios of Col3A1/Col1A2, TGF-beta3/TGF-beta1, MMP1/TIMP1, and MMP3/TIMP1 significantly increased (p < 0.01) in the healed tissue in the ASC-CM group. CONCLUSIONS: In conclusion, ASC-CM effectively accelerated the wound closure rate and enhanced the quality of healing, which might be through transforming wound dermal fibroblasts into the fetal counterparts. Therefore, the ASC-CM may have potential to be developed as a novel cell-free therapeutic for scarless wound healing.
DOI:10.1038/d41586-018-07430-wURLPMID:30464288 [本文引用: 1]
DOI:10.1002/ar.1091620401URLPMID:5701619 [本文引用: 1]
DOI:10.1002/ar.1091620402URLPMID:5701620 [本文引用: 1]
DOI:10.1007/s00438-010-0565-0URLPMID:20697743 [本文引用: 1]
Antlers of deer display the fastest and most robust bone development in the animal kingdom. Deposition of the minerals in the cartilage preceding ossification is a specific feature of the developing antler. We have cloned 28 genes which are upregulated in the cartilaginous section (called mineralized cartilage) of the developing (
DOI:10.1016/j.theriogenology.2020.06.042URLPMID:32947063 [本文引用: 1]
An alarming number of large mammalian species with low reproduction rates is threatened with extinction. As basic knowledge of reproductive physiology is currently lacking in many species, increasing the understanding of reproductive physiology is imperative and includes the development of novel artificial reproduction technologies. Despite the relatively comprehensive knowledge on molecular mechanisms underlying reproduction in livestock species such as cattle, pregnancy failures are likewise far from understood. Contrary to other wildlife species, the European roe deer (Capreolus capreolus) displays a remarkably high pregnancy rate. In parts, cattle and roe deer exhibit comparable features of preimplantation embryo development. Therefore, understanding the high fertility rate in the roe deer holds a great potential for cross-species knowledge gain. As the only known species among the artiodactylae, the roe deer displays a long period of embryonic diapause. The preimplantation blastocyst reaches a diameter of 1 mm only at around 4 months compared to around 13 days post estrus in cattle. The expanded blastocyst survives in a uterine microenvironment that contains a unique set of yet unidentified factors that allow embryonic stem cells to proliferate at low pace without impairing their developmental potential. Upon reactivation, intimate embryo-maternal communication comparable to those reported in cattle is thought to occur. In this review, current knowledge, parallels and differences of reproductive physiology in cattle and roe deer are reviewed. The roe deer is proposed as a unique model species to (1) enhance our knowledge of fertility processes, (2) define factors that support embryo survival for an extended period, (3) advance knowledge on embryonic stem cells, and (4) unravel potential implications for the development of novel strategies for artificial reproductive technologies.
DOI:10.1111/ahe.12315URLPMID:28960412 [本文引用: 1]
The embryonic stage of development is defined as the period between fertilization and the establishment of most of the organ systems by the end of this period. Development in this stage is rapid. In many mammalian species, particularly in humans, the interval between fertilization and implantation is exactly determined and continuous without intermission. However, European roe deer (Capreolus capreolus) embryos undergo a reversible retardation of development. This interesting reproduction strategy is called embryonic diapause (delayed implantation). After this period of embryonic arrest, development continues without further interruption. The aim of this study was to investigate embryonic development after diapause in European roe deer. Because of the embryonic diapause and the unknown date of fertilization, it was impossible to assign the embryos to a certain gestational age (days). This study describes normal stages of embryonic development mainly based on the external morphological traits of 56 well-preserved post-implantation roe deer embryos and attempts to assign the embryos to certain development stages. Carnegie stages of human embryos were used as an orientation for staging roe deer embryos. We observed a considerable range of variation of embryonic stages investigated until the end of January. We found post-implantation stages of embryonic development already at the end of December and foetuses at the end of January. Moreover, assigning the embryos to a particular stage of development allows the comparison between pairs of twins and triplets. We showed that twins and triplets were always at the same development level, despite the discrepancy in inter-twin and inter-triplet size.
DOI:10.1016/j.repbio.2019.04.003URLPMID:31147267 [本文引用: 1]
Embryonic diapause in the European roe deer includes a period of five months from August to December in which embryonic development is extremely decelerated. Following exit from diapause, the embryo rapidly elongates and subsequently implants. In diapausing carnivores and marsupials, resumption of embryonic growth is regulated by ovarian steroid hormones. In the roe deer, the role of steroid hormones is not known to date. In the present study, progesterone (P4), estradiol-17beta (E2) and total estrogens (Etot) were determined in blood plasma and endometrium of roe deer shot in the course of regular huntings between September and December. Steroid hormone concentrations were correlated to the corresponding size of the embryo derived from ex vivo uterine flushing and to the date of sampling. The mean plasma concentrations of P4 (5.4 +/- 0.2 ng/ml, mean +/- SE, N = 87), E2 (24.3 +/- 2.6 pg/ml, N = 86) and Etot (21.7 +/- 2.6 pg/ml, N = 78) remained constant over the sampling period and were not correlated to embryonic size. Likewise, endometrial concentrations of P4 (66.1 +/- 6.5 ng/g), E2 (284.0 +/- 24.43 pg/g) and, Etot (440.9 +/- 24.43 pg/g) showed no changes over time [corrected]. Therefore, it was concluded that ovarian steroid hormones do not play a determining role in resumption of embryonic growth following the period of diapause in the roe deer.
DOI:10.1111/asj.13289URLPMID:31486226 [本文引用: 1]
The aim was to evaluate in female roe deer: (a) PAG mRNA relative abundance in endometrial uterine tissue for determination of the duration of embryonic diapause, (b) mRNA relative abundance of progesterone, estradiol, and prolactin (P4, E2, and PRL) receptors (PGR, ESR, and PRLR) during diapause and after implantation in the endometrium; (c) concentration of P4, E2, and PRL in the blood, and (d) a noninvasive method of hormone detection by measurement of P4 and E2 concentrations in feces. A total of fifteen individuals were obtained post mortem during hunting seasons and divided into three experimental groups (November, December, January). The results did not reveal mRNA relative abundance for PAGs in the endometrium or detectable PAG concentrations in the serum of all examined females. Concentration of PRL and mRNA relative abundance for PRLR long isoform in the endometrium was the highest in January (p < .01). mRNA relative abundance for PGR, P4 concentration in the endometrium, serum, and feces was the highest in January (p < .01). Endometrial origin PRL and P4 may be responsible for the termination of this process and pregnancy development after implantation.
DOI:10.1093/biolre/ioz129URLPMID:31317179 [本文引用: 1]
Numerous intrauterine changes take place across species during embryo development. Following fertilization in July/August, the European roe deer (Capreolus capreolus) embryo undergoes diapause until embryonic elongation in December/January. Embryonic elongation prior to implantation is a common feature among ungulates. Unlike many other ruminants, the roe deer embryo does not secrete interferon-tau (IFNtau). This provides the unique opportunity to unravel IFNtau-independent signaling pathways associated with maternal recognition of pregnancy (MRP). This study aimed at identifying the cell-type-specific endometrial gene expression changes associated with the MRP at the time of embryo elongation that are independent of IFNtau in roe deer. The messenger RNA (mRNA) expression of genes known to be involved in embryo-maternal communication in cattle, pig, sheep, and mice was analyzed in laser capture microdissected (LMD) endometrial luminal, glandular epithelial, as well as stromal cells. The mRNA transcript abundances of the estrogen (ESR1), progesterone receptor (PGR), and IFNtau-stimulated genes were lower in the luminal epithelium in the presence of an elongated embryo compared to diapause. Retinol Binding Protein-4 (RBP4), a key factor involved in placentation, was more abundant in the luminal epithelium in the presence of an elongated embryo. The progesterone receptor localization was visualized by immunohistochemistry, showing an absence in the luminal epithelium and an overall lower abundance with time and thus prolonged progesterone exposure. Our data show a developmental stage-specific mRNA expression pattern in the luminal epithelium, indicating that these cells sense the presence of an elongated embryo in an IFNtau-independent manner.

{kind=link}
{kind=link}
{kind=link}
{kind=link}