 ,1,2,3
,1,2,3RAD51 regulates REV1 recruitment to DNA double-strand
break sites
Min Huang1,2, Yeran Yang1,2, Xiaoyan Sun1, Ting Zhang1, Caixia Guobreak sites
,1,2,3通讯作者:
编委: 朱卫国
收稿日期:2018-06-29修回日期:2018-10-19网络出版日期:2018-11-20
| 基金资助: |
Received:2018-06-29Revised:2018-10-19Online:2018-11-20
| Fund supported: |
作者简介 About authors
黄敏,博士研究生,专业方向:生物化学与分子生物学E-mail:huangmin@big.ac.cn。

摘要
关键词:
Abstract
Keywords:
PDF (2092KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄敏, 杨业然, 孙晓艳, 张婷, 郭彩霞. RAD51调控REV1参与DNA双链断裂修复[J]. 遗传, 2018, 40(11): 1007-1014 doi:10.16288/j.yczz.18-176
Min Huang, Yeran Yang, Xiaoyan Sun, Ting Zhang, Caixia Guo.
break sites
跨损伤DNA合成(translesion DNA synthesis, TLS)是哺乳动物细胞耐受DNA损伤的一种重要的方式[1],可防止复制叉崩解导致的细胞死亡。一般来说,当复制叉遇到受损的DNA时,高保真的DNA聚合酶无法继续完成复制继而发生停滞,随后跨损伤DNA聚合酶将高保真DNA复制酶置换下来,进而在损伤模板对侧整合核苷酸,然后由同一种或者另一种跨损伤DNA聚合酶继续延伸几个核苷酸,最后再由高保真的DNA复制酶负责完成DNA复制工作。由于跨损伤聚合酶的低保真性,一般认为,其能在DNA损伤模板对侧整合错误的碱基,从而引发基因组变异[1, 2]。迄今为止,已在哺乳动物中发现约有11种跨损伤DNA聚合酶,其中Polη、Polκ、REV1和Polι属于Y家族DNA聚合酶,它们在维持基因组完整性方面发挥了重要功能[1,2,3,4,5,6],其功能异常与肿瘤的发生、发展和耐药密切相关。
近年来研究表明,Y家族聚合酶除了参与TLS通路之外,还在其他一些DNA损伤修复通路中发挥作用[7]。例如,Polκ可在显微辐射(micro-irradiation)诱导的损伤位点处募集,参与调控氧化损伤诱发的单链和双链断裂修复[8];Polη也可参与双链断裂(DNA double-strand break, DSB)修复[9]。DSB是最严重的DNA损伤类型之一,如果不能及时修复将导致细胞死亡。DSB主要通过非同源末端连接(nonhomologous end joining, NHEJ)和同源重组(homologous recombination, HR)两条途径进行修复。Polη被报道能与HR通路中的重组酶RAD51相互作用,在RAD51调控下催化D-loop延伸[9]。2005年,REV1在鸡(Gallus domesticus) DT40细胞中被发现参与重组依赖的免疫球蛋白的基因转换[10]。随后REV1在出芽酵母(Saccharomyces cerevisiae)[11]和黑腹果蝇(Drosophila melanogaster)[12]也被报道有类似功能。与此同时,在人源细胞中的研究也发现,REV1与跨损伤聚合酶Polζ以复合体的形式参与DSB修复[13]。
随着研究的深入,REV1在HR通路中分子调控的具体机制也被逐渐揭示。Yang等[14]研究表明,RAD18能通过单泛素化的FANCD2 (FANCD2-mUb)调控REV1在UVA激光诱发的双链断裂损伤处的招募。REV1通过其泛素结合域(ubiquitin-binding motifs, UBMs)与FANCD2-mUb有较强的相互作用,被招募至DSBs;通过突变REV1的UBMs降低其与FANCD2-mUb的结合,则会导致REV1招募到DSBs的比例明显低于野生型。同时,BRCA1和BRCA2也能参与调控REV1的招募。细胞存活实验表明,敲除REV1或者敲低FANCD2的细胞均对可诱导DNA双链断裂的药物喜树碱(camptothecin, CPT)敏感性增强。此外,REV1还能通过稳定RAD51 filaments的方式来保护阻滞的复制叉处新生链不被降解。Wang等[15]发现miRNA-96通过靶向REV1和RAD51的mRNA抑制两者的蛋白表达水平,降低HR修复效率,同时对PARP抑制剂(AZD2281)和顺铂更加敏感。
由于REV1与RAD51存在相互作用,而RAD51在介导阻滞的复制叉处的重启以及HR修复过程中发挥了重要功能[16]。本文对RAD51与REV1在HR通路中的调控关系做了进一步研究,发现敲低RAD51会导致GFP-REV1的招募比例明显下降,表明RAD51可以调控REV1到UVA (λ=365 nm)激光诱导的DSB位点的招募即。有意思的是,REV1反过来也可以调控RAD51在CPT诱导的DSB位点的募集。RAD51C作为RAD51的同源物,虽然与REV1也存在相互作用,但是其并不调控REV1招募到DSB位点。
1 材料和方法
1.1 材料
HEK293T和U2OS细胞系均来自美国菌种保藏中心(American Type Culture Collection),MRC5细胞来自英国萨塞克斯大学Alan Lehmann实验室。这些细胞均置于37℃且有5% CO2的培养箱中培养,培养基为含有10%胎牛血清的DMEM。实验过程中所用siRNAs均购自于苏州吉玛公司,具体序列信息为siRAD51 (5′-GGCAGUAGAUGUGCAGAUA-3′),siREV1-1 (5′-GAACAGUGACGCAGGAAUATT-3′),siREV1-2 (5′-GCAUCAAAGCUGGACGACUTT-3′)[17],siNC (5′-UUCUCCGAACGUGUCACGU-3′)。靶向RAD51C的shRNA-RAD51C (TRCN00000146298和TRCN00000147658)及NC (shRNA-SHC002)均购于美国Open Biosystems公司。1.2 激光微辐射共聚焦显微镜检测GFP-REV1招募
本实验采用脉冲氮激光微辐射仪(Spectra-Physics, 365 nm, 10 Hz pulse),可诱发细胞核内DNA发生DSBs。仪器将荧光显微成像系统与微辐射系统相结合,可实时检测目的蛋白在损伤位点的招募情况。在35 mm的培养皿中铺适宜细胞密度的U2OS细胞,培养过夜,分别转染靶向RAD51的siRNA或者siNC oligos。第2天将细胞传代至激光切割专用培养皿(MatTek cultureware)中,待细胞贴壁完全后,转染GFP-REV1,24 h后,进行激光显微切割。每组统计25~30个细胞里GFP-REV1的招募情况,进行3次独立的生物学实验重复。1.3 免疫荧光检测RAD51的招募
在35 mm的培养皿中铺适宜细胞密度的U2OS细胞,培养过夜,分别转染靶向REV1的siRNA或者siNC oligos。第2天将细胞传代至盖玻片,培养过夜,CPT损伤处理或不作任何处理。收集细胞,先用PBS洗2遍,再用含0.5% Triton X-100的预抽缓冲液在室温预抽5 min,PBS洗3次。在4% PFA里室温条件下固定细胞15 min,PBS洗3遍后,在3% BSA里室温封闭1 h,再与RAD51抗体在4℃孵育过夜,PBST洗8次,与荧光二抗室温孵育1 h后,PBST洗8次,最后用含DAPI的封片剂封片[18]。荧光显微镜拍照统计RAD51的表达情况,每组样品至少统计200个细胞。1.4 检测REV1和SFB-RAD51C的相互作用
在6 cm的培养皿中铺适宜细胞密度的HEK293T细胞,培养过夜后转染SFB-RAD51C的全长或者其一系列删除不同区域的变体[19]。48 h后收集细胞,用4℃预冷的PBS洗涤细胞2次,离心收集细胞,用含有蛋白酶抑制剂的裂解缓冲液(50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1 mmol/L EDTA,1% Triton X-100)于4℃裂解细胞。然后低温离心,收集蛋白裂解液上清,与预洗过的S琼脂糖珠子于4℃孵育2 h,离心后再分别和等量的预先纯化好的His-mREV1蛋白(带His标签的小鼠REV1蛋白)[20]于低温孵育1 h,随后用裂解缓冲液分别快洗和慢洗(4℃旋转,10 min/次)3次。加入适量蛋白上样缓冲液,95℃加热5 min后离心,用于后续Western blot检测。1.5 建立RAD51C稳定敲低的MRC5细胞系
1.5.1 在MRC5细胞株中确定puromycin药物筛选的适宜浓度由于不同细胞株或者不同实验室的同种细胞株对puromycin的敏感性存在差异,因此在准备建立稳定细胞系前,需要摸索在3~5天内,杀死全部MRC5细胞的puromycin的最适宜浓度。将MRC5细胞以1×105/孔接种至6孔板,培养过夜后,将puromycin按照从0.5 μg/mL到5 μg/mL,每隔0.5 μg/mL递增的浓度加至不同培养孔中。期间,每天更换含有对应puromycin浓度的新鲜培养基。观察细胞死亡情况,最优puromycin浓度为3~5天内杀死全部细胞的最低puromycin浓度,用于后续稳定细胞系筛选。
1.5.2 筛选RAD51C稳定敲低的MRC5单克隆细胞株
转染前24 h,将HEK293T细胞接种于6 cm培养皿,使其第2天细胞密度在70%左右。将慢病毒包装质粒分别和shRNA-SHC002或者shRNA-RAD51C转染至HEK293T细胞,48 h和72 h后收集病毒上清,用于感染MRC5细胞。
将MRC5细胞提前1天铺于6 cm培养皿中,保证其被感染时的细胞密度约为70%。分别收集感染48和72 h后的病毒上清,用0.45 μm的滤器过滤去除细胞碎片,加入终浓度为8 μg/mL的polybrene,混合均匀。将收集的病毒上清分别感染MRC5细胞2次,在第一次感染48 h后,加入适宜浓度的puromycin进行药物筛选3天。
通过物筛选得到的混合克隆以适宜细胞数目接种至10 cm培养皿,待单克隆长至大小覆盖整个显微镜视野后,挑取单克隆至24孔板,并扩大培养。随后,取部分细胞进行Western blot鉴定,其余的继续培养。鉴定结果为阳性的单克隆,用去除支原体的试剂处理1周后,继续扩大培养并冻存,用于后期实验。
2 结果与分析
2.1 RAD51调控REV1在DSBs位点的招募
由于REV1能够通过稳定RAD51 filaments保护阻滞复制叉处的新生链不被降解,同时REV1和RAD51存在相互作用。为进一步研究二者间的调控关系,分别转染siNC或者siRAD51至U2OS细胞。48 h后,再转染GFP-REV1,然后对U2OS细胞进行UVA激光显微切割,检测GFP-REV1在UVA诱导的DSBs处的招募情况。结果显示,GFP-REV1能够被招募至UVA诱导的损伤位点(图1A)。同时,在siNC组GFP-REV1的招募比例为59.9%,敲低RAD51后,REV1的招募比例明显降低到34.7% (图1B)。与此同时,收集细胞裂解液利用Western blot进一步检测RAD51的敲低效果,结果证实RAD51的敲低效果明显(图1C)。以上结果表明,RAD51调控REV1在DSBs处的招募,即RAD51是GFP-REV1在UVA诱导的DSBs位点最适招募所必需的。图1
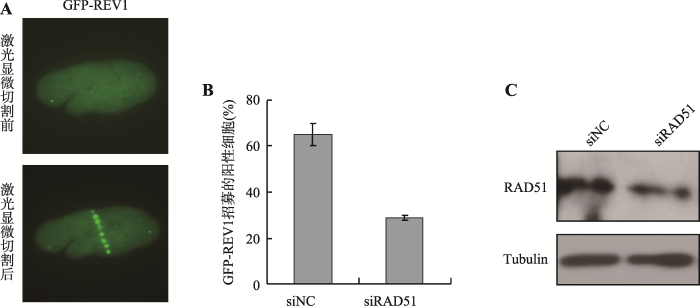 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1RAD51调控GFP-REV1到UVA诱导的DSBs位点的招募
A:U2OS细胞瞬时转染GFP-REV1。荧光检测结果显示GFP-REV1被招募至UVA诱导的DSBs损伤位点处。B:检测敲低RAD51对GFP-REV1招募的影响。数据为3次独立的生物学重复的统计结果。C:敲低RAD51的Western blot检测结果。Tubulin为内参对照。
Fig. 1RAD51 regulates the recruitment of GFP-REV1 to laser-induced DSBs
2.2 REV1调控RAD51在DSBs位点的招募
DNA损伤修复通路中很多信号分子间的关系并不是简单的单向上下游调控关系,因此本研究进一步检测了REV1是否调控RAD51在DSBs处的招募。在U2OS细胞中,分别转染siNC或者siREV1-1、siREV1-2,用双链断裂试剂CPT损伤处理后,检测RAD51的招募情况。结果发现,敲低REV1并用CPT处理后,RAD51的招募比例相较于NC组均明显下降(图2A)。免疫荧光的统计结果显示,相较于siNC转染组RAD51 foci的比例53.5%,siREV1-1和siREV1-2转染组RAD51 foci的比例明显分别下降至21.1%和16.6% (图2B)。同时,Western blot检测结果显示REV1敲低效果很好(图2C)。以上结果证明,REV1和RAD51存在相互调控的作用机制。图2
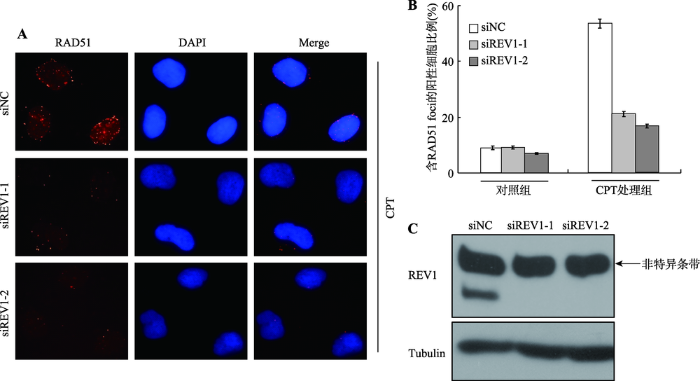 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2REV1促进CPT处理后RAD51在DSBs位点的招募
A:RAD51免疫荧光染色代表图。在U2OS细胞中瞬时敲低REV1,48 h后,用5 μmol/L CPT处理3 h,RAD51招募情况的免疫荧光代表图;B:含有RAD51 foci的阳性细胞的比例。在U2OS中敲低REV1后,不作任何损伤处理及CPT处理后,含RAD51 foci的细胞的统计结果,数据为3次独立的生物学重复的统计结果;C:Western blot检测REV1的敲低效果,Tubulin为内参对照。
Fig. 2REV1 facilitates the accumulation of RAD51 at DSBs after CPT treatment
2.3 REV1在DSBs位点的招募不依赖于RAD51C
2.3.1 REV1与RAD51C存在相互作用在HEK293T细胞中瞬时转染一系列SFB- RAD51C的删除RAD51C不同区域的突变体,48 h后,收集细胞并裂解,离心后收集到的上清与S琼脂糖珠子孵育后再次离心,再与纯化好的His-mREV1蛋白于低温孵育1 h,结果发现SFB-RAD51C相较于SFB空载,结合更多的REV1蛋白,表明RAD51C和REV1存在明显的相互作用(图3A)。同时,实验结果还表明RAD51C的N端是其结合REV1所必需的。
图3
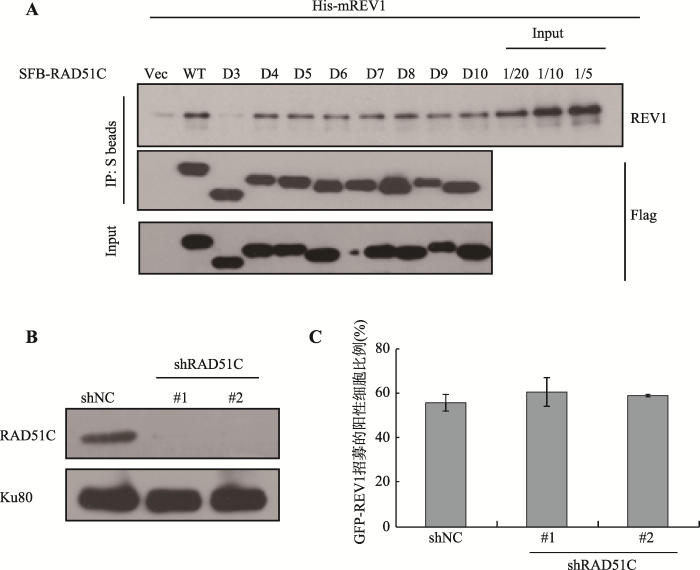 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3RAD51C结合REV1但是不调控REV1的招募
A:REV1和RAD51C存在相互作用。在HEK293T细胞中瞬时转染SFB载体或者SFB-RAD51C一系列删除其不同区域的变体,将蛋白裂解液与S beads孵育后离心,再与纯化好的His-mREV1于低温孵育1 h,检测REV1与前者的相互作用;B:检测稳定细胞系中RAD51C的蛋白水平。利用Western blot检测稳定敲低RAD51C,以及shNC的MRC5单克隆细胞系中RAD51C蛋白的表达情况,Ku80为内参对照;C:检测敲低RAD51C对REV1招募的影响。在稳定细胞系中瞬时转染GFP-REV1,统计GFP-REV1在UVA诱导的DSBs损伤位点处的招募,数据为3次独立的生物学重复的统计结果。
Fig. 3The recruitment of REV1 is independent on RAD51C, although they interact with each other
2.3.2 利用MRC5细胞构建稳定敲低RAD51C的细胞系
将慢病毒包装质粒和不靶向任何基因的质粒shRNA-SHC002或者靶向敲低RAD51C的质粒shRAD51C转染至HEK293T细胞,收集慢病毒上清,分别感染MRC5细胞。感染48 h后,加入puromycin进行药物筛选,以得到稳定敲低RAD51C的单克隆。收集部分单克隆细胞,进行Western blot验证,检测单克隆中RAD51C的表达情况。结果显示,shRAD51C的1#和2#克隆相较于shRNASHC002(即shNC)的1#克隆,RAD51C的表达明显下降,可用于后续实验(图3B)。
2.3.3 在稳定敲低RAD51C的细胞系中检测GFP-REV1的招募情况
在shNC-1#、shRAD51C-1#和-2#三株稳定细胞系中分别瞬时转染GFP-REV1,48 h后,对细胞进行激光显微切割,检测GFP-REV1的招募情况。结果显示,在shNC-1#稳定细胞系中,GFP-REV1的招募比例为59.1%。同时,在RAD51C稳定敲低的两株细胞系中,GFP-REV1的招募比例分别为66.7%和59.3% (图3C)。以上结果表明,REV1在DSBs位点的募集不依赖于RAD51C。
3 讨 论
细胞时刻会受到各种内源或者外源损伤因素的影响,造成基因组DNA的损伤,如果损伤不能被及时修复,会造成复制叉阻滞因而导致更为严重的DNA双链断裂,甚至细胞死亡。跨损伤合成利用TLS聚合酶可以在受损的碱基对侧整合碱基,保证复制继续进行[21]。近些年来,越来越多的证据表明TLS聚合酶也参与其他DNA损伤修复通路[7],其中Y家族聚合酶Polη,REV1都被报道参与调控HR通路[10, 22, 23]。一方面,REV1与Polζ以复合体的形式参与调控HR通路[13]。另一方面,REV1通过其UBM结构域,以依赖RAD18和FANCD2-mUb的方式,被招募至DSBs位点,同时REV1可通过稳定RAD51 filaments保护复制叉处新生链不被降解[14]。然而,REV1是如何被招募至DSBs具体的分子调控机制仍不是非常清楚。RAD51重组酶在HR介导的DSBs修复过程中发挥了重要功能[24]。一方面RAD51介导了复制叉崩解后的HR修复,另一方面它在复制叉短期阻滞后的复制重启中也发挥了重要功能[16]。同时,REV1和Polζ在HR中的共同作用依赖于上游RPA和RAD51[13],暗示着RAD51可能介导REV1在DSBs处的招募。本研究通过敲低RAD51,检测GFP-REV1到DSBs位点的招募,发现敲低RAD51后REV1的招募比例显著下降。同时,在REV1敲低细胞中,也对RAD51在应答双链断裂试剂CPT处理后的募集情况进行了检测。结果显示,在CPT处理后,RAD51在REV1敲低细胞中的募集均明显低于对照组。因此,REV1和RAD51不仅是单向调控的上下游关系,而是存在一个复杂的双向调控[25],即RAD51可调控REV1的招募,而REV1也可以调控RAD51的招募。类似地,以往的研究也支持在DNA损伤应答通路中,许多调控分子的关系都不是简单的单向线型调控,如BRCA2和RAD51彼此依赖,调控相互的蛋白表达水平[26],而PRP19可被ATR介导招募至损伤位点进而促进RPA的多泛素化,从而进一步前馈调控ATR-ATRIP的招募[27]。考虑到REV1与RAD51和BRCA1均具有很强的相互作用[14,28],并且BRCA1/PALB2/BRCA2复合体也调控RAD51的招募[29,30],其中BRCA2是介导RAD51被招募至单链DNA从而起始HR通路的关键蛋白[31,32,33],由此推测在起始HR通路激活过程中,REV1的招募依赖于上游RAD51,而REV1可能通过BRCA1/PALB2/ BRCA2复合体进一步前馈调控RAD51的招募。
RAD51C作为RAD51的同源物,与RAD51的其他同源物协同调控HR通路,并在HR通路的早期和晚期均发挥功能[34]。研究表明,在鸡DT40细胞和仓鼠细胞里,敲低RAD51C会导致RAD51的募集以及HR修复效率均减少[34]。因此,本研究首先证明了RAD51C和REV1存在相互作用,进一步在人源细胞中敲低RAD51C,检测了REV1的招募情况,结果却显示REV1的招募并不受RAD51C调控。关于RAD51C与REV1互作的生物学意义仍有待进一步研究。
本研究进一步加深了人们对REV1在HR调控通路中的作用机制的理解,即REV1和RAD51在HR通路中存在彼此相互调控的关系。RAD51调控REV1的招募,而REV1可能是通过某些蛋白,如BRCA1/PALB2/BRCA2复合体,进一步前馈调控RAD51的招募,但是具体两者间是如何调控仍需深入研究。
致谢
感谢英国萨塞克斯大学Alan Lehmann博士提供MRC5细胞;感谢浙江大学黄俊博士提供RAD51C质粒。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
.
URLPMID:22358330 [本文引用: 3]

Abstract The past 15 years have seen an explosion in our understanding of how cells replicate damaged DNA and how this can lead to mutagenesis. The Y-family DNA polymerases lie at the heart of this process, which is commonly known as translesion synthesis. This family of polymerases has unique features that enable them to synthesize DNA past damaged bases. However, as they exhibit low fidelity when copying undamaged DNA, it is essential that they are only called into play when they are absolutely required. Several layers of regulation ensure that this is achieved.
.
URL [本文引用: 2]
URLPMID:28279077 [本文引用: 1]
Life as we know it, simply would not exist without DNA replication. All living organisms utilize a complex machinery to duplicate their genomes and the central role in this machinery belongs to replicative DNA polymerases, enzymes that are specifically designed to copy DNA. 090008Hassle-free090009 DNA duplication exists only in an ideal world, while in real life, it is constantly threatened by a myriad of diverse challenges. Among the most pressing obstacles that replicative polymerases often cannot overcome by themselves are lesions that distort the structure of DNA. Despite elaborate systems that cells utilize to cleanse their genomes of damaged DNA, repair is often incomplete. The persistence of DNA lesions obstructing the cellular replicases can have deleterious consequences. One of the mechanisms allowing cells to complete replication is 090008Translesion DNA Synthesis (TLS)090009. TLS is intrinsically error-prone, but apparently, the potential downside of increased mutagenesis is a healthier outcome for the cell than incomplete replication. Although most of the currently identified eukaryotic DNA polymerases have been implicated in TLS, the best characterized are those belonging to the 090008Y-family090009 of DNA polymerases (pols 02·, 0201, 0202 and Rev1), which are thought to play major roles in the TLS of persisting DNA lesions in coordination with the B-family polymerase, pol 0209. In this review, we summarize the unique features of these DNA polymerases by mainly focusing on their biochemical and structural characteristics, as well as potential protein090009protein interactions with other critical factors affecting TLS regulation.
.
URLPMID:4018060 [本文引用: 1]
Abstract Y-Family DNA polymerases specialize in translesion synthesis, bypassing damaged bases that would otherwise block the normal progression of replication forks. Y-Family polymerases have unique structural features that allow them to bind damaged DNA and use a modified template base to direct nucleotide incorporation. Each Y-Family polymerase is unique and has different preferences for lesions to bypass and for dNTPs to incorporate. Y-Family polymerases are also characterized by a low catalytic efficiency, a low processivity, and a low fidelity on normal DNA. Recruitment of these specialized polymerases to replication forks is therefore regulated. The catalytic center of the Y-Family polymerases is highly conserved and homologous to that of high-fidelity and high-processivity DNA replicases. In this review, structural differences between Y-Family and A- and B-Family polymerases are compared and correlated with their functional differences. A time-resolved X-ray crystallographic study of the DNA synthesis reaction catalyzed by the Y-Family DNA polymerase human polymerase 畏 revealed transient elements that led to the nucleotidyl-transfer reaction.
.
URLPMID:29208956 [本文引用: 1]
The dominant polyglutamine (polyQ) disorders are a group of progressive and incurable neurodegenerative disorders, which are caused by unstable expanded CAG trinucleotide repeats in the coding regions of their respective causative genes. The most prevalent polyQ disorders worldwide are Huntington's disease and spinocerebellar ataxia type 3. Epigenetic mechanisms, such as DNA methylation,... [Show full abstract]
.
URLPMID:29590477 [本文引用: 1]
Abstract Translesion DNA synthesis (TLS) is one mode of DNA damage tolerance that uses specialized DNA polymerases to replicate damaged DNA. DNA polymerase 02· (Pol02·) is well known to facilitate TLS across ultraviolet (UV) irradiation and mutations in POLH are implicated in skin carcinogenesis. However, the basis for recruitment of Pol02· to stalled replication forks is not completely understood. In this study, we used an affinity purification approach to isolate a Pol02·-containing complex and have identified SART3, a pre-mRNA splicing factor, as a critical regulator to modulate the recruitment of Pol02· and its partner RAD18 after UV exposure. We show that SART3 interacts with Pol02· and RAD18 via its C-terminus. Moreover, SART3 can form homodimers to promote the Pol02·/RAD18 interaction and PCNA monoubiquitination, a key event in TLS. Depletion of SART3 also impairs UV-induced single-stranded DNA (ssDNA) generation and RPA focus formation, resulting in an impaired Pol02· recruitment and a higher mutation frequency and hypersensitivity after UV treatment. Notably, we found that several SART3 missense mutations in cancer samples lessen its stimulatory effect on PCNA monoubiquitination. Collectively, our findings establish SART3 as a novel Pol02·/RAD18 association regulator that protects cells from UV-induced DNA damage, which functions in a RNA binding-independent fashion.
.
URLPMID:17188030 [本文引用: 2]
The Y family of DNA polymerases plays crucial roles in carrying out translesion synthesis past damaged bases in DNA. Several recent papers suggest that they might have other roles as well in gene conversion, in nucleotide excision repair (NER), and in DNA replication under stressed conditions.
.
URLPMID:3636179 [本文引用: 1]
The Y-family of DNA polymerases support of translesion DNA synthesis (TLS) associated with stalled DNA replication by DNA damage. Recently, a number of studies suggest that some specialized TLS polymerases also support other aspects of DNA metabolism beyond TLS in vivo. Here we show that mouse polymerase kappa (Pol kappa) could accumulate at laser-induced sites of damage in vivo resembling polymerases eta and iota. The recruitment was mediated through Pol kappa C-terminus which contains the PCNA-interacting peptide, ubiquitin zinc finger motif 2 and nuclear localization signal. Interestingly, this recruitment was significantly reduced in MSH2-deficient LoVo cells and Rad18-depleted cells. We further observed that Pol kappa-deficient mouse embryo fibroblasts were abnormally sensitive to H2O2 treatment and displayed defects in both single-strand break repair and double-strand break repair. We speculate that Pol kappa may have an important role in strand break repair following oxidative stress in vivo. (C) 2013 Elsevier B.V. All rights reserved.
.
URLPMID:16337601 [本文引用: 2]
Stalled replication forks pose a serious threat to genome integrity. To overcome the catastrophic consequences associated with fork demise, translesion synthesis (TLS) polymerases such as polη promote DNA synthesis past lesions. Alternatively, a stalled fork may collapse and undergo repair by homologous recombination. By using fractionated cell extracts and purified recombinant proteins, we show that polη extends DNA synthesis from D loop recombination intermediates in which an invading strand serves as the primer. Extracts from XP-V cells, which are defective in polη, exhibit severely reduced D loop extension activity. The D loop extension activity of polη is unusual, as this reaction cannot be promoted by the replicative DNA polymerase δ or by other TLS polymerases such as polι. Moreover, we find that polη interacts with RAD51 recombinase and RAD51 stimulates polη-mediated D loop extension. Our results indicate a dual function for polη at stalled replication forks: the promotion of translesion synthesis and the reinitiation of DNA synthesis by homologous recombination repair.
.
URLPMID:1168817 [本文引用: 2]
In yeast, Rev1, Rev3, and Rev7 are involved in translesion synthesis over various kinds of DNA damage and spontaneous and UV-induced mutagenesis. Here, we disrupted Rev1, Rev3, and Rev7 in the chicken B-lymphocyte line DT40. REV1-/- REV3-/- REV7-/- cells showed spontaneous cell death, chromosomal instability/fragility, and hypersensitivity to various genotoxic treatments as observed in each of the single mutants. Surprisingly, the triple-knockout cells showed a suppressed level of sister chromatid exchanges (SCEs), which may reflect postreplication repair events mediated by homologous recombination, while each single mutant showed an elevated SCE level. Furthermore, REV1-/- cells as well as triple mutants showed a decreased level of immunoglobulin gene conversion, suggesting participation of Rev1 in a recombination-based pathway. The present study gives us a new insight into cooperative function of three Rev molecules and the Polzeta (Rev3-Rev7)-independent role of Rev1 in vertebrate cells.
.
URLPMID:16546083 [本文引用: 1]
DNA polymerase zeta (Polζ) and Rev1 contribute to the bypassing of DNA lesions, termed translesion DNA synthesis (TLS) . Polζ consists of two subunits, one encoded by (the catalytic subunit) and the other encoded by . Rev1 acts as a deoxycytidyl transferase, inserting dCMP opposite lesions. Polζ and Rev1 have been shown to operate in the same TLS pathway in the budding yeast [2]. Here, we show that budding yeast Polζ and Rev1 form a complex and associate together with double-strand breaks (DSBs). As a component of the Polζ-Rev1 complex, Rev1 plays a noncatalytic role in the association with DSBs. In budding yeast, the ATR-homolog Mec1 plays a central role in the DNA-damage checkpoint response . We further show that Mec1-dependent phosphorylation promotes the Polζ-Rev1 association with DSBs. Rev1 association with DSBs requires neither the function of the Rad24 checkpoint-clamp loader [5] nor the Rad6-Rad18-mediated ubiquitination of PCNA [3]. Our results reveal a novel role of Mec1 in the localization of the Polζ-Rev1 complex to DNA lesions and highlight a linkage of TLS polymerases to the checkpoint response.
URLPMID:3330096 [本文引用: 1]
Abstract In metazoans, the mechanism by which DNA is synthesized during homologous recombination repair of double-strand breaks is poorly understood. Specifically, the identities of the polymerase(s) that carry out repair synthesis and how they are recruited to repair sites are unclear. Here, we have investigated the roles of several different polymerases during homologous recombination repair in Drosophila melanogaster. Using a gap repair assay, we found that homologous recombination is impaired in Drosophila lacking DNA polymerase zeta and, to a lesser extent, polymerase eta. In addition, the Pol32 protein, part of the polymerase delta complex, is needed for repair requiring extensive synthesis. Loss of Rev1, which interacts with multiple translesion polymerases, results in increased synthesis during gap repair. Together, our findings support a model in which translesion polymerases and the polymerase delta complex compete during homologous recombination repair. In addition, they establish Rev1 as a crucial factor that regulates the extent of repair synthesis.
.
URLPMID:21926160 [本文引用: 3]
REV1 and DNA Polymerase ζ (REV3 and REV7) play important roles in translesion DNA synthesis (TLS) in which DNA replication bypasses blocking lesions. REV1 and Polζ have also been implicated in promoting repair of DNA double-stranded breaks (DSBs). However, the mechanism by which these two TLS polymerases increase tolerance to DSBs is poorly understood. Here we demonstrate that full-length human REV1, REV3 and REV7 interact in vivo (as determined by co-immunoprecipitation studies) and together, promote homologous recombination repair. Cells lacking REV3 were hypersensitive to agents that cause DSBs including the PARP inhibitor, olaparib. REV1, REV3 or REV7-depleted cells displayed increased chromosomal aberrations, residual DSBs and sites of HR repair following exposure to ionizing radiation. Notably, cells depleted of DNA polymerase η (Polη) or the E3 ubiquitin ligase RAD18 were proficient in DSB repair following exposure to IR indicating that Polη-dependent lesion bypass or RAD18-dependent monoubiquitination of PCNA are not necessary to promote REV1 and Polζ-dependent DNA repair. Thus, the REV1/Polζ complex maintains genomic stability by directly participating in DSB repair in addition to the canonical TLS pathway.
.
URLPMID:26187992 [本文引用: 3]
REV1 is a eukaryotic member of the Y-family of DNA polymerases involved in translesion DNA synthesis and genome mutagenesis. Recently, REV1 is also found to function in homologous recombination. However, it remains unclear how REV1 is recruited to the sites where homologous recombination is processed. Here, we report that loss of mammalian REV1 results in a specific defect in replication-associated gene conversion. We found that REV1 is targeted to laser-induced DNA damage stripes in a manner dependent on its ubiquitin-binding motifs, on RAD18, and on monoubiquitinated FANCD2 (FANCD2-mUb) that associates with REV1. Expression of a FANCD2-Ub chimeric protein in RAD18-depleted cells enhances REV1 assembly at laser-damaged sites, suggesting that FANCD2-mUb functions downstream of RAD18 to recruit REV1 to DNA breaks. Consistent with this suggestion we found that REV1 and FANCD2 are epistatic with respect to sensitivity to the double-strand break-inducer camptothecin. REV1 enrichment at DNA damage stripes also partially depends on BRCA1 and BRCA2, components of the FANCD2/BRCA supercomplex. Intriguingly, analogous to FANCD2-mUb and BRCA1/BRCA2, REV1 plays an unexpected role in protecting nascent replication tracts from degradation by stabilizing RAD51 filaments. Collectively these data suggest that REV1 plays multiple roles at stalled replication forks in response to replication stress.
.
URLPMID:22761336 [本文引用: 1]
Cell survival after DNA damage relies on DNA repair, the abrogation of which causes genomic instability. The DNA repair protein RAD51 and the trans-lesion synthesis DNA polymerase REV1 are required for resistance to DNA interstrand cross-linking agents such as cisplatin. In this study, we show that overexpression of miR-96 in human cancer cells reduces the levels of RAD51 and REV1 and impacts the cellular response to agents that cause DNA damage. MiR-96 directly targeted the coding region of RAD51 and the 3′-untranslated region of REV1. Overexpression of miR-96 decreased the efficiency of homologous recombination and enhanced sensitivity to the PARP inhibitor AZD2281 in vitro and to cisplatin both in vitro and in vivo. Taken together, our findings indicate that miR-96 regulates DNA repair and chemosensitivity by repressing RAD51 and REV1. As a therapeutic candidate, miR-96 may improve chemotherapeutic efficacy by increasing the sensitivity of cancer cells to DNA damage. Cancer Res; 72(16); 4037–46. 082012 AACR.
.
URL [本文引用: 2]
.
URLPMID:26795561 [本文引用: 1]
Translesion DNA synthesis (TLS) is a mode of DNA damage tolerance which plays an important role in genome mutagenesis and chromatin integrity maintenance. Proliferating cell nuclear antigen (PCNA) monoubiquitylation is one of the key factors for TLS pathway choice. So far, it remains unclear how the TLS pathway is elaborately regulated. Here, we report that TLS polymerase REV1 can promote PCNA monoubiquitylation after UV radiation. Further studies revealed that this stimulatory effect is mediated through the enhanced interaction between REV1 and ubiquitylated RAD18, which facilitates the release of nonubiquitylated RAD18 from ubiquitylated RAD18 trapping, after which RAD18 is recruited to chromatin for its TLS function. Furthermore, we found that this stimulatory effect could also be detected after exposure to hydroxyurea or mitomycin C, but not methyl methanesulfonate (MMS), which is in line with the fact that ubiquitylated RAD18 could not be detected after exposure to MMS.
.
URLPMID:25176633 [本文引用: 1]
Homologous recombination repair is initiated by nuclease-mediated DNA-end resection. Here, Dong et al. show that the human SRCAP chromatin remodeling complex facilitates CtIP-dependent DSB resection, on one hand, through a direct SRCAP-CtIP physical interaction and on the other hand via its role in promoting chromatin relaxation.
.
URLPMID:19396164 [本文引用: 1]
To maintain genome stability, cells respond to DNA damage by activating signaling pathways that govern cell cycle checkpoints and initiate DNA repair. Cell cycle checkpoint controls should somehow connect with DNA repair processes, however, exactly how such coordination occursin vivois largely unknown. Here we revealed a novel role of RAD18 as the integral component that translates the damage response signal to orchestrate homologous recombination (HR) repair. We show that RAD18 promotes HR in a manner strictly dependent upon its ability to be recruited to the sites of DNA breaks and this recruitment relies on a well-defined DNA damage-signaling pathway mediated by another E3 ligase RNF8. We further demonstrate that RAD18 functions as an adaptor to facilitate HR via a direct interaction with RAD51C. Together, our data uncovers RAD18 as a key factor that orchestrates HR repair via surveillance of the DNA damage signal.
.
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
.
URL [本文引用: 1]
.
URLPMID:16337602 [本文引用: 1]
Chicken B lymphocyte precursors and DT40 cells diversify their immunoglobulin-variable (IgV) genes through homologous recombination (HR)-mediated Ig gene conversion. To identify DNA polymerases that are involved in Ig gene conversion, we created DT40 clones deficient in DNA polymerase η (polη), which, in humans, is defective in the variant form of xeroderma pigmentosum (XP-V). Polη is an error-prone translesion DNA synthesis polymerase that can bypass UV damage-induced lesions and is involved in IgV hypermutation. Like XP-V cells, polη-disrupted ( polη) clones exhibited hypersensitivity to UV. Remarkably, polη cells showed a significant decrease in the frequency of both Ig gene conversion and double-strand break-induced HR when compared to wild-type cells, and these defects were reversed by complementation with human polη. Our findings identify a DNA polymerase that carries out DNA synthesis for physiological HR and provides evidence that a single DNA polymerase can play multiple cellular roles.
.
URL [本文引用: 1]
.
URLPMID:3127162 [本文引用: 1]
Camptothecin (CPT) and related chemotherapeutic drugs induce formation of DNA Topoisomerase I (Top1) covalent or cleavage complexes (Top1ccs) that block leading-strand DNA synthesis and elicit DNA Double Stranded Breaks (DSB) during S phase. The Fanconi Anemia (FA) pathway is implicated in tolerance of CPT-induced DNA damage yet the mechanism of FA pathway activation by Top1 poisons has not been studied. We show here that the FA core complex protein FANCA and monoubiquitinated FANCD2 (an effector of the FA pathway) are rapidly mobilized to chromatin in response to CPT treatment in several human cancer cell lines and untransformed primary human dermal fibroblasts. FANCD2 depletion using siRNA leads to impaired recovery from CPT-induced inhibition or DNA synthesis, persistence of H2AX (a DSB marker) and reduced cell survival following CPT treatment. The E3 ubiquitin ligase Rad18 is necessary for CPT-induced recruitment of FANCA and FANCD2 to chromatin. Moreover, Rad18-depletion recapitulates the DNA synthesis and survival defects of FANCD2-deficiency in CPT-treated cells. It is well-established that Rad18 promotes FA pathway activation and DNA damage tolerance in response to bulky DNA lesions via a mechanism involving PCNA monoubiquitination. In contrast, PCNA monoubiquitination is not involved in Rad18-mediated FA pathway activation or cell survival following acquisition of CPT-induced DSB. Moreover, while Rad18 is implicated in recombinational repair of DSB via an E3 ligase-independent mechanism, we demonstrate that Rad18 E3 ligase activity is essential for appropriate FA pathway activation and DNA damage tolerance after CPT treatment. Taken together, our results define a novel pathway of Rad18-dependent DSB repair that is dissociable from known Rad18-mediated DNA repair mechanisms based on its independence from PCNA ubiquitination and requirement for E3 ligase activity.
.
URL [本文引用: 1]
.
URLPMID:24332808 [本文引用: 1]
61PRP19 is identified as a sensor of RPA-ssDNA from a proteomic screen61PRP19 recognizes DNA damage via its interaction with RPA61PRP19 regulates ATR activation as a ubiquitin ligase61PRP19 promotes RPA ubiquitylation and ATRIP recruitment after DNA damage
.
URLPMID:23901102 [本文引用: 1]
Breast cancer gene 1 (BRCA1) deficient cells not only are hypersensitive to double-strand breaks but also are hypersensitive to UV irradiation and other agents that cause replication blockade; however, the molecular mechanisms behind these latter sensitivities are largely unknown. Here, we report that BRCA1 promotes cell survival by directly regulating the DNA damage tolerance pathway in response to agents that create cross-links in DNA. We show that BRCA1 not only promotes efficient mono- and polyubiquitination of proliferating cell nuclear antigen (PCNA) by regulating the recruitment of replication protein A, Rad18, and helicase-like transcription factor to chromatin but also directly recruits translesion polymerases, such as Polymerase eta and Rev1, to the lesions through protein-protein interactions. Our data suggest that BRCA1 plays a critical role in promoting translesion DNA synthesis as well as DNA template switching.
.
URLPMID:2750839 [本文引用: 1]
BRCA1 and BRCA2 are often mutated in familial breast and ovarian cancer. Both tumor suppressors play key roles in the DNA-damage response . However, it remains unclear whether these two tumor suppressor function together in the same DNA-damage response pathway. Here, we show that BRCA1 associates with BRCA2 through PALB2/FANCN, a major binding partner of BRCA2 [3]. The interaction between BRCA1 and BRCA2 is abrogated in PALB2-deficient Fanconi anemia cells and in the cells depleted of PALB2 by small interfering RNA. Moreover, we show that BRCA1 promotes the concentration of PALB2 and BRCA2 at DNA-damage sites and the interaction between BRCA1 and PALB2 is important for the homologous recombination repair. Taken together, our results indicate that BRCA1 is an upstream regulator of BRCA2 in the DNA-damage response, and PALB2 is the linker between BRCA1 and BRCA2.
.
URL [本文引用: 1]
Mutations in breast cancer susceptibility gene 1 and 2 (BRCA1 and BRCA2) predispose individuals to breast and ovarian cancer development. We previously reported an in vivo interaction between BRCA1 and BRCA2. However, the biological significance of their association is thus far undefined. Here, we report that PALB2, the partner and localizer of BRCA2, binds directly to BRCA1, and serves as the molecular scaffold in the formation of the BRCA1-PALB2-BRCA2 complex. The association between BRCA1 and PALB2 is primarily mediated via apolar bonding between their respective coiled-coil domains. More importantly, BRCA1 mutations identified in cancer patients disrupted the specific interaction between BRCA1 and PALB2. Consistent with the converging functions of the BRCA proteins in DNA repair, cells harboring mutations with abrogated BRCA1-PALB2 interaction resulted in defective homologous recombination (HR) repair. We propose that, via its direct interaction with PALB2, BRCA1 fine-tunes recombinational repair partly through its modulatory role in the PALB2-dependent loading of BRCA2-RAD51 repair machinery at DNA breaks. Our findings uncover PALB2 as the molecular adaptor between the BRCA proteins, and suggest that impaired HR repair is one of the fundamental causes for genomic instability and tumorigenesis observed in patients carrying BRCA1, BRCA2, or PALB2 mutations.
.
URLPMID:28698210 [本文引用: 1]
Abstract The apolipoprotein B mRNA editing enzyme catalytic polypeptide-like APOBEC3A and 3B have emerged as key mutation drivers in cancer. Here we show that APOBEC3A and 3B activities impose a unique type of replication stress by inducing abasic sites at replication forks. In contrast to cells under other types of replication stress, APOBEC3A-expressing cells were selectively sensitive to ATR inhibitors (ATRi), but not to a variety of DNA replication inhibitors and DNA-damaging drugs. In proliferating cells, APOBEC3A modestly elicited ATR but not ATM. ATR inhibition in APOBEC3A-expressing cells resulted in a surge of abasic sites at replication forks, revealing an ATR-mediated negative feedback loop during replication. The surge of abasic sites upon ATR inhibition associated with increased accumulation of single-stranded DNA, a substrate of APOBEC3A, triggering an APOBEC3A-driven feedforward loop that ultimately drove cells into replication catastrophe. In a panel of cancer cell lines, ATRi selectively induced replication catastrophe in those harboring high APOBEC3A and/or 3B activities, showing that APOBEC3A and 3B activities conferred susceptibility to ATRi. Our results define an APOBEC-driven replication stress in cancer cells that may offer an opportunity for ATR-targeted therapy. Copyright 2017, American Association for Cancer Research.
.
URLPMID:17549079 [本文引用: 1]
Nature Structural & Molecular Biology is an integrated forum for structural and molecular studies. The journal places a strong emphasis on functional and mechanistic understanding of how molecular components in a biological process work together. Structural data may provide such insights, but they are not a pre-requisite for publication in the journal.
.
URL [本文引用: 1]
.
URLPMID:2994284 [本文引用: 2]
Germline mutations in many of the genes that are involved in homologous recombination (HR)-mediated DNA double-strand break repair (DSBR) are associated with various human genetic disorders and cancer. RAD51 and RAD51 paralogs are important for HR and in the maintenance of genome stability. Despite the identification of five RAD51 paralogs over a decade ago, the molecular mechanism(s) by which RAD51 paralogs regulate HR and genome maintenance remains obscure. In addition to the known roles of RAD51C in early and late stages of HR, it also contributes to activation of the checkpoint kinase CHK2. One recent study identifies biallelic mutation in RAD51C leading to Fanconi anemia-like disorder. Whereas a second study reports monoallelic mutation in RAD51C associated with increased risk of breast and ovarian cancer. These reports show RAD51C is a cancer susceptibility gene. In this review, we focus on describing the functions of RAD51C in HR, DNA damage signaling and as a tumor suppressor with an emphasis on the new roles of RAD51C unveiled by these reports.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}