 ,1
,1Population genomics study of Vibrio alginolyticus
Hongyuan Zheng1, Lin Yan2, Chao Yang1,3, Yarong Wu1, Jingliang Qin1, Tongyu Hao1, Dajin Yang2, Yunchang Guo2, Xiaoyan Pei2, Tongyan Zhao1, Yujun Cui,1通讯作者: 崔玉军,博士,研究员,研究方向:微生物进化与基因组流行病学。E-mail:cuiyujun.new@gmail.com
编委: 高海春
收稿日期:2021-02-9修回日期:2021-03-16网络出版日期:2021-04-16
| 基金资助: |
Received:2021-02-9Revised:2021-03-16Online:2021-04-16
| Fund supported: |
作者简介 About authors
郑宏源,在读硕士研究生,专业方向:细菌基因组流行病学。E-mail:

摘要
溶藻弧菌(Vibrio alginolyticus)是一种能够对人类以及鱼、虾、贝类等水产品致病的弧菌,给人类健康带来威胁,也给水产养殖业造成巨大的经济损失。目前该物种基于全基因组的遗传多样性和重要遗传元件研究报道较少。本研究对采集自全国4个省份的68株溶藻弧菌进行高通量测序,获得全基因组序列,并结合113株公开发表的全球序列数据,利用fineSTRUCTURE软件、VFDB毒力因子库和CARD、ResFinder耐药数据库,对溶藻弧菌的种群结构和毒力、耐药因子分别进行解析。结果表明:溶藻弧菌可分为谱系1和谱系2。两个谱系在美洲和亚洲均有分布,但欧洲仅分离到谱系1菌株;共鉴定发现12个克隆群,其中一个克隆群内菌株存在跨洋传播现象。该物种携带tlh、OmpU、IlpA等多种不同功能的毒力因子;毒力因子在两个谱系间的分布无特异性,但存在地域间差异:其中欧洲菌株携带VP1611、vcrD、vopD和fleR/flrC的比率低于其他地区,而基因IlpA的携带率则明显高于其他地区,我国广西菌株中fleR/flrC基因携带率低于其他省份,且不携带IlpA。多个基因组携带blaCARB-42、tet(34)、tet(35)、parE、CRP、rsmA、TxR和fos等与多种抗生素耐受相关的基因,其中TxR和fos基因在谱系2中的出现频率远高于谱系1;此外,TxR基因在亚洲菌株中的携带率高于美洲和欧洲地区,而在我国四川菌株中的携带率则低于其他省份。在5个基因组中(VA24、VA28、2014V-1011、ZJ-T和Vb1833)观察到质粒或ICE等携带多种耐药基因的大片段。本研究通过群体基因组学的研究方法,揭示了溶藻弧菌的种群结构组成和毒力、耐药相关元件的分布,为进一步了解溶藻弧菌的遗传特征和致病机制提供必要基础,为该病原的监测、预防和控制工作提供科学支撑。
关键词:
Abstract
Vibrio alginolyticus is a Gram-negative bacillus that causes vibriosis to human and aquatic products, including fish, shrimp and shellfish. It poses a threat to public health and causes enormous economic losses to the aquaculture industry. However, research on genetic diversity and pathogenicity-related genetic elements based on whole genome is still lacking. In this study, sixty-eight strains of V. alginolyticus were collected from four provinces of China and the whole genome sequences were obtained. Combined with 113 publicly available genome sequences downloaded from NCBI, we inferred the population structure of V. alginolyticus by using fineSTRUCTURE software, and identified the virulence and antibiotic resistance factors using the VFDB, CARD and ResFinder database. The results indicated that V. alginolyticus included two main lineages, named Lineage 1 and Lineage 2. Both lineages distributed in America and Asia, but all the European genomes were classified into Lineage 1. A single cross-ocean transmission event was inferred from one of the 12 identified clonal groups in our dataset. V. alginolyticus genome contains a variety of virulence factors, such as tlh, OmpU, and IlpA, etc. The distribution of virulence factors revealed no lineage-specificity, but some of which revealed differences in their geographical distribution. A lower frequency of VP1611, vcrD, vopD, fleR/flrC and a higher frequency of IlpA were observed in genomes of Europe than other continents. In China, a lower frequency of fleR/flrC, and no IlpA were observed in genomes from Guangxi province. Among the identified antibiotic resistance genes, TxR and fos are significantly enriched in Lineage 2. In addition, TxR is more common in genomes from Asia, compared with the American and European genomes. But in China, the frequency of TxR in Sichuan genomes is much lower than in other provinces. We also found that large fragments of plasmids or ICEs that carried multiple drug resistance genes were present in five V. alginolyticus genomes (VA24, VA28, 2014V-1011, ZJ-T and Vb1833). Based on population genomics analysis, our study delineated the population structure, distribution of virulence and antibiotic resistance related factors of V. alginolyticus, which lays a foundation for future study of genetic characters and pathogenesis mechanism of this pathogen and will improve the works on monitoring, prevention and control of this pathogen.
Keywords:
PDF (1171KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郑宏源, 闫琳, 杨超, 武雅蓉, 秦婧靓, 郝彤宇, 杨大进, 郭云昌, 裴晓燕, 赵彤言, 崔玉军. 溶藻弧菌群体基因组学研究. 遗传[J], 2021, 43(4): 350-361 doi:10.16288/j.yczz.21-061
Hongyuan Zheng.
溶藻弧菌(Vibrio alginolyticus),是一种广泛存在于海洋和河口,可以从病人体内、海水、水产品等多种不同环境中分离得到的革兰氏阴性杆菌[1,2]。溶藻弧菌感染人体后可以引起皮肤感染、外耳道感染等多种疾病,但与霍乱弧菌(Vibrio cholerae)和副溶血弧菌(Vibrio parahaemolyticus)等食源性弧菌不同,临床上报导的溶藻弧菌感染病例均有海水暴露史,因此其主要感染途径是与海水的接触[3,4]。此外,溶藻弧菌可引起鱼、虾、贝类等水产品患病与大量死亡,带来巨大的经济损失[2,5,6]。美国COVIS (Cholera and Other Vibrio Illness Surveillance)和FoodNet (Foodborne Diseases Active Surveillance Network)两个监测数据库的数据表明:自2007年以来,溶藻弧菌已经跃升为第二常见的致病弧菌[7,8],为公共卫生及渔业经济带来巨大压力。深入研究溶藻弧菌遗传多样性和病原相关特征,将有助于该病原的精准检测和致病机制研究,对其监测和防控工作具有重要意义。
多年来,已有多种传统分子生物学方法应用于溶藻弧菌分型研究,例如核糖体分型、随机扩增多态性DNA (random amplified polymorphic DNA, RAPD)、肠杆菌基因间重复一致序列PCR (enterobacterial repetitive intergenic consensus PCR, ERIC- PCR)、低频限制性位点PCR (infrequent restriction site PCR, IRS-PCR)和脉冲场凝胶电泳(pulsed field gel electrophoresis, PFGE)等[9,10,11,12]。这些方法在溯源性研究等方面发挥了一定的作用,具有操作简便、获得结果较快等优势,但缺点是分辨率较低,实验结果不稳定,而且对实验条件要求较高。PFGE曾被认为是暴发溯源的金标准[13,14],但是这种分型方法需要严苛的实验条件,实验过程中操作导致的细微差别则可能得到不同的分型结果,不利于实验结果在不同实验室间的交流。目前尚无多位点可变数量串联重复序列分析(multilocus variable-number tandem repeat analysis, MLVA)和多位点序列分型(multilocus sequence typing, MLST)等多靶标分子分型方法应用于溶藻弧菌的研究报导,而基于全基因组层面的遗传多样性研究和种群结构鉴定的研究报道较少。另一方面,当前对于溶藻弧菌的毒力和耐药相关基因的研究,是利用已知其他弧菌物种的毒力和耐药基因特异性引物,通过PCR方法在部分菌株中检验相应基因是否存在。研究表明溶藻弧菌携带了一些副溶血弧菌等物种常见毒力因子,也表现出多种临床常见抗生素的耐受性,同时鉴定到部分菌株携带有耐药基因[1,15~21]。然而,溶藻弧菌全基因组层面毒力和耐药相关遗传因子分布的认识仍不够深入。
全基因组测序(whole genome sequencing, WGS)在细菌遗传多样性研究中有着最高的分辨率,而且已广泛运用在微生物溯源、传播及种群结构鉴定等各个方面[22,23,24,25]。本研究通过全基因组测序,利用fineSTRUCTURE方法[26]鉴定了溶藻弧菌的种群结构,并在此基础上研究了毒力、耐药相关因子及其在物种内不同菌株间的分布。
1 材料与方法
1.1 菌株来源
本研究共收集181株溶藻弧菌的全基因组序列,包括68株新测序的序列和113株公共基因组序列。新测序的菌株采样于国内四个省份(山东、湖北、四川、广西)的水产品、海水、水底沉积物等不同环境,采样时间均为2014年。公共序列下载于NCBI数据库(National Center for Biotechnology Information Search database,1.2 全基因组测序
使用细菌基因组DNA提取试剂盒(QIAGEN UltraClean? Microbial DNA Isolation kit),参照试剂盒说明书提取全基因组DNA。经检测质量合格后送测序公司(华大基因,深圳)进行细菌全基因组测序,测序平台为Illumina NovaSeq 6000,测序文库大小为350 bp,测序reads为双端150 bp,平均测序深度134X (94~188X)。1.3 质控及组装
使用软件Trimmomatic v0.38[27]将测序结果中低质量的数据进行过滤,然后利用软件shovill (1.4 变异检测和基因预测
单核苷酸多态性(single nucleotide polymorphism, SNP)位点的鉴定参照我们已发表的方法[28],以溶藻弧菌FA2(GCA_011801435.1)基因组完成图为参考序列,使用MUMmer软件包[29]将181株溶藻弧菌基因组序列分别比对到参考菌株基因组,获得核心基因组(所有菌株中均能找到对应碱基状态)SNP矩阵。为保证SNP鉴定结果的可靠性,位于重复区和测序质量值低的SNP 未纳入后续分析。采用Prokka软件[30]对各个菌株基因组序列进行基因预测,使用Roary软件[31]判断基因获得缺失。1.5 系统发育分析
使用TreeBest软件(1.6 种群结构鉴定
首先将SNP矩阵转换为FASTA格式,然后计算两两菌株间的SNP距离,为减小克隆结构的干扰,以10,000个SNP为阈值去除克隆信号,并从每个克隆群中随机选取一株菌作为该群的代表株,经过筛选最终得到129个代表株。将129株菌的SNP信息输入种群结构分析软件fineSTRUCTURE[26],设定默认参数,计算菌株间共祖率(coancestry),得到共祖率矩阵,再利用马尔可夫链蒙特卡罗(Markov Chain Monte Carlo, MCMC)算法实施矩阵的聚类分析,进而推断种群结构。1.7 毒力因子鉴定和耐药基因预测
使用BLASTn将所有菌株基因组数据与VFDB (Virulence Factor Database)[33]数据库核心库(setA,仅包含经试验验证过的毒力基因)进行比对,鉴定本研究中数据集的毒力因子;利用RGI v5.1.1[34]和ResFinder[35]两种软件分别基于CARD (Comprehensive Antibiotic Resistance Database,2 结果与分析
2.1 溶藻弧菌基因组特点
本研究在我国4个省份(山东、湖北、四川、广西)共分离了68株溶藻弧菌,加上公共数据113株,共181株菌基因组数据纳入分析。新测序的68株溶藻弧菌基因组序列,经过滤后组装,平均基因组大小为5.1 Mb (4.9~5.4 Mb),平均GC含量为44.6% (44.4%~44.9%)。整个数据集首先进行了泛基因组分析,共得到22,439个基因,其中核心基因2995个,附属基因19,444个。根据图1可以推测,随着菌株数量的增加,整个物种的泛基因数量呈上升趋势,而核心基因组则趋于稳定,说明整个物种的附属基因在逐渐增加,即溶藻弧菌的基因组是“开放”的,可以通过水平基因转移等方式不断获得新的基因。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1溶藻弧菌基因数量与基因组数量的关系
横轴为纳入分析的基因组数量,纵轴为基于数据集计算出的基因总数与核心基因数量。
Fig. 1Relationship between number of genes and number of genomes in V. alginolyticus
2.2 溶藻弧菌种群结构
通过变异检测,181株溶藻弧菌共鉴定出335,733个SNP。根据鉴定到的SNP,我们构建了整个数据集的系统发育树(图2A),结果显示溶藻弧菌的系统发育树和副溶血弧菌[24,37]等物种类似,呈现“轮盘状”结构(图2A),说明菌株间水平转移信号已将垂直遗传信号打乱,该物种属于高频重组弧菌。使用fineSTRUCTURE软件对溶藻弧菌的种群结构做进一步解析,最终将溶藻弧菌分成两个主要谱系(Lineage):谱系1和2 (图2B)。从图中可发现,在对角线上的少量种群中仍呈现明显的克隆信号,在我们已发表的相关研究中[24]提到,可通过进一步去除克隆群菌株以移除克隆信号干扰。本研究由于菌株数量相对较少,且进一步去除克隆信号后得到的结果仍分为同样两个谱系,故只展示第一次去除克隆信号的结果。图2
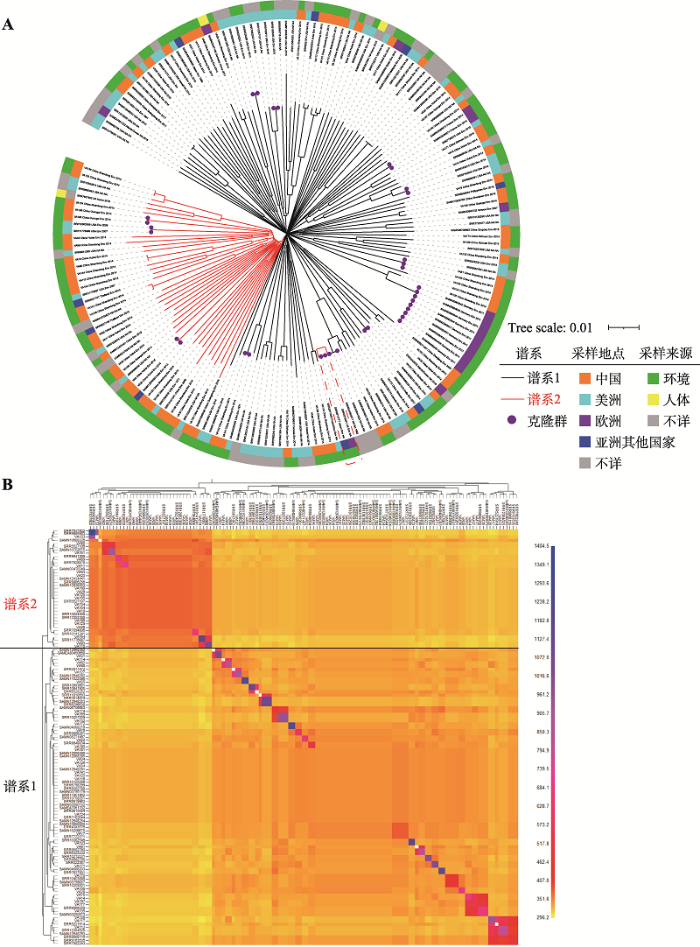 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2溶藻弧菌系统发育树与种群结构
A:181株菌基于核心基因组SNP构建的系统发育树。黑色、红色树枝分别代表谱系1和谱系2,树枝顶部紫色圆点代表鉴定到的12个克隆群(clonal groups, CGs),右下角红色虚线框代表的是发生跨洋传播的两株菌,内环代表不同的采样地点,外环代表不同采样来源。B:129株代表菌株基于fineSTRUCTURE软件的种群鉴定结果。列代表供体菌株(donor),行代表受体菌株(recipient),方格颜色表示供体向受体导入的序列片段数量,黑色线代表种群间的界线。
Fig. 2Phylogenic tree and population structure of V. alginolyticus
分析结果(表1)显示:谱系1中的美洲和中国菌株占比相当,而谱系2的中国菌株占比较高;谱系1菌株与谱系2菌株相比分布更为广泛,欧洲来源的17株菌仅出现在谱系1,提示谱系1在全球的扩散程度更高。
Table 1
表1
表1不同谱系中菌株分离地点统计
Table 1
| 中国 | 美洲 | 亚洲其他国家 | 欧洲 | 其他 | 总数 | |
|---|---|---|---|---|---|---|
| 谱系1 | 48*(35%#) | 58*(42%#) | 6*(4%#) | 17*(12%#) | 8*(6%#) | 137 |
| 谱系2 | 28*(64%#) | 10*(23%#) | 3*(7%#) | 0* | 3*(7%#) | 44 |
新窗口打开|下载CSV
参考我们已发表的研究方法[25],以SNP距离小于10为界鉴定出了近期传播或暴发的12个克隆群(CGs,图2A),共涉及34株菌。仅在其中一个克隆群中(图2A中虚线框),发现来自不同国家的菌株。相关菌株分别来自英国和日本(分离时间不详),这两株菌基因组间 SNP遗传距离为0,表明溶藻弧菌在上述两个国家之间发生了跨地域传播。背景信息显示这两株菌分别分离自蛤类和鱼类,提示跨国传播可能是由于水产品国际贸易导致的。本研究中溶藻弧菌跨洋传播现象的确定对我们将来的防治思路和策略有指导意义。
Table 2
表2
表2溶藻弧菌毒力因子及相关功能
Table 2
| 相关毒力基因 | 毒力因子编号 | 毒力因子名称 | 毒力因子功能 |
|---|---|---|---|
| flgD(100%) flgC(100%) flgB(100%) fliM(100%) fliG(100%) cheR(99%) cheW(99%) cheY(99%) flgF(99%) cheV(94%) flgE(84%) flgG(49%) fleR/flrC(45%) flhA(1%) | VF0519 | Flagella | Adhesin factor |
| vcrH(100%) vopB(99%) vscI(99%) exsD(99%) exsA(99%) vopQ(99%) vxsC(98%) vscD(98%) tyeA(97%) vscO(97%) vscU(90%) vcrD(86%) vscQ(82%) vopD(76%) vscN(2%) vscC(1%) | VF0408 | T3SS1 | Rapid apoptosis, Cell rounding, Osmotic lysis |
| tlh(100%) | TX170 | Thermolabile_hemolysin | Apoptosis, Membrane vesiculation, Necrosis |
| OmpU(98%) | VF0514 | OmpU | Porin, Iron balance, Vaccine candidate |
| VP1611(87%) | VF0512 | MAM7 | Multivalent adhesion molecule MAM7 |
| IlpA(26%) | VF0513 | IlpA | Immunogenic lipoprotein A |
| htpB(9%) | VF0159 | Hsp60 | Heat shock protein HtpB |
| ugd(1%) | VF0560 | Capsule | UDP-glucose 6-dehydrogenase |
| hcp-2(1%) | SS181 | VAS | T6SS substrate Hcp-2 |
新窗口打开|下载CSV
2.3 毒力因子的鉴定及分布
181个基因组中共鉴定出9类37个毒力因子(表2),其中鞭毛(flagella)与三型分泌系统(T3SS)相关基因最多,分别为14个和16个,占毒力因子总数的81%。鞭毛作为一种附着因子(adhesin factor),可介导溶藻弧菌在宿主表面的粘附;T3SS最初是在耶尔森氏菌属(Yersinia spp.)中发现的[38],而弧菌属(Vibrio spp.)中最早发现T3SS是在副溶血弧菌(血清型O3:K6, strain RIMD2210633)的两条染色体上,包含两套T3SS系统,分别命名为T3SS1和T3SS2[39],其中T3SS1在所有副溶血弧菌基因组中都存在,主要功能是诱导细胞自噬、细胞圆化和细胞裂解[40,41]。此外,我们还发现所有的溶藻弧菌(100%)都携带溶血素基因tlh。tlh是5类典型溶血素家族(TDH、HlyA、TLH、δ-VPH、HLX)[42]中的一种,实验表明这种基因在宿主中介导了凋亡、膜溶解、坏死等多种毒性表现[42,43]。98%的菌株携带膜外蛋白基因OmpU,这是溶藻弧菌的主要孔蛋白,也是一种重要的毒力因子,有研究已经证实OmpU在溶藻弧菌的铁代谢平衡中起到了重要作用,而且也是溶藻弧菌重要的疫苗研发靶标[44,45]。我们还分别在87%、26%和9%的溶藻弧菌中发现携带多价粘附分子基因VP1611、免疫原性脂蛋白基因IlpA、和热休克蛋白基因htpB。另有个别菌株表达荚膜基因ugd和T6SS亚单位hcp-2。在鉴定出的37个毒力因子中,其中23个在大于90%的菌株中携带, 8个在10%~90%的菌株中携带,其余6个仅在少数(<10%)菌株中携带。统计这8个分布差异性较大的毒力因子在不同谱系中、不同国家和地区间、中国不同省份的分布情况(表3),结合毒力因子在系统发育树上的分布(图3),未发现谱系1或谱系2的群特异性毒力因子;欧洲菌株中VP1611、vcrD、vopD和fleR/flrC的携带率低于其他国家和地区,而基因IlpA的携带率较高;除中国外的亚洲其他地区的菌株均不携带IlpA基因;此外,我国广西的菌株fleR/flrC基因的携带率低于其他省份,且均不携带IlpA基因。因本研究中的菌株采样量较小,相关遗传因子的地域分布特征仍需将来通过扩大菌株集进行验证。
Table 3
表3
表3部分毒力因子在不同谱系、不同国家和地区以及中国不同地点的分布统计
Table 3
| VP1611 (%) | vcrD (%) | flgE (%) | vscQ (%) | vopD (%) | flgG (%) | fleR/flrC (%) | IlpA (%) | |
|---|---|---|---|---|---|---|---|---|
| 谱系1 | 85 | 82 | 89 | 77 | 77 | 45 | 45 | 29 |
| 谱系2 | 93 | 95 | 68 | 98 | 73 | 61 | 43 | 16 |
| 中国 | 92 | 89 | 87 | 89 | 78 | 53 | 53 | 20 |
| 美洲 | 93 | 91 | 88 | 79 | 81 | 47 | 44 | 29 |
| 亚洲其他国家 | 89 | 89 | 56 | 78 | 89 | 44 | 44 | 0 |
| 欧洲 | 47 | 41 | 88 | 88 | 35 | 65 | 18 | 65 |
| 山东 | 90 | 90 | 77 | 95 | 74 | 56 | 62 | 21 |
| 湖北 | 100 | 100 | 94 | 76 | 82 | 47 | 41 | 24 |
| 四川 | 71 | 86 | 100 | 86 | 100 | 43 | 86 | 14 |
| 广西 | 100 | 80 | 100 | 100 | 60 | 40 | 20 | 0 |
新窗口打开|下载CSV
2.4 耐药基因的预测及分布
RGI和ResFinder均鉴定到32个耐药基因,经同源比对、去除冗余后汇总为31个耐药基因,分别介导溶藻弧菌对β内酰胺酶类、喹诺酮类、四环素类等多种常见抗生素的耐受(表4)。我们发现几乎所有菌株(>95%)普遍携带的耐药基因有:介导β内酰胺酶类耐受的blaCARB-42,介导四环素耐受的tet(34)和tet(35),介导喹诺酮类耐受的parE,以及调控耐药的基因CRP、rsmA。此外,我们发现介导四环素耐受的基因TxR在两个谱系中的分布存在显著差异(图3),谱系2和谱系1的菌株携带这种基因的比率分别为100%和12%。TxR是一种转录调节因子,与四环素耐药相关,而且另一个四环素耐药基因tet(35)耐药功能的正常发挥,依赖于TxR。我们还在9株溶藻弧菌中首次发现了一种和磷霉素耐受相关的基因fos,有趣的是其中8株菌(88.9%)属于谱系2。此外,经统计发现,TxR基因在中国和亚洲其他地区的菌株中的携带率(分别为45%和44%)高于美洲和欧洲菌株的携带率(分别为24%和6%);而我国四川菌株中该基因的携带率(14%)低于其他省(广西:60%,山东:49%,湖北:47%)。与毒力因子研究类似,对相关耐药因子的分布研究需要后期优化采样策略、扩大采样范围和样本量后进一步验证。
图3
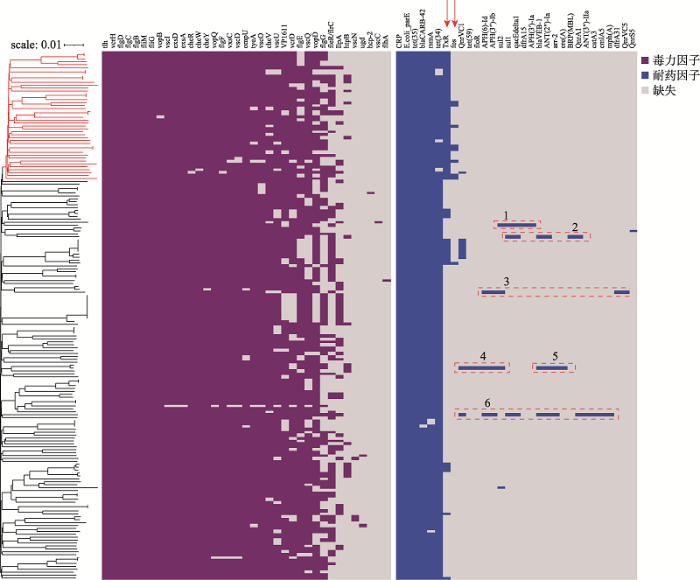 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3181株溶藻弧菌毒力与耐药因子分布热图
紫色代表携带毒力因子,蓝色代表携带耐药因子,灰色代表缺失。虚线框代
Fig. 3Heatmap of virulence and antibiotic resistance factors in V. alginolyticus
图3中,虚线框1代表的是菌株2014V-1011 (SAMN12648291)里的少数耐药基因,其中4个(APH(3')-Ia、sul1、qacEdelta1、dfrA15)位于该菌株一个尚未命名的质粒(CP046773.1)上。虚线框2和6中的基因分别代表新测序菌株VA28和VA24上的少数耐药基因,这些基因落在了2~3个组装片段上(contig),因此基于现有数据无法确认其在基因组上的位置是否相邻,但其中的sul1与qacEdelta1两个基因是Ⅰ类整合子(class 1 integron)3’端保守区域(conserved segments, 3’CS)的两个固有基因[46,47],另外虚线框1中的质粒片段上也有这两个基因,因此我们推断虚线框2和6中的少数耐药基因可能位于通过水平基因转移(horizontal gene transfer, HGT)而获得的Ⅰ类整合子片段或质粒片段上,这需要后期通过基因组完成图测序和分析进行确定。虚线框3中的基因位于菌株ZJ-T(SAMN05271497)染色体1上的相邻位置,是一个连续的大片段,我们截取这一段序列并在线BLAST比对后发现,该片段与溶藻弧菌的整合共轭元件(integrative conjugative elements, ICEs) ICEVal056-1 (accession number: KR231688.1),coverage和identity值分别为70%和99.98%,表明该菌株可能通过水平转移获得了一个携带多种耐药基因的大片段。虚线框4和5中的基因是位于菌株Vb1833 (SAMN15829766)染色体1上的两个大片段,我们分别截取了这两个片段在线BLAST后发现,比对结果中多为ICE片段或者质粒片段,这也提示菌株Vb1833 (SAMN15829766)在进化过程中,通过水平转移的方式获得了携带多种耐药基因的大片段。本研究在溶藻弧菌中发现了携带多种耐药基因的大片段,为今后溶藻弧菌所致疾病的治疗和防控提供了重要信息。
Table 4
表4
表4溶藻弧菌耐药基因与相关抗生素和耐药机制
Table 4
| 耐药基因 | 耐受抗生素 | 耐药机制 |
|---|---|---|
| blaCARB-42(99%) blaVEB-1(1%) | Beta-lactam resistance | Antibiotic inactivation |
| E.coli_parE(100%) QnrVC1(6%) QnrA1(1%) QnrS5(1%) QnrVC5(1%) | Quinolone resistance | Antibiotic target alteration (parE) Antibiotic target protection (qnr) |
| tet(35)(100%) tet(34)(98%) TxR(34%) tet(59)(1%) | Tetracycline resistance | Antibiotic efflux |
| ANT(2'')-Ia(2%) APH(3'')-Ib(2%) APH(6)-Id(2%) ANT(3'')-IIa(1%) APH(3')-Ia(1%) | Aminoglycoside antibiotic | Antibiotic inactivation |
| fos(5%) | Fosfomycin resistance | Antibiotic inactivation |
| sul1(2%) sul2(2%) | Sulfonamide antibiotic | Antibiotic target replacement |
| cmlA5(1%) floR(1%) catA3(1%) | Phenicol antibiotic | Antibiotic efflux (cmlA5, floR) Antibiotic inactivation (catA3) |
| Ere(A)(1%) mph(A)(1%) | Macrolide antibiotic | Antibiotic inactivation |
| arr-2(1%) | Rifamycin antibiotic | Antibiotic inactivation |
| dfrA15(1%) dfrA31(1%) | Trimethoprim resistant | Antibiotic target replacement |
| BRP(MBL)(1%) | Glycopeptide antibiotic | Antibiotic inactivation |
| CRP(100%) rsmA(98%) | ARG_regulator | Antibiotic efflux |
| qacEdelta1(2%) | Quaternary ammonium compounds | Antibiotic efflux |
新窗口打开|下载CSV
3 讨论
过去研究发现,副溶血弧菌的种群结构与不同海域密切相关[24]。但本研究现有数据集下,溶藻弧菌不同采样地点的菌株在整个系统发育树和不同种群间未呈现地域性聚集,而是呈现出“分散分布”,即:同一采样地点的菌株(如中国、美洲等)分散于两个不同的谱系中。根据群体遗传学理论,种群分化只在种群间迁徙率很低的情况下发生(每世代中只有一个迁徙或者更少)[24],当少量迁徙者(migrant)与当地种群共存时,会逐渐与当地种群交换遗传物质,进而变得与当地种群越来越相似。本研究中种群已经形成,但两个种群(谱系1和谱系2)中还是包含了不同地域来源的菌株,可能是迁徙者到达新环境后,尚未充分与当地种群交换遗传物质,依然保留着大量“家乡”种群的遗传特征。造成该现象的原因包括:跨地域迁徙发生在近期,进化时间相对较短;或者自身遗传特征限制了遗传物质在不同种群的菌株间进行交换;或者是不同种群溶藻弧菌生活在同一地区的不同微生境下,导致事实上的生态条件隔离。将来可通过开展生态调查和实验室研究以检验上述假说。本研究中,无论是从NCBI下载的公共数据,还是新测序的中国菌株,在采样地点上都存在一定的偏差。例如公共数据中,美国菌株占60%,而其他国家或大洲的菌株则相对较少,新测序的中国菌株也仅采集自中国的山东、湖北、四川和广西4个省份,其他省份数据缺失。采样偏差可能影响本研究中的某些结果。例如研究结果显示,同一谱系中(谱系1或谱系2)包含了不同大洲的菌株,提示历史上曾存在多次远距离迁徙事件,但本研究中我们仅在一个克隆群中观察到跨洋传播,可能是由于短时期迁徙事件频率低,或者是研究数据集不够完整所致。此外,种群结构研究中,发现欧洲菌株只存在于谱系1中;毒力、耐药因子分布研究中,发现某些毒力、耐药因子(VP1611、vcrD、vopD、fleR/flrC、TxR等)在不同的大洲以及中国不同的省份之间的分布存在差异。这些结果需要在将来通过完善采样策略、扩大采样范围、增加数据量等方式消除采样偏差造成的影响并进一步验证。
虽然本研究中鉴定到多种不同功能的毒力因子,但是除3株菌采集自病人外,其余均为环境菌株,因而难以将该病原的毒力因子与菌株致病性表型进行关联。此外,虽然本研究中鉴定到多种耐药相关基因,但部分鉴定结果可能为耐药基因的同源体(homology),而是否携带耐药基因同源体与菌株的临床耐药表型并不一定具有相关性,因为部分病原菌在临床上表现出对抗生素的耐受,是由染色体上的具体的基因突变导致的,包括点突变、基因片段获得缺失(Indel)等。另一方面,一个基因的表达与否还受到多种因素的调控,因此携带特定耐药基因(或同源体)的菌株是否表现出耐药表型,还要结合实验测定结果进一步研究。
综上所述,本研究利用全基因组测序技术和群体遗传学方法,对181株溶藻弧菌的遗传多样性和毒力、耐药特征进行了研究。研究结果表明溶藻弧菌是一种高频重组致病弧菌,由两个主要谱系组成,其中谱系1菌株的地域分布更为广泛。两个谱系菌株的地理分布特点和克隆群分析均表明该物种存在近期跨洋传播,但种群跨地域迁徙后尚未观察到种群融合。溶藻弧菌基因组包含多种不同功能的毒力因子,主要与溶藻弧菌自身代谢、对宿主细胞的粘附、裂解、免疫反应等功能相关,两个谱系中均未发现种群特异性毒力因子。此外,溶藻弧菌基因组既有多种核心耐药基因,也存在携带多种耐药基因的质粒或者ICE等移动遗传元件。本研究存在采样偏差以及缺乏表型实验结果等不足,但上述结果为更深入研究溶藻弧菌遗传特征和致病机制提供了基础数据,将加深对该病原的认识并促进其防控工作。此外,本研究中的研究思路和方法,也可供霍乱弧菌、河流弧菌(Vibrio fluvialis)等其他高频重组致病菌的群体基因组学研究参考,通过揭示相关病原菌的遗传多样性、致病因子、传播模式等特征,在溯源、防控和治疗等方面提供科学支撑并发挥积极作用。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1007/s11274-012-1147-6URLPMID:22918722 [本文引用: 2]

The antibiotic resistance patterns and the plasmids profiles of the predominant etiological agent responsible for vibriosis in Tunisia, V. alginolyticus were studied to contribute to control their spread in some Mediterranean aquaculture farms and seawater. The sixty-nine V. alginolyticus strains isolated from different marine Tunisian biotopes (bathing waters, aquaculture and conchylicole farms and a river connected to the seawater during the cold seasons) were multi-drug resistant with high resistance rate to ampicillin, kanamycin, doxycyclin, erythromycin, imipinem, and nalidixic acid. The multiple resistance index ranged from 0.3 to 0.7 for the isolates of Khenis, from 0.5 to 0.8 for those of Menzel Jmil, from 0.5 to 0.75 (Hergla) and from 0.3 to 0.7 for the isolates of Oued Soltane. The high value of antibiotic resistance index was recorded for the V. alginolyticus population isolated from the fish farm in Hergla (ARI = 0.672) followed by the population isolated from the conchylicole station of Menzel Jmil (ARI = 0.645). The results obtained by the MIC tests confirmed the resistance of the V. alginolyticus to ampicillin, erythromycin, kanamycin, cefotaxime, streptomycin and trimethoprim. Plasmids were found in 79.48 % of the strains analyzed and 30 different plasmid profiles were observed. The strains had a high difference in the size of plasmids varying between 0.5 and 45 kb. Our study reveals that the antibiotic-resistant bacteria are widespread in the aquaculture and conchylicole farm relatively to others strains isolated from seawater.
[本文引用: 2]
URLPMID:22027377 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1128/AEM.71.1.98-104.2005URLPMID:15640176 [本文引用: 1]
Two episodes of mortality of cultured carpet shell clams (Ruditapes decussatus) associated with bacterial infections were recorded during 2001 and 2002 in a commercial hatchery located in Spain. Vibrio alginolyticus was isolated as the primary organism from moribund clam larvae that were obtained during the two separate events. Vibrio splendidus biovar II, in addition to V. alginolyticus, was isolated as a result of a mixed Vibrio infection from moribund clam larvae obtained from the second mortality event. The larval mortality rates for these events were 62 and 73%, respectively. Mortality was also detected in spat. To our knowledge, this is the fist time that these bacterial species have been associated with larval and juvenile carpet shell clam mortality. The bacterial strains were identified by morphological and biochemical techniques and also by PCR and sequencing of a conserved region of the 16S rRNA gene. In both cases bacteria isolated in pure culture were inoculated into spat of carpet shell clams by intravalvar injection and by immersion. The mortality was attributed to the inoculated strains, since the bacteria were obtained in pure culture from the soft tissues of experimentally infected clams. V. alginolyticus TA15 and V. splendidus biovar II strain TA2 caused similar histological lesions that affected mainly the mantle, the velum, and the connective tissue of infected organisms. The general enzymatic activity of both live cells and extracellular products (ECPs), as evaluated by the API ZYM system, revealed that whole bacterial cells showed greater enzymatic activity than ECPs and that the activity of most enzymes ceased after heat treatment (100 degrees C for 10 min). Both strain TA15 and strain TA2 produced hydroxamate siderophores, although the activity was greater in strain TA15. ECPs from both bacterial species at high concentrations, as well as viable bacteria, caused significant reductions in hemocyte survival after 4 h of incubation, whereas no significant differences in viability were observed during incubation with heat-killed bacteria.
DOI:10.1128/AEM.01548-06URLPMID:17056701 [本文引用: 1]
A Vibrio strain isolated from Alaskan oysters and classified by its biochemical characteristics as Vibrio alginolyticus possessed a thermostable direct hemolysin-related hemolysin (trh) gene previously reported only in Vibrio parahaemolyticus. This trh-like gene was cloned and sequenced and was 98% identical to the trh2 gene of V. parahaemolyticus. This gene seems to be functional since it was transcriptionally active in early-stationary-phase growing cells. To our knowledge, this is the first report of V. alginolyticus possessing a trh gene.
DOI:10.1093/cid/cis243URLPMID:22572659 [本文引用: 1]
BACKGROUND: The Centers for Disease Control and Prevention monitors vibriosis through 2 surveillance systems: the nationwide Cholera and Other Vibrio Illness Surveillance (COVIS) system and the 10-state Foodborne Diseases Active Surveillance Network (FoodNet). COVIS conducts passive surveillance and FoodNet conducts active surveillance for laboratory-confirmed Vibrio infections. METHODS: We summarized Vibrio infections (excluding toxigenic V. cholerae O1 and O139) reported to COVIS and FoodNet from 1996 through 2010. For each system, we calculated incidence rates using US Census Bureau population estimates for the surveillance area. RESULTS: From 1996 to 2010, 7700 cases of vibriosis were reported to COVIS and 1519 to FoodNet. Annual incidence of reported vibriosis per 100,000 population increased from 1996 to 2010 in both systems, from 0.09 to 0.28 in COVIS and from 0.15 to 0.42 in FoodNet. The 3 commonly reported Vibrio species were V. parahaemolyticus, V. vulnificus, and V. alginolyticus; both surveillance systems showed that the incidence of each increased. In both systems, most hospitalizations and deaths were caused by V. vulnificus infection, and most patients were white men. The number of cases peaked in the summer months. CONCLUSIONS: Surveillance data from both COVIS and FoodNet indicate that the incidence of vibriosis increased from 1996 to 2010 overall and for each of the 3 most commonly reported species. Epidemiologic patterns were similar in both systems. Current prevention efforts have failed to prevent increasing rates of vibriosis; more effective efforts will be needed to decrease rates.
URLPMID:28202099 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:18586455 [本文引用: 1]
DOI:10.1016/s0195-6701(96)90055-9URLPMID:8666764 [本文引用: 1]
We assessed the discriminatory power of pulsed-field gel electrophoresis (PFGE) for the analysis of DNA restriction fragment length polymorphism (RFLP) in Pseudomonas aeruginosa. We determined DraI PFGE-RFLP of DNA of unrelated clinical and environmental strains, and clinical strains isolated from two intensive care units of the Besancon University Hospital. The typeability and reproducibility was 100%. The discriminatory index was 0.998, and the DNA patterns were stable in vitro and in vivo. There was a very low correlation between PFGE-RFLP and traditional typing methods. The typeability, reproducibility, the high discriminatory power and the stability of PFGE-RFLP make this a valuable method to be used in conjunction with serotyping. Further standardization and quantitative interpretation are possible and should lead to this technique becoming a library typing system.
DOI:10.1017/s0950268899002964URLPMID:10694154 [本文引用: 1]
A cluster of acute melioidosis cases occurred in a remote, coastal community in tropical Western Australia. Molecular typing of Burkholderia pseudomallei isolates from culture-confirmed cases and suspected environmental sources by pulsed-field gel electrophoresis (PFGE) of XbaI chromosomal DNA digests showed that a single PFGE type was responsible for five cases of acute infection in a community of around 300 during a 5 week period. This temporal and geographical clustering of acute melioidosis cases provided a unique opportunity to investigate the environmental factors contributing to this disease. B. pseudomallei isolated from a domestic tap at the home of an asymptomatic seroconverter was indistinguishable by PFGE. Possible contributing environmental factors included an unusually acid communal water supply, unrecordable chlorine levels during the probable exposure period, a nearby earth tremor, and gusting winds during the installation of new water and electricity supplies. The possible role of the potable water supply as a source of B. pseudomallei was investigated further.
URLPMID:16033522 [本文引用: 1]
DOI:10.2436/20.1501.01.277URLPMID:28504816
Vibrio alginolyticus has acquired increasing importance because this microorganism may be pathogenic to aquatic animals and humans. It has been reported that some V. alginolyticus strains carry virulence genes derived from pathogenic V. cholerae and V. parahaemolyticus strains. In this work V. alginolyticus was isolated from oyster samples acquired from a food-market in Mexico City. Thirty isolates were identified as V. alginolitycus. Strains showed beta-haemolysis and proteolytic activity and produced a capsule. Strains displayed swimming and swarming motility and 93.3% of them produced siderophores. Several genes encoding virulence factors were detected using PCR amplification. These included proA, wza, vopD, vopB, hcp, vasH and vgrG genes, which were present in all strains. Other genes had a variable representation: tdh (86.6%), lafA (96.6%), pvsA (62%) and pvuA (16%). The trh gene could not be amplified from any of the strains. The antimicrobial resistance profile revealed that more than 90% of the strains were resistant to beta-lactams antibiotics, 60% to cephalotin, 45% to amikacin, 16% to cephotaxime, and 10% to pefloxacin, while 100% were susceptible to ceftriaxone. The V. alginolyticus strains isolated from oysters showed multiple resistance to antibiotics and several virulence factors described in well-characterized pathogenic vibrios. [Int Microbiol 19(4):191-198 (2016)].
URLPMID:24459831
In this study, a total of 54 Vibrio alginolyticus strains were analyzed. The isolates were recovered from different compartments of the Ruditapes decussatus hatchery in the National Institute of Marine Sciences and Technologies, Monastir, Tunisia. All isolates were biochemically identified (API 20E and API ZYM strips), characterized by amplification of the Hsp-40 gene polymerase chain reaction (PCR) and analyzed by enterobacterial repetitive intergenic consensus (ERIC)-based genotyping to evaluate genetic relationship between the isolated strains. We also looked for the presence of ten V cholera virulence genes (toxRS, toxR, toxT toxS, tcpP, tcpA, ace, vpi, zot and ctxA) in the genomes of Vibrio isolates. The antibiotics susceptibility, exoenzymes production and in vitro cytotoxic activitiy against HeLa cell line were also carried out for all tested bacteria. Most of V alginolyticus isolates showed significant antimicrobial resistance rates to at least ten antibacterial agents. For most isolates, the minimum inhibitory concentration (MIC) data showed that tetracydclin and streptomycin were the most effective antibiotics. Construction of the phylogenetic dendogram showed that studied isolates were in general genetically heterogeneous; however some Vibrio strains were present in different structures of the R. decussatus hatchery. The V cholerae virulence genes investigation showed a wild distribution of toxS (49/54), toxaR (45/54) and toxT (22/54) genes among V alginolyticus strains isolated from the R. decussatus rearing system. Cytotoxic effects of several Vibrio extracellular products (28154) were also observed on HeLa cells.
URLPMID:28923608
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nmicrobiol.2015.8URLPMID:26877885 [本文引用: 1]
Burkholderia pseudomallei, a highly pathogenic bacterium that causes melioidosis, is commonly found in soil in Southeast Asia and Northern Australia(1,2). Melioidosis can be difficult to diagnose due to its diverse clinical manifestations and the inadequacy of conventional bacterial identification methods(3). The bacterium is intrinsically resistant to a wide range of antimicrobials, and treatment with ineffective antimicrobials may result in case fatality rates (CFRs) exceeding 70%(4,5). The importation of infected animals has, in the past, spread melioidosis to non-endemic areas(6,7). The global distribution of B. pseudomallei and burden of melioidosis, however, remain poorly understood. Here, we map documented human and animal cases, and the presence of environmental B. pseudomallei, and combine this in a formal modelling framework(8-10) to estimate the global burden of melioidosis. We estimate there to be 165,000 (95% credible interval 68,000-412,000) human melioidosis cases per year worldwide, of which 89,000 (36,000-227,000) die. Our estimates suggest that melioidosis is severely underreported in the 45 countries in which it is known to be endemic and that melioidosis is likely endemic in a further 34 countries which have never reported the disease. The large numbers of estimated cases and fatalities emphasise that the disease warrants renewed attention from public health officials and policy makers.
DOI:10.1038/nmicrobiol.2016.263URLPMID:28112723 [本文引用: 1]
The environmental bacterium Burkholderia pseudomallei causes an estimated 165,000 cases of human melioidosis per year worldwide and is also classified as a biothreat agent. We used whole genome sequences of 469 B. pseudomallei isolates from 30 countries collected over 79 years to explore its geographic transmission. Our data point to Australia as an early reservoir, with transmission to Southeast Asia followed by onward transmission to South Asia and East Asia. Repeated reintroductions were observed within the Malay Peninsula and between countries bordered by the Mekong River. Our data support an African origin of the Central and South American isolates with introduction of B. pseudomallei into the Americas between 1650 and 1850, providing a temporal link with the slave trade. We also identified geographically distinct genes/variants in Australasian or Southeast Asian isolates alone, with virulence-associated genes being among those over-represented. This provides a potential explanation for clinical manifestations of melioidosis that are geographically restricted.
URLPMID:31235840 [本文引用: 5]
DOI:10.1016/j.fm.2019.103270URLPMID:31421783 [本文引用: 2]
Vibrio parahaemolyticus is the leading bacterial cause of seafood-associated gastroenteritis worldwide. Moreover, infections and outbreaks caused by V. parahaemolyticus has kept increasing over the last two decades. In this study, we investigated the genetic diversity, virulence factors and farm-to-table spread pattern of V. parahaemolyticus by analyzing 383 genomes of food-associated isolates. These strains were isolated from diverse sample types from six provinces of China in 2014, being classified into three tiers of the farm-to-table spread process: food production, circulation and consumption. The genetic diversity of V. parahaemolyticus in different classifications, including geographical location, sample type, source and spread tier, was similar, as the median number of pairwise SNPs within each classification was between 33,013 and 33,659. Specifically, there was no clear boundaries in genetic diversity of the isolates from inland vs. coastal provinces, as well as of those from freshwater vs. seawater products. Moreover, the virulence genes and genomic islands were only found in a small number of isolates, indicating a low disease risk of the food-associated isolates in this study. By further exploring 28 recently emerged clonal groups, we identified seven farm-to-table spread events, showing a common pattern of single-source radial spread accompanied with occasional gene gain/loss events. Generally speaking, our work highlighted the colonization of V. parahaemolyticus in inland provinces and freshwater environment, and provided a snapshot of the farm-to-table spread pattern of V. parahaemolyticus food-associated isolates. Our results showed the feasibility of tracking the farm-to-table spread of foodborne pathogen, which would help construct the whole genome sequencing-based molecular tracking network in the future.
URLPMID:22291602 [本文引用: 2]
DOI:10.1093/bioinformatics/btu170URLPMID:24695404 [本文引用: 1]
MOTIVATION: Although many next-generation sequencing (NGS) read preprocessing tools already existed, we could not find any tool or combination of tools that met our requirements in terms of flexibility, correct handling of paired-end data and high performance. We have developed Trimmomatic as a more flexible and efficient preprocessing tool, which could correctly handle paired-end data. RESULTS: The value of NGS read preprocessing is demonstrated for both reference-based and reference-free tasks. Trimmomatic is shown to produce output that is at least competitive with, and in many cases superior to, that produced by other tools, in all scenarios tested. AVAILABILITY AND IMPLEMENTATION: Trimmomatic is licensed under GPL V3. It is cross-platform (Java 1.5+ required) and available at http://www.usadellab.org/cms/index.php?page=trimmomatic CONTACT: usadel@bio1.rwth-aachen.de SUPPLEMENTARY INFORMATION: Supplementary data are available at Bioinformatics online.
URLPMID:23271803 [本文引用: 1]
DOI:10.1002/cpbi.86URLPMID:31756036 [本文引用: 1]
MetaboAnalyst (https://www.metaboanalyst.ca) is an easy-to-use web-based tool suite for comprehensive metabolomic data analysis, interpretation, and integration with other omics data. Since its first release in 2009, MetaboAnalyst has evolved significantly to meet the ever-expanding bioinformatics demands from the rapidly growing metabolomics community. In addition to providing a variety of data processing and normalization procedures, MetaboAnalyst supports a wide array of functions for statistical, functional, as well as data visualization tasks. Some of the most widely used approaches include PCA (principal component analysis), PLS-DA (partial least squares discriminant analysis), clustering analysis and visualization, MSEA (metabolite set enrichment analysis), MetPA (metabolic pathway analysis), biomarker selection via ROC (receiver operating characteristic) curve analysis, as well as time series and power analysis. The current version of MetaboAnalyst (4.0) features a complete overhaul of the user interface and significantly expanded underlying knowledge bases (compound database, pathway libraries, and metabolite sets). Three new modules have been added to support pathway activity prediction directly from mass peaks, biomarker meta-analysis, and network-based multi-omics data integration. To enable more transparent and reproducible analysis of metabolomic data, we have released a companion R package (MetaboAnalystR) to complement the web-based application. This article provides an overview of the main functional modules and the general workflow of MetaboAnalyst 4.0, followed by 12 detailed protocols: (c) 2019 by John Wiley & Sons, Inc. Basic Protocol 1: Data uploading, processing, and normalization Basic Protocol 2: Identification of significant variables Basic Protocol 3: Multivariate exploratory data analysis Basic Protocol 4: Functional interpretation of metabolomic data Basic Protocol 5: Biomarker analysis based on receiver operating characteristic (ROC) curves Basic Protocol 6: Time-series and two-factor data analysis Basic Protocol 7: Sample size estimation and power analysis Basic Protocol 8: Joint pathway analysis Basic Protocol 9: MS peaks to pathway activities Basic Protocol 10: Biomarker meta-analysis Basic Protocol 11: Knowledge-based network exploration of multi-omics data Basic Protocol 12: MetaboAnalystR introduction.
DOI:10.1093/bioinformatics/btu153URLPMID:24642063 [本文引用: 1]
UNLABELLED: The multiplex capability and high yield of current day DNA-sequencing instruments has made bacterial whole genome sequencing a routine affair. The subsequent de novo assembly of reads into contigs has been well addressed. The final step of annotating all relevant genomic features on those contigs can be achieved slowly using existing web- and email-based systems, but these are not applicable for sensitive data or integrating into computational pipelines. Here we introduce Prokka, a command line software tool to fully annotate a draft bacterial genome in about 10 min on a typical desktop computer. It produces standards-compliant output files for further analysis or viewing in genome browsers. AVAILABILITY AND IMPLEMENTATION: Prokka is implemented in Perl and is freely available under an open source GPLv2 license from http://vicbioinformatics.com/.
DOI:10.1093/bioinformatics/btv421URLPMID:26198102 [本文引用: 1]
UNLABELLED: A typical prokaryote population sequencing study can now consist of hundreds or thousands of isolates. Interrogating these datasets can provide detailed insights into the genetic structure of prokaryotic genomes. We introduce Roary, a tool that rapidly builds large-scale pan genomes, identifying the core and accessory genes. Roary makes construction of the pan genome of thousands of prokaryote samples possible on a standard desktop without compromising on the accuracy of results. Using a single CPU Roary can produce a pan genome consisting of 1000 isolates in 4.5 hours using 13 GB of RAM, with further speedups possible using multiple processors. AVAILABILITY AND IMPLEMENTATION: Roary is implemented in Perl and is freely available under an open source GPLv3 license from http://sanger-pathogens.github.io/Roary CONTACT: roary@sanger.ac.uk SUPPLEMENTARY INFORMATION: Supplementary data are available at Bioinformatics online.
DOI:10.1093/nar/gkw290URLPMID:27095192 [本文引用: 1]
Interactive Tree Of Life (http://itol.embl.de) is a web-based tool for the display, manipulation and annotation of phylogenetic trees. It is freely available and open to everyone. The current version was completely redesigned and rewritten, utilizing current web technologies for speedy and streamlined processing. Numerous new features were introduced and several new data types are now supported. Trees with up to 100,000 leaves can now be efficiently displayed. Full interactive control over precise positioning of various annotation features and an unlimited number of datasets allow the easy creation of complex tree visualizations. iTOL 3 is the first tool which supports direct visualization of the recently proposed phylogenetic placements format. Finally, iTOL's account system has been redesigned to simplify the management of trees in user-defined workspaces and projects, as it is heavily used and currently handles already more than 500,000 trees from more than 10,000 individual users.
DOI:10.1093/nar/gky1080URLPMID:30395255 [本文引用: 1]
The virulence factor database (VFDB, http://www.mgc.ac.cn/VFs/) is devoted to providing the scientific community with a comprehensive warehouse and online platform for deciphering bacterial pathogenesis. The various combinations, organizations and expressions of virulence factors (VFs) are responsible for the diverse clinical symptoms of pathogen infections. Currently, whole-genome sequencing is widely used to decode potential novel or variant pathogens both in emergent outbreaks and in routine clinical practice. However, the efficient characterization of pathogenomic compositions remains a challenge for microbiologists or physicians with limited bioinformatics skills. Therefore, we introduced to VFDB an integrated and automatic pipeline, VFanalyzer, to systematically identify known/potential VFs in complete/draft bacterial genomes. VFanalyzer first constructs orthologous groups within the query genome and preanalyzed reference genomes from VFDB to avoid potential false positives due to paralogs. Then, it conducts iterative and exhaustive sequence similarity searches among the hierarchical prebuilt datasets of VFDB to accurately identify potential untypical/strain-specific VFs. Finally, via a context-based data refinement process for VFs encoded by gene clusters, VFanalyzer can achieve relatively high specificity and sensitivity without manual curation. In addition, a thoroughly optimized interactive web interface is introduced to present VFanalyzer reports in comparative pathogenomic style for easy online analysis.
DOI:10.1093/nar/gkz935URLPMID:31665441 [本文引用: 1]
The Comprehensive Antibiotic Resistance Database (CARD; https://card.mcmaster.ca) is a curated resource providing reference DNA and protein sequences, detection models and bioinformatics tools on the molecular basis of bacterial antimicrobial resistance (AMR). CARD focuses on providing high-quality reference data and molecular sequences within a controlled vocabulary, the Antibiotic Resistance Ontology (ARO), designed by the CARD biocuration team to integrate with software development efforts for resistome analysis and prediction, such as CARD's Resistance Gene Identifier (RGI) software. Since 2017, CARD has expanded through extensive curation of reference sequences, revision of the ontological structure, curation of over 500 new AMR detection models, development of a new classification paradigm and expansion of analytical tools. Most notably, a new Resistomes & Variants module provides analysis and statistical summary of in silico predicted resistance variants from 82 pathogens and over 100 000 genomes. By adding these resistance variants to CARD, we are able to summarize predicted resistance using the information included in CARD, identify trends in AMR mobility and determine previously undescribed and novel resistance variants. Here, we describe updates and recent expansions to CARD and its biocuration process, including new resources for community biocuration of AMR molecular reference data.
DOI:10.1093/jac/dkaa345URLPMID:32780112 [本文引用: 1]
OBJECTIVES: WGS-based antimicrobial susceptibility testing (AST) is as reliable as phenotypic AST for several antimicrobial/bacterial species combinations. However, routine use of WGS-based AST is hindered by the need for bioinformatics skills and knowledge of antimicrobial resistance (AMR) determinants to operate the vast majority of tools developed to date. By leveraging on ResFinder and PointFinder, two freely accessible tools that can also assist users without bioinformatics skills, we aimed at increasing their speed and providing an easily interpretable antibiogram as output. METHODS: The ResFinder code was re-written to process raw reads and use Kmer-based alignment. The existing ResFinder and PointFinder databases were revised and expanded. Additional databases were developed including a genotype-to-phenotype key associating each AMR determinant with a phenotype at the antimicrobial compound level, and species-specific panels for in silico antibiograms. ResFinder 4.0 was validated using Escherichia coli (n = 584), Salmonella spp. (n = 1081), Campylobacter jejuni (n = 239), Enterococcus faecium (n = 106), Enterococcus faecalis (n = 50) and Staphylococcus aureus (n = 163) exhibiting different AST profiles, and from different human and animal sources and geographical origins. RESULTS: Genotype-phenotype concordance was >/=95% for 46/51 and 25/32 of the antimicrobial/species combinations evaluated for Gram-negative and Gram-positive bacteria, respectively. When genotype-phenotype concordance was <95%, discrepancies were mainly linked to criteria for interpretation of phenotypic tests and suboptimal sequence quality, and not to ResFinder 4.0 performance. CONCLUSIONS: WGS-based AST using ResFinder 4.0 provides in silico antibiograms as reliable as those obtained by phenotypic AST at least for the bacterial species/antimicrobial agents of major public health relevance considered.
DOI:10.1371/journal.pntd.0008046URLPMID:32069325 [本文引用: 1]
Non-toxigenic Vibrio cholerae isolates have been found associated with diarrheal disease globally, however, the global picture of non-toxigenic infections is largely unknown. Among non-toxigenic V. cholerae, ctxAB negative, tcpA positive (CNTP) isolates have the highest risk of disease. From 2001 to 2012, 71 infectious diarrhea cases were reported in Hangzhou, China, caused by CNTP serogroup O1 isolates. We sequenced 119 V. cholerae genomes isolated from patients, carriers and the environment in Hangzhou between 2001 and 2012, and compared them with 850 publicly available global isolates. We found that CNTP isolates from Hangzhou belonged to two distinctive lineages, named L3b and L9. Both lineages caused disease over a long time period with usually mild or moderate clinical symptoms. Within Hangzhou, the spread route of the L3b lineage was apparently from rural to urban areas, with aquatic food products being the most likely medium. Both lineages had been previously reported as causing local endemic disease in Latin America, but here we show that global spread of them has occurred, with the most likely origin of L3b lineage being in Central Asia. The L3b lineage has spread to China on at least three occasions. Other spread events, including from China to Thailand and to Latin America were also observed. We fill the missing links in the global spread of the two non-toxigenic serogroup O1 V. cholerae lineages that can cause human infection. The results are important for the design of future disease control strategies: surveillance of V. cholerae should not be limited to ctxAB positive strains.
DOI:10.7554/eLife.54136URLPMID:32195663 [本文引用: 1]
Investigating fitness interactions in natural populations remains a considerable challenge. We take advantage of the unique population structure of Vibrio parahaemolyticus, a bacterial pathogen of humans and shrimp, to perform a genome-wide screen for coadapted genetic elements. We identified 90 interaction groups (IGs) involving 1,560 coding genes. 82 IGs are between accessory genes, many of which have functions related to carbohydrate transport and metabolism. Only 8 involve both core and accessory genomes. The largest includes 1,540 SNPs in 82 genes and 338 accessory genome elements, many involved in lateral flagella and cell wall biogenesis. The interactions have a complex hierarchical structure encoding at least four distinct ecological strategies. One strategy involves a divergent profile in multiple genome regions, while the others involve fewer genes and are more plastic. Our results imply that most genetic alliances are ephemeral but that increasingly complex strategies can evolve and eventually cause speciation.
DOI:10.1080/21505594.2017.1414134URLPMID:29252102 [本文引用: 1]
Vibrio alginolyticus is a Gram-negative bacterium that is an opportunistic pathogen of both marine animals and people. Its pathogenesis likely involves type III secretion system (T3SS) mediated induction of rapid apoptosis, cell rounding and osmotic lysis of infected eukaryotic cells. Herein, we report that effector proteins, Val1686 and Val1680 from V. alginolyticus, were responsible for T3SS-mediated death of fish cells. Val1686 is a Fic-domain containing protein that not only contributed to cell rounding by inhibiting Rho guanosine triphosphatases (GTPases), but was requisite for the induction of apoptosis because the deletion mutant (Deltaval1686) was severely weakened in its ability to induce cell rounding and apoptosis in fish cells. In addition, Val1686 alone was sufficient to induce cell rounding and apoptosis as evidenced by the transfection of Val1686 into fish cells. Importantly, the Fic-domain essential for cell rounding activity was equally important to activation of apoptosis of fish cells, indicating that apoptosis is a downstream event of Val1686-dependent GTPase inhibition. V. alginolyticus infection likely activates JNK and ERK pathways with sequential activation of caspases (caspase-8/-10, -9 and -3) and subsequent apoptosis. Val1680 contributed to T3SS-dependent lysis of fish cells in V. alginolyticus, but did not induce autophagy as has been reported for its homologue (VopQ) in V. parahaemolyticus. Together, Val1686 and Val1680 work together to induce apoptosis, cell rounding and cell lysis of V. alginolyticus-infected fish cells. These findings provide new insights into the mechanism of cell death caused by T3SS of V. alginolyticus.
DOI:10.1016/S0140-6736(03)12659-1URLPMID:12620739 [本文引用: 1]
BACKGROUND: Vibrio parahaemolyticus, a gram-negative marine bacterium, is a worldwide cause of food-borne gastroenteritis. V parahaemolyticus strains of a few specific serotypes, probably derived from a common clonal ancestor, have lately caused a pandemic of gastroenteritis. The organism is phylogenetically close to V cholerae, the causative agent of cholera. METHODS: The whole genome sequence of a clinical V parahaemolyticus strain RIMD2210633 was established by shotgun sequencing. The coding sequences were identified by use of Gambler and Glimmer programs. Comparative analysis with the V cholerae genome was undertaken with MUMmer. FINDINGS: The genome consisted of two circular chromosomes of 3288558 bp and 1877212 bp; it contained 4832 genes. Comparison of the V parahaemolyticus genome with that of V cholerae showed many rearrangements within and between the two chromosomes. Genes for the type III secretion system (TTSS) were identified in the genome of V parahaemolyticus; V cholerae does not have these genes. INTERPRETATION: The TTSS is a central virulence factor of diarrhoea-causing bacteria such as shigella, salmonella, and enteropathogenic Escherichia coli, which cause gastroenteritis by invading or intimately interacting with intestinal epithelial cells. Our results suggest that V parahaemolyticus and V cholerae use distinct mechanisms to establish infection. This finding explains clinical features of V parahaemolyticus infections, which commonly include inflammatory diarrhoea and in some cases systemic manifestations such as septicaemia, distinct from those of V cholerae infections, which are generally associated with non-inflammatory diarrhoea.
[本文引用: 1]
[本文引用: 1]
DOI:10.3354/dao02225URLPMID:20662368 [本文引用: 2]
Hemolysin is a putative pathogenicity factor in many bacterial pathogens. In this study, a DNA fragment containing the open reading frame (1254 bp) of the thermolabile hemolysin gene (tlh) from Vibrio alginolyticus V05 was amplified and cloned into the expression plasmid pET-24d(+). The deduced amino acid sequence of the thermolabile hemolysin (TLH) shared 94 and 83% identity with the lecithin-dependent hemolysin (LDH)/TLH of V. parahaemolyticus and V. harveyi thermolabile hemolysin (VHH), respectively. The sequence analysis also indicated that it contained a GDSL lipase domain like VHH. The recombinant protein with a predicted molecular mass of 47.2 kDa was expressed in the Escherichia coli strain BL21 (DE3) as a His-tag fused protein. TLH purified by the nickel-nitrilotriacetic acid (Ni-NTA) His-Bind Resin method showed phospholipase activity on an egg yolk emulsion plate and hemolytic activity against flounder erythrocytes with a specific activity of 18 hemolytic units microg(-1). The addition of divalent cations at different concentrations decreased hemolytic activity of the purified TLH, but monavalent cations did not affect hemolytic activity. The hemolytic activity of TLH was also markedly inhibited by protein modification reagents, i.e. beta-mercaptoethanol, phenylmethylsulfonyl fluoride, and 5,5'-dithio-bis(2-nitrobenzoic acid). Moreover, TLH was toxic to zebrafish when injected intraperitoneally, with a median lethal dose (LD50) of 0.8 microg protein g(-1) fish. This work shows that TLH could potentially be developed as a vaccine and used as a diagnostic tool for vibriosis.
[本文引用: 1]
DOI:10.3354/dao02206URLPMID:20597431 [本文引用: 1]
The outer membrane proteins (OMPs) of the marine aquatic animal pathogen Vibrio alginolyticus play an important role in the virulence of the bacterium and are potential candidates for vaccine development. In this study, the major 35.6 kDa OMP of V. alginolyticus was isolated by gel excision from the crude OMP fraction from V. alginolyticus. The sequence of the first 27 amino acid residues from the N-terminal end of the protein is ATV YKD GGT ELL VGG RVE FRG DFI GSD, which has high homology with OmpU proteins from other Vibrio spp. (92%). Lutjanus erythropterus were vaccinated with OmpU, and immunogenicity was confirmed by subsequent western blotting. Enzyme-linked immunosorbent assay (ELISA) analysis demonstrated that OmpU produced an observable antibody response in all sera of the vaccinated fish. L. erythropterus vaccinated with OmpU produced specific antibodies, and were highly resistant to infection with virulent V. alginolyticus. These results indicate that OmpU is an effective vaccine candidate against V. alginolyticus for L. erythropterus.
DOI:10.1016/j.micres.2019.126350URLPMID:31629270 [本文引用: 1]
Outer membrane protein U (OmpU) is a major porin from Vibrio alginolyticus and has been considered a vaccine candidate against infection by V. alginolyticus. After pre-incubated with polyclonal antibody against rOmpU, V. alginolyticus showed a 78% decrease in extracellular iron level, suggesting that interruption of OmpU could increase intracellular iron level. The mRNA expression of ompU under iron-limited conditions was determined using real-time reverse transcriptase PCR. The mRNA level of ompU was downregulated to 0.27-, 0.036- and 0.019-fold after the addition of the iron chelator 2,2'-bipyridyl for 10, 30 and 60 min, respectively. In addition, the promoter of ompU contained a ferric uptake regulator (Fur) binding site, which revealed the potential regulation of ompU by Fur and iron. Fur from V. alginolyticus was purified and used for electrophoretic mobility shift assay. The result showed that in the absence of Fe(2+), purified recombinant Fur could specifically bind to the promoter DNA of ompU, while in the presence of Fe(2+), the binding of Fur and the promoter DNA was suppressed. Our study preliminarily explored the function of OmpU in iron balance in V. alginolyticus, and these findings were helpful in understanding iron metabolism in V. alginolyticus.
DOI:10.1146/annurev-genet-102209-163504URLPMID:20707672 [本文引用: 1]
Integrons are genetic elements able to acquire and rearrange open reading frames (ORFs) embedded in gene cassette units and convert them to functional genes by ensuring their correct expression. They were originally identified as a mechanism used by Gram-negative bacteria to collect antibiotic resistance genes and express multiple resistance phenotypes in synergy with transposons. More recently, their role has been broadened with the discovery of chromosomal integron (CI) structures in the genomes of hundreds of bacterial species. This review focuses on the resources carried in these elements, on their unique recombination mechanisms, and on the different mechanisms controlling the cassette dynamics. We discuss the role of the toxin/antitoxin (TA) cassettes for the stabilization of the large cassette arrays carried in the larger CIs, known as superintegrons. Finally, we explore the central role played by single-stranded DNA in the integron cassette dynamics in light of the recent discovery that the integron integrase expression is controlled by the SOS response.
URLPMID:29434398 [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}