2003年,Przeworski等[55]首次运用ABC算法推测有利突变的选择初始时间。作者使用溯祖模拟产生相关模拟数据,再分别利用分离位点数(the number of segregating sites, S)、Tajima’sD、SFS和单倍型种类数(the number of distinct haplotypes in the sample, H)等统计量对这些模拟数据进行分析。作者发现随着统计量数目的增加,选择初始时间估计值的后验概率分布曲线斜率逐渐增大,即估计时间结果更为准确[53]。为了了解生活在美国白沙国家公园的两种蜥蜴(Sceloporus cowlesi和Aspidoscelis inornata)由于环境变化而产生适应的遗传机制,Laurent等[56]使用ABC算法结合21种统计量对与肤色相关的MC1R基因进行了选择初始时间估计,结果发现Sceloporus cowlesi群体中的估计时间较早(约1200年前),而在Aspidoscelis inornata群体中的估计时间则稍晚(约900年前)。虽然两者的估计时间不一致,但其肤色都是在白沙形成之后(约7000年前)改变的。
约在10,000年前,猪在近东和中国被驯化。驯化过程中,猪受到自然选择和人工选择的共同作用,在外形、繁殖力、生长发育和环境适应性等方面表现出显著改变[63]。目前在中国家猪中已发现多个与驯化性状相关的基因受到选择。例如, 与神经系统相关的SLC5A5基因、与免疫应答相关的MARCH1基因以及与生长相关的MSTN 基因等[86]。但是,目前仍缺乏被选择基因选择初始时间的研究报道。梁作翔[53]使用ABC算法估计了部分中国家猪中与驯化性状相关基因的选择压力和选择初始时间,并比较了不同基因的选择初始时间差异。结果表明,与神经系统相关的SLC5A5基因(约8,473.32年前)和与生长相关的MSTN基因(约1,587.75年前)之间的选择初始时间相差甚远。经过估计多个基因选择初始时间后, 其认为中国家猪的驯化是一个不断复杂变化的过程, 与同一驯化性状相关的基因既包括早期选择又包括近期选择,持续不同的驯化过程影响着这些性状的改变。最近的研究中,Chen等[87]利用266只欧亚野猪和家猪的全基因组测序数据,得到了法系长白猪(French Large White, FLW)与中国猪之间的渗入比例,并通过ALDER软件计算得到法系长白猪在约200~300年前与华南猪(Southern Chinese pigs, SCN)和华东猪(Eastern Chinese pigs, ECN)发生过基因交流事件。
4.2 家鸡
家鸡驯化于8000~10,000年前,是最早驯化的鸟类之一。在地理隔离和人工选择等因素作用下,家鸡成为表型和用途最为丰富的驯化动物之一,包括给人类提供食物的蛋鸡和肉鸡、用来娱乐打斗的斗鸡(gamecock)及用于观赏消遣的观赏鸡等。Rubin等[88]首次对9个鸡群体进行了全基因组测序(whole genome sequencing, WGS),其中包括红色原鸡(Red Junglefowl)、肉鸡及蛋鸡。根据标准化杂合度(Z- transformations of the pooled heterozygosity, ZHP)方法发现TSHR基因在驯化中受到了强烈选择。进一步研究发现TSHR基因有一个非同义突变发生在外显子区域,可能与鸡季节性发情相关。李明洲课题组利用D统计量检测了红色原鸡和10种家鸡之间的基因渗入,发现红色原鸡与藏鸡之间存在更多的渗入区域,可能跟野生红色原鸡与山区散养的藏鸡经常混在一起生活所导致。该研究还发现藏鸡长期生活在高海拔地区的过程中,获得了适应高海拔环境的能力,例如其红细胞数量、血氧亲和力和血红蛋白浓度都有所增加,而通过θπ、ZFST(Z-transformations of FST)和Tajima’sD检验等方法进行选择信号分析得到,NT5C1A和HEYL基因在参与藏鸡适应高海拔输送氧的方面可能起作用。该研究也发现藏鸡中GTP酶(GTPase)的调控活性显著增加,这表明能量代谢对于维持藏鸡体温的重要性[89]。然而,基因渗入除了发生在野生鸡种和地方鸡种外,商业品种也会有基因渗入到家鸡当中。张春媛等[90]通过比较群体单倍型相似性发现在我国商业鸡种的基因渗入到了地方鸡种基因组中,而由于我国遗传育种保护工作起步较晚,很多方面缺少有效监管,导致基因渗入事件的发生,可能对我国家鸡遗传资源造成了基因污染。
群体遗传学用于动物驯化研究,极大地扩展了研究的内容,并推动了学科的发展。一些经典的研究内容,如基因组选择信号和基因交流信号的检测已经相对比较成熟,而对群体历史及时间估计等方面的方法研究可做的工作还有很多。对于群体历史推断,不同的方法各具优点,例如在缺失先验模型的情况下推断种群有效群体大小历史变化和种群间分化时间时,可以利用PSMC、MSMC2及SMC++等方法,不过这类方法没有过多考虑群体之间的基因流事件,其历史真实性会受到影响[96];SFS方法对重构复杂的多群体历史变化和推测群体之间的迁徙事件具有很好的效果;推测近期历史事件则可以利用基于LD或者IBD (identical by descent)的方法来得到较为准确的时间。为了更准确的估算结果则需要充分考虑可能发生的历史事件,构建更全面的模型进行推断。基于恰当模型量化特定群体历史的方式逐渐成为一种主流,然而模型的适合度固然重要,但是其稳定可行性却更为重要。一个模型若要在任何参数上都与真实的群体历史相匹配的概率极其微小,我们需要带着实际科学问题去设计不同的模型来研究我们最感兴趣的问题,通过利用赤池信息量(Akaike information criterion, AIC)或贝叶斯信息量(Bayesian information criterion, BIC)等标准来筛选与实际数据最吻合的模型。
ZederMA. Core questions in domestication research Proc Natl Acad Sci USA, 2015, 112(11): 3191- 3198. [本文引用: 1]
GambleC, DaviesW, PettittP, RichardsM. Climate change and evolving human diversity in Europe during the Last Glacial Philos Trans R Soc Lond B Biol Sci, 2004, 359( 1442): 243- 254. [本文引用: 1]
DiamondJ. Evolution, consequences and future of plant and animal domestication Nature, 2002,418(6898):700- 707. [本文引用: 1]
EvershedRP, PayneS, SherrattAG, CopleyMS, CoolidgeJ, Urem-KotsuD, KotsakisK, OzdoğanM, OzdoğanAE, NieuwenhuyseO, AkkermansPMMG, BaileyD, AndeescuRR, CampbellS, FaridS, HodderI, YalmanN, OzbaşaranM, BiçakciE, GarfinkelY, LevyT, BurtonMM. Earliest date for milk use in the Near East and southeastern Europe linked to cattle herding Nature, 2008, 455(7212): 528- 531. [本文引用: 1]
OutramAK, StearNA, BendreyR, OlsenS, KasparovA, ZaibertV, ThorpeN, EvershedRP. The earliest horse harnessing and milking Science, 2009,323(5919):1332- 1335. [本文引用: 1]
DanielLH, AndrewGC. Principles of population genetics. 4th ed. Sinauer Associates, Sunderland, MA, 2006. [本文引用: 1]
ShiY, LiHP. Population genomics: from classical statistics to supervised learning Sci Sin Vitae, 2019, 49(4): 445- 455. [本文引用: 6]
ZhengZQ. Population structure and genetic introgression from wild relatives in worldwide goat populations [Dissertation]. Northwest A&F University, 2019. [本文引用: 3]
WangGD, ZhaiWW, YangHC, WangL, ZhongL, LiuYH, FanRX, YinTT, ZhuCL, PoyarkovAD, IrwinDM, HytönenMK, LohiH, WuCI, SavolainenP, ZhangYP. Out of southern East Asia: the natural history of domestic dogs across the world Cell Res, 2016, 26(1): 21- 33. [本文引用: 2]
PetersJ, LebrasseurO, DengH, LarsonG. Holocene cultural history of Red jungle fowl ( Gallus gallus ) and its domestic descendant in East Asia Quaternary Sci Rev, 2016,142:102- 119. [本文引用: 1]
LiH, DurbinR. Inference of human population history from individual whole-genome sequences Nature, 2011, 475(7357): 493- 496. [本文引用: 1]
SchiffelsS, DurbinR. Inferring human population size and separation history from multiple genome sequences Nat Genet, 2014,46(8):919- 925. [本文引用: 1]
TerhorstJ, KammJA, SongYS. Robust and scalable inference of population history from hundreds of unphased whole genomes Nat Genet, 2017, 49(2): 303- 309. [本文引用: 3]
QanbariS, StromTM, HabererG, WeigendS, GheyasAA, TurnerF, BurtDW, PreisingerR, GianolaD, SimianerH. A high resolution genome-wide scan for significant selective sweeps: an application to pooled sequence data in laying chickens PLoS One, 2012, 7( 11): e49525. [本文引用: 1]
LiHP, StephanW.Inferring the demographic history and rate of adaptive substitution in Drosophila. PLoS Genet, 2006, 2( 10): e166. [本文引用: 1]
GutenkunstRN, HernandezRD, WilliamsonSH, BustamanteCD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data PLoS Genet, 2009, 5( 10): e1000695. [本文引用: 2]
ExcoffierL, DupanloupI, Huerta-SánchezE, SousaVC, FollM. Robust demographic inference from genomic and SNP data PLoS Genet, 2013, 9( 10): e1003905. [本文引用: 3]
BarbatoM, Orozco-terWengelP, TapioM, BrufordMW. Snep: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data Front Genet, 2015,6:109. [本文引用: 2]
WangJL. Estimation of effective population sizes from data on genetic markers Philos Trans R Soc Lond B Biol Sci, 2005,360( 1459):1395- 1409. [本文引用: 1]
HillWG. Estimation of effective population size from data on linkage disequilibrium Genet Res, 1981,38(3):209- 216. [本文引用: 1]
CorbinLJ, LiuAYH, BishopSC, WoolliamsJA. Estimation of historical effective population size using linkage disequilibria with marker data J Anim Breed Genet, 2012,129(4):257- 270. [本文引用: 1]
WegmannD, LeuenbergerC, NeuenschwanderS, ExcoffierL. Abctoolbox: A versatile toolkit for approximate Bayesian computations BMC Bioinformatics, 2010,11(116). [本文引用: 1]
SanchezT, CuryJ, CharpiatG, JayF. Deep learning for population size history inference: design, comparison and combination with approximate Bayesian computation Mol Ecol Resour, 2020. [本文引用: 1]
Tiago Do Prado Paim, PatríciaI, SamuelRP, AlexandreRC, Concepta Margaret Mcmanus Pimentel. Detection and evaluation of selection signatures in sheep Pesqui Agropecu Bras, 2018,53(5):527- 539. [本文引用: 1]
XueZYY, SongXW, WuLH, WangLZ, CuiJA, SunZJ, ZhangZ, MaYL. The identification methods of selection signatures in livestock and its statistical problems Acta Vet Et Zootech Sin, 2018, 49(6): 1099- 1107. [本文引用: 4]
PenningsPS, HermissonJ. Soft sweeps II—molecular population genetics of adaptation from recurrent mutation or migration Mol Biol Evol, 2006,23(5):1076- 1084. [本文引用: 1]
HermissonJ, PenningsPS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation Genetics, 2005,169(4):2335- 2352. [本文引用: 1]
SuzukiY. Statistical methods for detecting natural selection from genomic data Genes Genet Syst, 2010,85(6):359- 376. [本文引用: 1]
NielsenR. Molecular signatures of natural selection Annu Rev Genet, 2005,39:197- 218. [本文引用: 1]
LohmuellerKE, BustamanteCD, ClarkAG. Detecting directional selection in the presence of recent admixture in African-Americans Genetics, 2011,187(3):823- 835. [本文引用: 1]
WangYZ, ZhaoYQ. Research progress of genomic signature of selection and its detection methods Acta Ecol Anim Domas, 2019, 40(5): 1- 6. [本文引用: 1]
de Simoni Gouveia JJ, daSilva MVGB, PaivaSR, de Oliveira SMP. Identification of selection signatures in livestock species Genet Mol Biol, 2014, 37(2): 330- 342. [本文引用: 1]
ChenH, PattersonN, ReichD. Population differentiation as a test for selective sweeps Genome Res, 2010,20(3):393- 402. [本文引用: 1]
OleksykTK, SmithMW, O'BrienSJ. Genome-wide scans for footprints of natural selection Philos Trans R Soc Lond B Biol Sci, 2010, 365( 1537): 185- 205. [本文引用: 1]
GrossmanSR, ShlyakhterI, KarlssonEK, ByrneEH, MoralesS, FriedenG, HostetterE, AngelinoE, GarberM, ZukO, LanderES, SchaffnerSF, SabetiPC. A composite of multiple signals distinguishes causal variants in regions of positive selection Science, 2010, 327(5967): 883- 886. [本文引用: 1]
UtsunomiyaYT, PérezO'Brien AM, SonstegardTS, VanTassell CP, doCarmo AS, MészárosG, SölknerJ, GarciaJF. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome- wide scan methods PLoS One, 2013,8(5): e64280. [本文引用: 1]
RandhawaIAS, KhatkarMS, ThomsonPC, RaadsmaHW. Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep BMC Genet, 2014,15:34. [本文引用: 1]
MaY, DingX, QanbariS, WeigendS, ZhangQ, SimianerH. Properties of different selection signature statistics and a new strategy for combining them Heredity(Edinb), 2015,115(5):426- 436. [本文引用: 1]
MalaspinasAS, MalaspinasO, EvansSN, SlatkinM. Estimating allele age and selection coefficient from time-serial data Genetics, 2012, 192(2): 599- 607. [本文引用: 1]
PrzeworskiM. Estimating the time since the fixation of a beneficial allele Genetics, 2003,164(4):1667- 1676. [本文引用: 1]
LaurentS, PfeiferSP, SettlesML, HunterSS, HardwickKM, OrmondL, SousaVC, JensenJD, RosenblumEB. The population genomics of rapid adaptation: disentangling signatures of selection and demography in white sands lizards Mol Ecol, 2016, 25(1): 306- 323. [本文引用: 1]
MedinaP, ThornlowB, NielsenR, Corbett-DetigR. Estimating the timing of multiple admixture pulses during local ancestry inference Genetics, 2018,210(3):1089- 1107. [本文引用: 1]
ChengCH, HuangY. Construction and application of phylogenetic network Entomotaxonomia, 2008, 30(3): 215- 221. [本文引用: 1]
DobneyK, LarsonG. Genetics and animal domestication: New windows on an elusive process J Zool, 2006, 269(2): 261- 271. [本文引用: 1]
MarshallFB, DobneyK, DenhamT, CaprilesJM. Evaluating the roles of directed breeding and gene flow in animal domestication Proc Natl Acad Sci USA, 2014, 111(17): 6153- 6158. [本文引用: 1]
WinklerCA, NelsonGW, SmithMW. Smith. Admixture mapping comes of age. Annu Rev Genomics Hum Genet, 2010,11:65- 89. [本文引用: 1]
RacimoF, SankararamanS, NielsenR, Huerta-SánchezE. Evidence for archaic adaptive introgression in humans Nat Rev Genet, 2015,16(6):359- 371. [本文引用: 1]
AiHS, FangXD, YangB, HuangZY, ChenH, MaoLK, ZhangF, ZhangL, CuiLL, HeWM, YangJ, YaoXM, ZhouLS, HanLJ, LiJ, SunSL, XieXH, LaiBX, SuY, LuY, YangH, HuangT, DengWJ, NielsenR, RenJ, HuangLS. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing Nat Genet, 2015,47(3):217- 225. [本文引用: 2]
LiangSY, ZhouZK, HouSS. The research progress of farm animal genomics based on sequencing technologies Hereditas(Beijing), 2017, 39(4): 276- 292. [本文引用: 1]
PattersonN, MoorjaniP, LuoY, MallickS, RohlandN, ZhanYP, GenschoreckT, WebsterT, ReichD. Ancient admixture in human history Genetics, 2012,192(3):1065- 1093. [本文引用: 2]
ReichD, ThangarajK, PattersonN, PriceAL, SinghL. Reconstructing Indian population history Nature, 2009,461(7263):489- 494. [本文引用: 2]
PeaseJB, HahnMW. Detection and polarization of introgression in a five-taxon phylogeny Syst Biol, 2015,64(4):651- 662. [本文引用: 1]
ZhengYC, JankeA. Gene flow analysis method, the D -statistic, is robust in a wide parameter space BMC Bioinformatics, 2018, 19( 1): 10. [本文引用: 3]
FrantzLAF, SchraiberJG, MadsenO, MegensHJ, BosseM, PaudelY, SemiadiG, MeijaardE, LiN, CrooijmansRPMA, ArchibaldAL, SlatkinM, SchookLB, LarsonG, GroenenMAM.Genome sequencing reveals fine scale diversification and reticulation history during speciation in Sus. Genome Biol, 2013,14(9): R107. [本文引用: 1]
SandersonJ, SudoyoH, KarafetTM, HammerMF, CoxMP. Reconstructing past admixture processes from local genomic ancestry using wavelet transformation Genetics, 2015, 200(2): 469- 481. [本文引用: 1]
PugachI, MatveyevR, WollsteinA, KayserM, StonekingM. Dating the age of admixture via wavelet transform analysis of genome-wide data Genome Biol, 2011, 12( 2): R19. [本文引用: 1]
XuSH, HuangW, QianJ, JinL. Analysis of genomic admixture in Uyghur and its implication in mapping strategy Am J Hum Genet, 2008,82(4):883- 894. [本文引用: 1]
PriceAL, TandonA, PattersonN, BarnesKC, RafaelsN, RuczinskiI, BeatyTH, MathiasR, ReichD, MyersS. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations PLoS Genet, 2009,5(6): e1000519. [本文引用: 1]
ChimusaER, DefoJ, ThamiPK, AwanyD, MulisaDD, AllaliI, GhazalH, MoussaA, MazanduGK. Dating admixture events is unsolved problem in multi-way admixed populations Brief Bioinform, 2020, 21(2): 144- 155. [本文引用: 6]
MoorjaniP, PattersonN, HirschhornJN, KeinanA, HaoL, AtzmonG, BurnsE, OstrerH, PriceAL, ReichD. The history of African gene flow into Southern Europeans, Levantines, and Jews PLoS Genet, 2011,7(4): e1001373. [本文引用: 3]
LohPR, LipsonM, PattersonN, MoorjaniP, PickrellJK, ReichD, BergerB. Inferring admixture histories of human populations using linkage disequilibrium Genetics, 2013,193(4):1233- 1254. [本文引用: 2]
ZhouY, YuanK, YuY, NiX, XieP, XingEP, XuS. Inference of multiple-wave population admixture by modeling decay of linkage disequilibrium with polynomial functions Heredity (Edinb), 2017,118(5):503- 510.
NiXM, YuanK, YangX, FengQD, GuoW, MaZM, XuSH. Inference of multiple-wave admixtures by length distribution of ancestral tracks Heredity (Edinb), 2018, 121(1): 52- 63.
HellenthalG, BusbyGBJ, BandG, WilsonJF, CapelliC, FalushD, MyersS. A genetic atlas of human admixture history Science, 2014, 343(6172): 747- 751. [本文引用: 1]
NiXM, YangX, GuoW, YuanK, ZhouY, MaZM, XuSH. Length distribution of ancestral tracks under a general admixture model and its applications in population history inference Sci Rep, 2016,6:20048.
Corbett-DetigR, NielsenR. A hidden Markov model approach for simultaneously estimating local ancestry and admixture time using next generation sequence data in samples of arbitrary ploidy PLoS Genet, 2017, 13( 1): e1006529.
LawsonDJ, HellenthalG, MyersS, FalushD. Inference of population structure using dense haplotype data PLoS Genet, 2012, 8( 1): e1002453. [本文引用: 1]
GalaverniM, CanigliaR, PaganiL, FabbriE, BoattiniA, RandiE. Disentangling timing of admixture, patterns of introgression, and phenotypic indicators in a hybridizing wolf population Mol Biol Evol, 2017,34(9):2324- 2339. [本文引用: 1]
ZhuYL, LiWB, YangB, ZhangZY, AiHS, RenJ, HuangLS. Signatures of selection and interspecies introgression in the genome of Chinese domestic pigs Genome Biol Evol, 2017, 9(10): 2592- 2603. [本文引用: 1]
ChenH, HuangM, YangB, WuZP, DengZ, HouY, RenJ, HuangLS. Introgression of Eastern Chinese and Southern Chinese haplotypes contributes to the improvement of fertility and immunity in European modern pigs Gigascience, 2020,9(3): giaa014. [本文引用: 1]
RubinCJ, ZodyMC, ErikssonJ, MeadowsJRS, SherwoodE, WebsterMT, JiangL, IngmanM, SharpeT, KaS, HallböökF, BesnierF, CarlborgO, Bed'homB, Tixier-BoichardM, JensenP, SiegelP, Lindblad-TohK, AnderssonL. Whole-genome resequencing reveals loci under selection during chicken domestication Nature, 2010,464(7288):587- 591. [本文引用: 1]
TerhorstJ, KammJA, SongYS. Robust and scalable inference of population history from hundreds of unphased whole genomes Nat Genet, 2017, 49(2): 303- 309. [本文引用: 1]
OrmondL, FollM, EwingGB, PfeiferSP, JensenJD. Inferring the age of a fixed beneficial allele Mol Ecol, 2016, 25(1): 157- 169. [本文引用: 1]
FrantzLA, SchraiberJG, MadsenO, MegensHJ, CaganA, BosseM, PaudelY, CrooijmansRPMA, LarsonG, GroenenMAM. Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes Nat Genet, 2015, 47(10): 1141- 1148. [本文引用: 1]
FreudenthalJA, AnkenbrandMJ, GrimmDG, KorteA. GWAS-Flow: A GPU accelerated framework for efficient permutation based genome-wide association studies BioRxiv, 2019,1:783100. [本文引用: 1]
BuLN, WangQ, GuWJ, YangRF, ZhuD, SongZ, LiuXJ, ZhaoYQ. Improving read alignment through the generation of alternative reference via iterative strategy Sci Rep, 2020, 10( 1): 18712. [本文引用: 1]
 ,1, 赵毅强
,1, 赵毅强
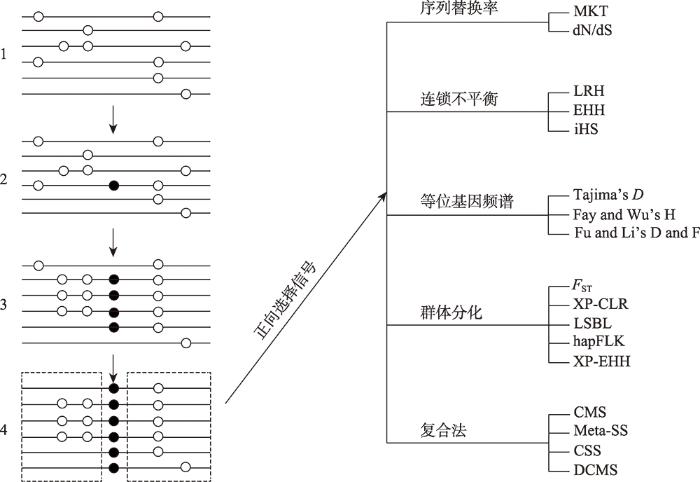 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}