 ,2,3,*, 王艇,1,*
,2,3,*, 王艇,1,*Intron Loss and Molecular Evolution Rate of rpoC1 in Ferns
Yang Peng1, Yingjuan Su,2,3,*, Ting Wang,1,*通讯作者:
责任编辑: 白羽红
收稿日期:2019-06-6接受日期:2020-03-24网络出版日期:2020-05-01
| 基金资助: |
Corresponding authors:
Received:2019-06-6Accepted:2020-03-24Online:2020-05-01

摘要
关键词:
Abstract
Keywords:
PDF (1410KB)摘要页面多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
引用本文
彭阳, 苏应娟, 王艇. 蕨类植物rpoC1内含子缺失及其分子进化速率. 植物学报, 2020, 55(3): 287-298 doi:10.11983/CBB19105
Peng Yang, Su Yingjuan, Wang Ting.
rpoC1 (RNA polymerase beta' subunit)基因编码RNA聚合酶β°亚基, 属于遗传系统基因(genetic system genes)。在转录过程中, RNA聚合酶β°亚基与DNA模板结合, 与β-β°复合体共同构成RNA合成的催化中心。因此rpoC1基因在转录过程中具有重要作用。内含子在调节基因表达和可变剪接(alternative splicing)中扮演重要角色, 一个基因可以因此而编码多种不同的蛋白质。研究发现, 芽殖酵母(Saccharomyces cerevisiae)基因组中内含子的存在促进饥饿条件下细胞存活, 其通过抑制TORC1和PKA途径核糖体蛋白基因的营养感应来增强对饥饿的抗性(Parenteau et al., 2019; Morgan et al., 2019)。rpoC1基因内含子丢失发生在许多谱系中, 被认为是仙人掌亚科(Cactoideae) (Wallace and Cota, 1996)与番杏科(Aizoaceae)舟叶花族和弥生花族(Thiede et al., 2007)的一种近裔共性(synapomorphy)特征。
蕨类植物是维管植物的第二大类群, 现存约12 000种, 主要分布于中低海拔的热带、亚热带地区。蕨类植物起源于距今约3.59亿年的泥盆纪, 经历过3次“物种辐射”(Lovis, 1977; Rothwel, 1987)。本研究的海金沙属(Lygodium)是海金沙科的基部类群(Schuettpelz and Pryer, 2007)。海金沙属为陆生攀缘植物, 根状茎有毛而无鳞片, 叶呈单轴型, 叶脉通常分离, 孢子囊生于小脉顶端且形如梨状, 孢子为四面体。目前, 海金沙属包含约45种, 中国有10种(中国科学院中国植物志编辑委员会, 1959)。最古老的海金沙化石源于侏罗纪时期(Taylor et al., 2009)。蕨类植物rpoC1基因绝大部分位于叶绿体基因组大单拷贝区, 大小为2 800 bp左右。Kim等(2014)检测了有限的蕨类样本, 发现在蕨类植物中rpoC1基因内含子丢失仅发生在海金沙属中。经统计, 本研究的64种蕨类植物仅海金沙属内含子缺失。
分子进化速率最早由Zuckerkandl和Pauling (1962)提出, 进化速率一般定义为单位时间内分子在每个位点上(核苷酸或氨基酸)发生的替换数。对于基因的编码区和非编码区而言, 它们具有不同的核苷酸替换模式。非编码区由于其核苷酸的替换对翻译后的氨基酸序列影响较小, 故一般直接采用总核苷酸的替换速率作为其替换速率; 编码区则将替换速率分为两种类型: 同义替换率(synonymous substitution rate, dS)和非同义替换率(nonsynonymous substitution rate, dN)。通过比较每一个非同义位点上的非同义替换率和每一个同义位点上的同义替换率可以揭示正选择或负选择作用(positive or negative selection)。若dS大于dN, 说明该DNA处于负选择作用下; 若dN大于dS, 说明该DNA处于正选择作用下。在核苷酸水平上, 替换率包括转换率(transition rates)和颠换率(transversion rates)。一般情况下, 核苷酸转换率大于颠换率。
基于构建的系统发育树和密码子替换模型可对不同支系和不同氨基酸位点的适应性进化进行极大似然法检测。由于不同氨基酸位点功能可能有所不同, 故不同位点选择压力存在差异。Yang (1998)提出检测适应性进化的3种模型, 即分支模型(branch model)、位点模型(site model)和分支-位点模型(branch-site model)。这3种模型均通过似然比检验(likelihood ratio test, LRT)检测不同模型之间的差异, 然后进行显著性分析, 从而确定选择模型。
目前, 关于蕨类植物rpoC1基因内含子丢失的研究相对较少, 其对分子进化速率的影响尚不明确。本研究通过检测rpoC1基因的选择位点并分析rpoC1基因的替换速率, 解析rpoC1基因内含子丢失与分子进化速率的关系。
1 材料与方法
1.1 序列数据
选取包含25个科的64种蕨类植物(表1), 其序列数据来自NCBI (National Center for Biotechnology Information)的GenBank数据库(https://www.ncbi.nlm. nih.gov/)。除毛杆蕨(Callistopteris apiifolia)、Ceratopteris richardii和Polypodium glycyrrhiza叶绿体基因组数据不全外, 其余61种蕨类植物均选取叶绿体基因组全序列。此外, 为获得海金沙(Lygodium japonicum)以及南国田字草(Marsilea crenata)的叶绿体基因组数据, 于中国科学院武汉植物园采集样本, 使用DNeasy Plant Mini Kit (QIAGEN)试剂盒提取幼叶总DNA, 并通过HiSeq 2000测序系统(Illumina)测序(Gao et al., 2013)。将测序、组装和注释完成的序列上传至NCBI。Table 1
表1
表1植物材料名称和叶绿体基因组序列GenBank登录号
Table 1
| Family name | Species | GenBank accession number | Length (bp) |
|---|---|---|---|
| Aspleniaceae | Asplenium pekinense | KY427331 | 152479 |
| As. prolongatum | KY427332 | 151115 | |
| Hymenasplenium unilaterale | KY427350 | 151723 | |
| Athyriaceae | Athyrium anisopterum | NC_035738 | 151284 |
| At. opacum | KY427335 | 150979 | |
| At. sheareri | KY427330 | 151068 | |
| At. sinense | KY427333 | 151319 | |
| Deparia lancea | KY427338 | 151011 | |
| De. pycnosora | KY427339 | 151126 | |
| De. viridifrons | KY427340 | 150939 | |
| Diplazium bellum | KY427343 | 151601 | |
| Di. dilatatum | KY427344 | 151114 | |
| Di. dushanense | KY427345 | 150179 | |
| Di. striatum | KY427346 | 150779 | |
| Di. unilobum | KY427347 | 127840 | |
| Blechnaceae | Austroblechnum melanocaulon | KY427334 | 150202 |
| Woodwardia unigemmata | NC_028543 | 153717 | |
| Cibotiaceae | Cibotium barometz | MH105066 | 166027 |
| Cyatheaceae | Alsophila podophylla | MG262389 | 166151 |
| Al. spinulosa | NC_012818 | 156661 | |
| Cystopteridaceae | Cystopteris chinensis | KY427337 | 151269 |
| Family name | Species | GenBank accession number | Length (bp) |
| Dennstaedtiaceae | Pteridium aquilinum | NC_014348 | 152362 |
| Diplaziopsidaceae | Diplaziopsis cavaleriana | KY427341 | 151934 |
| Dip. javanica | KY427342 | 151496 | |
| Homalosorus pycnocarpos | KY427349 | 152159 | |
| Dryopteridaceae | Cyrtomium devexiscapulae | NC_028542 | 151684 |
| C. falcatum | NC_028705 | 151628 | |
| C. fortunei | MG913607 | 151699 | |
| Dryopteris decipiens | KY427348 | 150987 | |
| Dr. fragrans | KX418656 | 151978 | |
| Equisetaceae | Equisetum arvense | NC_014699 | 133309 |
| E. hyemale | NC_020146 | 131760 | |
| Hymenophyllaceae | Callistopteris apiifolia | MH265125 | 144918 |
| Hymenophyllum holochilum | MH265124 | 142214 | |
| Vandenboschia speciosa | NC_041000 | 146874 | |
| Hypodematiaceae | Hypodematium crenatum | KY427351 | 149794 |
| Lygodiaceae | Lygodium japonicum | NC_022136 | 157260 |
| L. microphyllum | NC_039378 | 158891 | |
| Marattiaceae | Angiopteris angustifolia | NC_026300 | 153596 |
| An. evecta | NC_008829 | 153901 | |
| Marsileaceae | Marsilea crenata | NC_022137 | 151628 |
| Pilularia americana | KY863504 | 153076 | |
| Onocleaceae | Matteuccia struthiopteris | KY427353 | 151003 |
| Onoclea sensibilis | KY427354 | 148395 | |
| Ophioglossaceae | Botrychium ternatum | KM817789 | 139127 |
| Helminthostachys zeylanica | KM817788 | 145120 | |
| Mankyua chejuensis | NC_017006 | 146221 | |
| Osmundaceae | Osmundastrum cinnamomeum | NC_024157 | 142812 |
| Polypodiaceae | Lepisorus clathratus | NC_035739 | 156998 |
| Polypodium glycyrrhiza | KP136832 | 129223 | |
| Psilotaceae | Psilotum nudum | NC_003386 | 138829 |
| Pteridaceae | Adiantum shastense | MG432483 | 150414 |
| Ceratopteris richardii | KM052729 | 148444 | |
| Myriopteris lindheimeri | NC_014592 | 155770 | |
| Rhachidosoraceae | Rhachidosorus consimilis | KY427356 | 152642 |
| Schizaeaceae | Schizaea elegans | KX258660 | 156603 |
| S. pectinata | KX258661 | 156392 | |
| Thelypteridaceae | Ampelopteris prolifera | KY427329 | 151772 |
| Christella appendiculata | NC_035842 | 151571 | |
| Macrothelypteris torresiana | KY427352 | 151130 | |
| Pseudophegopteris aurita | KY427355 | 149917 | |
| Stegnogramma sagittifolia | KY427357 | 151132 | |
| Woodsiaceae | Woodsia macrochlaena | KY427358 | 150987 |
| W. polystichoides | KY427359 | 150685 |
新窗口打开|下载CSV
将从NCBI下载的叶绿体基因组全序列或部分序列数据导入Genious Prime 2019软件中, 分别提取rbcL、matK及rpoC1基因序列。用BioEdit7.0.5.3软件(Hall, 1999) Accessory Application模块中的ClustalW Multiple Alignment进行序列多重比对, 并手工校正(附录1)。分别建立64种蕨类植物的rbcL数据集、matK数据集、rbcL与matK的串联数据集以及rpoC1数据集。其中, rpoC1数据集包括rpoC1基因数据集、rpoC1外显子数据集和rpoC1氨基酸数据集。
1.2 系统发育树的构建
根据已有的系统发育树, 对rbcL编码序列(coding sequence, CDS)与matK编码序列的串联数据集用最大似然法(maximum likelihood, ML)构建的系统发育树进行手工校正, 并根据Smith等(2006)和The Pteridophyte Phylogeny Group (2016)的系统发育树对该树进行部分修改。该树将用于后续分子进化分析。用jModelTest2.1.10软件(Darriba et al., 2012)进行核苷酸替换模型选择。根据赤池信息准则(Akaike Information Criterion, AIC)选取最佳模型(TVM+I+G) (附录2) (Akaike, 1974)。在PhyML-3.1_win32.exe (Guindon et al., 2010)应用程序中导入已比对完成的序列文件, 设置最佳模型的相关参数。其中, 自展值设为1 000, C值设为6, 然后运行程序。用FigTree v1.4.3软件对已构建的系统发育树进行编辑和美化。
1.3 分子进化速率分析
使用HyPhy2.2.4 (hypothesis testing using phylogenies)软件分析海金沙属和其它62种蕨类植物的rpoC1外显子的转换率(transition rate, trst)、颠换率(transversion rate, trsv)、转换率/颠换率(trst/trsv)、替换数(number of substitutions, Es)、转换数(number of transition, Et)、颠换数(number of transversion, Ev)、同义替换率(synonymous substitution rate, dS)、非同义替换率(nonsynonymous substitution rate, dN)以及dN/dS (Pond and Muse, 2005)。转换率、颠换率以及两者比值的分析用局部参数下核苷酸HKY85替换模型; 同义替换率、非同义替换率以及ω值(dN/dS)分析选用局部参数下密码子MG94× HKY85_3×4模型。将dN和dS分别约束, 分析不同约束条件下的似然比检验(LRT)所得到的P值, 探究海金沙属rpoC1基因的替换速率是否明显大于其它蕨类植物。使用SPSS16软件中的曼-惠特尼(Mann-Whitney)秩和检验对实验统计结果进行差异显著性分析, 比较海金沙属和其它蕨类植物在替换速率方面的差异。
1.4 选择压力分析
使用HyPhy软件中的密码子MG94×HKY85×3_4×2_ Rates模型, 通过设定Rate Het参数以及调用类别处理器(categories processor)执行后验贝叶斯(posterior Bayes)分析。其中贝叶斯因子(后验概率与先验概率的比值)设定为大于20, 检测不同位点经受的选择压力。1.5 适应性进化分析
采用基于机理式模型的PAML4.9软件中的codeml程序分析海金沙属rpoC1基因的适应性进化(Yang, 1997, 2007)。分析时采用分支间可变ω模型、位点间可变ω模型和分支-位点模型。分支间可变ω模型(Yang, 1998)允许在不同支系上ω值不同。其中, 单一比率(one-ratio, M0)模型假定系统发育树上的各支系ω相同; 二比率(two-ratio, MT)模型设定前景支和背景支, 前景支和背景支分别具有不同的ω值; 自由比率(free-ratio, MF)模型假定系统发育树上各支系ω值不同。
位点间可变ω模型(Nielsen and Yang, 1998; Yang et al., 2000)允许不同位点上ω值不同, 包含7种密码子替换模型, 分别为M0 (单一比率)、M1a (近中性)、M2a (选择)、M3 (离散)、M7 (beta)、M8 (beta & ω)和M8a (beta & ωs=1)。这7种密码子模型可分为4套嵌套模型, 即M2a和其零假设模型M1a (筛选正选择位点)、M3和其零假设模型M0 (检测各位点ω是否一致)、M8和其零假设模型M7 (筛选正选择位点, 易产生假阳性)以及M8和其零假设模型M8a (有助于降低假阳性)。分别对4套嵌套模型进行LRT运算, 比较不同模型间的差异显著性。在PAML软件中运行2种贝叶斯方法鉴定正选择位点: NEB (native empirical Bayes) (Nielsen and Yang, 1998; Yang et al., 2000)和BEB (Bayes empirical Bayes) (Yang, 2005)。前者使用极大似然估计(maximum likelihood estimates, MLEs)参数(如比例p和比率ω)来检验位点后验概率, 未考虑取样误差, 产生的假阳性高; 后者可以通过引用贝叶斯检验处理取样误差, 故在筛选正选择位点时使用后者, 但BEB只可在M2a和M8下运行(Yang, 1998; Yang and Neilson, 2002; Zhang et al., 2005)。
分支-位点模型(Zhang et al., 2005)将系统发育树分为前景支和背景支, 不仅位点间ω值变化, 同时支系间ω值也变化。该模型主要用于检测前景支中正选择作用对部分位点的影响, 仅允许前景支中出现正选择位点。本研究采用该模型下的MA (model A)中检验2 (Model=2, NSsites=2)筛选正选择位点。设置以海金沙属分支为前景支的Ma0, 其ω值固定为1; 设置以海金沙属分支为前景支的Ma1, 其ω为估计值。MA与位点模型中M1a进行差异显著性比较。若差异显著, 说明前景支在进化进程中受到正选择作用或受到的选择约束较宽松。
筛选出来的正选择位点根据SWISS-MODEL (https://www.swissmodel.expasy.org/)数据库比对结果, 绘制正选择位点的空间位置图。
2 结果与讨论
2.1 64种蕨类植物系统发育树
参考Smith等(2006)的蕨类植物分类系统和The Pteridophyte Phylogeny Group (2016)的系统发育树, 选取rbcL编码序列(CDS)与matK编码序列的串联数据集, 在PhyML软件下用最大似然法(ML)构建系统发育树。由于matK和rbcL编码序列串联构建的系统发育树与目前认可的蕨类系统树存在细微差异, 为保证构建的系统发育树与目前认可的蕨类系统树基本一致, 在手工校正过程中, 改变了部分物种的系统发育位置, 使其拓扑结构发生了一定改变; 且本系统发育树并非用于系统发育分析, 其作用主要是为后续分子进化分析提供树文件。构建的系统发育树如图1所示。
图1
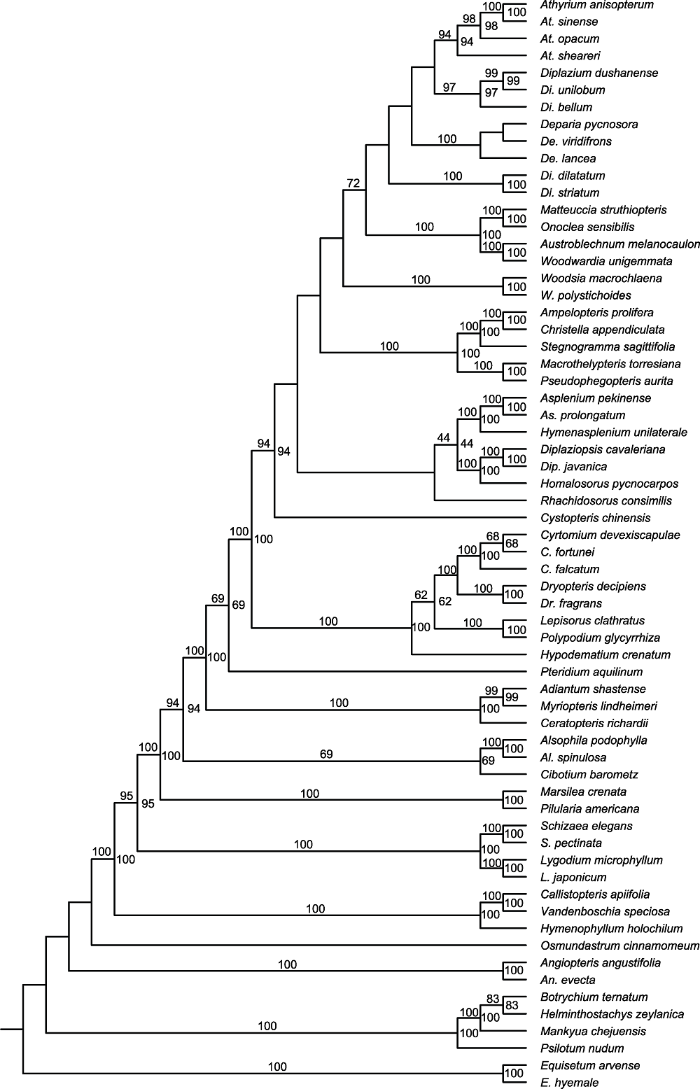 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图164种蕨类植物的系统发育树
Figure 1Phylogenetic tree of 64 ferns species
2.2 rpoC1基因的进化速率
使用HyPhy软件局部参数下核苷酸HKY85替换模型和密码子MG94×HKY85_3×4模型计算海金沙属和其它蕨类rpoC1外显子的进化速率(附录3), 大部分蕨类dN值小于dS, trsv值小于trst。用似然比检验判断海金沙属和其它蕨类的替换速率是否存在显著差异。似然比检验的P值及Mann-Whitney检验值如表2所示。其中, 海金沙属rpoC1 CDS的trst均值和trsv/trst均值大于其它蕨类植物。在似然比检验中, 仅有trst、trsv以及dN的P值小于0.05, 说明海金沙属rpoC1 CDS在转换率、颠换率以及非同义替换上与其它蕨类植物有显著差异。Mann-Whitney秩和检验值均无显著差异, 与似然比检验结果相差较大, 推测原因是海金沙属样本量少(仅2个), 秩和检验误差大。Table 2
表2
表2海金沙属与其它蕨类植物rpoC1编码序列(CDS)进化速率检验
Table 2
| Et | Ev | trsv/trst | trst | trsv | dN | dS | ω | |
|---|---|---|---|---|---|---|---|---|
| Lygodium | 0.014 | 0.004 | 0.118 | 0.579 | 0.007 | 0.022 | 0.147 | 0.144 |
| Other ferns | 0.022 | 0.005 | 0.096 | 0.088 | 0.010 | 0.045 | 0.201 | 0.256 |
| P value | 1.000 | 1.000 | 1.000 | 0.008 | 0.036 | 0.000 | 0.517 | 1.000 |
| Mann-Whitney | 0.834 | 0.778 | 0.362 | 0.834 | 0.778 | 0.502 | 0.923 | 0.316 |
新窗口打开|下载CSV
2.3 rpoC1基因的选择压力分析
用HyPhy软件Rate Het参数下密码子MG94×HKY85× 3_4×2_Rates模型进行正选择位点和负选择位点筛选。在贝叶斯因子大于20的条件下, 筛选出正选择位点3个, 负选择位点541个(表3)。3个正选择位点的贝叶斯因子均在35-50之间(图2A); 541个负选择位点的贝叶斯因子最大超过2.4×1012 (图2B), 说明筛选出来的位点均受到了较强的选择作用。结合64种蕨类植物ω值, 发现除大囊岩蕨(Woodsia macrochlaena)、Schizaea elegans、尖裂荚果蕨(Matteuccia struthiopteris)和毛柄双盖蕨(Diplazium dilatatum)外, 其余蕨类植物ω值均小于0.5。因此, 相较于正选择作用, rpoC1基因所受到的负选择压力更大。由此表明, RPOC1蛋白在长期的进化过程中比较保守。Table 3
表3
表3rpoC1基因正选择和负选择位点
Table 3
| Amino acid sites | |
|---|---|
| Positive selection sites | 156, 184, 568 |
| Negative selection sites | 1-3, 6-12, 14-21, 23-26, 28-32, 35-37, 39-42, 44-54, 56, 58-63, 66-74, 76, 79-82, 84, 86-102, 104-107, 109-128, 130-136, 138, 139, 141-144, 147-155, 158-173, 180-181, 183, 186-191, 193-195, 197-198, 200-208, 211, 213-218, 223, 225, 228-230, 232-235, 237, 241-244, 246-247, 249-258, 260-276, 278-291, 294-296, 298-301, 303-315, 318-337, 339-344, 346-368, 370-391, 393-400, 402-411, 413-414, 416-417, 419-434, 436-437, 439-440, 442, 444-445, 447-449, 453-455, 457-480, 482-492, 494-499, 501-526, 528-538, 540-550, 553-556, 558-561, 566-567, 575, 577, 579, 582-583, 586-588, 590-591, 593, 597-600, 603-613, 620, 622, 625-626, 628, 630, 632-635, 639, 642-644, 646, 648-649, 654, 660, 663-668, 671-674, 676, 677, 679-682, 687-689, 694, 697, 710 |
新窗口打开|下载CSV
图2
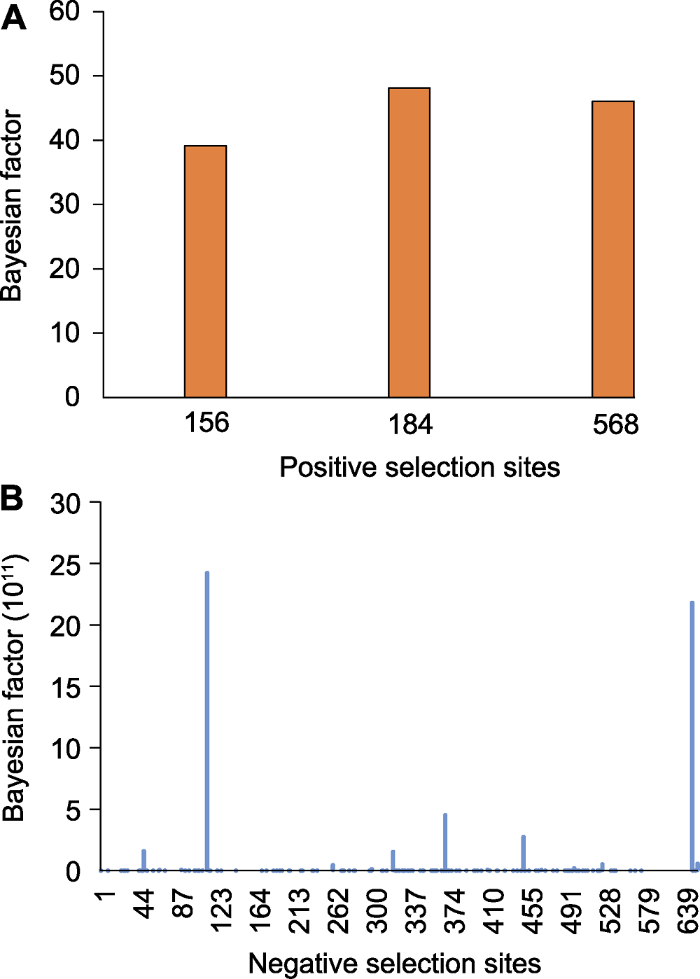 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2rpoC1氨基酸残基正(A)负(B)选择位点
Figure 2Positive (A) and negative (B) selection sites of rpoC1 amino acid residues
2.4 rpoC1基因的适应性进化
使用PAML软件codeml程序下的分支模型、位点模型和分支-位点模型分析rpoC1基因的适应性进化。在分支模型下进行单一比率和以海金沙属为前景支的二比率检验, 得到单一比率似然值为-33 984.018 106, 二比率似然值为-33 971.070 759 (表4)。将单一比率和二比率进行似然值检验, 得到P值为3.606×10-7 (表5), 表明单一比率模型和二比率模型差异显著, 海金沙属与其它蕨类植物之间ω值差异显著。单一比率模型和自由比率模型在自由度为125、二倍对数似然值为169.474 0下进行卡方检验, P值为0.005, 表明不同支系间的ω值差异显著。Table 4
表4
表4rpoC1基因在不同模型下的参数估计值和对数似然值
Table 4
| Model | Np | ? | Parameter estimate | Positive selection sites | |
|---|---|---|---|---|---|
| Branch model | M0 | 128 | -33984.0181 | ω=0.18862 | Not allowed |
| MA | 129 | -33971.0708 | ω1=0.19344, ω2=0.07154 | Not allowed | |
| Site model | Model 1a (M1a) | 129 | -33025.1095 | P0=0.77581, ω0=0.09604 | Not allowed |
| P1=0.22419, ω1=1.00000 | |||||
| Model 2a (M2a) | 131 | -33003.8671 | P0=0.77323, ω0=0.09782 | 687P**, 697S** | |
| P1=0.20583, ω1=1.00000 | 692S**, 700A* | ||||
| P2=0.02094, ω2=2.53148 | |||||
| Model 3 (M3) | 132 | -32770.6265 | P0=0.45915, ω0=0.02383 | None | |
| P1=0.39593, ω1=0.23422 | |||||
| P2=0.14492, ω2=0.91531 | |||||
| Model 7 (M7) | 129 | -32768.3591 | P=0.36168, q=1.11178 | Not allowed | |
| Model 8 (M8) | 131 | -32715.0671 | P0=0.95310, P=0.46388 | 578V*, 579Y*, 686S* | |
| q=2.02806, P1=0.04690 | 687P**, 689T*, 692S** | ||||
| ω=1.62513 | 693I*, 696T*, 697S**, 700A* | ||||
| Model 8a (M8a) | 130 | -32735.2130 | P0=0.91188, P=0.48169 | None | |
| q=2.38635, P1=0.08812 | |||||
| ω=1.00000 | |||||
| Branch-site model | Ma0 | 130 | -33025.1095 | P0=0.77581, P1=0.22419 | Not allowed |
| P2a+P2b=0.00000, ω2=1.00000 | |||||
| Ma1 | 131 | -33025.1095 | P0=0.77581, P1=0.22419 | None | |
| P2a+P2b=0.00000, ω2=1.00000 |
新窗口打开|下载CSV
Table 5
表5
表5PAML4.9软件中不同模型的似然比值检验统计量(2Δ?)
Table 5
| Model comparison | Df. | 2Δl | P value |
|---|---|---|---|
| M0 & MA | 1 | 25.8947 | 3.606×10-7 |
| M1a & M2a | 2 | 42.4848 | 5.950×10-10 |
| M0 & M3 | 4 | 2426.7832 | 0 |
| M7 & M8 | 2 | 106.5838 | 0 |
| M8a & M8 | 1 | 40.2917 | 2.187×10-10 |
| M0 & MF | 125 | 169.4740 | 4.998×10-3 |
新窗口打开|下载CSV
模型M8a的似然值?=-32 735.212 985, 与模型M8相比2倍对数似然值2Δ?=2×(-32 715.067 141+ 32 735.212 985)=40.291 7, 由χ2分布(Df.=1)得出P= 2.187×10-10, 即P值远小于0.05, 因此模型M8a被极显著地拒绝。模型M2a和M8的假定条件中均允许ω值大于1, PAML分析结果均显示rpoC1基因中存在正选择位点。与它们各自对应的零假设模型M1a和M7相比, 模型M2a比M1a多2个参数, 它们的LRT统计量2Δ?=42.484 8, 卡方检验P值为5.950×10-10。模型M8与M7相比, 在自由度为2的条件下, 2Δ?=106.583 8。在95%水平上, 模型M2a鉴定出4个氨基酸位点(687P、692S、697S和700A)受到正选择, 其中687P、692S和697S后验概率超过99%; 模型M8鉴定出10个氨基酸位点(578V、579Y、686S、687P、689T、692S、693I、696T、697S和700A)受到正选择, 其中687P、692S和697S后验概率超过99%。
2.5 RPOC1蛋白质三维结构
将海金沙RPOC1蛋白质序列导入SWISS-MODEL中, 通过与结核分枝杆菌(Mycobacterium tuberculosis) RNAP启动子蛋白质(SMTL ID: 6ee8.1)序列进行比对, 基于同源建模原理得到其蛋白质结构图(图3)。其中, HyPhy软件筛选出来的正选择位点156T、184F及568I如图3所示。异亮氨酸(isoleucine, I)和苯丙氨酸(phenylalanine, F)均为非极性R基氨基酸。156T和184F正选择位点靠近DNA结合结构域, 周围大部分氨基酸为非极性R基氨基酸(疏水氨基酸), 568I正选择位点周围大部分是不带电荷的极性R基氨基酸。图3
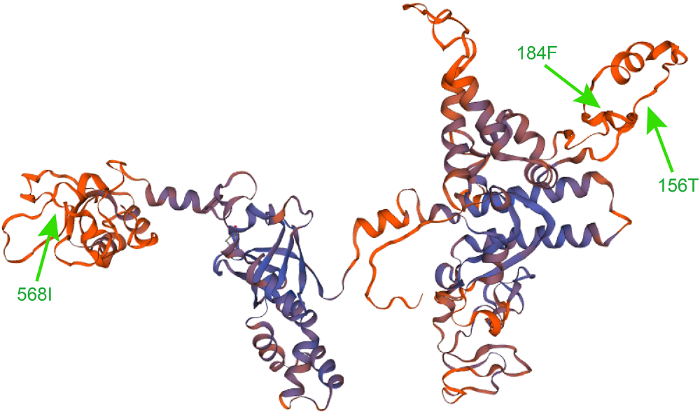 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3海金沙RPOC1蛋白质的三维结构
Figure 3Three-dimensional structure of RPOC1 protein in Lygodium japonicum
2.6 讨论
2.6.1 rpoC1基因内含子缺失及与进化速率的关系目的基因的选择是分子系统学研究最关键的环节之一。由于不同基因行使的功能不同, 它们的进化速率也存在一定差异。对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列。目前, 对atpB (Korall et al., 2006)、rps4 (Guillon, 2004)以及rbcL基因(Dubuisson, 1997)已研究得较为深入, 而对rpoC1基因的研究较少。对于内含子缺失的研究, 由于我们仅分析了海金沙属与其它蕨类植物的进化速率差异, 且海金沙属叶绿体基因组全序列只公布了2个物种(海金沙和小叶海金沙(L. microphyllum)), 因此, 在样本量以及对比分析上存在一定不足。后续研究可以增加海金沙属物种的叶绿体基因组测序分析, 扩大研究对象数目, 比较海金沙属与其它属之间的进化速率差异, 以获得更加准确的结果。rpoC1基因内含子缺失的属对应的分子进化速率与其它蕨类植物之间有一定的区别, 但并不能完全说明rpoC1基因内含子缺失事件必然对分子进化速率有影响, 其与海金沙属进化速率高可能存在关联。因此, 在后续研究中可以深入分析。rpoC1基因内含子缺失在植物中较为常见, 牻牛儿苗科(Geraniaceae) (Weng et al., 2014)、禾本科(Poaceae) (Katayama and Ogihara, 1993)、西番莲科(Passifloraceae) (Hansen et al., 2006)、豆科(Fabaceae) (Downie et al., 1998)、草海桐科(Goodeniaceae) (Downie et al., 1996)、仙人掌科(Cactaceae) (Wallace and Cota, 1996)以及番杏科(Aizoaceae) (Thiede et al., 2007)中均存在该事件。rpoC1基因内含子存在于被子植物的共同祖先中, 随后在禾本科和柱状仙人掌亚科的谱系中独立丢失(Downie et al., 1996)。而真核生物基因组中的内含子密度变化超过3个数量级, 因此在进化过程中必然发生大量的内含子增加和/或损失(He et al., 2017)。Downie等(1998)研究了65种苜蓿属(Medicago)质体rpoC1内含子, 结果表明有17个物种缺少内含子, 而其中有3种在内含子含量方面是异质的, 且rpoC1内含子在该群体的进化过程中独立丢失至少3次。目前, 关于rpoC1基因内含子丢失与分子进化速率的关系研究较少, 部分研究集中在rpoC1基因内含子丢失与系统发育关系的重建上。Hansen等(2006)发现rpoC1内含子缺失对西番莲属系统发育树构建有一定的影响。
在似然比检验中, trst、trsv以及dN的P值小于0.05, 说明海金沙属rpoC1 CDS在转换率、颠换率以及非同义替换率上与其它蕨类有显著差异; 但Mann- Whitney检验值显示无显著差异, 与似然比检验结果相差较大, 推测原因是海金沙属样本量少(仅2个), 秩和检验误差大。此外, rpoC1基因的转换率、颠换率、同义替换率和非同义替换率在不同类群之间差异较大。桫椤类和合囊蕨类的分支短, 进化速率相对较慢; 铁角蕨类和凤尾蕨类分支长, 进化速率相对较快。海金沙目中莎草蕨科Schizaea elegans的颠换率、非同义替换率和ω值非常高, 其有可能正在经历正选择作用。
2.6.2 rpoC1正选择位点
HyPhy软件筛选出的正选择位点与PAML软件筛选出来的位点不同, 可能是由于软件筛选原理和方法有差异。HyPhy要求贝叶斯因子大于20; PAML则要求后验概率大于95%或99%。此外, PAML位点模型需要所有谱系的平均dN大于dS, 该谱系中的正选择位点才可被筛选到, 因此正选择位点可能会由于谱系dN小于dS而无法被检测出来。rpoC1基因多处于负选择作用下, 负选择位点更加保守, 有利于维持蛋白质结构和功能的稳定(Hong et al., 2008)。由于筛选出来的正选择位点空间结构在SWISS-MODEL构建的模型中置信度较低, 因此, 对其所处二级结构以及结构域的分析需要进一步建模。相较于中性突变, 正选择发生频率低, 一般只发生在序列的少数位点或某个短的历史时期, 尤其是正选择作用发生在进化早期且已经被固定下来(Perutz, 1983; Newcomb et al., 1997)。因此, 检测正选择位点相对困难。正选择位点对于探究该基因适应性进化和其蛋白质功能位点有重要意义。此外, 正选择位点的确定还可以用于分析蛋白质序列的适应性进化。
附录1 64种蕨类植物经手工校正后rpoC1 CDS序列
Appendix 1 The rpoC1 CDS sequence of 64 ferns after manual correction附录2 jModelTest2.1.10基于AIC得到的matK和rbcL串联集核苷酸模型参数
Appendix 2 Nucleotide model parameters of matK and rbcL tandem set based on AIC of jModelTest2.1.10附录3rpoC1基因分子进化速率
Appendix 3 Molecular evolution rate of rpoC1 genehttp://www.chinbullbotany.com/fileup/1674-3466/PDF/t19-105.pdf
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
URLPMID:22847108 [本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1006/mpev.1997.0414URL [本文引用: 1]
DOI:10.1093/gbe/evt099URLPMID:23821521 [本文引用: 1]

Previous studies have shown that core leptosporangiates, the most species-rich group of extant ferns (monilophytes), have a distinct plastid genome (plastome) organization pattern from basal fern lineages. However, the details of genome structure transformation from ancestral ferns to core leptosporangiates remain unclear because of limited plastome data available. Here, we have determined the complete chloroplast genome sequences of Lygodium japonicum (Lygodiaceae), a member of schizaeoid ferns (Schizaeales), and Marsilea crenata (Marsileaceae), a representative of heterosporous ferns (Salviniales). The two species represent the sister and the basal lineages of core leptosporangiates, respectively, for which the plastome sequences are currently unavailable. Comparative genomic analysis of all sequenced fern plastomes reveals that the gene order of L. japonicum plastome occupies an intermediate position between that of basal ferns and core leptosporangiates. The two exons of the fern ndhB gene have a unique pattern of intragenic copy number variances. Specifically, the substitution rate heterogeneity between the two exons is congruent with their copy number changes, confirming the constraint role that inverted repeats may play on the substitution rate of chloroplast gene sequences.
[本文引用: 1]
URLPMID:20525638 [本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
URLPMID:18487149 [本文引用: 1]
DOI:10.1007/BF00352016URLPMID:8431958 [本文引用: 1]
The distribution of structural rearrangements of the chloroplast genome found in grass cpDNA in comparison to that of tobacco was systematically checked in the cpDNAs of representative monocots. The physical map of lily cpDNA, which shares a key position in the diversity of monocotyledonous plants, was constructed to assess whether three inversions found in grass cpDNA are common in monocots. Specific probes for the detection of (1) intron loss in the rpoC1 gene, (2) insertional sequence gain in rpoC2, (3) deletion of ORF2280 in the inverted repeats, (4) non-reciprocal translocation of rpl23, and (5) rearrangements of ORF512, were hybridized to cpDNAs of lily, onion, spiderwort, two turf grasses, and wheat. The existence of intervening sequences in the rpoC1 and rpoC2 genes was also confirmed by PCR analysis. All markers used in the study revealed that structural rearrangements of the chloroplast genome were restricted to grasses, indicating that drastic structural alterations of the chloroplast genome had occurred in the ancestor(s) of grasses. These results also suggest that structural analysis of the chloroplast genome is applicable to the phylogenetic reconstruction of related plants.
URLPMID:24823358 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/s41586-018-0828-1URLPMID:30651636 [本文引用: 1]
Spliceosomal introns are ubiquitous non-coding RNAs that are typically destined for rapid debranching and degradation. Here we describe 34 excised introns in Saccharomyces cerevisiae that-despite being rapidly degraded in log-phase growth-accumulate as linear RNAs under either saturated-growth conditions or other stresses that cause prolonged inhibition of TORC1, which is a key integrator of growth signalling. Introns that become stabilized remain associated with components of the spliceosome and differ from other spliceosomal introns in having a short distance between their lariat branch point and 3' splice site, which is necessary and sufficient for their stabilization. Deletion of these unusual introns is disadvantageous in saturated conditions and causes aberrantly high growth rates in yeast that are chronically challenged with the TORC1 inhibitor rapamycin. The reintroduction of native or engineered stable introns suppresses this aberrant rapamycin response. Thus, excised introns function within the TOR growth-signalling network of S. cerevisiae and, more generally, excised spliceosomal introns can have biological functions.
DOI:10.1073/pnas.94.14.7464URLPMID:9207114 [本文引用: 1]
Resistance to organophosphorus (OP) insecticides is associated with decreased carboxylesterase activity in several insect species. It has been proposed that the resistance may be the result of a mutation in a carboxylesterase that simultaneously reduces its carboxylesterase activity and confers an OP hydrolase activity (the
URLPMID:9539414 [本文引用: 2]
Several codon-based models for the evolution of protein-coding DNA sequences are developed that account for varying selection intensity among amino acid sites. The
DOI:10.1038/s41586-018-0859-7URLPMID:30651641 [本文引用: 1]
Introns are ubiquitous features of all eukaryotic cells. Introns need to be removed from nascent messenger RNA through the process of splicing to produce functional proteins. Here we show that the physical presence of introns in the genome promotes cell survival under starvation conditions. A systematic deletion set of all known introns in budding yeast genes indicates that, in most cases, cells with an intron deletion are impaired when nutrients are depleted. This effect of introns on growth is not linked to the expression of the host gene, and was reproduced even when translation of the host mRNA was blocked. Transcriptomic and genetic analyses indicate that introns promote resistance to starvation by enhancing the repression of ribosomal protein genes that are downstream of the nutrient-sensing TORC1 and PKA pathways. Our results reveal functions of introns that may help to explain their evolutionary preservation in genes, and uncover regulatory mechanisms of cell adaptations to starvation.
URLPMID:6400645 [本文引用: 1]
URLPMID:15509596 [本文引用: 1]
DOI:10.1002/j.1537-2197.1987.tb08628.xURL [本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1007/BF02221558URLPMID:8595674 [本文引用: 2]
The deletion of an approximately 700-bp intron in the chloroplast-encoded gene rpoC1 was shown in 21 representative species of the subfamily Cactoideae of the angiosperm family Cactaceae. Members of the subfamilies Pereskioideae and Opuntioideae were found to possess the intron, as did members of the related families Aizoaceae, Basellaceae, Didiereaceae, Phytolaccaceae, and Portulacaceae. These results support a monophyletic origin for the most-speciose subfamily of the cactus family, and represent a first report of the loss of this intron in dicots.
URLPMID:24336877 [本文引用: 1]
URLPMID:9367129 [本文引用: 1]
DOI:10.1093/oxfordjournals.molbev.a025957URLPMID:9580986 [本文引用: 3]
An excess of nonsynonymous substitutions over synonymous ones is an important indicator of positive selection at the molecular level. A lineage that underwent Darwinian selection may have a nonsynonymous/synonymous rate ratio (dN/dS) that is different from those of other lineages or greater than one. In this paper, several codon-based likelihood models that allow for variable dN/dS ratios among lineages were developed. They were then used to construct likelihood ratio tests to examine whether the dN/dS ratio is variable among evolutionary lineages, whether the ratio for a few lineages of interest is different from the background ratio for other lineages in the phylogeny, and whether the dN/dS ratio for the lineages of interest is greater than one. The tests were applied to the lysozyme genes of 24 primate species. The dN/dS ratios were found to differ significantly among lineages, indicating that the evolution of primate lysozymes is episodic, which is incompatible with the neutral theory. Maximum-likelihood estimates of parameters suggested that about nine nonsynonymous and zero synonymous nucleotide substitutions occurred in the lineage leading to hominoids, and the dN/dS ratio for that lineage is significantly greater than one. The corresponding estimates for the lineage ancestral to colobine monkeys were nine and one, and the dN/dS ratio for the lineage is not significantly greater than one, although it is significantly higher than the background ratio. The likelihood analysis thus confirmed most, but not all, conclusions Messier and Stewart reached using reconstructed ancestral sequences to estimate synonymous and nonsynonymous rates for different lineages.
URLPMID:15689528 [本文引用: 1]
DOI:10.1093/molbev/msm088URLPMID:17483113 [本文引用: 1]
PAML, currently in version 4, is a package of programs for phylogenetic analyses of DNA and protein sequences using maximum likelihood (ML). The programs may be used to compare and test phylogenetic trees, but their main strengths lie in the rich repertoire of evolutionary models implemented, which can be used to estimate parameters in models of sequence evolution and to test interesting biological hypotheses. Uses of the programs include estimation of synonymous and nonsynonymous rates (d(N) and d(S)) between two protein-coding DNA sequences, inference of positive Darwinian selection through phylogenetic comparison of protein-coding genes, reconstruction of ancestral genes and proteins for molecular restoration studies of extinct life forms, combined analysis of heterogeneous data sets from multiple gene loci, and estimation of species divergence times incorporating uncertainties in fossil calibrations. This note discusses some of the major applications of the package, which includes example data sets to demonstrate their use. The package is written in ANSI C, and runs under Windows, Mac OSX, and UNIX systems. It is available at -- (http://abacus.gene.ucl.ac.uk/software/paml.html).
DOI:10.1093/oxfordjournals.molbev.a004148URLPMID:12032247 [本文引用: 1]
The nonsynonymous (amino acid-altering) to synonymous (silent) substitution rate ratio (omega = d(N)/d(S)) provides a measure of natural selection at the protein level, with omega = 1, >1, and <1, indicating neutral evolution, purifying selection, and positive selection, respectively. Previous studies that used this measure to detect positive selection have often taken an approach of pairwise comparison, estimating substitution rates by averaging over all sites in the protein. As most amino acids in a functional protein are under structural and functional constraints and adaptive evolution probably affects only a few sites at a few time points, this approach of averaging rates over sites and over time has little power. Previously, we developed codon-based substitution models that allow the omega ratio to vary either among lineages or among sites. In this paper we extend previous models to allow the omega ratio to vary both among sites and among lineages and implement the new models in the likelihood framework. These models may be useful for identifying positive selection along prespecified lineages that affects only a few sites in the protein. We apply those branch-site models as well as previous branch- and site-specific models to three data sets: the lysozyme genes from primates, the tumor suppressor BRCA1 genes from primates, and the phytochrome (PHY) gene family in angiosperms. Positive selection is detected in the lysozyme and BRCA genes by both the new and the old models. However, only the new models detected positive selection acting on lineages after gene duplication in the PHY gene family. Additional tests on several data sets suggest that the new models may be useful in detecting positive selection after gene duplication in gene family evolution.
DOI:10.1093/oxfordjournals.molbev.a026245URLPMID:11018152 [本文引用: 2]
Maximum-likelihood models of codon substitution were used to analyze sperm lysin genes of 25 abalone (HALIOTIS:) species to identify lineages and amino acid sites under diversifying selection. The models used the nonsynonymous/synonymous rate ratio (omega = d(N)/d(S)) as an indicator of selective pressure and allowed the ratio to vary among lineages or sites. Likelihood ratio tests suggested significant variation in selective pressure among lineages. The variable selective pressure provided an explanation for the previous observation that the omega ratio is >1 in comparisons of closely related species and <1 in comparisons of distantly related species. Computer simulations demonstrated that saturation of nonsynonymous substitutions and constraint on lysin structure were unlikely to account for the observed pattern. Lineages linking closely related sympatric species appeared to be under diversifying selection, while lineages separating distantly related species from different geographic locations were associated with low evolutionary rates. The selective pressure indicated by the omega ratio was found to vary greatly among amino acid sites in lysin. Sites under potential diversifying selection were identified. Ancestral lysins were inferred to trace the route of evolution at individual sites and to provide lysin sequences for future laboratory studies.
DOI:10.1093/molbev/msi237URLPMID:16107592 [本文引用: 2]
Detecting positive Darwinian selection at the DNA sequence level has been a subject of considerable interest. However, positive selection is difficult to detect because it often operates episodically on a few amino acid sites, and the signal may be masked by negative selection. Several methods have been developed to test positive selection that acts on given branches (branch methods) or on a subset of sites (site methods). Recently, Yang, Z., and R. Nielsen (2002. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19:908-917) developed likelihood ratio tests (LRTs) based on branch-site models to detect positive selection that affects a small number of sites along prespecified lineages. However, computer simulations suggested that the tests were sensitive to the model assumptions and were unable to distinguish between relaxation of selective constraint and positive selection (Zhang, J. 2004. Frequent false detection of positive selection by the likelihood method with branch-site models. Mol. Biol. Evol. 21:1332-1339). Here, we describe a modified branch-site model and use it to construct two LRTs, called branch-site tests 1 and 2. We applied the new tests to reanalyze several real data sets and used computer simulation to examine the performance of the two tests by examining their false-positive rate, power, and robustness. We found that test 1 was unable to distinguish relaxed constraint from positive selection affecting the lineages of interest, while test 2 had acceptable false-positive rates and appeared robust against violations of model assumptions. As test 2 is a direct test of positive selection on the lineages of interest, it is referred to as the branch-site test of positive selection and is recommended for use in real data analysis. The test appeared conservative overall, but exhibited better power in detecting positive selection than the branch-based test. Bayes empirical Bayes identification of amino acid sites under positive selection along the foreground branches was found to be reliable, but lacked power.
[本文引用: 1]
1
1959
... 蕨类植物是维管植物的第二大类群, 现存约12 000种, 主要分布于中低海拔的热带、亚热带地区.蕨类植物起源于距今约3.59亿年的泥盆纪, 经历过3次“物种辐射”(
A new look at the statistical model identification
1
1974
... 用jModelTest2.1.10软件(
jModelTest 2: more models, new heuristics and parallel computing
1
2012
... 用jModelTest2.1.10软件(
Multiple independent losses of the plastid rpoC1 intron in Medicago (Fabaceae) as inferred from phylogenetic analyses of nuclear ribosomal DNA internal transcribed spacer sequences
2
1998
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
... ).
Multiple independent losses of the rpoC1 intron in angiosperm chloroplast DNA’s
2
1996
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
... 基因内含子存在于被子植物的共同祖先中, 随后在禾本科和柱状仙人掌亚科的谱系中独立丢失(
rbcL sequences: a promising tool for the molecular systematics of the fern genus Trichomanes (Hymenophyllaceae)?
1
1997
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
Plastome sequences of Lygodium japonicum and Marsilea crenata reveal the genome organization transformation from basal ferns to core Leptosporangiates
1
2013
... 选取包含25个科的64种蕨类植物(
Phylogeny of Horsetails (Equisetum) based on the chloroplast rps4 gene and adjacent noncoding sequences
1
2004
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0
1
2010
... 用jModelTest2.1.10软件(
BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/ 98/NT
1
1999
... 将从NCBI下载的叶绿体基因组全序列或部分序列数据导入Genious Prime 2019软件中, 分别提取rbcL、matK及rpoC1基因序列.用BioEdit7.0.5.3软件(
Phylogenetic relationships and chromosome number evolution in Passiflora
2
2006
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
... 基因内含子丢失与系统发育关系的重建上.
Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies
1
2017
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
Complete sequence determination and phylogenetic analysis of FKN among seven higher primates including homonids and old world monkeys
1
2008
... HyPhy软件筛选出的正选择位点与PAML软件筛选出来的位点不同, 可能是由于软件筛选原理和方法有差异.HyPhy要求贝叶斯因子大于20; PAML则要求后验概率大于95%或99%.此外, PAML位点模型需要所有谱系的平均dN大于dS, 该谱系中的正选择位点才可被筛选到, 因此正选择位点可能会由于谱系dN小于dS而无法被检测出来.rpoC1基因多处于负选择作用下, 负选择位点更加保守, 有利于维持蛋白质结构和功能的稳定(
Structural alterations of the chloroplast genome found in grasses are not common in monocots
1
1993
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
Chloroplast genome evolution in early diverged leptosporangiate ferns
1
2014
... 蕨类植物是维管植物的第二大类群, 现存约12 000种, 主要分布于中低海拔的热带、亚热带地区.蕨类植物起源于距今约3.59亿年的泥盆纪, 经历过3次“物种辐射”(
On the phylogenetic position of Cystodium: it’s not a tree fern—it’s a polypod!
1
2006
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
Evolutionary patterns and processes in ferns
1
1978
... 蕨类植物是维管植物的第二大类群, 现存约12 000种, 主要分布于中低海拔的热带、亚热带地区.蕨类植物起源于距今约3.59亿年的泥盆纪, 经历过3次“物种辐射”(
Excised linear introns regulate growth in yeast
1
2019
... rpoC1 (RNA polymerase beta' subunit)基因编码RNA聚合酶β°亚基, 属于遗传系统基因(genetic system genes).在转录过程中, RNA聚合酶β°亚基与DNA模板结合, 与β-β°复合体共同构成RNA合成的催化中心.因此rpoC1基因在转录过程中具有重要作用.内含子在调节基因表达和可变剪接(alternative splicing)中扮演重要角色, 一个基因可以因此而编码多种不同的蛋白质.研究发现, 芽殖酵母(Saccharomyces cerevisiae)基因组中内含子的存在促进饥饿条件下细胞存活, 其通过抑制TORC1和PKA途径核糖体蛋白基因的营养感应来增强对饥饿的抗性(
A single amino acid substitution converts a carboxylesterase to an organophosphorus hydrolase and confers insecticide resistance on a blowfly
1
1997
... HyPhy软件筛选出的正选择位点与PAML软件筛选出来的位点不同, 可能是由于软件筛选原理和方法有差异.HyPhy要求贝叶斯因子大于20; PAML则要求后验概率大于95%或99%.此外, PAML位点模型需要所有谱系的平均dN大于dS, 该谱系中的正选择位点才可被筛选到, 因此正选择位点可能会由于谱系dN小于dS而无法被检测出来.rpoC1基因多处于负选择作用下, 负选择位点更加保守, 有利于维持蛋白质结构和功能的稳定(
Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene
2
1998
... 位点间可变ω模型(
... =1).这7种密码子模型可分为4套嵌套模型, 即M2a和其零假设模型M1a (筛选正选择位点)、M3和其零假设模型M0 (检测各位点ω是否一致)、M8和其零假设模型M7 (筛选正选择位点, 易产生假阳性)以及M8和其零假设模型M8a (有助于降低假阳性).分别对4套嵌套模型进行LRT运算, 比较不同模型间的差异显著性.在PAML软件中运行2种贝叶斯方法鉴定正选择位点: NEB (native empirical Bayes) (
Introns are mediators of cell response to starvation
1
2019
... rpoC1 (RNA polymerase beta' subunit)基因编码RNA聚合酶β°亚基, 属于遗传系统基因(genetic system genes).在转录过程中, RNA聚合酶β°亚基与DNA模板结合, 与β-β°复合体共同构成RNA合成的催化中心.因此rpoC1基因在转录过程中具有重要作用.内含子在调节基因表达和可变剪接(alternative splicing)中扮演重要角色, 一个基因可以因此而编码多种不同的蛋白质.研究发现, 芽殖酵母(Saccharomyces cerevisiae)基因组中内含子的存在促进饥饿条件下细胞存活, 其通过抑制TORC1和PKA途径核糖体蛋白基因的营养感应来增强对饥饿的抗性(
Species adaptation in a protein molecule
1
1983
... HyPhy软件筛选出的正选择位点与PAML软件筛选出来的位点不同, 可能是由于软件筛选原理和方法有差异.HyPhy要求贝叶斯因子大于20; PAML则要求后验概率大于95%或99%.此外, PAML位点模型需要所有谱系的平均dN大于dS, 该谱系中的正选择位点才可被筛选到, 因此正选择位点可能会由于谱系dN小于dS而无法被检测出来.rpoC1基因多处于负选择作用下, 负选择位点更加保守, 有利于维持蛋白质结构和功能的稳定(
HyPhy: hypothesis testing using phylogenies
1
2005
... 使用HyPhy2.2.4 (hypothesis testing using phylogenies)软件分析海金沙属和其它62种蕨类植物的rpoC1外显子的转换率(transition rate, trst)、颠换率(transversion rate, trsv)、转换率/颠换率(trst/trsv)、替换数(number of substitutions, Es)、转换数(number of transition, Et)、颠换数(number of transversion, Ev)、同义替换率(synonymous substitution rate, dS)、非同义替换率(nonsynonymous substitution rate, dN)以及dN/dS (
Complex paleozoic filicales in the evolutionary radiation of ferns
1
1987
... 蕨类植物是维管植物的第二大类群, 现存约12 000种, 主要分布于中低海拔的热带、亚热带地区.蕨类植物起源于距今约3.59亿年的泥盆纪, 经历过3次“物种辐射”(
Fern phylogeny inferred from 400 leptosporangiate species and three plastid genes
1
2007
... 蕨类植物是维管植物的第二大类群, 现存约12 000种, 主要分布于中低海拔的热带、亚热带地区.蕨类植物起源于距今约3.59亿年的泥盆纪, 经历过3次“物种辐射”(
A classification for extant ferns
2
2006
... 根据已有的系统发育树, 对rbcL编码序列(coding sequence, CDS)与matK编码序列的串联数据集用最大似然法(maximum likelihood, ML)构建的系统发育树进行手工校正, 并根据
... 参考
1
2009
... 蕨类植物是维管植物的第二大类群, 现存约12 000种, 主要分布于中低海拔的热带、亚热带地区.蕨类植物起源于距今约3.59亿年的泥盆纪, 经历过3次“物种辐射”(
A community-derived classification for extant lycophytes and ferns
2
2016
... 根据已有的系统发育树, 对rbcL编码序列(coding sequence, CDS)与matK编码序列的串联数据集用最大似然法(maximum likelihood, ML)构建的系统发育树进行手工校正, 并根据
... 参考
Phylogenetic implication of the chloroplast rpoC1 intron loss in the Aizoaceae (Caryophyllales)
2
2007
... rpoC1 (RNA polymerase beta' subunit)基因编码RNA聚合酶β°亚基, 属于遗传系统基因(genetic system genes).在转录过程中, RNA聚合酶β°亚基与DNA模板结合, 与β-β°复合体共同构成RNA合成的催化中心.因此rpoC1基因在转录过程中具有重要作用.内含子在调节基因表达和可变剪接(alternative splicing)中扮演重要角色, 一个基因可以因此而编码多种不同的蛋白质.研究发现, 芽殖酵母(Saccharomyces cerevisiae)基因组中内含子的存在促进饥饿条件下细胞存活, 其通过抑制TORC1和PKA途径核糖体蛋白基因的营养感应来增强对饥饿的抗性(
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
An intron loss in the chloroplast gene rpoC1 supports a monophyletic origin for the subfamily Cactoideae of the Cactaceae
2
1996
... rpoC1 (RNA polymerase beta' subunit)基因编码RNA聚合酶β°亚基, 属于遗传系统基因(genetic system genes).在转录过程中, RNA聚合酶β°亚基与DNA模板结合, 与β-β°复合体共同构成RNA合成的催化中心.因此rpoC1基因在转录过程中具有重要作用.内含子在调节基因表达和可变剪接(alternative splicing)中扮演重要角色, 一个基因可以因此而编码多种不同的蛋白质.研究发现, 芽殖酵母(Saccharomyces cerevisiae)基因组中内含子的存在促进饥饿条件下细胞存活, 其通过抑制TORC1和PKA途径核糖体蛋白基因的营养感应来增强对饥饿的抗性(
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates
1
2014
... 目的基因的选择是分子系统学研究最关键的环节之一.由于不同基因行使的功能不同, 它们的进化速率也存在一定差异.对于不同分类阶元的系统发育和分子进化研究一般选用不同的DNA序列.目前, 对atpB (
PAML: a program package for phylogenetic analysis by maximum likelihood
1
1997
... 采用基于机理式模型的PAML4.9软件中的codeml程序分析海金沙属rpoC1基因的适应性进化(
Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution
3
1998
... 基于构建的系统发育树和密码子替换模型可对不同支系和不同氨基酸位点的适应性进化进行极大似然法检测.由于不同氨基酸位点功能可能有所不同, 故不同位点选择压力存在差异.
... 分支间可变ω模型(
... 位点间可变ω模型(
Bayes empirical Bayes inference of amino acid sites under positive selection
1
2005
... 位点间可变ω模型(
PAML 4: phylogenetic analysis by maximum likelihood
1
2007
... 采用基于机理式模型的PAML4.9软件中的codeml程序分析海金沙属rpoC1基因的适应性进化(
Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages
1
2002
... 位点间可变ω模型(
Maximum- likelihood analysis of molecular adaptation in abalone sperm lysin reveals variable selective pressures among lineages and sites
2
2000
... 位点间可变ω模型(
... ;
Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level
2
2005
... 位点间可变ω模型(
... 分支-位点模型(
Molecular disease, evolution, and genic heterogeneity
1
1962
... 分子进化速率最早由
备案号: 京ICP备16067583号-21
版权所有 © 2021 《植物学报》编辑部
地址:北京香山南辛村20号 邮编:100093
电话:010-62836135 010-62836131 E-mail:cbb@ibcas.ac.cn
本系统由北京玛格泰克科技发展有限公司设计开发

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}