 ,*西北农林科技大学生命科学学院, 杨凌 712100
,*西北农林科技大学生命科学学院, 杨凌 712100Origin and Evolution of Soybean Protein-coding Genes
Kang Tang, Ruolin Yang,*College of Life Sciences, Northwest A&F University, Yangling 712100, China通讯作者:
收稿日期:2018-08-14接受日期:2018-12-10网络出版日期:2019-07-01
| 基金资助: |
Corresponding authors:
Received:2018-08-14Accepted:2018-12-10Online:2019-07-01

摘要
关键词:
Abstract
Keywords:
PDF (2715KB)摘要页面多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
引用本文
唐康, 杨若林. 大豆蛋白编码基因起源与进化. 植物学报, 2019, 54(3): 316-327 doi:10.11983/CBB18176
Tang Kang, Yang Ruolin.
基因组变异是生物表型多样性的主要来源, 而新基因的出现是基因组进化及物种间遗传差异的重要因素之一(Long et al., 2003; Kaessmann, 2010; Chen et al., 2013)。新基因起源是一个动态变化过程, 物种中的每个基因都“诞生”于某个特定的进化节点, 即大部分基因只出现在特定的种系或物种中, 这些新基因最初以很快的速度进化, 直到被整合到原始基因网络中, 表明这些基因可能与物种形成或适应性有关(Lynch and Conery, 2000; Long et al., 2003; Kaessmann, 2010; Chen et al., 2013)。
真核生物中, 基因复制(gene duplication, GD)是新基因的主要来源, 可作为研究新功能进化的原材料, 也是增加基因家族大小的重要因素(Ohno, 1970; Zhang, 2003; 孙红正和葛颂, 2010)。基因复制后, 由于拷贝数的增加会使该基因先经历短暂的选择压放松期, 此时拷贝序列将累积突变, 如果发生了有害突变, 则会导致基因功能减弱, 最终被清除出基因组; 也可能由于遗传漂变的原因, 使得假基因化的某一拷贝得以在群体中保留, 而突变与假基因化是导致基因家族减小的重要原因(Albalat and Ca?estro, 2016)。基因某一拷贝上偶尔也会累积有利突变, 在达尔文正选择作用下驱动该拷贝进化出新的生物学功能, 即所谓的新功能化(neo-functionalization), 而另一拷贝则执行原有的功能(Zhang, 2003)。此外, 对于多功能的基因还有可能出现复制产生的2个拷贝以功能互补的方式各自承担起父本基因的部分角色, 即亚功能化(sub-functionalization) (Zhang, 2003)。基因复制的机制主要包括全基因组复制(whole genome duplication, WGD)、片段重复(segmental duplication)、串联重复(tandem duplication)和转座诱导重复(trans- poson-induced duplication) (Freeling, 2009; Panchy et al., 2016), 每种机制对重复基因的功能、进化命运及基因组结构产生不同的影响。此外, 基因的从头起源能产生新的基因家族, 其中非编码区的从头演变是新基因出现的重要模式(Tautz and Domazet-Lo?o, 2011)。
随着高通量技术的快速发展, 有来自不同分类群的100多种植物相继完成全基因组测序(Michael and Jackson, 2013; Michael and VanBuren, 2015), 海量的基因组序列和功能数据为我们在全基因组水平研究基因进化模式带来了前所未有的契机。基因出现法被用于研究单个物种或特定生物类群的基因基于系统发育水平的进化起源(Domazet-Lo?o et al., 2007; Cai et al., 2009; Quint et al., 2012; Guo, 2013)。 该方法根据被调查物种基因在一组亲缘关系由近及远的代表物种基因组中是否有序列相似的同源基因, 将基因映射到不同的系统发育层级, 称为phylostratigraphy或phylostrata (PS), 代表相关基因大致的起源时间区间。目前, 该方法已广泛应用于追溯基因的进化历程, 及研究基因的起源时间与一些重要生物学过程(如胚胎发育)的关系(Quint et al., 2012)。
豆科是被子植物中物种数目最多的家族之一, 有20 000多个种(Doyle and Luckow, 2003)。大豆(Glycine max)作为该科非常重要的经济作物, 其基因组已经完成测序(Schmutz et al., 2010)。除大豆外, 本研究还选择了18种具有代表性的被子植物, 包括1种基部被子植物、5种单子叶植物和12种双子叶植物, 通过将大豆蛋白编码基因与这些基因组进行序列比对, 对大豆基因的起源时间进行推测, 并在此基础上, 对不同年龄的基因家族(和基因)进行比较分析: (1) 基因家族大小分布模式以及不同拷贝数基因家族的相对比例; (2) 基因序列水平的进化速率和选择压大小; (3) 基因在转录及转录后水平的特征, 包括在不同组织不同发育阶段的表达水平、表达模式以及选择性剪切等。综上, 我们通过对19个被子植物进行比较基因组学研究, 以期揭示大豆基因在不同起源时期不同复制状态下的进化动态。
1 材料与方法
1.1 材料
1.1.1 基因组序列、蛋白组序列及注释数据除大豆(Glycine max (Linn.) Merr.)外, 我们还选择了18种具有代表性且已完成测序的被子植物(图1A), 其基因组序列文件、蛋白组序列文件、GTF文件及CDS序列文件下载自Ensembl Plants v.33和Phytozome v.11数据库。这19种被子植物包括1种基部被子植物无油樟(Amborella trichopoda)、5种单子叶植物和13种双子叶植物。其中单子叶植物包含1种凤梨科植物(菠萝(Ananas comosus))、4种禾本科植物(水稻(Oryza sativa)、二穗短柄草(Brachypodium distachyon)、高粱(Sorghum bicolor)及玉米(Zea mays)); 双子叶植物中有2种茄科植物(马铃薯(Solanum tuberosum)和番茄(S. lycopersicum))、1种葡萄科植物(葡萄(Vitis vinifera))、1种杨柳科植物(毛果杨(Populus trichocarpa))、1种锦葵科植物(雷蒙德氏棉(Gossypium raimondii))、1种番木瓜科植物(番木瓜(Carica papaya))、2种十字花科植物(拟南芥(Arabidopsis thaliana)和琴叶拟南芥(A. lyrata))、1种葫芦科植物(黄瓜(Cucumis sativus))、1种蔷薇科植物(碧桃(Prunus persica))及3种豆科植物(蒺藜苜蓿(Medicago truncatula)、大豆和菜豆(Phaseolus vulgaris))。
1.1.2 大豆转录组数据
大豆28个不同发育阶段组织样本的RNA-seq数据从NCBI SRA数据库筛选得到, 其项目编号为SRP038111。28个样本包括: 萌发阶段的子叶、根、茎和叶芽; 三叶期的子叶、茎、叶芽和叶; 发芽分化阶段的叶芽、叶、花和芽分生组织; 开花期收集了3种花型, 花芽、花(2个样本)和开花后5天的花; 种子萌发后2周、3周和4周的荚粒及3周、4周和5周豆荚; 萌发后大约3周、5周、6周、8周和10周的种子; 衰老期的叶。
1.2 方法
1.2.1 直系同源基因家族的鉴定为了获得高质量的蛋白质序列数据以鉴定基因的同源关系, 我们对上述19种被子植物的蛋白质组数据按以下条件进行过滤: (1) 去除长度小于50个氨基酸残基的蛋白质; (2) 对于由可变剪切产生的多个转录本所翻译的蛋白质, 只保留每个基因最长转录本对应的蛋白质。过滤之后, 所有19个物种的共641 473条蛋白质序列作为输入数据提交至OrthoMCL v2.0.9 (Li et al., 2003)进行蛋白聚类。OrthoMCL运行中的2个关键步骤是: (1) All-against-all BLASTP, 即用blastp v2.6.0将每个蛋白与所有其它蛋白进行比对(E-value<1×10-6), 产生原始的blast输出; (2) 使用马尔科夫聚类算法(Markov Cluster algorithm, MCL)对解析的blast结果构建马尔科夫矩阵, 然后产生最终的基因家族(Enright et al., 2002)。MCL聚类时的重要参数(膨胀系数)设为1.5。本研究中我们共鉴定到34 010个直系同源基因家族。
图1
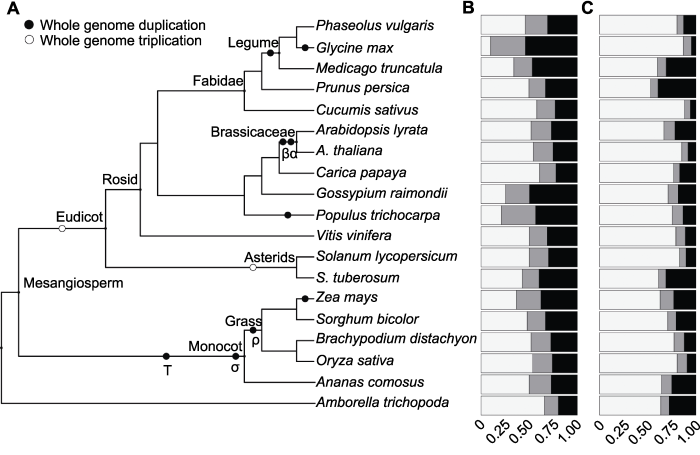 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图119种被子植物基因家族大小分布
(A) 系统发育树代
Figure 1Gene family size distribution of 19 angiosperm species
(A) Phylogenetic tree showing the relationships between the 19 angiosperm species used in this study; (B) Homologous gene family sizes; (C) Gene family sizes of orphan genes. The colors indicate the proportions of genes, white for singletons, grey for two-genes and black for multigenes.
1.2.2 大豆基因的起源分析
我们参考Domazet-Lo?o等(2007)的方法对大豆蛋白编码基因进行起源时间预测。具体而言, 根据OrthoMCL聚类结果, 对于每个大豆蛋白基因, 我们按照其出现在上述18种植物基因组中的分布模式, 将其起源时间范围指定到以下7个系统发育层级之一。这7个基因分类为: (1) Soybean (PS7), 含有大豆特有的基因; (2) Phaseoleae (PS6), 起源于大豆与菜豆的最近共同祖先(most recent common ancestor, MRCA); (3) Legume (PS5), 起源自豆科植物大豆、菜豆与苜蓿的共同祖先; (4) Rosid (PS4), 在豆科植物与葡萄的共同祖先起源; (5) Eudicot (PS3), 在蔷薇目植物与茄科植物的最近共同祖先中起源; (6) Mesangiosperm (PS2), 在双子叶植物与单子叶植物的MRCA中起源; (7) Angiosperm (PS1), 基因出现在核心被子植物与无油樟的MRCA中。
1.2.3 选择压分析
为了估算出大豆中基因的进化速率, 我们从OrthoMCL的聚类结果中提取出大豆与菜豆的直系同源基因对; 用ClustalW v2.1软件(Larkin et al., 2007)进行直系同源蛋白比对; 再利用PAL2NAL v14.0 (Suyama et al., 2006)以比对好的蛋白序列为指导, 将相应的核苷酸编码序列进行比对; 最后将上述序列文件提交至PAML v4.9b (Yang, 2007)程序以计算每对直系同源基因间的非同义替换速率(dN)、同义替换速率(dS)及选择压值(dN/dS)。
1.2.4 基因表达分析
RNA-seq产生的原始fastq格式文件从NCBI SRA数据库中筛选获得, 然后采用Trimmomatic v0.36软件 (Bolger et al., 2014)去除原始读段(raw reads)两端低质量序列, 并保证读段最小长度为50 bp。我们进一步使用Hisat v2.1.0软件(Kim et al., 2015)将clean reads比对到大豆(Williams 82)参考基因组(v1.0.33)上, 最大内含子长度设为参考基因模型(gene model)的最大内含子长(由R包GenomicFeatures计算得出), 其它参数均使用软件默认值。本研究只保留能比对到参考基因组的最好结果。最后使用StringTie v1.3.3b软件(Pertea et al., 2015)对每个样本进行转录组构建, 并计算出标准化后的基因及转录水平。
本研究中, 基因在28个被调查的不同发育阶段组织样本的至少1个样本中满足FPKM值大于1, 即认为该基因已表达, 并使用上述已表达基因在28个样本中的FPKM值的中值来估计基因总体的表达水平。
1.2.5 基因表达的组织和发育阶段特异性指数(τ)
指数τ定义为:
$\tau =\frac{\mathop{\sum }_{i=1}^{N}\left( 1-\frac{log({{x}_{i}}+1)}{log({{x}_{max}}+1)} \right)}{N-1}$
其中, N代表参与计算的不同发育阶段的所有组织样本的数量, xi代表基因在每个样本中的表达量, xmax代表基因在所有样本中的最大表达量。τ值在0-1范围内, 一般将τ≥0.85的基因作为组织和发育阶段特异性表达基因, 而τ<0.15则视为持家基因(Yanai et al., 2005)。
1.2.6 基因选择性剪切事件的鉴定
利用Cufflinks (Trapnell et al., 2010)程序中的Cuffcompare将组装好的转录本与参考基因模型(gene model)做对比, 并进行命名。其中class codes为 “c”、“j”、“=”、“e”或“o”的转录本被提取出来(Merkin et al., 2012)。利用ASTALAVISTA v 4.0软件
1.2.7 基因GO注释和功能分析
使用在线工具GOSlim (
2 结果与分析
2.1 基因家族大小分布
利用OrthoMCL软件(Li et al., 2003), 我们共获得19个物种的34 010个直系同源基因家族, 物种间基因家族总数变异较大, 从11 407个(无油樟)至16 957个(拟南芥)不等。根据基因家族大小, 我们将基因归为单拷贝基因(singleton)、两拷贝基因(two-genes)或多拷贝基因(multigenes, 家族大小≥3), 发现各物种基因组中不同拷贝数的基因家族之间的相对比例各不相同。如表1和图1B所示, 与其它物种相比, 大豆中两拷贝(36.2%)和多拷贝(53.9%)基因家族占很大比例, 这可能与大豆基因组发生过2次最近的全基因组复制事件(分别为大约5 900万和1 300万年前)有关(Schmutz et al., 2010)。我们初步统计了上述19种植物中orphan基因占总蛋白编码基因数目的相对比例, 结果显示各物种特有的基因在总的蛋白编码基因中的比例差异显著, 菜豆中仅为9.2%, 而无油樟中高达45.5%, 其中大豆基因组中orphan基因所占比例为21.1%。单拷贝orphan基因占总orphan基因的比例在物种间变异不大(表2; 图1C), 大豆中该比例为87.1%, 表明orphan基因主要以单拷贝形式存在。
2.2 大豆基因起源
在54 174个大豆基因中, 97.3% (52 733个)被定位到7个主要关注的系统发育层级(表3; 图2A)。本研究显示, 超过58.7%的基因定位到PS1, 表明一半以上的大豆蛋白编码基因至少起源于无油樟与核心被子植物分歧之前, 同时有超过21.7%的基因为大豆特异性基因(PS7) (表3; 图2B)。有趣的是, 起源晚的基因更多以单拷贝形式存在, 单拷贝基因占PS1到PS7中基因总数的比例逐渐从6.4%上升至87.1% (图2C)。对于定位到7个不同系统发育层级的52 733个基因, 73.4% (38 730个)的基因具有GO注释(图2D)。有趣的是, 基因起源时间与包含GO注释信息的基因比例之间存在很强的负相关关系, 起源于PS1-PS7的基因中, 分别有81.4%-56.9%不等的基因被注释。
Table 1
表1
表119种被子植物中直系同源基因家族(及基因)数目
Table 1
| Species | Singletons | Two-gene families | Multigene families | Total gene families | Maximum gene family size |
|---|---|---|---|---|---|
| Amborella trichopoda | 9823 | 1061(2122) | 523(2935) | 11407 | 207 |
| Ananas comosus | 9059 | 2087(4174) | 1007(4916) | 12153 | 124 |
| Oryza sativa | 11966 | 2269(4538) | 1167(5805) | 15402 | 64 |
| Brachypodium distachyon | 11455 | 2264(4528) | 1209(6066) | 14928 | 50 |
| Sorghum bicolor | 12663 | 2529(5058) | 1399(8749) | 16591 | 416 |
| Zea mays | 10277 | 3568(7136) | 1964(10539) | 15809 | 297 |
| Solanum tuberosum | 11592 | 2390(4780) | 1399(10741) | 15381 | 1051 |
| S. lycopersicum | 12210 | 2448(4896) | 1371(7277) | 16029 | 72 |
| Vitis vinifera | 10408 | 1931(3862) | 1104(6483) | 13443 | 100 |
| Populus trichocarpa | 6550 | 5476(10952) | 2337(13368) | 14363 | 108 |
| Gossypium raimondii | 7582 | 3700(7400) | 2960(14806) | 14242 | 90 |
| Carica papaya | 10776 | 1505(3010) | 667(3948) | 12948 | 194 |
| Arabidopsis thaliana | 13278 | 2485(4970) | 1194(6144) | 16957 | 125 |
| A. lyrata | 12767 | 2605(5210) | 1327(6596) | 16699 | 67 |
| Cucumis sativus | 10152 | 1691(3382) | 795(4038) | 12638 | 38 |
| Prunus persica | 10822 | 1876(3752) | 1106(7192) | 13804 | 217 |
| Medicago truncatula | 9936 | 2812(5624) | 1948(13673) | 14696 | 308 |
| Glycine max | 4241 | 7735(15470) | 4206(23027) | 16182 | 153 |
| Phaseolus vulgaris | 11324 | 2873(5746) | 1430(7626) | 15569 | 132 |
新窗口打开|下载CSV
Table 2
表2
表219种被子植物中的orphan基因家族(及基因)数目
Table 2
| Species | Singletons | Two-gene families | Multigene families | Species-specific genes | Maximum gene family size |
|---|---|---|---|---|---|
| Amborella trichopoda | 7892 | 547(1094) | 502(3447) | 12433 | 105 |
| Ananas comosus | 5685 | 483(966) | 297(2224) | 8875 | 94 |
| Oryza sativa | 10774 | 686(1372) | 292(1224) | 13370 | 29 |
| Brachypodium distachyon | 3485 | 235(470) | 125(548) | 4503 | 15 |
| Sorghum bicolor | 5682 | 350(700) | 254(1644) | 8026 | 103 |
| Zea mays | 7253 | 813(1626) | 552(2643) | 11522 | 65 |
| Solanum tuberosum | 7278 | 471(942) | 376(3688) | 11908 | 163 |
| S. lycopersicum | 7836 | 308(616) | 177(950) | 9402 | 51 |
| Vitis vinifera | 7238 | 445(890) | 229(1006) | 9134 | 44 |
| Populus trichocarpa | 7923 | 593(1186) | 281(1398) | 10507 | 31 |
| Gossypium raimondii | 5495 | 408(816) | 293(1406) | 7717 | 26 |
| Carica papaya | 7680 | 307(614) | 224(1653) | 9947 | 88 |
| Arabidopsis thaliana | 2751 | 105(210) | 57(261) | 3222 | 21 |
| A. lyrata | 5413 | 461(922) | 366(1759) | 8094 | 83 |
| Cucumis sativus | 3458 | 125(250) | 54(223) | 3931 | 13 |
| Prunus persica | 3347 | 242(484) | 195(2483) | 6314 | 838 |
| Medicago truncatula | 12763 | 962(1924) | 820(6524) | 21211 | 145 |
| Glycine max | 9961 | 476(952) | 118(523) | 11436 | 23 |
| Phaseolus vulgaris | 2013 | 85(170) | 58(318) | 2501 | 19 |
新窗口打开|下载CSV
Table 3
表3
表3定位到每个系统发育层级的大豆基因家族(和基因)数目
Table 3
| Phylostratum internode | Genes (%) | Singletons | Two-genes | Multigenes |
|---|---|---|---|---|
| Angiosperm (PS1) | 30932(58.7%) | 1982 | 5150(10300) | 3400(18650) |
| Mesangiosperm (PS2) | 4057(7.7%) | 508 | 708(1416) | 359(2133) |
| Eudicot (PS3) | 2356(4.5%) | 303 | 521(1042) | 206(1011) |
| Rosid (PS4) | 582(1.1%) | 109 | 181(362) | 31(111) |
| Legume (PS5) | 1780(3.4%) | 460 | 452(904) | 87(416) |
| Phaseoleae (PS6) | 1590(3.0%) | 568 | 400(800) | 49(222) |
| Soybean (PS7) | 11436(21.7%) | 9961 | 476(952) | 118(523) |
新窗口打开|下载CSV
图2
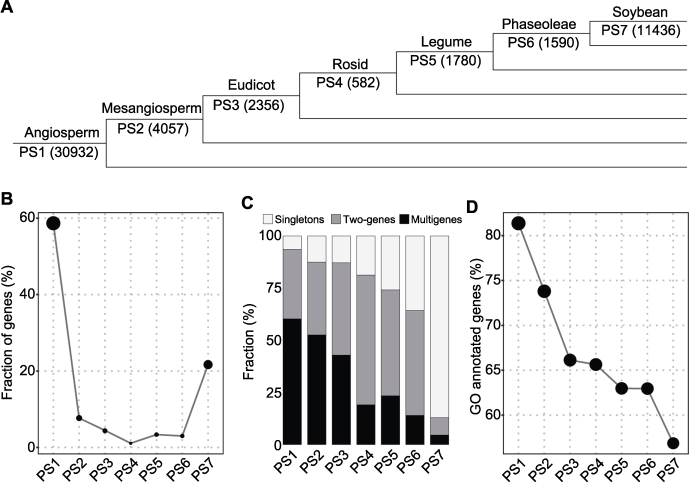 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2大豆基因起源
(A) 不同起源节点(PS1-PS7)基因数目; (B) 基因比例; (C) 基因拷贝数状态; (D) 基因GO注释
Figure 2Origination of soybean genes
(A) Numbers in parenthesis denote the number of genes per phylostratum (PS1-PS7); (B) Gene fraction; (C) Gene copy status; (D) Gene Ontology annotation
2.3 大豆基因的进化速率及所受选择压大小
为了分析大豆基因在进化过程中所受选择约束和进化速率, 我们对大豆与菜豆直系同源基因对的dN、dS和dN/dS值进行计算。如图3A所示, 起源时期不同的基因尽管同义位点的核苷酸进化速率(dS在0.345 8 (PS1)-0.398 4 (PS6)之间)差异不大; 但从dN/dS来看, 不同组的基因所受到的选择压存在显著差异, 其dN/dS值的中值分布在0.194 5 (PS1)-0.358 0 (PS6)之间。因此, 本研究显示, 古老基因受到的选择压较大, 而新起源基因受到的选择压较小, 这可能是由于古老基因中富集了更多的被子植物生存所必需的基因。与这一推测相吻合的是, 在PS1-PS4中, 单拷贝基因比多拷贝基因受到的选择压要小, 这表明重要基因在基因组中进化出更多拷贝数以提高基因组鲁棒性(robustness)的趋势。非同义替换速率(dN值)也是衡量基因进化速率的重要因素。如图3C所示, dN值的中值范围在
0.0767 5 (PS1)-0.138 3 (PS6)之间。与此同时, 基因在不同拷贝状态下的进化速率不同, 其中多拷贝基因dS和dN值的中值最大, 单拷贝基因次之, 两拷贝基因最小(图3B, C)。
图3
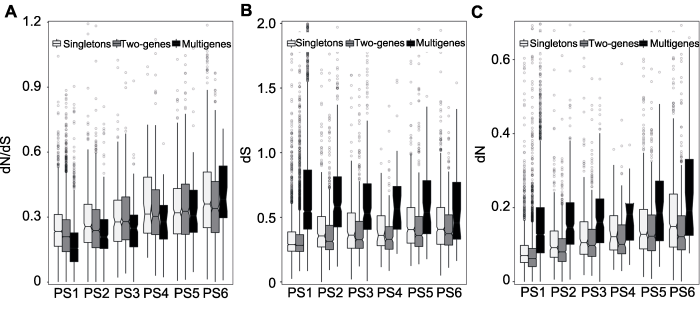 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3大豆基因分歧程度通过大豆与菜豆同源基因对来评估选择压(dN/dS)
(A)、同义替换率(dS) (B)和非同义替换率(dN) (C)。
Figure 3Divergence degrees of soybean genes Estimated between soybean and common bean selection pressure (dN/dS)
(A), synonymous substitution rate (dS) (B) and nonsynonymous substitution rate (dN) (C).
2.4 大豆基因表达
对于起源时间指定到7个不同系统发育层级的52 733个大豆基因, 其中有42 591个(80.1%)在至少1个组织样本中表达(Shen et al., 2014)。起源于PS1-PS7的基因中所表达的基因比例差别较大, 其中在PS1中表达的比例(93.2%)最高, 而PS7中表达的比例(49.5%)最低(图4A)。与此现象一致的是, 从基因总体表达水平来看, PS1中最高, 而PS7中最低(图4B)。我们进一步比较了不同年龄基因表达的组织和发育阶段特异性。结果显示, 起源较早的基因趋于广谱表达, 而起源较近的基因则具有较高组织和发育阶段特异性表达(图4C)。有意思的是, 从整体水平来看, 两拷贝基因具有较高的基因表达水平及组织和发育阶段特异性(图4B, C)。
图4
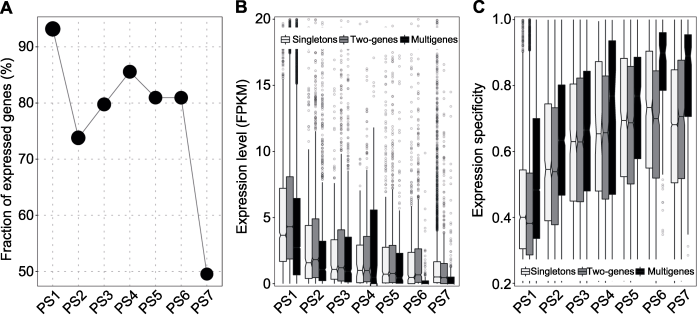 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4大豆基因表达
(A) 已表达基因; (B) 表达水平; (C) 表达特异性
Figure 4Expression of soybean genes
(A) Expressed genes; (B) Expression level; (C) Expression specificity
2.5 大豆选择性剪切
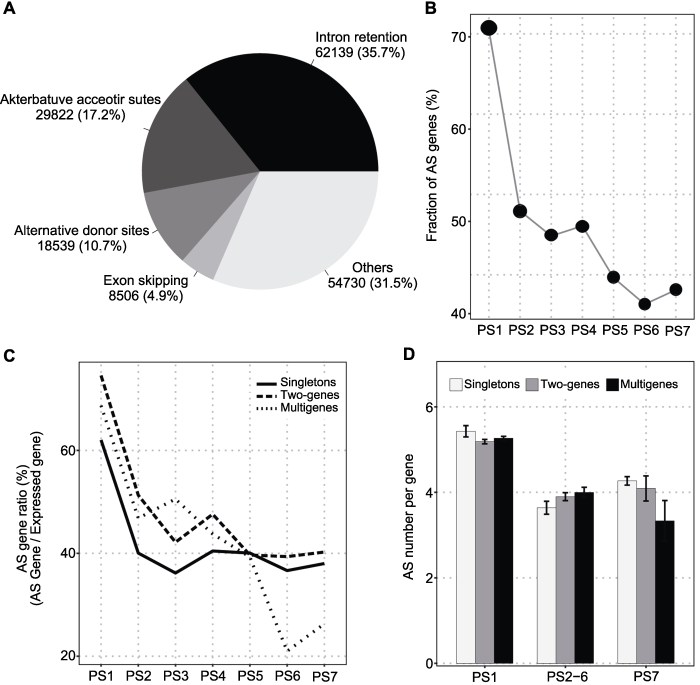
为探明大豆基因复制事件与选择性剪切事件之间的进化关系, 我们对28个样本的转录组数据进行分析, 发现有61.3% (26 114个)的已表达基因发生选择性剪切, 这一比例与前人研究结果一致(Shen et al., 2014), 同时发现内含子保留(intron retention, IR)是最主要的剪切类型, 占35.7% (图5A)。总体水平上, 基因的起源时间与发生选择性剪切基因的比例之间存在负相关, 发生选择性剪切的基因比例从36.4%-70.8%不等, 其中PS6的比例最低, 而PS1的比例最高(图5B)。有趣的是, 与单拷贝基因和多拷贝基因相比, 两拷贝基因更趋向于发生选择性剪切, 在不同起源阶段, 发生选择性剪切的两拷贝基因占总体两拷贝基因的比例介于39.4% (PS6)-74.6% (PS1)之间; 起源较早的基因中, 以多拷贝形式存在的基因发生选择性剪切的比例要高于单拷贝基因,起源较晚的基因趋势与之相反(图5C)。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5大豆基因的选择性剪切(AS)
(A) 选择性剪切事件; (B) 发生选择性剪切的基因比例; (C) 不同拷贝数状态下发生选择性剪切的基因; (D) 每个基因发生选择性剪切事件的数目
Figure 5Alternative splicing (AS) of soybean genes
(A) AS event; (B) AS genes ratio; (C) AS genes for different copy status; (D) AS number per gene
本研究中, 我们发现不同起源时期基因发生选择性剪切的数目具有差异。如图5D所示, 起源较早基因(PS1)其选择性剪切数目较多, 而起源较晚的基因发生选择性剪切的数目较少。与此同时, 在不同起源时期, 选择性剪切数目与基因家族大小之间的关系明显不同, 具体表现为: 在起源早期(PS1), 平均每个基因发生选择性剪切事件的数目较多且与基因家族大小没有显著关联; 在起源中期(PS2-6), 选择性剪切数目与基因家族大小呈正相关; 而在起源晚期(PS7), 选择性剪切数目与基因家族大小呈负相关。这可能与基因复制后不同的选择性剪切进化模式有关。
3 讨论
相比前人的研究(Guo, 2013; Jiao and Paterson, 2014), 我们选择的19个植物物种中包含了无油樟, 其被普遍认为是被子植物中最早分化出来的一个进化分支(图1A)。无油樟基因组已经完成测序(Amborella Genome Project, 2013), 这为祖先被子植物基因家族大小的重构提供了重要参考, 也有利于推断早期起源的基因家族(基因)更加精细的起源时间。借助这一关键物种基因组数据, 本研究阐明高达58.7%的大豆蛋白编码基因起源于被子植物多样化之前。本研究通过OrthoMCL (Li et al., 2003)聚类共获得34 010个直系同源基因家族, 其中所有这些物种共有的基因家族为6 122个, 这些家族可能代表了“核心”被子植物功能基因。我们挑选拟南芥基因组中每个基因家族中的一个代表性基因, 以此作为参考进行核心基因的功能富集分析, 结果显示核心基因的功能主要与氧化还原、跨膜转运和植物组织发育等重要生物学过程相关(图6)。此外, 我们发现大豆比其它所有代表种的基因组具有更高比例的两拷贝(30.3%)和多拷贝(43.5%)基因, 这可能与大豆基因组发生过2次最近的WGD事件有关, 即发生在大约5 900万年前的蝶形花亚科起源处的复制事件和大约1 300万年前大豆属特有的复制事件(Schmutz et al., 2010)。
图6
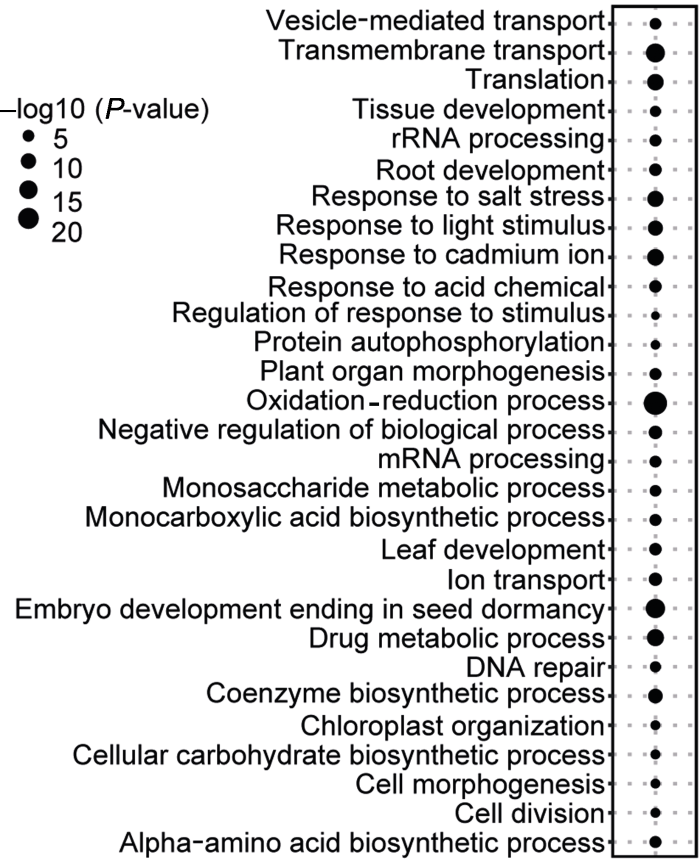 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6核心被子植物基因的功能富集分析
Figure 6Functional enrichment analyses of the core angiosperm genes
本研究中, 大豆基因组中较古老基因受到更强的选择压且进化速率较慢(图3), 这与拟南芥中的研究结果一致(Guo, 2013)。有意思的是, 在起源早期基因(PS1-PS4)中, 多拷贝基因所占比例较高(在19.1% (PS4)-60.3% (PS1)之间)且受到较大的选择压, 这表明古老基因中的复制基因可能在序列上受到更强的功能约束, 从而更容易保留下来。不同复制机制(Panchy et al., 2016)产生的基因拷贝保留下来的程度具有差异。一般来说, WGD产生的基因拷贝更倾向于保留下来。例如, WGD同时使所有基因的数量增加1倍, 而出于维持剂量平衡(dosage balance)的需要导致参与编码大分子复合体或处在同一生物学网络的基因优先保留(Tasdighian et al., 2017), 这些基因一般处在强的负选择压下。而非WGD产生的复制基因一般会导致剂量不平衡, 从而发生假基因化或丢失。相较于WGD能大量增加基因数目, 非WGD可能会将新拷贝与祖先调控特征分开, 并将它们置于新的基因组环境中, 这可能更容易产生新的表达模式和新的功能(Jiao and Paterson, 2014)。据此我们推断古老基因中的拷贝更多以WGD形式产生并保留下来。
与基因复制相同, 选择性剪切也被认为是提高物种转录组和蛋白组多样性的主要机制(Keren et al., 2010)。随着二代测序技术的发展, 研究显示植物中也存在大量的选择性剪切事件(Reddy et al., 2013)。然而, 选择性剪切和基因复制在增加蛋白组多样性的方式及进化模式上具有明显差异。基因复制之后选择性剪切的模式是如何受到影响的? 前人已经提出3种模型来解释基因组复制和选择性剪切之间的关系(Reddy et al., 2013): (1) 独立模型(independent model), 即基因家族大小与选择性剪切数量之间相互独立; (2) 功能共享模型(function-sharing model), 其预测基因家族大小与选择性剪切数量之间呈反向相关性; (3) 促进选择性剪切模型(accelerated AS model), 其推测每个基因选择性剪切事件数量的增加是由于每个旁系同源基因的放松选择压力引起。与人(Homo sapiens)和小鼠(Mus musculus)基因组中的研究结果一致(Chen et al., 2011), 不同年龄大豆基因在基因复制后的选择性剪切进化模式不同。本研究中, 我们发现起源早期(PS1)基因的选择性剪切数量与基因家族大小没有显著关联, 起源中期(PS2-6)基因的选择性剪切数量与基因家族大小呈正相关; 而在起源晚期(PS7), 选择性剪切数量与基因家族大小呈负相关(图5D)。这表明起源早期、中期和晚期基因可能分别符合独立模型、促进选择性剪切模型和功能共享模型。
综上, 大豆基因组中不同起源时间的基因具有不同的特征。大多数基因(58.7%)起源于被子植物多样化之前, 较古老基因通常以多拷贝状态存在, 功能注释较完整, 受到更大的选择压且进化速率更小, 更趋向于广谱表达且表达量较高, 有更多比例的基因发生选择性剪切且具有更多的剪切事件。此外, 不同复制状态下基因的特征也具有显著差异, 其中以两拷贝状态存在的基因进化速率较慢、表达水平较高, 越不趋向于特异性表达且更容易发生选择性剪切。本文首次分析了大豆基因在不同起源阶段的进化特征, 对理解大豆基因组的宏进化过程具有重要意义。
(责任编辑: 朱亚娜)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.3969/j.issn.1674-3466.2010.01.002URL [本文引用: 1]

基因重复是普遍存在的生物学现象, 是基因组和遗传系统多样化的重要推动力量, 在生物进化过程中发挥着极其重要的作用。基因重复有何利弊, 基因发生重复后, 2个重复子拷贝的保留在基因功能方面是否存在偏好性, 子拷贝在表达和进化速率上如何分化, 以及重复基因为什么会被保留下来一直是进化生物学领域研究的热点问题之一。该文对以上重复基因研究的热点问题进行了介绍, 并对重复基因的进化机制和理论模型及其近年来的一些主要研究进展进行了综述。
[本文引用: 1]
DOI:10.1126/science.1241089URLPMID:24357323 [本文引用: 1]
Amborella trichopoda is strongly supported as the single living species of the sister lineage to all other extant flowering plants, providing a unique reference for inferring the genome content and structure of the most recent common ancestor (MRCA) of living angiosperms. Sequencing the Amborella genome, we identified an ancient genome duplication predating angiosperm diversification, without evidence of subsequent, lineage-specific genome duplications. Comparisons between Amborella and other angiosperms facilitated reconstruction of the ancestral angiosperm gene content and gene order in the MRCA of core eudicots. We identify new gene families, gene duplications, and floral protein-protein interactions that first appeared in the ancestral angiosperm. Transposable elements in Amborella are ancient and highly divergent, with no recent transposon radiations. Population genomic analysis across Amborella's native range in New Caledonia reveals a recent genetic bottleneck and geographic structure with conservation implications.
DOI:10.1093/bioinformatics/btu170URLPMID:4103590 [本文引用: 1]
Abstract MOTIVATION: Although many next-generation sequencing (NGS) read preprocessing tools already existed, we could not find any tool or combination of tools that met our requirements in terms of flexibility, correct handling of paired-end data and high performance. We have developed Trimmomatic as a more flexible and efficient preprocessing tool, which could correctly handle paired-end data. RESULTS: The value of NGS read preprocessing is demonstrated for both reference-based and reference-free tasks. Trimmomatic is shown to produce output that is at least competitive with, and in many cases superior to, that produced by other tools, in all scenarios tested. AVAILABILITY AND IMPLEMENTATION: Trimmomatic is licensed under GPL V3. It is cross-platform (Java 1.5+ required) and available at http://www.usadellab.org/cms/index.php?page=trimmomatic CONTACT: usadel@bio1.rwth-aachen.de SUPPLEMENTARY INFORMATION: Supplementary data are available at Bioinformatics online. The Author 2014. Published by Oxford University Press.
DOI:10.1093/gbe/evp013URLPMID:20333184 [本文引用: 1]
A number of studies have showed that recently created genes differ from the genes created in deep evolutionary past in many aspects. Here, we determined the age of emergence and propensity for gene loss (PGL) of all human protein鈥揷oding genes and compared disease genes with non-disease genes in terms of their evolutionary rate, strength of purifying selection, mRNA expression, and genetic redundancy. The older and the less prone to loss, non-disease genes have been evolving 1.5- to 3-fold slower between humans and chimps than young non-disease genes, whereas Mendelian disease genes have been evolving very slowly regardless of their ages and PGL. Complex disease genes showed an intermediate pattern. Disease genes also have higher mRNA expression heterogeneity across multiple tissues than non-disease genes regardless of age and PGL. Young and middle-aged disease genes have fewer similar paralogs as non-disease genes of the same age. We reasoned that genes were more likely to be involved in human disease if they were under a strong functional constraint, expressed heterogeneously across tissues, and lacked genetic redundancy. Young human genes that have been evolving under strong constraint between humans and chimps might also be enriched for genes that encode important primate or even human-specific functions.
DOI:10.1038/nrg3521URLPMID:4236023 [本文引用: 2]
Abstract During the course of evolution, genomes acquire novel genetic elements as sources of functional and phenotypic diversity, including new genes that originated in recent evolution. In the past few years, substantial progress has been made in understanding the evolution and phenotypic effects of new genes. In particular, an emerging picture is that new genes, despite being present in the genomes of only a subset of species, can rapidly evolve indispensable roles in fundamental biological processes, including development, reproduction, brain function and behaviour. The molecular underpinnings of how new genes can develop these roles are starting to be characterized. These recent discoveries yield fresh insights into our broad understanding of biological diversity at refined resolution.
DOI:10.1186/1471-2164-12-S3-S16URLPMID:22369477 [本文引用: 1]
Background Gene duplication provides resources for developing novel genes and new functions while retaining the original functions. In addition, alternative splicing could increase the complexity of expression at the transcriptome and proteome level without increasing the number of gene copy in the genome. Duplication and alternative splicing are thought to work together to provide the diverse functions or expression patterns for eukaryotes. Previously, it was believed that duplication and alternative splicing were negatively correlated and probably interchangeable. Results We look into the relationship between occurrence of alternative splicing and duplication at different time after duplication events. We found duplication and alternative splicing were indeed inversely correlated if only recently duplicated genes were considered, but they became positively correlated when we took those ancient duplications into account. Specifically, for slightly or moderately duplicated genes with gene families containing 2 - 7 paralogs, genes were more likely to evolve alternative splicing and had on average a greater number of alternative splicing isoforms after long-term evolution compared to singleton genes. On the other hand, those large gene families (contain at least 8 paralogs) had a lower proportion of alternative splicing, and fewer alternative splicing isoforms on average even when ancient duplicated genes were taken into consideration. We also found these duplicated genes having alternative splicing were under tighter evolutionary constraints compared to those having no alternative splicing, and had an enrichment of genes that participate in molecular transducer activities. Conclusions We studied the association between occurrences of alternative splicing and gene duplication. Our results implicate that there are key differences in functions and evolutionary constraints among singleton genes or duplicated genes with or without alternative splicing incidences. It implies that the gene duplication and alternative splicing may have different functional significance in the evolution of speciation diversity.
DOI:10.1016/j.tig.2007.08.014URLPMID:18029048 [本文引用: 1]
Macroevolutionary trends traditionally are studied by fossil analysis, comparative morphology or evo-devo approaches. With the availability of genome sequences and associated data from an increasing diversity of taxa, it is now possible to add an additional level of analysis: genomic phylostratigraphy. As an example of this approach, we use a phylogenetic framework and embryo expression data from Drosophila to show that grouping genes by their phylogenetic origin can uncover footprints of important adaptive events in evolution.
DOI:10.1104/pp.102.018150URL [本文引用: 1]
http://www.plantphysiol.org/cgi/doi/10.1104/pp.102.018150
URLPMID:11917018 [本文引用: 1]
Abstract Detection of protein families in large databases is one of the principal research objectives in structural and functional genomics. Protein family classification can significantly contribute to the delineation of functional diversity of homologous proteins, the prediction of function based on domain architecture or the presence of sequence motifs as well as comparative genomics, providing valuable evolutionary insights. We present a novel approach called TRIBE-MCL for rapid and accurate clustering of protein sequences into families. The method relies on the Markov cluster (MCL) algorithm for the assignment of proteins into families based on precomputed sequence similarity information. This novel approach does not suffer from the problems that normally hinder other protein sequence clustering algorithms, such as the presence of multi-domain proteins, promiscuous domains and fragmented proteins. The method has been rigorously tested and validated on a number of very large databases, including SwissProt, InterPro, SCOP and the draft human genome. Our results indicate that the method is ideally suited to the rapid and accurate detection of protein families on a large scale. The method has been used to detect and categorise protein families within the draft human genome and the resulting families have been used to annotate a large proportion of human proteins.
DOI:10.1093/nar/gkm311URLPMID:17485470 [本文引用: 1]
n the process of establishing more and more complete annotations of eukaryotic genomes, a constantly growing number of alternative splicing (AS) events has been reported over the last decade. Consequently, the increasing transcript coverage also revealed the real complexity of some variations in the exon-intron structure between transcript variants and the need for computational tools to address 'complex' AS events. ASTALAVISTA (alternative splicing transcriptional landscape visualization tool) employs an intuitive and complete notation system to univocally identify such events. The method extracts AS events dynamically from custom gene annotations, classifies them into groups of common types and visualizes a comprehensive picture of the resulting AS landscape. Thus, ASTALAVISTA can characterize AS for whole transcriptome data from reference annotations (GENCODE, REFSEQ, ENSEMBL) as well as for genes selected by the user according to common functional/structural attributes of interest: http://genome.imim.es/astalavista.
DOI:10.1146/annurev.arplant.043008.092122URL [本文引用: 1]
DOI:10.1111/tpj.12089URLPMID:23216999 [本文引用: 3]
Gene family size variation is an important mechanism that shapes the natural variation for adaptation in various species. Despite its importance, the pattern of gene family size variation in green plants is still not well understood. In particular, the evolutionary pattern of genes and gene families remains unknown in the model plant Arabidopsis thaliana in the context of green plants. In this study, eight representative genomes of green plants are sampled to study gene family evolution and characterize the origination of A. thaliana genes, respectively. Four important insights gained are that: (i) the rate of gene gains and losses is about 0.001359 per gene every million years, similar to the rate in yeast, Drosophila, and mammals; (ii) some gene families evolved rapidly with extreme expansions or contractions, and 2745 gene families present in all the eight species represent the ore proteome of green plants; (iii) 70% of A. thaliana genes could be traced back to 450 million years ago; and (iv) intriguingly, A. thaliana genes with early origination are under stronger purifying selection and more conserved. In summary, the present study provides genome-wide insights into evolutionary history and mechanisms of genes and gene families in green plants and especially in A. thaliana.
DOI:10.1098/rstb.2013.0355URLPMID:4071528 [本文引用: 2]
The occurrence of polyploidy in land plant evolution has led to an acceleration of genome modifications relative to other crown eukaryotes and is correlated with key innovations in plant evolution. Extensive genome resources provide for relating genomic changes to the origins of novel morphological and physiological features of plants. Ancestral gene contents for key nodes of the plant family tree are inferred. Pervasive polyploidy in angiosperms appears likely to be the major factor generating novel angiosperm genes and expanding some gene families. However, most gene families lose most duplicated copies in a quasi-neutral process, and a few families are actively selected for single-copy status. One of the great challenges of evolutionary genomics is to link genome modifications to speciation, diversification and the morphological and/or physiological innovations that collectively compose biodiversity. Rapid accumulation of genomic data and its ongoing investigation may greatly improve the resolution at which evolutionary approaches can contribute to the identification of specific genes responsible for particular innovations. The resulting, more 'particulate' understanding of plant evolution, may elevate to a new level fundamental knowledge of botanical diversity, including economically important traits in the crop plants that sustain humanity.
DOI:10.1101/gr.101386.109URL [本文引用: 2]
DOI:10.1038/nrg2776URLPMID:20376054 [本文引用: 1]
Over the past decade, it has been shown that alternative splicing (AS) is a major mechanism for the enhancement of transcriptome and proteome diversity, particularly in mammals. Splicing can be found in species from bacteria to humans, but its prevalence and characteristics vary considerably. Evolutionary studies are helping to address questions that are fundamental to understanding this important process: how and when did AS evolve? Which AS events are functional? What are the evolutionary forces that shaped, and continue to shape, AS? And what determines whether an exon is spliced in a constitutive or alternative manner? In this Review, we summarize the current knowledge of AS and evolution and provide insights into some of these unresolved questions.
DOI:10.1038/nmeth.3317URLPMID:4655817 [本文引用: 1]
Abstract HISAT (hierarchical indexing for spliced alignment of transcripts) is a highly efficient system for aligning reads from RNA sequencing experiments. HISAT uses an indexing scheme based on the Burrows-Wheeler transform and the Ferragina-Manzini (FM) index, employing two types of indexes for alignment: a whole-genome FM index to anchor each alignment and numerous local FM indexes for very rapid extensions of these alignments. HISAT's hierarchical index for the human genome contains 48,000 local FM indexes, each representing a genomic region of 09080464,000 bp. Tests on real and simulated data sets showed that HISAT is the fastest system currently available, with equal or better accuracy than any other method. Despite its large number of indexes, HISAT requires only 4.3 gigabytes of memory. HISAT supports genomes of any size, including those larger than 4 billion bases.
DOI:10.1093/bioinformatics/btm404URL [本文引用: 1]
DOI:10.1101/gr.1224503URL [本文引用: 3]
[本文引用: 2]
DOI:10.1126/science.290.5494.1151URLPMID:11073452 [本文引用: 1]
Gene duplication has generally been viewed as a necessary source of material for the origin of evolutionary novelties, but it is unclear how often gene duplicates arise and how frequently they evolve new functions. Observations from the genomic databases for several eukaryotic species suggest that duplicate genes arise at a very high rate, on average 0.01 per gene per million years. Most duplicated genes experience a brief period of relaxed selection early in their history, with a moderate fraction of them evolving in an effectively neutral manner during this period. However, the vast majority of gene duplicates are silenced within a few million years, with the few survivors subsequently experiencing strong purifying selection. Although duplicate genes may only rarely evolve new functions, the stochastic silencing of such genes may play a significant role in the passive origin of new species.
DOI:10.1126/science.1228186URLPMID:23258891 [本文引用: 1]
Abstract Most mammalian genes produce multiple distinct messenger RNAs through alternative splicing, but the extent of splicing conservation is not clear. To assess tissue-specific transcriptome variation across mammals, we sequenced complementary DNA from nine tissues from four mammals and one bird in biological triplicate, at unprecedented depth. We find that while tissue-specific gene expression programs are largely conserved, alternative splicing is well conserved in only a subset of tissues and is frequently lineage-specific. Thousands of previously unknown, lineage-specific, and conserved alternative exons were identified; widely conserved alternative exons had signatures of binding by MBNL, PTB, RBFOX, STAR, and TIA family splicing factors, implicating them as ancestral mammalian splicing regulators. Our data also indicate that alternative splicing often alters protein phosphorylatability, delimiting the scope of kinase signaling.
DOI:10.3835/plantgenome2013.03.0001inURL [本文引用: 1]
Crop yields at summit positions of rolling landscapes often are lower than backslope yields. The differences in plant response may be the result of many different factors. We examined corn (Zea mays L.) plant productivity, gene expression, soil water, and nutrient availability in two landscape positions located in historically high (backslope) and moderate (summit and shoulder) yielding zones to gain insight into plant response differences. Growth characteristics, gene expression, and soil parameters (water and N and P content) were determined at the V12 growth stage of corn. At tassel, plant biomass, N content, C-13 isotope discrimination (Delta), and soil water was measured. Soil water was 35% lower in the summit and shoulder compared with the lower backslope plots. Plants at the summit had 16% less leaf area, biomass, and N and P uptake at V12 and 30% less biomass at tassel compared with plants from the lower backslope. Transcriptome analysis at V12 indicated that summit and shoulder-grown plants had 496 downregulated and 341 upregulated genes compared with backslope-grown plants. Gene set and subnetwork enrichment analyses indicated alterations in growth and circadian response and lowered nutrient uptake, wound recovery, pest resistance, and photosynthetic capacity in summit and shoulder-grown plants. Reducing plant populations, to lessen demands on available soil water, and applying pesticides, to limit biotic stress, may ameliorate negative water stress responses.
DOI:10.1016/j.pbi.2015.02.002URLPMID:25703261 [本文引用: 1]
The availability of plant reference genomes has ushered in a new era of crop genomics. More than 100 plant genomes have been sequenced since 2000, 63% of which are crop species. These genome sequences provide insight into architecture, evolution and novel aspects of crop genomes such as the retention of key agronomic traits after whole genome duplication events. Some crops have very large, polyploid, repeat-rich genomes, which require innovative strategies for sequencing, assembly and analysis. Even low quality reference genomes have the potential to improve crop germplasm through genome-wide molecular markers, which decrease expensive phenotyping and breeding cycles. The next stage of plant genomics will require draft genome refinement, building resources for crop wild relatives, resequencing broad diversity panels, and plant ENCODE projects to better understand the complexities of these highly diverse genomes.
[本文引用: 1]
DOI:10.1104/pp.16.00523URLPMID:27288366 [本文引用: 2]
Ancient duplication events and a high rate of retention of extant pairs of duplicate genes have contributed to an abundance of duplicate genes in plant genomes. These duplicates have contributed to the evolution of novel functions, such as the production of floral structures, induction of disease resistance, and adaptation to stress. Additionally, recent whole-genome duplications that have occurred in the lineages of several domesticated crop species, including wheat (Triticum aestivum), cotton (Gossypium hirsutum), and soybean (Glycine max), have contributed to important agronomic traits, such as grain quality, fruit shape, and flowering time. Therefore, understanding the mechanisms and impacts of gene duplication will be important to future studies of plants in general and of agronomically important crops in particular. In this review, we survey the current knowledge about gene duplication, including gene duplication mechanisms, the potential fates of duplicate genes, models explaining duplicate gene retention, the properties that distinguish duplicate from singleton genes, and the evolutionary impact of gene duplication.
DOI:10.1038/nbt.3122URLPMID:25690850 [本文引用: 1]
Abstract Methods used to sequence the transcriptome often produce more than 200 million short sequences. We introduce StringTie, a computational method that applies a network flow algorithm originally developed in optimization theory, together with optional de novo assembly, to assemble these complex data sets into transcripts. When used to analyze both simulated and real data sets, StringTie produces more complete and accurate reconstructions of genes and better estimates of expression levels, compared with other leading transcript assembly programs including Cufflinks, IsoLasso, Scripture and Traph. For example, on 90 million reads from human blood, StringTie correctly assembled 10,990 transcripts, whereas the next best assembly was of 7,187 transcripts by Cufflinks, which is a 53% increase in transcripts assembled. On a simulated data set, StringTie correctly assembled 7,559 transcripts, which is 20% more than the 6,310 assembled by Cufflinks. As well as producing a more complete transcriptome assembly, StringTie runs faster on all data sets tested to date compared with other assembly software, including Cufflinks.
DOI:10.1038/nature11394URLPMID:22951968 [本文引用: 2]
Abstract Animal and plant development starts with a constituting phase called embryogenesis, which evolved independently in both lineages. Comparative anatomy of vertebrate development--based on the Meckel-Serrs law and von Baer's laws of embryology from the early nineteenth century--shows that embryos from various taxa appear different in early stages, converge to a similar form during mid-embryogenesis, and again diverge in later stages. This morphogenetic series is known as the embryonic 'hourglass', and its bottleneck of high conservation in mid-embryogenesis is referred to as the phylotypic stage. Recent analyses in zebrafish and Drosophila embryos provided convincing molecular support for the hourglass model, because during the phylotypic stage the transcriptome was dominated by ancient genes and global gene expression profiles were reported to be most conserved. Although extensively explored in animals, an embryonic hourglass has not been reported in plants, which represent the second major kingdom in the tree of life that evolved embryogenesis. Here we provide phylotranscriptomic evidence for a molecular embryonic hourglass in Arabidopsis thaliana, using two complementary approaches. This is particularly significant because the possible absence of an hourglass based on morphological features in plants suggests that morphological and molecular patterns might be uncoupled. Together with the reported developmental hourglass patterns in animals, these findings indicate convergent evolution of the molecular hourglass and a conserved logic of embryogenesis across kingdoms.
DOI:10.1105/tpc.113.117523URL [本文引用: 2]
DOI:10.1038/nature08957URL [本文引用: 3]
DOI:10.1105/tpc.114.122739URLPMID:24681622 [本文引用: 2]
Alternative splicing (AS) is common in higher and plays an important role in gene posttranscriptional regulation. It has been suggested that AS varies dramatically among species, tissues, and duplicated gene families of different sizes. However, the genomic forces that govern AS variation remain poorly understood. Here, through genome-wide identification of AS events in the () genome using high-throughput sequencing of 28 samples from different developmental stages, we found that more than 63% of multiexonic genes underwent AS. More AS events occurred in the younger developmental stages than in the older developmental stages for the same type of tissue, and the four main AS types, exon skipping, intron retention, alternative donor sites, and alternative acceptor sites, exhibited different characteristics. Global computational analysis demonstrated that the variations of AS frequency and AS types were significantly correlated with the changes of gene features and gene transcriptional level. Further investigation suggested that the decrease of AS within the genome-wide duplicated genes were due to the diminution of intron length, exon number, and transcriptional level. Altogether, our study revealed that a large number of genes were alternatively spliced in the genome and that variations in gene structure and transcriptional level may play important roles in regulating AS.
DOI:10.1093/nar/gkl315URLPMID:16845082 [本文引用: 1]
PAL2NAL is a web server that constructs a multiple codon alignment from the corresponding aligned protein sequences. Such codon alignments can be used to evaluate the type and rate of nucleotide substitutions in coding DNA for a wide range of evolutionary analyses, such as the identification of levels of selective constraint acting on genes, or to perform DNA-based phylogenetic studies. The server takes a protein sequence alignment and the corresponding DNA sequences as input. In contrast to other existing applications, this server is able to construct codon alignments even if the input DNA sequence has mismatches with the input protein sequence, or contains untranslated regions and polyA tails. The server can also deal with frame shifts and inframe stop codons in the input models, and is thus suitable for the analysis of pseudogenes. Another distinct feature is that the user can specify a subregion of the input alignment in order to specifically analyze functional domains or exons of interest. The PAL2NAL server is available at http://www.bork.embl.de/pal2nal.
DOI:10.1105/tpc.17.00313URLPMID:29061868 [本文引用: 1]
In several organisms, particular functional categories of genes, such as regulatory and complex-forming genes, are preferentially retained after whole-genome multiplications but rarely duplicate through small-scale duplication, a pattern referred to as reciprocal retention. This peculiar duplication behavior is hypothesized to stem from constraints on the dosage balance between the genes concerned and their interaction context. However, the evidence for a relationship between reciprocal retention and dosage balance sensitivity remains fragmentary. Here, we identified which gene families are most strongly reciprocally retained in the angiosperm lineage and studied their functional and evolutionary characteristics. Reciprocally retained gene families exhibit stronger sequence divergence constraints and lower rates of functional and expression divergence than other gene families, suggesting that dosage balance sensitivity is a general characteristic of reciprocally retained genes. Gene families functioning in regulatory and signaling processes are much more strongly represented at the top of the reciprocal retention ranking than those functioning in multiprotein complexes, suggesting that regulatory imbalances may lead to stronger fitness effects than classical stoichiometric protein complex imbalances. Finally, reciprocally retained duplicates are often subject to dosage balance constraints for prolonged evolutionary times, which may have repercussions for the ease with which genome multiplications can engender evolutionary innovation.
DOI:10.1038/nrg3053URLPMID:21878963 [本文引用: 1]
Gene evolution has long been thought to be primarily driven by duplication and rearrangement mechanisms. However, every evolutionary lineage harbours orphan genes that lack homologues in other lineages and whose evolutionary origin is only poorly understood. Orphan genes might arise from duplication and rearrangement processes followed by fast divergence; however, de novo evolution out of non-coding genomic regions is emerging as an important additional mechanism. This process appears to provide raw material continuously for the evolution of new gene functions, which can become relevant for lineage-specific adaptations.
DOI:10.1038/nbt.1621URLPMID:20436464 [本文引用: 1]
High-throughput mRNA sequencing (RNA-Seq) promises simultaneous transcript discovery and abundance estimation. However, this would require algorithms that are not restricted by prior gene annotations and that account for alternative transcription and splicing. Here we introduce such algorithms in an open-source software program called Cufflinks. To test Cufflinks, we sequenced and analyzed >430 million paired 75-bp RNA-Seq reads from a mouse myoblast cell line over a differentiation time series. We detected 13,692 known transcripts and 3,724 previously unannotated ones, 62% of which are supported by independent expression data or by homologous genes in other species. Over the time series, 330 genes showed complete switches in the dominant transcription start site (TSS) or splice isoform, and we observed more subtle shifts in 1,304 other genes. These results suggest that Cufflinks can illuminate the substantial regulatory flexibility and complexity in even this well-studied model of muscle development and that it can improve transcriptome-based genome annotation.
DOI:10.1093/bioinformatics/bti042URLPMID:15388519 [本文引用: 1]
Genes are often characterized dichotomously as either housekeeping or single-tissue specific. We conjectured that crucial functional information resides in genes with midrange profiles of expression.
DOI:10.1093/molbev/msm088URLPMID:17483113 [本文引用: 1]
Abstract PAML, currently in version 4, is a package of programs for phylogenetic analyses of DNA and protein sequences using maximum likelihood (ML). The programs may be used to compare and test phylogenetic trees, but their main strengths lie in the rich repertoire of evolutionary models implemented, which can be used to estimate parameters in models of sequence evolution and to test interesting biological hypotheses. Uses of the programs include estimation of synonymous and nonsynonymous rates (d(N) and d(S)) between two protein-coding DNA sequences, inference of positive Darwinian selection through phylogenetic comparison of protein-coding genes, reconstruction of ancestral genes and proteins for molecular restoration studies of extinct life forms, combined analysis of heterogeneous data sets from multiple gene loci, and estimation of species divergence times incorporating uncertainties in fossil calibrations. This note discusses some of the major applications of the package, which includes example data sets to demonstrate their use. The package is written in ANSI C, and runs under Windows, Mac OSX, and UNIX systems. It is available at -- (http://abacus.gene.ucl.ac.uk/software/paml.html).
DOI:10.1016/S0169-5347(03)00033-8URL [本文引用: 3]
The importance of gene duplication in supplying raw genetic material to biological evolution has been recognized since the 1930s. Recent genomic sequence data provide substantial evidence for the abundance of duplicated genes in all organisms surveyed. But how do newly duplicated genes survive and acquire novel functions, and what role does gene duplication play in the evolution of genomes and organisms? Detailed molecular characterization of individual gene families, computational analysis of genomic sequences and population genetic modeling can all be used to help us uncover the mechanisms behind the evolution by gene duplication.
重复基因的进化——回顾与进展
1
2010
... 真核生物中, 基因复制(gene duplication, GD)是新基因的主要来源, 可作为研究新功能进化的原材料, 也是增加基因家族大小的重要因素(
Evolution by gene loss
1
2016
... 真核生物中, 基因复制(gene duplication, GD)是新基因的主要来源, 可作为研究新功能进化的原材料, 也是增加基因家族大小的重要因素(
The Amborella genome and the evolution of flowering plants
1
2013
... 相比前人的研究(
Trimmomatic: a flexible trimmer for Illumina sequence data
1
2014
... RNA-seq产生的原始fastq格式文件从NCBI SRA数据库中筛选获得, 然后采用Trimmomatic v0.36软件 (
Similarly strong purifying selection acts on human disease genes of all evolutionary ages
1
2009
... 随着高通量技术的快速发展, 有来自不同分类群的100多种植物相继完成全基因组测序(
New genes as drivers of phenotypic evolution
2
2013
... 基因组变异是生物表型多样性的主要来源, 而新基因的出现是基因组进化及物种间遗传差异的重要因素之一(
... ;
Interrogation of alternative splicing events in duplicated genes during evolution
1
2011
... 与基因复制相同, 选择性剪切也被认为是提高物种转录组和蛋白组多样性的主要机制(
A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages
1
2007
... 随着高通量技术的快速发展, 有来自不同分类群的100多种植物相继完成全基因组测序(
The rest of the iceberg. Legume diversity and evolution in a phylogenetic context
1
2003
... 豆科是被子植物中物种数目最多的家族之一, 有20 000多个种(
An efficient algorithm for large-scale detection of protein families
1
2002
... 为了获得高质量的蛋白质序列数据以鉴定基因的同源关系, 我们对上述19种被子植物的蛋白质组数据按以下条件进行过滤: (1) 去除长度小于50个氨基酸残基的蛋白质; (2) 对于由可变剪切产生的多个转录本所翻译的蛋白质, 只保留每个基因最长转录本对应的蛋白质.过滤之后, 所有19个物种的共641 473条蛋白质序列作为输入数据提交至OrthoMCL v2.0.9 (
ASTALAVISTA: dynamic and flexible analysis of alternative splicing events in custom gene datasets
1
2007
... 利用Cufflinks (
Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition
1
2009
... 真核生物中, 基因复制(gene duplication, GD)是新基因的主要来源, 可作为研究新功能进化的原材料, 也是增加基因家族大小的重要因素(
Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes
3
2013
... 随着高通量技术的快速发展, 有来自不同分类群的100多种植物相继完成全基因组测序(
... 相比前人的研究(
... 本研究中, 大豆基因组中较古老基因受到更强的选择压且进化速率较慢(
Polyploidy-associated genome modifications during land plant evolution
2
2014
... 相比前人的研究(
... 本研究中, 大豆基因组中较古老基因受到更强的选择压且进化速率较慢(
Origins, evolution, and phenotypic impact of new genes
2
2010
... 基因组变异是生物表型多样性的主要来源, 而新基因的出现是基因组进化及物种间遗传差异的重要因素之一(
... ;
Alternative splicing and evolution: diversification, exon definition and function
1
2010
... 与基因复制相同, 选择性剪切也被认为是提高物种转录组和蛋白组多样性的主要机制(
HISAT: a fast spliced aligner with low memory requirements
1
2015
... RNA-seq产生的原始fastq格式文件从NCBI SRA数据库中筛选获得, 然后采用Trimmomatic v0.36软件 (
Clustal W and Clustal X version 2.0
1
2007
... 为了估算出大豆中基因的进化速率, 我们从OrthoMCL的聚类结果中提取出大豆与菜豆的直系同源基因对; 用ClustalW v2.1软件(
OrthoMCL: identification of ortholog groups for eukaryotic genomes
3
2003
... 为了获得高质量的蛋白质序列数据以鉴定基因的同源关系, 我们对上述19种被子植物的蛋白质组数据按以下条件进行过滤: (1) 去除长度小于50个氨基酸残基的蛋白质; (2) 对于由可变剪切产生的多个转录本所翻译的蛋白质, 只保留每个基因最长转录本对应的蛋白质.过滤之后, 所有19个物种的共641 473条蛋白质序列作为输入数据提交至OrthoMCL v2.0.9 (
... 利用OrthoMCL软件(
... 本研究通过OrthoMCL (
The origin of new genes: glimpses from the young and old
2
2003
... 基因组变异是生物表型多样性的主要来源, 而新基因的出现是基因组进化及物种间遗传差异的重要因素之一(
... ;
The evolutionary fate and consequences of duplicate genes
1
2000
... 基因组变异是生物表型多样性的主要来源, 而新基因的出现是基因组进化及物种间遗传差异的重要因素之一(
Evolutionary dynamics of gene and isoform regulation in mam- malian tissues
1
2012
... 利用Cufflinks (
The first 50 plant genomes
1
2013
... 随着高通量技术的快速发展, 有来自不同分类群的100多种植物相继完成全基因组测序(
Progress, challenges and the future of crop genomes
1
2015
... 随着高通量技术的快速发展, 有来自不同分类群的100多种植物相继完成全基因组测序(
Evolution by Gene Duplication
1
1970
... 真核生物中, 基因复制(gene duplication, GD)是新基因的主要来源, 可作为研究新功能进化的原材料, 也是增加基因家族大小的重要因素(
Evolution of gene duplication in plants
2
2016
... 真核生物中, 基因复制(gene duplication, GD)是新基因的主要来源, 可作为研究新功能进化的原材料, 也是增加基因家族大小的重要因素(
... 本研究中, 大豆基因组中较古老基因受到更强的选择压且进化速率较慢(
StringTie enables improved reconstruction of a transcriptome from RNA-seq reads
1
2015
... RNA-seq产生的原始fastq格式文件从NCBI SRA数据库中筛选获得, 然后采用Trimmomatic v0.36软件 (
A transcriptomic hourglass in plant embryogenesis
2
2012
... 随着高通量技术的快速发展, 有来自不同分类群的100多种植物相继完成全基因组测序(
... ). 该方法根据被调查物种基因在一组亲缘关系由近及远的代表物种基因组中是否有序列相似的同源基因, 将基因映射到不同的系统发育层级, 称为phylostratigraphy或phylostrata (PS), 代表相关基因大致的起源时间区间.目前, 该方法已广泛应用于追溯基因的进化历程, 及研究基因的起源时间与一些重要生物学过程(如胚胎发育)的关系(
Complexity of the alternative splicing landscape in plants
2
2013
... 与基因复制相同, 选择性剪切也被认为是提高物种转录组和蛋白组多样性的主要机制(
... ).然而, 选择性剪切和基因复制在增加蛋白组多样性的方式及进化模式上具有明显差异.基因复制之后选择性剪切的模式是如何受到影响的? 前人已经提出3种模型来解释基因组复制和选择性剪切之间的关系(
Genome sequence of the palaeopolyploid soybean
3
2010
... 豆科是被子植物中物种数目最多的家族之一, 有20 000多个种(
... 利用OrthoMCL软件(
... 本研究通过OrthoMCL (
Global dissection of alternative splicing in paleopolyploid soybean
2
2014
... 对于起源时间指定到7个不同系统发育层级的52 733个大豆基因, 其中有42 591个(80.1%)在至少1个组织样本中表达(
... 为探明大豆基因复制事件与选择性剪切事件之间的进化关系, 我们对28个样本的转录组数据进行分析, 发现有61.3% (26 114个)的已表达基因发生选择性剪切, 这一比例与前人研究结果一致(
PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments
1
2006
... 为了估算出大豆中基因的进化速率, 我们从OrthoMCL的聚类结果中提取出大豆与菜豆的直系同源基因对; 用ClustalW v2.1软件(
Reciprocally retained genes in the angiosperm lineage show the hallmarks of dosage balance sensitivity
1
2017
... 本研究中, 大豆基因组中较古老基因受到更强的选择压且进化速率较慢(
The evolutionary origin of orphan genes
1
2011
... 真核生物中, 基因复制(gene duplication, GD)是新基因的主要来源, 可作为研究新功能进化的原材料, 也是增加基因家族大小的重要因素(
Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation
1
2010
... 利用Cufflinks (
Genome-wide midrange transcription profiles reveal expression level re- lationships in human tissue specification
1
2005
... 其中, N代表参与计算的不同发育阶段的所有组织样本的数量, xi代表基因在每个样本中的表达量, xmax代表基因在所有样本中的最大表达量.τ值在0-1范围内, 一般将τ≥0.85的基因作为组织和发育阶段特异性表达基因, 而τ<0.15则视为持家基因(
PAML 4: phylogenetic analysis by maximum likelihood
1
2007
... 为了估算出大豆中基因的进化速率, 我们从OrthoMCL的聚类结果中提取出大豆与菜豆的直系同源基因对; 用ClustalW v2.1软件(
Evolution by gene duplication: an update
3
2003
... 真核生物中, 基因复制(gene duplication, GD)是新基因的主要来源, 可作为研究新功能进化的原材料, 也是增加基因家族大小的重要因素(
... ).基因某一拷贝上偶尔也会累积有利突变, 在达尔文正选择作用下驱动该拷贝进化出新的生物学功能, 即所谓的新功能化(neo-functionalization), 而另一拷贝则执行原有的功能(
... ).此外, 对于多功能的基因还有可能出现复制产生的2个拷贝以功能互补的方式各自承担起父本基因的部分角色, 即亚功能化(sub-functionalization) (
备案号: 京ICP备16067583号-21
版权所有 © 2021 《植物学报》编辑部
地址:北京香山南辛村20号 邮编:100093
电话:010-62836135 010-62836131 E-mail:cbb@ibcas.ac.cn
本系统由北京玛格泰克科技发展有限公司设计开发

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}