Abstract Nucleosomes are the basic unit of the three-dimensional structure of chromatin. It is now widely accepted that the positioning and occupancy of nucleosomes play important roles in fundamental genomic processes such as DNA transcription, replication and repair. Among the methods used to provide genome-wide nucleosomal positions and occupancy levels, MNase-seq has proven to be highly effective. Indeed, with this method, the nucleosomal landscapes of a variety of organisms have now been investigated, revealing both commonalities and differences. In this review, we first introduce the technical principles underlying MNase-seq, focusing on details essential to precisely resolve nucleosome positioning and occupancy. We then describe recent advances with this method, as well as future perspectives of its role in chromatin biology, with a particular focus of uncovering mechanistic insights of many disease process. Keywords:nucleosome;chromatin structure;chromatin remodeling;next-generation sequencing (NGS);micrococcal nuclease
PDF (744KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 邓玮杭, 李鑫辉. MNase-seq与核小体定占位研究. 遗传[J], 2020, 42(12): 1143-1155 doi:10.16288/j.yczz.20-178 Weihang Deng. Resolving nucleosomal positioning and occupancy with MNase-seq. Hereditas(Beijing)[J], 2020, 42(12): 1143-1155 doi:10.16288/j.yczz.20-178
MNase-seq的优点在于技术难度较低,具有较高的分辨率,且数据处理相对简单。较高的分辨率得益于MNase的酶切特性,MNase处理染色质可以高效去除连接DNA,得到的DNA片段末端正是包裹组蛋白的DNA两端。相比用超声打断DNA的常规方法,MNase处理DNA可以获得长度较均一的DNA片段,从而得到核小体更为精确的位置坐标。然而,由于不同DNA片段对MNase酶切的敏感性不同,这一酶切效率的差异使得MNase-seq测序片段的末端不能准确反映核小体边缘的位置,因此测序数据的处理分析尤为关键。MNase-seq数据处理过程主要为数据预处理及质量控制、序列比对、核小体定位与占位分析以及数据的可视化[12]。在序列比对后,研究者们先后运用多种算法(如iNPS[13]、DiNuP[14]等)分析MNase-seq测序数据,解析全基因组核小体定位精确图谱或对差异化占位核小体进行分析。其中,Chen等[13]建立了iNPS算法,该算法在NPS(nucleosome positioning from sequencing)的基础上增加了“核小体边界信号调整”与“相邻核小体合并或分离”步骤,它比通用的NPS算法识别核小体边界信号的能力更强,因此可以多检测到约60%的核小体。该算法具有更高的检测准确性和稳健性,因此有利于下游数据分析。核小体定位的理论预测同样具有较好的研究前景,研究者们利用核小体DNA/连接DNA的序列特性、碱基二联体周期信号等建立数学模型(如Segal模型[3]、N-Score模型[15]等)对核小体定位进行预测。近年来,MNase-seq测序数据分析方法的进步使得该技术日趋成熟,模型的改进与优化使得人们对核小体定位预测的准确性不断提高,MNase-seq技术目前已广泛应用于各种研究场景。
A: MNase-seq; B: Array-seq; C: MNase-ChIP-seq; D: CUT&RUN。 Fig. 1MNase-seq and its derivative technologies
除MNase-seq及其衍生技术以外,染色质免疫共沉淀测序技术(chromatin immunoprecipitation sequencing, ChIP-seq)、染色质开放性测序技术(assay for transposase-accessible chromatin with high throughput sequencing, ATAC-seq)、DNase I超敏感位点测序(DNase I hypersensitive site sequencing, DNase-seq)以及核小体占位及甲基化测序(nucleosome occupancy and methylome sequencing, NOMe-seq)等技术也在解析核小体定位及染色质结构及功能的研究中起到重要作用。表1对这些技术进行了总结比较。
Table 1 表1 表1研究核小体、染色质结构的常用技术 Table 1Technology for the study of nucleosome and chromatin structures
LugerK, M?derAW, RichmondRK, SargentDF, RichmondTJ . Crystal structure of the nucleosome core particle at 2.8 ? resolution Nature, 1997,389(6648):251-260. [本文引用: 1]
ZhouKD, GaullierG, LugerK . Nucleosome structure and dynamics are coming of age Nat Struct Mol Biol, 2019,26(1):3-13. [本文引用: 1]
SegalE, Fondufe-MittendorfY, ChenL, Th?str?mA, FieldY, MooreIK, WangJZ, WidomJ . A genomic code for nucleosome positioning Nature, 2006,442(7104):772-778. [本文引用: 4]
NollM . Subunit structure of chromatin Nature, 1974,251(5472):249-251. [本文引用: 1]
LohrD, KovacicRT, Van HoldeKE . Quantitative analysis of the digestion of yeast chromatin by staphylococcal nuclease Biochemistry, 1977,16(3):463-471. [本文引用: 1]
FanXC, MoqtaderiZ, JinY, ZhangY, LiuXS, StruhlK . Nucleosome depletion at yeast terminators is not intrinsic and can occur by a transcriptional mechanism linked to 3’-end formation Proc Natl Acad Sci USA, 2010,107(42):17945-17950. [本文引用: 1]
TsompanaM, BuckMJ . Chromatin accessibility: a window into the genome Epigenetics Chromatin, 2014,7(1):33. [本文引用: 2]
ZhangWL, JiangJM . Application of MNase-Seq in the global mapping of nucleosome positioning in plants Methods Mol Biol, 2018,1830:353-366. [本文引用: 1]
GaffneyDJ , McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, Pitchard JK. Controls of nucleosome positioning in the human genome PLoS Genet, 2012,8(11):e1003036. [本文引用: 2]
ZhangZH, PughBF . High-resolution genome-wide mapping of the primary structure of chromatin Cell, 2011,144(2):175-186. [本文引用: 1]
KleinDC, HainerSJ . Genomic methods in profiling DNA accessibility and factor localization Chromosome Res, 2020,28(1):69-85. [本文引用: 2]
ChenWZ, LiuY, ZhuSS, GreenCD, WeiG, HanJDJ . Improved nucleosome-positioning algorithm iNPS for accurate nucleosome positioning from sequencing data Nat Commun, 2014,5:4909. [本文引用: 2]
FuK, TangQZ, FengJX, LiuXS, ZhangY . DiNuP: a systematic approach to identify regions of differential nucleosome positioning Bioinformatics, 2012,28(15):1965-1971. [本文引用: 1]
YuanGC, LiuJS . Genomic sequence is highly predictive of local nucleosome depletion PLoS Comput Biol, 2008,4(1):e13. [本文引用: 1]
WalM, PughBF . Genome-wide mapping of nucleosome positions in yeast using high-resolution MNase ChIP-Seq Methods Enzymol, 2012,513:233-250. [本文引用: 1]
OcampoJ, CuiF, ZhurkinVB, ClarkDJ . The proto-chromatosome: A fundamental subunit of chromatin? Nucleus, 2016,7(4):382-387. [本文引用: 1]
RizzoJM, SinhaS . Analyzing the global chromatin structure of keratinocytes by MNase-seq Methods Mol Biol, 2014,1195:49-59.
CuiKR, ZhaoKJ . Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq Methods Mol Biol, 2012,833:413-419.
MeyerCA, LiuXS . Identifying and mitigating bias in next-generation sequencing methods for chromatin biology Nat Rev Genet, 2014,15(11):709-721.
ParkPJ . ChIP-seq: advantages and challenges of a maturing technology Nat Rev Genet, 2009,10(10):669-680.
MardisER . ChIP-seq: welcome to the new frontier Nat Methods, 2007,4(8):613-614.
BuenrostroJD, WuBJ, ChangHY, GreenleafWJ. ATAC-seq: A method for assaying chromatin accessibility genome-Wide Curr Protoc Mol Biol, 2015, 109: 21.29.1-21.29.9.
BuenrostroJD, GiresiPG, ZabaLC, ChangHY, GreenleafWJ . Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position Nat Methods, 2013,10(12):1213-1218.
SchepAN, BuenrostroJD, DennySK, SchwartzK, SherlockG, GreenleafWJ . Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions Genome Res, 2015,25(11):1757-1770.
KellyTK, LiuYP, LayFD, LiangGN, BermanBP, JonesPA . Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules Genome Res, 2012,22(12):2497-2506.
KrebsAR, ImanciD, HoernerL, GaidatzisD, BurgerL, SchübelerD. Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters Mol Cell, 2017,67(3): 411-422.e4.
SkenePJ, HenikoffJG, HenikoffS . Targeted in situ genome-wide profiling with high efficiency for low cell numbers Nat Protoc, 2018,13(5):1006-1019. [本文引用: 1]
HainerSJ, Bo?kovi?A, McCannellKN, RandoOJ, Fazzio TG. Profiling of pluripotency factors in single cells and early embryos Cell, 2019, 177(5): 1319-1329.e11. [本文引用: 1]
GaoWW, LaiBB, NiB, ZhaoKJ . Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scMNase-seq Nat Protoc, 2020,15(1):68-85. [本文引用: 1]
BaldiS . Nucleosome positioning and spacing: from genome-wide maps to single arrays Essays Biochem, 2019,63(1):5-14. [本文引用: 1]
BaldiS, KorberP, BeckerPB . Beads on a string- nucleosome array arrangements and folding of the chromatin fiber Nat Struct Mol Biol, 2020,27(2):109-118. [本文引用: 3]
LeeW, TilloD, BrayN, MorseRH, DavisRW, HughesTR, NislowC . A high-resolution atlas of nucleosome occupancy in yeast Nat Genet, 2007,39(10):1235-1244. [本文引用: 1]
SchonesDE, CuiKR, CuddapahS, RohTY, BarskiA, WangZB, WeiG, ZhaoKJ . Dynamic regulation of nucleosome positioning in the human genome Cell, 2008,132(5):887-898. [本文引用: 2]
ValouevA, IchikawaJ, TonthatT, StuartJ, RanadeS, PeckhamH, ZengK, MalekJA, CostaG, McKernan K, Sidow A, Fire A, Johnson SM. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning Genome Res, 2008,18(7):1051-1063. [本文引用: 1]
LaiWKM, PughBF . Understanding nucleosome dynamics and their links to gene expression and DNA replication Nat Rev Mol Cell Biol, 2017,18(9):548-562. [本文引用: 3]
AhmadK, HenikoffS . The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly Mol Cell, 2002,9(6):1191-1200. [本文引用: 1]
SarmaK, ReinbergD . Histone variants meet their match Nat Rev Mol Cell Biol, 2005,6(2):139-149. [本文引用: 1]
RamachandranS, ZentnerGE, HenikoffS . Asymmetric nucleosomes flank promoters in the budding yeast genome Genome Res, 2015,25(3):381-390. [本文引用: 1]
KaplanN, MooreIK, Fondufe-MittendorfY, GossettAJ, TilloD, FieldY, LeProustEM, HughesTR, LiebJD, WidomJ, SegalE,. The DNA-encoded nucleosome organization of a eukaryotic genome Nature, 2009,458(7236):362-366. [本文引用: 2]
AlbertI, MavrichTN, TomshoLP, QiJ, ZantonSJ, SchusterSC, PughBF . Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome Nature, 2007,446(7135):572-576. [本文引用: 2]
CuiF, ChenLL, LoVerso PR, ZhurkinVB. Prediction of nucleosome rotational positioning in yeast and human genomes based on sequence-dependent DNA anisotropy BMC Bioinformatics, 2014,15(1):313. [本文引用: 1]
de DieuleveultM, YenKY, HmitouI, DepauxA, BoussouarF, Bou DarghamD, JounierS, HumbertclaudeH, RibierreF, BaulardC, FarrellNP, ParkB, KeimeC, CarrièreL, BerlivetS, GutM, GutL, WernerM, DeleuzeJF, OlasoR, AudeJC, ChantalatS, PughBF, GérardM . Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells Nature, 2016,530(7588):113-116. [本文引用: 1]
HoL, CrabtreeGR . Chromatin remodelling during development Nature, 2010,463(7280):474-484. [本文引用: 1]
DuttaA, GogolM, KimJH, SmolleM, VenkateshS, GilmoreJ, FlorensL, WashburnMP, WorkmanJL . Swi/Snf dynamics on stress-responsive genes is governed by competitive bromodomain interactions Genes Dev, 2014,28(20):2314-2330. [本文引用: 1]
Ribeiro-SilvaC, VermeulenW, LansH . SWI/SNF: Complex complexes in genome stability and cancer DNA Repair, 2019,77:87-95. [本文引用: 1]
ArasS, SaladiSV, BasuroyT, MaratheHG, LorèsP, dela Serna IL,. BAF60A mediates interactions between the microphthalmia-associated transcription factor and the BRG1-containing SWI/SNF complex during melanocyte differentiation J Cell Physiol, 2019,234(7):11780-11791. [本文引用: 1]
OppikoferM, BaiTY, GanYT, HaleyB, LiuP, SandovalW, CiferriC, CochranAG . Expansion of the ISWI chromatin remodeler family with new active complexes EMBO Rep, 2017,18(10):1697-1706. [本文引用: 1]
LevendoskyRF, BowmanGD . Asymmetry between the two acidic patches dictates the direction of nucleosome sliding by the ISWI chromatin remodeler eLife, 2019,8:e45472. [本文引用: 1]
CairnsBR . The logic of chromatin architecture and remodelling at promoters Nature, 2009,461(7261):193-198. [本文引用: 3]
OcampoJ, CherejiRV, ErikssonPR, ClarkDJ . The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res, 2016,44(10):4625-4635. [本文引用: 1]
LeeCK, ShibataY, RaoB, StrahlBD, LiebJD . Evidence for nucleosome depletion at active regulatory regions genome-wide Nat Genet, 2004,36(8):900-905. [本文引用: 1]
SekingerEA, MoqtaderiZ, StruhlK . Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast Mol Cell, 2005,18(6):735-748. [本文引用: 1]
BernsteinBE, LiuCL, HumphreyEL, PerlsteinEO, SchreiberSL . Global nucleosome occupancy in yeast Genome Biol, 2004,5(9):R62. [本文引用: 1]
YuanGC, LiuYJ, DionMF, SlackMD, WuLF, AltschulerSJ, RandoOJ . Genome-scale identification of nucleosome positions in S. cerevisiae Science, 2005,309(5734):626-630. [本文引用: 2]
RandoOJ, AhmadK . Rules and regulation in the primary structure of chromatin Curr Opin Cell Biol, 2007,19(3):250-256. [本文引用: 1]
MavrichTN, IoshikhesIP, VentersBJ, JiangCZ, TomshoLP, QiJ, SchusterSC, AlbertI, PughBF . A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome Genome Res, 2008,18(7):1073-1083. [本文引用: 1]
KubikS, BruzzoneMJ, ChallalD, DreosR, MattarocciS, BucherP, LibriD, ShoreD . Opposing chromatin remodelers control transcription initiation frequency and start site selection Nat Struct Mol Biol, 2019,26(8):744-754. [本文引用: 1]
KubikS, O’DuibhirE, de JongeWJ, MattarocciS, AlbertB, FalconeJL, BruzzoneMJ, HolstegeFCP, ShoreD. Sequence-directed action of RSC remodeler and general regulatory factors modulates +1 nucleosome position to facilitate transcription Mol Cell, 2018, 71(1): 89-102.e5. [本文引用: 1]
ValouevA, JohnsonSM, BoydSD, SmithCL, FireAZ, SidowA . Determinants of nucleosome organization in primary human cells Nature, 2011,474(7352):516-520. [本文引用: 2]
BoegerH, GriesenbeckJ, StrattanJS, KornbergRD . Removal of promoter nucleosomes by disassembly rather than sliding in vivo Mol Cell, 2004,14(5):667-673. [本文引用: 1]
ErtelF, Dirac-SvejstrupAB, HertelCB, BlaschkeD, SvejstrupJQ, KorberP . In vitro reconstitution of PHO5 promoter chromatin remodeling points to a role for activator-nucleosome competition in vivo Mol Cell Biol, 2010,30(16):4060-4076. [本文引用: 1]
ShivaswamyS, BhargavaP . Positioned nucleosomes due to sequential remodeling of the yeast U6 small nuclear RNA chromatin are essential for its transcriptional activation J Biol Chem, 2006,281(15):10461-10472. [本文引用: 1]
ShivaswamyS, BhingeA, ZhaoYJ, JonesS, HirstM, IyerVR . Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation PLoS Biol, 2008,6(3):e65. [本文引用: 1]
KulaevaOI, HsiehFK, ChangHW, LuseDS, StuditskyVM . Mechanism of transcription through a nucleosome by RNA polymerase II Biochim Biophys Acta, 2013,1829(1):76-83. [本文引用: 1]
RejaR, VinayachandranV, GhoshS, PughBF . Molecular mechanisms of ribosomal protein gene coregulation Genes Dev, 2015,29(18):1942-1954. [本文引用: 1]
VogelauerM, WuJ, SukaN, GrunsteinM . Global histone acetylation and deacetylation in yeast Nature, 2000,408(6811):495-498. [本文引用: 1]
BernsteinBE, HumphreyEL, ErlichRL, SchneiderR, BoumanP, LiuJS, KouzaridesT, SchreiberSL . Methylation of histone H3 Lys 4 in coding regions of active genes Proc Natl Acad Sci USA, 2002,99(13):8695-8700. [本文引用: 1]
KlemmSL, ShiponyZ, WJ. Chromatin accessibility and the regulatory epigenome Nat Rev Genet, 2019,20(4):207-220. [本文引用: 1]
DuYH, LiuZP, CaoXK, ChenXL, ChenZY, ZhangXB, ZhangXQ, JiangCZ . Nucleosome eviction along with H3K9ac deposition enhances Sox2 binding during human neuroectodermal commitment Cell Death Differ, 2017,24(6):1121-1131. [本文引用: 1]
DaneshpajoohM, BacosK, BysaniM, BaggeA, Ottosson LaaksoE, VikmanP, EliassonL, MulderH, LingC . HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells Diabetologia, 2017,60(1):116-125. [本文引用: 1]
VallianatosCN, RainesB, PorterRS, BonefasKM, WuMC, GarayPM, ColletteKM, SeoYA, DouY, KeeganCE, Tronson NC, IwaseS,. Mutually suppressive roles of KMT2A and KDM5C in behaviour, neuronal structure, and histone H3K4 methylation Commun Biol, 2020,3(1):278. [本文引用: 1]
Sasidharan NairV, El SalhatH, TahaRZ, JohnA, AliBR, ElkordE . DNA methylation and repressive H3K9 and H3K27 trimethylation in the promoter regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 genes in human primary breast cancer Clin Epigenetics, 2018,10:78. [本文引用: 1]
BarskiA, CuddapahS, CuiKR, RohTY, SchonesDE, WangZB, WeiG, ChepelevL, ZhaoKJ . High-resolution profiling of histone methylations in the human genome Cell, 2007,129(4):823-837. [本文引用: 1]
GiaimoBD, FerranteF, Herchenr?therA, HakeSB, BorggrefeT . The histone variant H2A.Z in gene regulation Epigenetics Chromatin, 2019,12(1):37. [本文引用: 1]
BagchiDN, BattenhouseAM, ParkD, IyerVR . The histone variant H2A.Z in yeast is almost exclusively incorporated into the +1 nucleosome in the direction of transcription Nucleic Acids Res, 2020,48(1):157-170. [本文引用: 1]
RanjanA, NguyenVQ, LiuS, WisniewskiJ, KimJM, TangXN, MizuguchiG, ElalaouiE, NickelsTJ, JouV, EnglishBP, ZhengQS, LukE, LavisLD, LionnetT, WuC,. Live-cell single particle imaging reveals the role of RNA polymerase II in histone H2A.Z eviction eLife, 2020,9:e55667. [本文引用: 1]
SharmaS, KellyTK, JonesPA . Epigenetics in cancer Carcinogenesis, 2010,31(1):27-36. [本文引用: 2]
WangHF, FuC, DuJ, WangHS, HeR, YinXF, LiHX, LiX, WangHX, LiK, ZhengL, LiuZC, QiuYR . Enhanced histone H3 acetylation of the PD-L1 promoter via the COP1/c-Jun/HDAC3 axis is required for PD-L1 expression in drug-resistant cancer cells J Exp Clin Cancer Res, 2020,39(1):29. [本文引用: 1]
KumarA, KumariN, SharmaU, RamS, SinghSK, KakkarN, KaushalK, PrasadR . Reduction in H3K4me patterns due to aberrant expression of methyltransferases and demethylases in renal cell carcinoma: prognostic and therapeutic implications Sci Rep, 2019,9(1):8189. [本文引用: 1]
WebberLP, WagnerVP, CurraM, VargasPA, MeurerL, CarrardVC, SquarizeCH, CastilhoRM, MartinsMD . Hypoacetylation of acetyl-histone H3 (H3K9ac) as marker of poor prognosis in oral cancer Histopathology, 2017,71(2):278-286. [本文引用: 1]
 ,上海交通大学生物医学工程学院,上海 200240
,上海交通大学生物医学工程学院,上海 200240
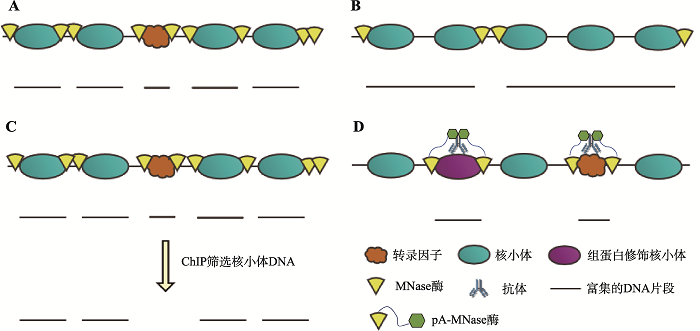 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT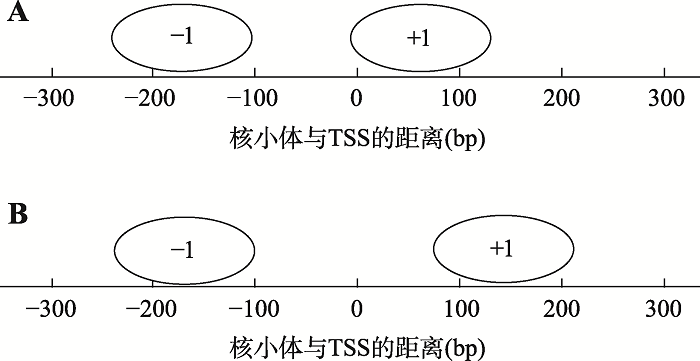 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}