王影 1 , 2 , * , ,
李相敢 2 ,
邱丽娟 1 , * , 1 中国农业科学院作物科学研究所, 北京 1000812 先正达生物科技(中国)有限公司, 北京 102206Wang Ying 1 , 2 , * , ,
Li Xianggan 2 ,
Qiu Lijuan 1 , * , 1 Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China2 Syngenta Biotechnology China Co, LTD, Beijing 102206, China引用本文 王影 ,
李相敢 ,
邱丽娟 . CRISPR/Cas9基因组定点编辑中脱靶现象的研究进展. 植物学报, 2018, 53(4): 528-541
贡献者
* 通讯作者。E-mail: ying.wang@syngenta.com * 通讯作者。E-mail: qiulijuan@caas.cn 接受日期:2018-01-3接受日期:2018-03-29网络出版日期:2018-07-1
-->Copyright
2018《植物学报》编辑部
Contributors
* Authors for correspondence. E-mail: ying.wang@syngenta.com * Authors for correspondence. E-mail: qiulijuan@caas.cn History
Received :
Accepted :
Online :
摘要: 近年来, CRISPR定点编辑技术发展迅猛, 在动物、植物和微生物中均得到广泛应用。其中, 备受关注的脱靶现象也是研究的热点, 迄今已取得了重要进展。该文介绍了脱靶现象的产生原理及体内和体外检测脱靶现象的方法, 评价了通过改进sgRNA设计和优化CRISPR系统等来降低脱靶率的方法。在植物基因组定点编辑过程中, 应适时检测脱靶现象, 提高脱靶检测的精确度和准确度。 关键词 :
CRISPR/Cas9 ;
定点编辑 ;
脱靶 Abstract :
CRISPR/Cas9 technology has rapidly developed in recent years. It is widely used for study of animals, plants and microorganisms. Among all developments, the use of the technology for the off-target issue has progressed, with important breakthroughs for this hot topic. This paper elucidates the principle of off-target generation and the method for detecting off target both in vitro and in vivo . Also, how to reduce off-targets by improving single-guide RNA design and optimizing the CRISPR system is evaluated. Further more, detecting off-targets at the right time and improving the precision and accuracy of off-target detection are proposed for plant site-specific genome editing. Key words :
CRISPR/Cas9 ;
site specific editing ;
off-target 近年来, CRISPR定点编辑技术迅猛发展, 在动物、植物、微生物和人类(
Homo sapiens )细胞中应用广泛(
Jiang et al., 2013 ;
Li et al., 2013 ;
Wang et al., 2014 ,
2017 )。该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用。然而, CRISPR系统作为细菌中的获得性免疫系统(
Deveau et al., 2010 ;
Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配。在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象。脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用。自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况。从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展。
Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA。2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(
Fu et al., 2013 )。他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变。这一发现迅速引起了科研人员的关注。Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(
Hsu et al., 2013 )。他们在人
EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性。结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列。同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割。关于植物细胞中CRISPR/Cas9脱靶现象的研究较少。现有文献显示, 植物中的脱靶切割频率较低。例如,
Belhaj等(2013) 在烟草(
Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割。朱健康实验室在水稻(
Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异。结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(
Zhang et al., 2014 )。同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(
Xu et al., 2014 )。本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性。
1 脱靶现象的产生细菌来源的CRISPR/Cas9系统在真核细胞中行使定点切割的功能, 需要3个重要的元件: Cas9蛋白质、向导RNA (crRNA)和转运激活RNA (
trans -activating crRNA, tracrRNA)。向导RNA和转运激活RNA可以组装成一个元件, 称为单一向导RNA (sgRNA)。向导RNA的前20个碱基依靠碱基互补配对原则识别目标基因, Cas9蛋白和sgRNA复合体首先识别目标基因中的PAM序列, 从而完成与目标基因的结合。在整个目标基因的识别过程中, sgRNA和PAM发挥非常重要的作用。脱靶的产生也很大程度上来自对目标基因识别的准确性降低。sgRNA在与目标基因互补配对过程中允许1-5个碱基的错配, 从而导致目标以外的相似序列被编辑(
Fu et al., 2013 )。sgRNA中的前20个负责互补配对的碱基中, 靠近PAM端的8-12个碱基被认为是核心序列, 在识别特异性上起重要作用(Patrick et al., 2013;
Pattanayak et al., 2013 )。也有报道显示, 通过染色质免疫共沉淀测序(ChIP-seq)的方法检测到核心序列是靠近PAM端1-5个碱基(
Wu et al., 2014 )。但ChIP-seq方法由于是利用没有切割功能的Cas9去识别并结合目标基因组中的序列, 只能反映识别和结合的情况, 无法推测切割的情况, 因此会高估脱靶位点的数量。PAM序列对CRISPR/Cas9识别和切割的准确性也产生较大影响。起初人们认为NGG是PAM序列(N代表A\T\G\C), 但经过研究发现Cas9不仅可以识别NGG, 还可以识别NRG (R代表A或G), 但NRG的切割频率仅为NGG的1/5 (
Hsu et al., 2013 )。尽管识别NRG的频率远低于NGG, 但仍然存在脱靶的可能性。植物中关于这方面的研究很少,
Meng等(2018) 在水稻中验证了野生型的spCas9能够识别NGG和NAG, 且经过对若干目标位点进行验证, 表明无论PAM序列是NGG还是NAG, 均能完成高效率的编辑。因此, 在选择目标位点时应考虑PAM序列。除sgRNA和PAM之外, Cas9的切割方式也影响该系统的精确度。原始的Cas9含有2个负责切割的元件, 即HNH区和RuvC区, 分别负责切割2条DNA链, 产生DNA双链断裂。整个切割过程由1个Cas9蛋白完成。研究表明, 若将1个Cas9蛋白中HNH区的氨基酸突变, 将另1个Cas9蛋白的RuvC区氨基酸突变, 然后将2个突变后的Cas9蛋白及2个目标位点相距较近的sgRNA共同转入细胞中, 从而产生DNA双链断裂, 能够降低脱靶频率(
Cho et al., 2014 )。
2 脱靶的检测方法近年来, 脱靶的检测方法发展迅速。人们不仅希望可以检测到所有可能的脱靶突变, 还需要检测方法简单易操作。本文将介绍几种较为成熟的体外和体内检测方法。
2.1 体内检测
2.1.1 软件预测与测序 软件预测与测序是最早被使用的脱靶检测方法, 即通过生物学软件搜索出基因组中潜在的可能脱靶位点, 然后从基因组中PCR扩增这些可能的脱靶序列, 进一步测序验证是否被编辑。常用的脱靶位点预测工具包括CRISPR Design (http://crispr.mit.edu/)、E-CRIS- PR (www.e-crispr.org/ECRISPR/designcrispr) (
Hsu et al., 2014 )、Cas-OFF-inder (http://www.rgenome. net) (
Bae et al., 2014 )、Target Finder及CRISPR Design Tool (http://www.broadinstitute.org/mpg/cr- ispr_design/) (
Doench et al., 2016 )。由于此方法只能检测预测得到的可能序列, 因此会存在遗漏, 不能完全反映整个基因组中的脱靶编辑情况。
2.1.2 T7E1检测方法 T7E1是T7核酸内切酶1的简称, 该酶能够识别并切割DNA双链中不完全匹配的碱基序列, 产生DNA双链断裂。人们利用T7E1的这一特点, 检测特定位点基因编辑的效果, 进而检测脱靶编辑。与软件预测+测序和全基因组深度测序类似, 该方法同样需要使用脱靶位点预测工具得到可能的脱靶位点。所不同的是应用T7E1的酶切检测代替了测序, 节省了时间和成本(
Cradick et al., 2013 )。
2.1.3 全基因组深度测序 全基因组深度测序也是一种检测脱靶的方法, 但此方法成本高, 数据分析复杂, 所得结论仍需要通过PCR和Sanger测序做进一步验证(
Veres et al., 2014 ;
Smith et al., 2014 )。全基因组测序方法的关键是选择合适的对照基因组, 将自然的突变背景尽量去除, 然后通过数据分析, 寻找含有PAM基序的突变位点, 最后再通过PCR和Sanger测序进行验证。如此得到的可能脱靶序列数量较大, 还需利用辅助软件在其中找到那些与目标序列相似的序列进行PCR和Sanger测序验证, 因此会存在遗漏现象。由于测序的精确度问题, 该方法只能用于检测高频率的脱靶现象, 而对于低频率事件则无法检出。
2.1.4 GUIDE-seq GUIDE-seq (genome-wide unbiased identification of DSBs enabled by sequencing)方法由Tsai等(2015)创建。此方法是用两端磷酸化修饰的双链脱氧核糖核苷酸序列(double-stranded oligodeoxynucleotides, dsODNs)与Cas9/sgRNA一同转染进靶细胞, 并整合到基因组中的DNA双链断裂(DSB)处, 这些DSB大部分来自CRISPR/cas9的切割。然后将整合后的基因组DNA随机切割成片段, 在片段的5'端加上相同的标签, 再以单链的加尾序列和dsODNs作为引物进行二代测序, 从而找到插入位置, 即可能的脱靶切割位点(
图1 )。此方法灵敏度较高, 能检测到细胞中0.1%的突变。此外, 在定位切割位点的过程中, 可以通过测序得到的序列数目推测CRISPR/Cas9的切割活性。但由于细胞转染的局限, 使其只能应用于易转化的细胞。此后, 应用GUIDE-seq方法成功检测CRI- SPR/Cas9系统中的脱靶切割屡见报道, 且评价较好(
Friedland et al., 2015 ;
Kleinstiver et al., 2015a ,
2015b ;
Lee et al., 2016 )。Tsai等开发了用于GUIDE- seq测序结果分析的软件(https://github.com/aryee- lab/umi), 并公开供广大研究者使用。
图1 https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-4-528/img_1.png <b>图1</b> GUIDE-seq和非整合病毒载体(IDLV)方法示意图<br/><b>Figure 1</b> Schematic diagram of genome-wide unbiased identification of DSBs enabled by sequencing (GUIDE-seq) and integrase-defective lentiviral vectors (IDLV) Figure 1 https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-4-528/img_1.png <b>图1</b> GUIDE-seq和非整合病毒载体(IDLV)方法示意图<br/><b>Figure 1</b> Schematic diagram of genome-wide unbiased identification of DSBs enabled by sequencing (GUIDE-seq) and integrase-defective lentiviral vectors (IDLV) 在新窗口打开 下载 下载原图ZIP 生成PPT 图1
GUIDE-seq和非整合病毒载体(IDLV)方法示意图
Figure 1
Schematic diagram of genome-wide unbiased identification of DSBs enabled by sequencing (GUIDE-seq) and integrase-defective lentiviral vectors (IDLV)
2.1.5 非整合病毒载体 慢病毒载体(lentiviral vectors)能够高效率地插入目标细胞基因组。但非整合病毒载体(integrase-defec- tive lentiviral vectors, IDLV)能够高效进入目标细胞却无法整合到基因组。只有在基因组中存在DNA双链断裂时, 才能整合进切口处(
Gabriel et al., 2011 )。这一整合特性使其得以应用到基因组编辑的脱靶检测(
图1 ) (
Wang et al., 2015 ;
Osborn et al., 2016 )。
Wang等(2015) 研究表明IDLV的检测精度达1%, 并检测到CRISPR/Cas9的脱靶位点与目标位点之间的差异高达1-13个碱基。
Osborn等(2016) 应用IDLV方法在人类T-细胞中检测出TALEN、CRISPR和Meg- Tal三种基因编辑工具的脱靶频率, 结果在TALEN和CRISPR的实验中所有IDLV检测到的脱靶位点均没有被切割。在Meg-Tal实验中确认了所有IDLV实验产生的脱靶位点均被切割产生变异。这2项研究均证明IDLV方法可以无偏见地检测全基因组中的脱靶位点, 但精确度只有1%。
2.1.6 高通量全基因组易位测序 高通量全基因组易位测序(high-throughput genome- wide translocation sequencing, HTGTS)是在2011年首次被应用于检测Meganuclease所产生的DNA双链断裂和染色体重组的方法(
Chiarle et al., 2011 )。如
图2 所示, 该方法是利用已知的DNA双链断裂与其它断裂DNA的融合, 并结合接头PCR (adapter-PCR)或环化PCR (circulation-PCR)扩增来检测DNA双链断裂。随着CRISPR/Cas9技术的发展, 对DNA双链断裂的检测需求与日俱增, 在最初的HTGTS方法基础上, 人们开发出了更高通量的检测脱靶的方法(
Frock et al., 2015 )。其原理是用LAM-PCR代替最初使用的环化PCR和接头PCR。然后结合槽式PCR和Tag-PCR扩增出基因组中所有发生断裂的片段。但无论新旧方法, 都是建立在已知DNA双链断裂位置的基础上扩增与之结合的其它断裂片段。由于已知断裂片段只能与距其较近的不同染色体上的DNA断裂片段融合, 距之较远或者同一染色体上的片段以及那些没有与已知断裂片段结合的片段则无法被检出。
图2 https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-4-528/img_2.png <b>图2</b> 高通量全基因组易位测序(HTGTS)的简化流程(改自<xref ref-type="bibr" rid="b15-1674-3466-53-4-528">Frock et al., 2015</xref>)<br/><b>Figure 2</b> Simplified process of high-throughput genomewide translocation sequencing (HTGTS) (modified from <xref ref-type="bibr" rid="b15-1674-3466-53-4-528">Frock et al., 2015</xref>) Figure 2 https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-4-528/img_2.png <b>图2</b> 高通量全基因组易位测序(HTGTS)的简化流程(改自<xref ref-type="bibr" rid="b15-1674-3466-53-4-528">Frock et al., 2015</xref>)<br/><b>Figure 2</b> Simplified process of high-throughput genomewide translocation sequencing (HTGTS) (modified from <xref ref-type="bibr" rid="b15-1674-3466-53-4-528">Frock et al., 2015</xref>) 在新窗口打开 下载 下载原图ZIP 生成PPT 图2
高通量全基因组易位测序(HTGTS)的简化流程(改自
Frock et al., 2015 )
Figure 2
Simplified process of high-throughput genomewide translocation sequencing (HTGTS) (modified from
Frock et al., 2015 )
2.1.7 BLESS BLESS (direct in situ breaks labeling, enrichment on streptavidin, and next-generation sequencing)是基于对DNA双链断裂的检测来推测脱靶位点的方法(
Crosetto et al., 2013 )。Cas9或其它核酸酶切割DNA后产生DNA双链断裂, 在细胞中将产生断裂的DNA末端处用生物素标记, 然后将基因组打碎并加上另一个接头, 通过PCR扩增富集生物素标记的片段, 经二代测序得到脱靶位点。该方法克服了特定的核酸酶限制, 凡是能够产生双链断裂的系统, 均可以使用此方法检测突变。但由于其需要分离细胞核并将基因组中的双链断裂进行生物素标记, 操作较复杂, 且仅能够检测标记的特定时期产生的双链断裂, 无法检测还未发生或已经修复的断裂。
2.2 体外检测
2.2.1 染色质免疫共沉淀测序法 染色质免疫共沉淀测序法(chromatin immunoprecipitation sequencing, ChIP-seq)是利用dCas9- sgRNA (deactivated Cas9-sgRNA)与靶标和非靶标位点的结合, 并通过免疫共沉淀的方法分离出与dCas9-sgRNA结合的序列并加以测序, 从而推测可能的脱靶位点(
图3 ) (
Kuscu et al., 2014 ;
Duan et al., 2014 )。这种方法能够覆盖全基因组, 但由于只检测基因组中与Cas9结合的位点, 没有考虑Cas9的切割活性, 因此其准确度较低, 容易过度估计脱靶效率。
Cencic等(2014) 对43个通过ChIP-seq预测的脱靶位点进行验证, 发现只有1个目标位点和1个脱靶位点被切割, 其它41个预测位点均没有产生切割和突变。另据报道, 应用dCas9和免疫共沉淀方法预测12个sgRNA的潜在脱靶位点, 其中, 根据sgRNA的不同, 脱靶位点的数量由10到1 000不等。经过有活性的Cas9蛋白和相应sgRNA组合对基因组进行编辑, 结果显示大部分预测的脱靶位点都是可被切割的, 只是切割频率低于目标位点(
Kuscu et al., 2014 )。因此, 用该方法预测的脱靶位点与实际的脱靶位点可能存在很大差异。其主要原因是由于使用了无活性的dCas9而不是有活性的Cas9, 因此切割活性无法体现。
图3 https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-4-528/img_3.png <b>图3</b> 染色质免疫共沉淀测序法(ChIP-seq)流程示意图<br/><b>Figure 3</b> Schematic diagram of chromatin immunoprecipitation sequencing (ChIP-seq) Figure 3 https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-4-528/img_3.png <b>图3</b> 染色质免疫共沉淀测序法(ChIP-seq)流程示意图<br/><b>Figure 3</b> Schematic diagram of chromatin immunoprecipitation sequencing (ChIP-seq) 在新窗口打开 下载 下载原图ZIP 生成PPT 图3
染色质免疫共沉淀测序法(ChIP-seq)流程示意图
Figure 3
Schematic diagram of chromatin immunoprecipitation sequencing (ChIP-seq)
2.2.2 数字基因组测序 数字基因组测序(digenome sequencing, Digenome- seq)是一种高效率、高灵敏度的全面检测脱靶的方法。这种方法充分利用了计算机的分析能力, 将基因组中Cas9切割产生的5'末端序列与其它测序片段区分开, 并最终通过不同的连接方式推测脱靶位点。Cas9切割产生的序列为平末端的直接连接, 而其它测序片段则为粘性连接模式(
Kim et al., 2015 )。这种方法的灵敏度可达0.1%, 但一些存在混合切割模式的位点和测序深度不足的位点仍会被遗漏。
Kim等(2016) 通过改进算法, 将之前的只检测平末端序列, 改进为假设Cas9能够产生1-2个碱基的突出, 大大提高了检测的灵敏度。经验证, Digenome-seq可同时检测10个sgRNA所产生的脱靶位点(
Kim et al., 2016 )。
2.2.3 FIND-seq FIND-seq (full interrogation of nuclease dsbs and sequencing)是
Joung和Tsai (2017a) 发明的一种检测由Cas9产生的DNA双链断裂的方法。此方法充分利用了发夹接头和PCR技术, 首先将基因组DNA打碎成500 bp左右的片段, 然后在片段两端加上5'单加尾发夹接头, 去除溶液中所有核酸外切酶后, 加入Cas9蛋白和sgRNA, 对目标序列和脱靶序列进行切割。凡被切割的DNA片段都只有一端带有发夹接头, 然后再给这些被切割的序列加上3'单加尾发夹接头。打开发夹结构后, 通过PCR扩增带有2种单加尾发夹接头的序列进行测序(
图4 )。此方法灵敏度较高, 其中也有一些环节会产生个别遗漏, 如在加尾的过程中, 不是所有的DNA片段都能在两端加上发夹接头。
图4 https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-4-528/img_4.png <b>图4</b> FIND-seq和Circle-seq流程图<br/><b>Figure 4</b> Process of FIND-seq and Circle-seq Figure 4 https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-4-528/img_4.png <b>图4</b> FIND-seq和Circle-seq流程图<br/><b>Figure 4</b> Process of FIND-seq and Circle-seq 在新窗口打开 下载 下载原图ZIP 生成PPT 图4
FIND-seq和Circle-seq流程图
Figure 4
Process of FIND-seq and Circle-seq
2.2.4 Circle-seq Circle-seq是Joung和Tsai研发的检测基因编辑中脱靶突变的方法(
Joung and Tsai, 2017b ;
Tsai et al., 2017 )。该方法通过对剪切后的基因组片段进行环化, 然后用Cas9切割环化后的基因组片段, 所有能够被切割的环均被切开, 为线型, 再对线性DNA片段进行二代测序, 从而得到可能的脱靶位点(
图4 )。与其它检测方法相比, 此方法能够检测到更多低频率的可能脱靶位点。例如, 在测定人的
HBB 基因中相同的目标序列时, Circle-seq方法比Digenome-seq方法多检测出156个潜在脱靶位点, 经确认其中有29个位点是真实的脱靶位点。为了方便Circle-seq方法的应用, 研究人员还开发了公开的数据分析软件(python package circleseq), 使用者可直接得到1个含有脱靶位点的表格及比对后的潜在脱靶序列。
2.2.5 SITE-seq 与Circle-seq和FIND-Seq相似, SITE-seq (the selective enrichment and identification of tagged genomic DNA ends by sequencing)通过Cas9蛋白体外切割目标基因组, 在切口处加接头和生物素标记, 再将基因组通过酶切等方法切割成可测序的500 bp左右的片段, 然后加上另外一个接头, 通过生物素筛选和PCR富集由Cas9蛋白切割产生的片段DNA, 经过NGS测序并筛选出峰值所对应的序列, 再用开发好的算法推测可能的切割位点。以
VEGFA 和
FANCF 基因为例, 所有GUIDE-seq、HTGTS和Digenome-seq检测出的脱靶位点均能被SITE-seq检出, 且用SITE- seq方法检测出11个新的脱靶位点(
Cameron et al., 2017 )。
2.3 体内检测和体外检测方法比较上述几种方法总体可以分为体外检测和体内检测2种。无论是体外检测还是体内检测均存在优点和不足(
表1 )。软件预测+测序、深度测序、T7E1检测、GUIDE-seq、HTGTS、IDLV和BLESS均属于体内检测。其中软件预测+测序、深度测序和T7E1检测均需通过软件预测潜在的脱靶位点, 且不能覆盖全基因组。GUIDE-seq、HTGTS、IDLV和BLESS无需软件预测, 能够覆盖全基因组且灵敏度相对较高, 但是GUIDE-seq和IDLV存在细胞转染和精确度不高的问题。HTGTS检测法通量高, 但由于原理的限制存在漏检现象。BLESS方法的重大突破是克服了核酸酶的限制, 但由于只能检测一个固定时期发生的切割, 漏检率较高。ChIP-seq、Digenome-seq、Circle-Seq、FIND-seq和Site-seq属于体外检测。ChIP-seq方法应用dCas9与靶序列结合, 使其无法真实反映Cas9的切割活性, 假阳性较多。Digenome-seq虽然可以覆盖全基因组且灵敏度较高, 但其需要深度测序产生大量的数据再加以分析, 从而限制了其对于多个sgRNA的高通量检测。但改进后的Digenome-seq方法克服了其对多个sgRNA检测的局限。Circle-seq、FIND- seq和Site-seq方法能够较为全面地覆盖基因组, 且灵敏度较高(<0.1%), 但由于是体外检测, 其结果与体内检测结果可能存在差别, 同时由于各自的技术原理限制, 均存在一定的漏检现象。
表1 Table 1 表1 表1 脱靶的体内和体外检测方法对比
Table 1 Advantage and disadvantage of
in vivo and
in vitro off-target detection methods
检测方法 优点 缺点 软件预测+测序 简单, 易操作 无法覆盖全基因组, 只检测软件预测的序列 深度测序 简单, 易操作 成本高, 数据分析复杂 T7E1检测 成本低, 速度快 需辅助软件预测脱靶位点, 无法覆盖全基因组 GUIDE-seq 精确度高(0.1%), 全基因组检测 受细胞转染的限制, dsODNs的整合效率影响结果 HTGTS 全基因组覆盖 只能检测到与断裂片段结合的脱靶位点, 存在遗漏 IDLV 全基因组覆盖, 无偏见检测脱靶位点 精确度较低(1%) BLESS 摆脱了特定核酸酶的限制, 可检测任何酶所产生的DSB 操作复杂, 只能检测特定时期所产生的突变 ChIP-seq 全基因组检测 体外检测, 未考虑切割频率, 准确度低 Digenome-seq 高效率, 高灵敏度(0.1%), 全基因组检测 体外检测, 成本高, 分析难度大 Circle-seq 全基因组检测, 灵敏度高, 提供分析平台 体外检测, 准确度不高 FIND-seq 全基因组检测, 灵敏度高 体外检测, 存在个别遗漏 Site-seq 全基因组检测, 灵敏度高 体外检测, 测序结果分析需要特定的算法
表1
脱靶的体内和体外检测方法对比
Table 1
Advantage and disadvantage of
in vivo and
in vitro off-target detection methods
3 减少脱靶的方法3.1 改进sgRNA的设计由于CRISPR系统是通过碱基互补配对识别目标位点, 因此目标位点的选择对于减少脱靶尤为重要。研究表明, 为减少脱靶, 目标位点至少需要与基因组中其它位点存在2-3个碱基的错配(
Cho et al., 2014 )。目前, 研究人员已开发出许多脱靶预测和sgRNA设计软件(
表2 )。人们可以根据具体需求选择合适的软件设计sgRNA和预测脱靶可能性。另外, 使用较短(16-18个碱基)的间隔序列, 能够降低脱靶率(
Fu et al., 2014 )。经过不断总结经验, 研究人员发现在设计sgRNA时遵循一定规律可以降低脱靶率, 如在设计sgRNA时要选择基因组中特异的序列或尽可能增加所选择的目标序列与基因组中其它序列的错配碱基数目; 靠近PAM序列的1-6个碱基最好是基因组中完全特异的; 另外也要考虑PAM序列, 在设计sgRNA时除比较sgRNA的序列是否特异外, 还要在基因组中查找是否存在同样的序列位于除NGG以外的其它PAM序列的下游, 如NAG和NGC等(
Doench et al., 2016 ;
Meng et al., 2018 )。
表2 Table 2 表2 表2 sgRNA设计软件汇总
Table 2 Summary of sgRNA design tools
设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点。可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点。可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索。可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计。能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
表2
sgRNA设计软件汇总
Table 2
Summary of sgRNA design tools
3.2 CRISPR系统的优化在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%。通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(
Slaymaker et al., 2016 )。根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式。
Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平。该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(
Kleinstiver et al., 2015b ,
2016 )。Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体。
Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白。
Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率。另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(
Shen et al., 2014 ;
Havlicek et al., 2017 )。一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(
Shin et al., 2017 ;
Harrington et al., 2017 )。
4 植物细胞中的脱靶检测脱靶给植物基因组改造带来的负面效应要远低于动物。许多研究显示, 植物基因编辑产生的植株中未检测到脱靶现象(
Pan et al., 2016 ;
Tian et al., 2017 ;
Zong et al., 2017 ;
Chen et al., 2017 )。也有研究表明, CRISPR系统在植物基因组编辑中产生大量脱靶编辑, 需要通过使用Cas9突变体和其它方法才能降低脱靶频率(
Zhang et al., 2017b ), 该研究是在拟南芥(
Arabidopsis thaliana )中进行的, 而在拟南芥的其它报道中并未发现高脱靶频率现象, 这可能与选择的目标位点有关(
Jiang et al., 2014 )。另有研究表明, 在应用Cas9突变体降低脱靶频率的同时, 若选择与目标序列完全一致的20个碱基作为gRNA, 则可以大大提高定点编辑的频率(
Zhang et al., 2017a )。因此, 在选择目标位点时应尽量选择基因组中特异的序列以减小脱靶的可能性, 过多的脱靶将大大影响优良性状选育的商业化进程。
本文提到的几种检测脱靶切割的方法, 在植物中并不能全部适用。如Guide-seq方法中, 需要dsODN转染目标细胞或基因组, 再根据dsODN的插入位置推测脱靶位点。将dsODN导入植物细胞的过程中, 非常容易导致大量的随机插入, 从而给数据分析带来较大困难。检测植物细胞中的脱靶位点需要选择合适的检测方法。比如选择一种或几种脱靶检测方法推测潜在脱靶位点, 再结合PCR和测序技术加以验证, 找到真正的脱靶位点。目前大多数植物基因编辑均采用先预测可能脱靶位点, 再通过T7E1、PCR和测序的方法来检测脱靶(
Pan et al., 2016 ;
Zhang et al., 2017a ,
2017b ;
Zong et al., 2017 )。
脱靶检测的时间也同样重要。由于Cas9不停地切割靶序列, 人们在完成目标位点编辑后要尽早地将Cas9分离出去, 得到1个不含有Cas9和sgRNA的突变株。该突变株可进一步用于性状鉴定等商业化过程, 因此在哪个时期进行脱靶检测对于产品研发和成本控制有重要影响。
5 研究展望5.1 提高脱靶检测的准确度和精确度现有的脱靶检测技术均侧重于改进其检测的精度, 在检测的准确度方面提高较少, 检出的假阳性结果较多, 需要大量的测序确认工作来区分假阳性结果。这无疑增加了研究人员的工作量和研究成本。在今后的研究中, 建议在改进检测精度的同时提高检测的准确度。已有的检测方法中, GUIDE-seq的准确度相对较高, 在某些检测中可达到79% (
Kim et al., 2016 )。但GUIDE-seq受细胞转染的限制, 其应用范围较小。Digenome-seq和Circle-seq都是很好的检测方法, 检测的精确度高, 但准确度却不高。例如, 在Digeno- me-seq检测中, 有74个位点被检出脱靶编辑, 经过验证, 只有5个是真正的脱靶编辑位点; 在Circle-seq检测中, 仅有20%左右的潜在脱靶位点被证实。因此, 提高检测的准确度也是目前需要解决的问题之一。
CRISPR/Cas9技术的快速应用促使基因组定点编辑更加简单易行。脱靶技术研究在短短几年内取得了快速发展, 仅脱靶检测方面就研发出了十几种甚至更多的新方法。这些新方法的应用无疑为基因组定点编辑及以后的基因组相关研究奠定了基础。目前开发的检测技术精确度在0.01%-1%之间, 一般为0.1%, 这个精确度对于精准编辑来说还不够, 人们还需进一步改进检测方法和系统, 如增加测序深度、提高所用试剂和标记的灵敏度等, 以期能够达到脱靶检测精确度在0.01%以内。这将为今后的定点编辑技术发展提供更有力的技术支持。
5.2 利用现代分子育种技术移除脱靶突变脱靶突变的存在可能会影响基因组编辑在植物中的应用, 减缓了编辑产生的优良性状投放市场的进度。在作物育种过程中, 分子标记的应用十分广泛, 提高了育种的精准性, 并加快了育种速度。在大豆(
Gly- cine max )的成熟期育种过程中, 已通过确定成熟期相关基因和开发分子标记的方法加快育种进程(
Li et al., 2017 )。分子标记技术在玉米(
Zea mays )中也得到了广泛的应用, 目前已有55 K的SNP标记应用到玉米辅助育种(
Xu et al., 2017 )。移除脱靶突变、保留目标位点的最佳选择便是定向回交, 结合分子标记辅助育种方法能够加速筛选, 缩短遗传背景纯化周期。一方面, 可以通过全基因组分子标记检测, 保留目标位点, 加速脱靶突变移除进程; 另一方面, 可以根据本文所述的脱靶检测方法推测脱靶位点, 开发相应的分子标记, 在回交或杂交的过程中移除脱靶位点。分子标记在育种中应用较为成熟, 通过分子标记移除脱靶位点与育种中纯化品种十分相似。因此分子标记技术在植物基因组定点编辑中将发挥助力作用并得到广泛应用。
5.3 运用基因编辑手段研究植物基因功能时的脱靶问题随着CRISPR技术的不断发展, 其在植物基因功能研究中的应用也日益广泛。大量研究表明, 植物基因编辑过程中脱靶频率很低, 只有少数文献报道发现高频率的脱靶现象。拟南芥非生物胁迫相关基因编辑的研究中, 通过CRISPR/sgRNA产生3个基因的突变体并验证了基因的功能。实验表明, 通过软件预测或基因的同源性来推测潜在的脱靶位点, 并检测了突变体中的脱靶情况, 结果未发现脱靶编辑(
Osakabe et al., 2016 )。水稻耐寒基因编辑的脱靶研究表明, 通过BLASTN找到的潜在脱靶位点, 在突变体中均未检测到突变(
Shen et al., 2017 )。综合其它植物基因编辑文献对脱靶的报道, 大多数研究都是通过软件预测潜在的脱靶位点, 然后定向检测预测所得的可能脱靶序列(
Peterson et al., 2016 ;
Kaya et al., 2016 )。这种方式无疑可以一定程度地检测脱靶效应, 但软件的选择对检测结果影响很大。因此, 广大科研工作者在今后的研究中, 可以用试验结合软件预测来选择潜在的脱靶位点(如FIND-seq、Circle-seq和SITE-seq), 从而提高脱靶检测的准确性。对于那些用于产品生产的突变体, 要结合全基因组深度测序进一步筛查脱靶编辑。 当然, 许多实验中没有检测到脱靶现象, 也与实验之前精心选择Cas9蛋白及目标位点有密切关系。本文介绍了一些在实验设计过程中减少产生脱靶的方法, 广大****可以根据实验需求参考借鉴。
The authors have declared that no competing interests exist. 参考文献 文献选项 原文顺序 文献年度倒序 文中引用次数倒序 被引期刊影响因子 [1] Bae S Park J Kim JS 2014 ). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases.Bioinformatics 30, 1473 -1475 .[2] Belhaj K Chaparro-Garcia A Kamoun S Nekrasov V 2013 ). Plant genome editing made easy: targeted muta- genesis in model and crop plants using the CRISPR/Cas system.Plant Methods 9, 39 .[3] Cameron P Fuller CK Donohoue PD Jones BN Thom- pson MS Carter MM Gradia S Vidal B Garner E Slorach EM Lau E Banh LM Lied AM Edwards LS Settle AH Capurso D Llaca V Deschamps S Cigan M Young JK May AP 2017 ). Mapping the genomic landscape of CRISPR-Cas9 cleavage.Nat Methods 14, 600 -606 .[4] Cencic R Miura H Malina A Robert F Ethier S Schme- ing TM Dostie J Pelletier J 2014 ). Protospacer adjacent motif (PAM)-distal sequences engage CRISPR Cas9 DNA target cleavage.PLoS One 9, e109213 .[5] Chari R Mali P Moosburner M Church GM 2015 ). Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach.Nat Methods 12, 823 -826 .[6] Chen XG Lu XK Shu N Wang S Wang JJ Wang DL Guo LX Ye WW 2017 ). Targeted mutagenesis in cotton (Gossypium hirsutum L.) using the CRISPR/Cas9 system. Sci Rep 44304 .[7] Chiarle R Zhang Y Frock RL Lewis SM Molinie B Ho YJ Myers DR Choi VW Compagno M Malkin DJ Neuberg D Monti S Giallourakis CC Gostissa M Alt FW 2011 ). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107 -119 .[8] Cho SW Kim S Kim Y Kweon J Kim HS Bae S Kim JS 2014 ). Analysis of off-target effects of CRISPR/Cas- derived RNA-guided endonucleases and nickases.Genome Res 24, 132 -141 .[9] Cradick TJ Fine EJ Antico CJ Bao G 2013 ). CRISPR/ Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 9584 -9592 .[10] Crosetto N Mitra A Silva MJ Bienko M Dojer N Wang Q Karaca E Chiarle R Skrzypczak M Ginalski K Pasero P Rowicka M Dikic I 2013 ). Nucleotide-resolution DNA double-strand break mapping by next-generation sequen- cing.Nat Methods 10, 361 -365 .[11] Deveau H Garneau JE Moineau S 2010 ). CRISPR/Cas system and its role in phage-bacteria interactions.Annu Rev Microbiol 64, 475 -493 .[12] Doench JG Fusi N Sullender M Hegde M Vaimberg EW Donovan KF Smith I Tothova Z Wilen C Orchard R Virgin HW Listgarten J Root DE 2016 ). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9.Nat Biotechnol 34, 184 -191 .[13] Duan JZ Lu GQ Xie Z Lou ML Luo J Guo L Zhang Y 2014 ). Genome-wide identification of CRISPR/Cas9 off- targets in human genome.Cell Res 24, 1009 -1012 .[14] Friedland AE Baral R Singhal P Loveluck K Shen S Sanchez M Marco E Gotta GM Maeder ML Kennedy EM Kornepati AV Sousa A Collins MA Jayaram H Cullen BR BumcrotEmail D 2015 ). Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol 257 .[15] Frock RL Hu JZ Meyers RM Ho YJ Kii E Alt FW 2015 ). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases.Nat Biotechnol 33, 179 -186 .[16] Fu YF Foden JA Khayter C Maeder ML Reyon D Joung JK Sander JD 2013 ). High-frequency off-target mutag- enesis induced by CRISPR-Cas nucleases in human cells.Nat Biotechnol 31, 822 -826 .[17] Fu YF Sander JD Reyon D Cascio VM Joung JK 2014 ). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs.Nat Biotechnol 32, 279 -284 .[18] Gabriel R Lombardo A Arens A Miller JC Genovese P Kaeppel C Nowrouzi A Bartholomae CC Wang JB Friedman G Holmes MC Gregory PD Glimm H Schmidt M Naldini L von Kalle C 2011 ). An unbiased genome-wide analysis of zinc-finger nuclease specificity.Nat Biotechnol 29, 816 -823 .[19] Harrington LB Doxzen KW Ma E Liu JJ Knott GJ Edraki A Garcia B Amrani N Chen JS Cofsky JC Kranzusch PJ Sontheimer EJ Davidson AR Maxwell KL Doudna JA 2017 ). A broad-spectrum inhibitor of CRISPR-Cas9.Cell 170, 1224 -1233 .[20] Havlicek S Shen Y Alpagu Y Bruntraeger MB Zufir NBM Phuah ZY Fu ZY Dunn NR Stanton LW 2017 ). Re-engineered RNA-guided fokI-nucleases for improved genome editing in human cells.Mol Ther 25, 342 -355 .[21] Hsu PD Lander ES Zhang F 2014 ). Development and applications of CRISPR-Cas9 for genome engineering.Cell 157, 1262 -1278 .[22] Hsu PD Scott DA Weinstein JA Ran FA Konermann S Agarwala V Li YQ Fine EJ Wu XB Shalem O Cradick TJ Marraffini LA Bao G Zhang F 2013 ). DNA targeting specificity of RNA-guided Cas9 nucleases.Nat Biotechnol 31, 827 -832 .[23] Jiang WY Bikard D Cox D Zhang F Marraffini LA 2013 ). RNA-guided editing of bacterial genomes using CRISPR- Cas systems.Nat Biotechnol 31, 233 -239 .[24] Jiang WZ Yang B Weeks DP 2014 ). Efficient CRISPR/ Cas9-mediated gene editing in Arabidopsis thaliana and inheritance of modified genes in the T2 and T3 generations. PLoS One e99225 .[25] Jinek M Chylinski K Fonfara I Hauer M Doudna JA Charpentier E 2012 ). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity.Science 337, 816 -821 .[26] Joung KJ Tsai S 2017 a). Full interrogation of nuclease DSBS and sequencing (find-seq). USA, 20170073747 . 2017-03-16.[27] Joung KJ Tsai S 2017 b). Comprehensive in vitro reporting of cleavage events by sequencing (circle-seq). USA, 9850- 484 . 2017-12-26.[28] Kaya H Mikami M Endo A Endo M Toki S 2016 ). Highly specific targeted mutagenesis in plants using Staphylococcus aureus Cas9. Sci Rep 26871 .[29] Kim D Bae S Park J Kim E Kim S Yu HR Hwang J Kim JI Kim JS 2015 ). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells.Nat Methods 12, 237 -243 .[30] Kim D Kim S Kim S Park J Kim JS 2016 ). Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq.Genome Res 26, 406 -415 .[31] Kleinstiver BP Pattanayak V Prew MS Tsai SQ Nguyen NT Zheng ZL Joung JK 2016 ). High-fidelity CRISPR- Cas9 nucleases with no detectable genome-wide off-target effects.Nature 529, 490 -495 .[32] Kleinstiver BP Prew MS Tsai SQ Nguyen NT Topkar VV Zheng ZL Joung JK 2015 a). Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 1293 -1298 .[33] Kleinstiver BP Prew MS Tsai SQ Topkar VV Nguyen NT Zheng ZL Gonzales APW Li ZY Peterson RT Yeh JRJ Aryee MJ Joung JK 2015 b). Engineered CRISPR- Cas9 nucleases with altered PAM specificities.Nature 523, 481 -485 .[34] Kuscu C Arslan S Singh R Thorpe J Adli M 2014 ). Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease.Nat Biotechnol 32, 677 -683 .[35] Lee CM Cradick TJ Fine EJ Bao G 2016 ). Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing.Mol Ther 24, 475 -487 .[36] Lee JK Jeong E Lee J Jung M Shin E Kim YH Lee K Kim D Kim S Kim JS 2017 ). Directed evolution of CRISPR- Cas9 to increase its specificity.bioRxiv (1 ), 237040 .[37] Li JC Wang XB Song WW Huang XY Zhou J Zeng HY Sun S Jia HC Li WB Zhou XA Li SZ Chen PY Wu CX Guo Y Han TF Qiu LJ 2017 ). Genetic variation of maturity groups and four E genes in the Chinese soybean mini core collection. PLoS One e0172106 .[38] Li W Teng F Li TD Zhou Q 2013 ). Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems.Nat Biotechnol 31, 684 -686 .[39] Lozano-Juste J Cutler SR 2014 ). Plant genome engineering in full bloom.Trends Plant Sci 19, 284 -287 .[40] Mali P Yang LH Esvelt KM Aach J Guell M DiCarlo JE Norville JE Church GM 2013 ). RNA-guided human genome engineering via Cas9.Science 339, 823 -826 .[41] Meng XB Hu XX Liu Q Song XG Gao CX Li JY Wang KJ 2018 ). Robust genome editing of CRISPR-Cas9 at NAG PAMs in rice.Sci China Life Sci 61, 122 -125 .[42] Montague TG Cruz JM Gagnon JA Church GM Valen E 2014 ). CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing.Nucleic Acids Res 42, W401 -W407 .[43] Osakabe Y Watanabe T Sugano SS Ueta R Ishihara R Shinozaki K Osakabe K 2016 ). Optimization of CRISPR/ Cas9 genome editing to modify abiotic stress responses in plants.Sci Rep 6, 26685 .[44] Osborn MJ Webber BR Knipping F Lonetree CL Tennis N DeFeo AP McElroy AN Starker CG Lee C Merkel S Lund TC Kelly-Spratt KS Jensen MC Voytas DF von Kalle C Schmidt M Gabriel R Hippen KL Miller JS Scharenberg AM Tolar J Blazar BR 2016 ). Eval- uation of TCR gene editing achieved by TALENs, CRISPR/ Cas9, and megaTAL nucleases.Mol Ther 24, 570 -581 .[45] Pan CT Ye L Qin L Liu X He YJ Wang J Chen LF Lu G 2016 ). CRISPR/Cas9-mediated efficient and heritable targeted mutagenesis in tomato plants in the first and later generations.Sci Rep 6, 24765 .[46] Pattanayak V Lin S Guilinger JP Ma E Doudna JA Liu DR 2013 ). High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity.Nat Biotechnol 31, 839 -843 .[47] Peterson BA Haak DC Nishimura MT Teixeira PJPL James SR Dangl JL Nimchuk ZL 2016 ). Genome-wide assessment of efficiency and specificity in CRISPR/Cas9 mediated multiple site targeting in Arabidopsis.PLoS One 11, e0162169 .[48] Pourcel C Drevet C 2013 ). Occurrence, diversity of CRISPR- Cas systems and genotyping implications. In: Barrangou R, van der Oost J, eds. CRISPR-Cas Systems . Berlin: Springer. pp. 33 -59 .[49] Prykhozhij SV Rajan V Gaston D Berman JN 2015 ). CRISPR multitargeter: a web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences.PLoS One 10, e0119372 .[50] Sampson TR Weiss DS 2013 ). Alternative roles for CRISPR/Cas systems in bacterial pathogenesis.PLoS Pa- thog 9, e1003621 .[51] Shen B Zhang WS Zhang J Zhou JK Wang JY Chen L Wang L Hodgkins A Iyer V Huang XX Skarnes WC 2014 ). Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects.Nat Methods 11, 399 -402 .[52] Shen CX Que ZQ Xia YM Tang N Li D He RH Cao ML 2017 ). Knock out of the annexin gene OsAnn3 via CRISPR/Cas9-mediated genome editing decreased cold tolerance in rice. J Plant Biol 539 -547 .[53] Shin J Jiang FG Liu JJ Bray NL Rauch BJ Baik SH Nogales E Bondy-Denomy J Corn JE Doudna JA 2017 ). Disabling Cas9 by an anti-CRISPR DNA mimic.Sci Adv 3, e1701620 .[54] Slaymaker IM Gao LY Zetsche B Scott DA Yan WX Zhang F 2016 ). Rationally engineered Cas9 nucleases with improved specificity.Science 351, 84 -88 .[55] Smith C Gore A Yan W Abalde-Atristain L Li Z He CX Wang Y Brodsky RA Zhang K Cheng LZ Ye ZH 2014 ). Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs.Cell Stem Cell 15, 12 -13 .[56] Tian SW Jiang LJ Gao Q Zhang J Zong M Zhang HY Ren Y Guo SG Gong GY Liu F Xu Y 2017 ). Efficient CRISPR/Cas9-based gene knockout in watermelon.Plant Cell Rep 36, 399 -406 .[57] Tsai SQ Nguyen NT Malagon-Lopez J Topkar VV Aryee MJ Joung JK 2017 ). CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 607 -614 .[58] Tsai SQ Zheng Zl Nguyen NT Liebers M Topkar VV Thapar V Wyvekens N Khayter C Iafrate AJ Le LP Aryee MJ Joung JK 2015 ). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases.Nat Biotechnol 33, 187 -197 .[59] Veres A Gosis BS Ding QR Collins R Ragavendran A Brand H Erdin S Cowan CA Talkowski ME Musunuru K 2014 ). Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing.Cell Stem Cell 15, 27 -30 .[60] Wang T Wei JJ Sabatini DM Lander ES 2014 ). Genetic screens in human cells using the CRISPR-Cas9 system.Science 343, 80 -84 .[61] Wang XL Wang YB Wu XW Wang JH Wang YJ Qiu ZJ Chang T Huang H Lin RJ Yee JK 2015 ). Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors.Nat Biotechnol 33, 175 -178 .[62] Wang Y Geng LZ Yuan ML Wei J Jin C Li M Yu K Zhang Y Jin HB Wang E Chai ZJ Fu XD Li XG 2017 ). Deletion of a target gene in indica rice via CRISPR/Cas9. Plant Cell Rep 1333 -1343 .[63] Wu XB Scott DA Kriz AJ Chiu AC Hsu PD Dadon DB Cheng AW Trevino AE Konermann S Chen SD Jaenisch R Zhang F Sharp PA 2014 ). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells.Nat Biotechnol 32, 670 -676 .[64] Xu C Ren YH Jian YQ Guo ZF Zhang Y Xie CX Fu JJ Wang HW Wang GY Xu YB Li P Zou C 2017 ). Deve- lopment of a maize 55 K SNP array with improved genome coverage for molecular breeding.Mol Breed 37, 20 .[65] Xu RF Li H Qin RY Wang L Li L Wei PC Yang JB 2014 ). Gene targeting using the Agrobacterium tumefaciens -mediated CRISPR-Cas system in rice. Rice 5 .[66] Zhang DB Zhang HW Li TD Chen KL Qiu JL Gao CX 2017 a). Perfectly matched 20-nucleotide guide RNA sequences enable robust genome editing using high-fidelity SpCas9 nucleases.Genome Biol 18, 191 .[67] Zhang H Zhang JS Wei PL Zhang BT Gou F Feng ZY Mao YF Yang L Zhang H Xu NF Zhu JK 2014 ). The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation.Plant Biotechnol J 12, 797 -807 .[68] Zhang Q Xing HL Wang ZP Zhang HY Yang F Zhou Y Wang XC Chen QJ 2017 b). High-frequency off-target mutagenesis induced by CRISPR/Cas9 in Arabidopsis and its prevention by improving specificity of the tools.bioRxiv (1 ), 203489 .[69] Zong Y Wang YP Li C Zhang R Chen KL Ran YD Qiu JL Wang DW Gao CX 2017 ). Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion.Nat Biotechnol 35, 438 -440 .
2
2014
... 软件预测与测序是最早被使用的脱靶检测方法, 即通过生物学软件搜索出基因组中潜在的可能脱靶位点, 然后从基因组中PCR扩增这些可能的脱靶序列, 进一步测序验证是否被编辑.常用的脱靶位点预测工具包括CRISPR Design (http://crispr.mit.edu/)、E-CRIS- PR (www.e-crispr.org/ECRISPR/designcrispr) (
Hsu et al., 2014 )、Cas-OFF-inder (http://www.rgenome. net) (
Bae et al., 2014 )、Target Finder及CRISPR Design Tool (http://www.broadinstitute.org/mpg/cr- ispr_design/) (
Doench et al., 2016 ).由于此方法只能检测预测得到的可能序列, 因此会存在遗漏, 不能完全反映整个基因组中的脱靶编辑情况. ...
... Summary of sgRNA design tools
设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...VEGFA 和FANCF 基因为例, 所有GUIDE-seq、HTGTS和Digenome-seq检测出的脱靶位点均能被SITE-seq检出, 且用SITE- seq方法检测出11个新的脱靶位点(Cameron et al., 2017 ). ...图3 ) (Kuscu et al., 2014 ; Duan et al., 2014 ).这种方法能够覆盖全基因组, 但由于只检测基因组中与Cas9结合的位点, 没有考虑Cas9的切割活性, 因此其准确度较低, 容易过度估计脱靶效率.Cencic等(2014) 对43个通过ChIP-seq预测的脱靶位点进行验证, 发现只有1个目标位点和1个脱靶位点被切割, 其它41个预测位点均没有产生切割和突变.另据报道, 应用dCas9和免疫共沉淀方法预测12个sgRNA的潜在脱靶位点, 其中, 根据sgRNA的不同, 脱靶位点的数量由10到1 000不等.经过有活性的Cas9蛋白和相应sgRNA组合对基因组进行编辑, 结果显示大部分预测的脱靶位点都是可被切割的, 只是切割频率低于目标位点(Kuscu et al., 2014 ).因此, 用该方法预测的脱靶位点与实际的脱靶位点可能存在很大差异.其主要原因是由于使用了无活性的dCas9而不是有活性的Cas9, 因此切割活性无法体现. ... 设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Pan et al., 2016 ; Tian et al., 2017 ; Zong et al., 2017 ; Chen et al., 2017 ).也有研究表明, CRISPR系统在植物基因组编辑中产生大量脱靶编辑, 需要通过使用Cas9突变体和其它方法才能降低脱靶频率(Zhang et al., 2017b ), 该研究是在拟南芥(Arabidopsis thaliana )中进行的, 而在拟南芥的其它报道中并未发现高脱靶频率现象, 这可能与选择的目标位点有关(Jiang et al., 2014 ).另有研究表明, 在应用Cas9突变体降低脱靶频率的同时, 若选择与目标序列完全一致的20个碱基作为gRNA, 则可以大大提高定点编辑的频率(Zhang et al., 2017a ).因此, 在选择目标位点时应尽量选择基因组中特异的序列以减小脱靶的可能性, 过多的脱靶将大大影响优良性状选育的商业化进程. ...Chiarle et al., 2011 ).如图2 所示, 该方法是利用已知的DNA双链断裂与其它断裂DNA的融合, 并结合接头PCR (adapter-PCR)或环化PCR (circulation-PCR)扩增来检测DNA双链断裂.随着CRISPR/Cas9技术的发展, 对DNA双链断裂的检测需求与日俱增, 在最初的HTGTS方法基础上, 人们开发出了更高通量的检测脱靶的方法(Frock et al., 2015 ).其原理是用LAM-PCR代替最初使用的环化PCR和接头PCR.然后结合槽式PCR和Tag-PCR扩增出基因组中所有发生断裂的片段.但无论新旧方法, 都是建立在已知DNA双链断裂位置的基础上扩增与之结合的其它断裂片段.由于已知断裂片段只能与距其较近的不同染色体上的DNA断裂片段融合, 距之较远或者同一染色体上的片段以及那些没有与已知断裂片段结合的片段则无法被检出. ...trans -activating crRNA, tracrRNA).向导RNA和转运激活RNA可以组装成一个元件, 称为单一向导RNA (sgRNA).向导RNA的前20个碱基依靠碱基互补配对原则识别目标基因, Cas9蛋白和sgRNA复合体首先识别目标基因中的PAM序列, 从而完成与目标基因的结合.在整个目标基因的识别过程中, sgRNA和PAM发挥非常重要的作用.脱靶的产生也很大程度上来自对目标基因识别的准确性降低.sgRNA在与目标基因互补配对过程中允许1-5个碱基的错配, 从而导致目标以外的相似序列被编辑(Fu et al., 2013 ).sgRNA中的前20个负责互补配对的碱基中, 靠近PAM端的8-12个碱基被认为是核心序列, 在识别特异性上起重要作用(Patrick et al., 2013; Pattanayak et al., 2013 ).也有报道显示, 通过染色质免疫共沉淀测序(ChIP-seq)的方法检测到核心序列是靠近PAM端1-5个碱基(Wu et al., 2014 ).但ChIP-seq方法由于是利用没有切割功能的Cas9去识别并结合目标基因组中的序列, 只能反映识别和结合的情况, 无法推测切割的情况, 因此会高估脱靶位点的数量.PAM序列对CRISPR/Cas9识别和切割的准确性也产生较大影响.起初人们认为NGG是PAM序列(N代表A\T\G\C), 但经过研究发现Cas9不仅可以识别NGG, 还可以识别NRG (R代表A或G), 但NRG的切割频率仅为NGG的1/5 (Hsu et al., 2013 ).尽管识别NRG的频率远低于NGG, 但仍然存在脱靶的可能性.植物中关于这方面的研究很少, Meng等(2018) 在水稻中验证了野生型的spCas9能够识别NGG和NAG, 且经过对若干目标位点进行验证, 表明无论PAM序列是NGG还是NAG, 均能完成高效率的编辑.因此, 在选择目标位点时应考虑PAM序列.除sgRNA和PAM之外, Cas9的切割方式也影响该系统的精确度.原始的Cas9含有2个负责切割的元件, 即HNH区和RuvC区, 分别负责切割2条DNA链, 产生DNA双链断裂.整个切割过程由1个Cas9蛋白完成.研究表明, 若将1个Cas9蛋白中HNH区的氨基酸突变, 将另1个Cas9蛋白的RuvC区氨基酸突变, 然后将2个突变后的Cas9蛋白及2个目标位点相距较近的sgRNA共同转入细胞中, 从而产生DNA双链断裂, 能够降低脱靶频率(Cho et al., 2014 ). ...Cho et al., 2014 ).目前, 研究人员已开发出许多脱靶预测和sgRNA设计软件(表2 ).人们可以根据具体需求选择合适的软件设计sgRNA和预测脱靶可能性.另外, 使用较短(16-18个碱基)的间隔序列, 能够降低脱靶率(Fu et al., 2014 ).经过不断总结经验, 研究人员发现在设计sgRNA时遵循一定规律可以降低脱靶率, 如在设计sgRNA时要选择基因组中特异的序列或尽可能增加所选择的目标序列与基因组中其它序列的错配碱基数目; 靠近PAM序列的1-6个碱基最好是基因组中完全特异的; 另外也要考虑PAM序列, 在设计sgRNA时除比较sgRNA的序列是否特异外, 还要在基因组中查找是否存在同样的序列位于除NGG以外的其它PAM序列的下游, 如NAG和NGC等(Doench et al., 2016 ; Meng et al., 2018 ). ...Cradick et al., 2013 ). ...Crosetto et al., 2013 ).Cas9或其它核酸酶切割DNA后产生DNA双链断裂, 在细胞中将产生断裂的DNA末端处用生物素标记, 然后将基因组打碎并加上另一个接头, 通过PCR扩增富集生物素标记的片段, 经二代测序得到脱靶位点.该方法克服了特定的核酸酶限制, 凡是能够产生双链断裂的系统, 均可以使用此方法检测突变.但由于其需要分离细胞核并将基因组中的双链断裂进行生物素标记, 操作较复杂, 且仅能够检测标记的特定时期产生的双链断裂, 无法检测还未发生或已经修复的断裂. ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...Hsu et al., 2014 )、Cas-OFF-inder (http://www.rgenome. net) (Bae et al., 2014 )、Target Finder及CRISPR Design Tool (http://www.broadinstitute.org/mpg/cr- ispr_design/) (Doench et al., 2016 ).由于此方法只能检测预测得到的可能序列, 因此会存在遗漏, 不能完全反映整个基因组中的脱靶编辑情况. ...Cho et al., 2014 ).目前, 研究人员已开发出许多脱靶预测和sgRNA设计软件(表2 ).人们可以根据具体需求选择合适的软件设计sgRNA和预测脱靶可能性.另外, 使用较短(16-18个碱基)的间隔序列, 能够降低脱靶率(Fu et al., 2014 ).经过不断总结经验, 研究人员发现在设计sgRNA时遵循一定规律可以降低脱靶率, 如在设计sgRNA时要选择基因组中特异的序列或尽可能增加所选择的目标序列与基因组中其它序列的错配碱基数目; 靠近PAM序列的1-6个碱基最好是基因组中完全特异的; 另外也要考虑PAM序列, 在设计sgRNA时除比较sgRNA的序列是否特异外, 还要在基因组中查找是否存在同样的序列位于除NGG以外的其它PAM序列的下游, 如NAG和NGC等(Doench et al., 2016 ; Meng et al., 2018 ). ... 设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...图3 ) (Kuscu et al., 2014 ; Duan et al., 2014 ).这种方法能够覆盖全基因组, 但由于只检测基因组中与Cas9结合的位点, 没有考虑Cas9的切割活性, 因此其准确度较低, 容易过度估计脱靶效率.Cencic等(2014) 对43个通过ChIP-seq预测的脱靶位点进行验证, 发现只有1个目标位点和1个脱靶位点被切割, 其它41个预测位点均没有产生切割和突变.另据报道, 应用dCas9和免疫共沉淀方法预测12个sgRNA的潜在脱靶位点, 其中, 根据sgRNA的不同, 脱靶位点的数量由10到1 000不等.经过有活性的Cas9蛋白和相应sgRNA组合对基因组进行编辑, 结果显示大部分预测的脱靶位点都是可被切割的, 只是切割频率低于目标位点(Kuscu et al., 2014 ).因此, 用该方法预测的脱靶位点与实际的脱靶位点可能存在很大差异.其主要原因是由于使用了无活性的dCas9而不是有活性的Cas9, 因此切割活性无法体现. ...图1 ).此方法灵敏度较高, 能检测到细胞中0.1%的突变.此外, 在定位切割位点的过程中, 可以通过测序得到的序列数目推测CRISPR/Cas9的切割活性.但由于细胞转染的局限, 使其只能应用于易转化的细胞.此后, 应用GUIDE-seq方法成功检测CRI- SPR/Cas9系统中的脱靶切割屡见报道, 且评价较好(Friedland et al., 2015 ; Kleinstiver et al., 2015a , 2015b ; Lee et al., 2016 ).Tsai等开发了用于GUIDE- seq测序结果分析的软件(https://github.com/aryee- lab/umi), 并公开供广大研究者使用. ...Chiarle et al., 2011 ).如图2 所示, 该方法是利用已知的DNA双链断裂与其它断裂DNA的融合, 并结合接头PCR (adapter-PCR)或环化PCR (circulation-PCR)扩增来检测DNA双链断裂.随着CRISPR/Cas9技术的发展, 对DNA双链断裂的检测需求与日俱增, 在最初的HTGTS方法基础上, 人们开发出了更高通量的检测脱靶的方法(Frock et al., 2015 ).其原理是用LAM-PCR代替最初使用的环化PCR和接头PCR.然后结合槽式PCR和Tag-PCR扩增出基因组中所有发生断裂的片段.但无论新旧方法, 都是建立在已知DNA双链断裂位置的基础上扩增与之结合的其它断裂片段.由于已知断裂片段只能与距其较近的不同染色体上的DNA断裂片段融合, 距之较远或者同一染色体上的片段以及那些没有与已知断裂片段结合的片段则无法被检出. ...Frock et al., 2015 ) ...Frock et al., 2015 ) ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...trans -activating crRNA, tracrRNA).向导RNA和转运激活RNA可以组装成一个元件, 称为单一向导RNA (sgRNA).向导RNA的前20个碱基依靠碱基互补配对原则识别目标基因, Cas9蛋白和sgRNA复合体首先识别目标基因中的PAM序列, 从而完成与目标基因的结合.在整个目标基因的识别过程中, sgRNA和PAM发挥非常重要的作用.脱靶的产生也很大程度上来自对目标基因识别的准确性降低.sgRNA在与目标基因互补配对过程中允许1-5个碱基的错配, 从而导致目标以外的相似序列被编辑(Fu et al., 2013 ).sgRNA中的前20个负责互补配对的碱基中, 靠近PAM端的8-12个碱基被认为是核心序列, 在识别特异性上起重要作用(Patrick et al., 2013; Pattanayak et al., 2013 ).也有报道显示, 通过染色质免疫共沉淀测序(ChIP-seq)的方法检测到核心序列是靠近PAM端1-5个碱基(Wu et al., 2014 ).但ChIP-seq方法由于是利用没有切割功能的Cas9去识别并结合目标基因组中的序列, 只能反映识别和结合的情况, 无法推测切割的情况, 因此会高估脱靶位点的数量.PAM序列对CRISPR/Cas9识别和切割的准确性也产生较大影响.起初人们认为NGG是PAM序列(N代表A\T\G\C), 但经过研究发现Cas9不仅可以识别NGG, 还可以识别NRG (R代表A或G), 但NRG的切割频率仅为NGG的1/5 (Hsu et al., 2013 ).尽管识别NRG的频率远低于NGG, 但仍然存在脱靶的可能性.植物中关于这方面的研究很少, Meng等(2018) 在水稻中验证了野生型的spCas9能够识别NGG和NAG, 且经过对若干目标位点进行验证, 表明无论PAM序列是NGG还是NAG, 均能完成高效率的编辑.因此, 在选择目标位点时应考虑PAM序列.除sgRNA和PAM之外, Cas9的切割方式也影响该系统的精确度.原始的Cas9含有2个负责切割的元件, 即HNH区和RuvC区, 分别负责切割2条DNA链, 产生DNA双链断裂.整个切割过程由1个Cas9蛋白完成.研究表明, 若将1个Cas9蛋白中HNH区的氨基酸突变, 将另1个Cas9蛋白的RuvC区氨基酸突变, 然后将2个突变后的Cas9蛋白及2个目标位点相距较近的sgRNA共同转入细胞中, 从而产生DNA双链断裂, 能够降低脱靶频率(Cho et al., 2014 ). ...Cho et al., 2014 ).目前, 研究人员已开发出许多脱靶预测和sgRNA设计软件(表2 ).人们可以根据具体需求选择合适的软件设计sgRNA和预测脱靶可能性.另外, 使用较短(16-18个碱基)的间隔序列, 能够降低脱靶率(Fu et al., 2014 ).经过不断总结经验, 研究人员发现在设计sgRNA时遵循一定规律可以降低脱靶率, 如在设计sgRNA时要选择基因组中特异的序列或尽可能增加所选择的目标序列与基因组中其它序列的错配碱基数目; 靠近PAM序列的1-6个碱基最好是基因组中完全特异的; 另外也要考虑PAM序列, 在设计sgRNA时除比较sgRNA的序列是否特异外, 还要在基因组中查找是否存在同样的序列位于除NGG以外的其它PAM序列的下游, 如NAG和NGC等(Doench et al., 2016 ; Meng et al., 2018 ). ...Gabriel et al., 2011 ).这一整合特性使其得以应用到基因组编辑的脱靶检测(图1 ) (Wang et al., 2015 ; Osborn et al., 2016 ).Wang等(2015) 研究表明IDLV的检测精度达1%, 并检测到CRISPR/Cas9的脱靶位点与目标位点之间的差异高达1-13个碱基.Osborn等(2016) 应用IDLV方法在人类T-细胞中检测出TALEN、CRISPR和Meg- Tal三种基因编辑工具的脱靶频率, 结果在TALEN和CRISPR的实验中所有IDLV检测到的脱靶位点均没有被切割.在Meg-Tal实验中确认了所有IDLV实验产生的脱靶位点均被切割产生变异.这2项研究均证明IDLV方法可以无偏见地检测全基因组中的脱靶位点, 但精确度只有1%. ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Hsu et al., 2014 )、Cas-OFF-inder (http://www.rgenome. net) (Bae et al., 2014 )、Target Finder及CRISPR Design Tool (http://www.broadinstitute.org/mpg/cr- ispr_design/) (Doench et al., 2016 ).由于此方法只能检测预测得到的可能序列, 因此会存在遗漏, 不能完全反映整个基因组中的脱靶编辑情况. ... 设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...trans -activating crRNA, tracrRNA).向导RNA和转运激活RNA可以组装成一个元件, 称为单一向导RNA (sgRNA).向导RNA的前20个碱基依靠碱基互补配对原则识别目标基因, Cas9蛋白和sgRNA复合体首先识别目标基因中的PAM序列, 从而完成与目标基因的结合.在整个目标基因的识别过程中, sgRNA和PAM发挥非常重要的作用.脱靶的产生也很大程度上来自对目标基因识别的准确性降低.sgRNA在与目标基因互补配对过程中允许1-5个碱基的错配, 从而导致目标以外的相似序列被编辑(Fu et al., 2013 ).sgRNA中的前20个负责互补配对的碱基中, 靠近PAM端的8-12个碱基被认为是核心序列, 在识别特异性上起重要作用(Patrick et al., 2013; Pattanayak et al., 2013 ).也有报道显示, 通过染色质免疫共沉淀测序(ChIP-seq)的方法检测到核心序列是靠近PAM端1-5个碱基(Wu et al., 2014 ).但ChIP-seq方法由于是利用没有切割功能的Cas9去识别并结合目标基因组中的序列, 只能反映识别和结合的情况, 无法推测切割的情况, 因此会高估脱靶位点的数量.PAM序列对CRISPR/Cas9识别和切割的准确性也产生较大影响.起初人们认为NGG是PAM序列(N代表A\T\G\C), 但经过研究发现Cas9不仅可以识别NGG, 还可以识别NRG (R代表A或G), 但NRG的切割频率仅为NGG的1/5 (Hsu et al., 2013 ).尽管识别NRG的频率远低于NGG, 但仍然存在脱靶的可能性.植物中关于这方面的研究很少, Meng等(2018) 在水稻中验证了野生型的spCas9能够识别NGG和NAG, 且经过对若干目标位点进行验证, 表明无论PAM序列是NGG还是NAG, 均能完成高效率的编辑.因此, 在选择目标位点时应考虑PAM序列.除sgRNA和PAM之外, Cas9的切割方式也影响该系统的精确度.原始的Cas9含有2个负责切割的元件, 即HNH区和RuvC区, 分别负责切割2条DNA链, 产生DNA双链断裂.整个切割过程由1个Cas9蛋白完成.研究表明, 若将1个Cas9蛋白中HNH区的氨基酸突变, 将另1个Cas9蛋白的RuvC区氨基酸突变, 然后将2个突变后的Cas9蛋白及2个目标位点相距较近的sgRNA共同转入细胞中, 从而产生DNA双链断裂, 能够降低脱靶频率(Cho et al., 2014 ). ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...Pan et al., 2016 ; Tian et al., 2017 ; Zong et al., 2017 ; Chen et al., 2017 ).也有研究表明, CRISPR系统在植物基因组编辑中产生大量脱靶编辑, 需要通过使用Cas9突变体和其它方法才能降低脱靶频率(Zhang et al., 2017b ), 该研究是在拟南芥(Arabidopsis thaliana )中进行的, 而在拟南芥的其它报道中并未发现高脱靶频率现象, 这可能与选择的目标位点有关(Jiang et al., 2014 ).另有研究表明, 在应用Cas9突变体降低脱靶频率的同时, 若选择与目标序列完全一致的20个碱基作为gRNA, 则可以大大提高定点编辑的频率(Zhang et al., 2017a ).因此, 在选择目标位点时应尽量选择基因组中特异的序列以减小脱靶的可能性, 过多的脱靶将大大影响优良性状选育的商业化进程. ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...Joung和Tsai (2017a) 发明的一种检测由Cas9产生的DNA双链断裂的方法.此方法充分利用了发夹接头和PCR技术, 首先将基因组DNA打碎成500 bp左右的片段, 然后在片段两端加上5'单加尾发夹接头, 去除溶液中所有核酸外切酶后, 加入Cas9蛋白和sgRNA, 对目标序列和脱靶序列进行切割.凡被切割的DNA片段都只有一端带有发夹接头, 然后再给这些被切割的序列加上3'单加尾发夹接头.打开发夹结构后, 通过PCR扩增带有2种单加尾发夹接头的序列进行测序(图4 ).此方法灵敏度较高, 其中也有一些环节会产生个别遗漏, 如在加尾的过程中, 不是所有的DNA片段都能在两端加上发夹接头. ...Joung and Tsai, 2017b ; Tsai et al., 2017 ).该方法通过对剪切后的基因组片段进行环化, 然后用Cas9切割环化后的基因组片段, 所有能够被切割的环均被切开, 为线型, 再对线性DNA片段进行二代测序, 从而得到可能的脱靶位点(图4 ).与其它检测方法相比, 此方法能够检测到更多低频率的可能脱靶位点.例如, 在测定人的HBB 基因中相同的目标序列时, Circle-seq方法比Digenome-seq方法多检测出156个潜在脱靶位点, 经确认其中有29个位点是真实的脱靶位点.为了方便Circle-seq方法的应用, 研究人员还开发了公开的数据分析软件(python package circleseq), 使用者可直接得到1个含有脱靶位点的表格及比对后的潜在脱靶序列. ...Osakabe et al., 2016 ).水稻耐寒基因编辑的脱靶研究表明, 通过BLASTN找到的潜在脱靶位点, 在突变体中均未检测到突变(Shen et al., 2017 ).综合其它植物基因编辑文献对脱靶的报道, 大多数研究都是通过软件预测潜在的脱靶位点, 然后定向检测预测所得的可能脱靶序列(Peterson et al., 2016 ; Kaya et al., 2016 ).这种方式无疑可以一定程度地检测脱靶效应, 但软件的选择对检测结果影响很大.因此, 广大科研工作者在今后的研究中, 可以用试验结合软件预测来选择潜在的脱靶位点(如FIND-seq、Circle-seq和SITE-seq), 从而提高脱靶检测的准确性.对于那些用于产品生产的突变体, 要结合全基因组深度测序进一步筛查脱靶编辑. 当然, 许多实验中没有检测到脱靶现象, 也与实验之前精心选择Cas9蛋白及目标位点有密切关系.本文介绍了一些在实验设计过程中减少产生脱靶的方法, 广大****可以根据实验需求参考借鉴. ...Kim et al., 2015 ).这种方法的灵敏度可达0.1%, 但一些存在混合切割模式的位点和测序深度不足的位点仍会被遗漏.Kim等(2016) 通过改进算法, 将之前的只检测平末端序列, 改进为假设Cas9能够产生1-2个碱基的突出, 大大提高了检测的灵敏度.经验证, Digenome-seq可同时检测10个sgRNA所产生的脱靶位点(Kim et al., 2016 ). ...Kim et al., 2015 ).这种方法的灵敏度可达0.1%, 但一些存在混合切割模式的位点和测序深度不足的位点仍会被遗漏.Kim等(2016) 通过改进算法, 将之前的只检测平末端序列, 改进为假设Cas9能够产生1-2个碱基的突出, 大大提高了检测的灵敏度.经验证, Digenome-seq可同时检测10个sgRNA所产生的脱靶位点(Kim et al., 2016 ). ...Kim et al., 2016 ). ...Kim et al., 2016 ).但GUIDE-seq受细胞转染的限制, 其应用范围较小.Digenome-seq和Circle-seq都是很好的检测方法, 检测的精确度高, 但准确度却不高.例如, 在Digeno- me-seq检测中, 有74个位点被检出脱靶编辑, 经过验证, 只有5个是真正的脱靶编辑位点; 在Circle-seq检测中, 仅有20%左右的潜在脱靶位点被证实.因此, 提高检测的准确度也是目前需要解决的问题之一. ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...图1 ).此方法灵敏度较高, 能检测到细胞中0.1%的突变.此外, 在定位切割位点的过程中, 可以通过测序得到的序列数目推测CRISPR/Cas9的切割活性.但由于细胞转染的局限, 使其只能应用于易转化的细胞.此后, 应用GUIDE-seq方法成功检测CRI- SPR/Cas9系统中的脱靶切割屡见报道, 且评价较好(Friedland et al., 2015 ; Kleinstiver et al., 2015a , 2015b ; Lee et al., 2016 ).Tsai等开发了用于GUIDE- seq测序结果分析的软件(https://github.com/aryee- lab/umi), 并公开供广大研究者使用. ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...图1 ).此方法灵敏度较高, 能检测到细胞中0.1%的突变.此外, 在定位切割位点的过程中, 可以通过测序得到的序列数目推测CRISPR/Cas9的切割活性.但由于细胞转染的局限, 使其只能应用于易转化的细胞.此后, 应用GUIDE-seq方法成功检测CRI- SPR/Cas9系统中的脱靶切割屡见报道, 且评价较好(Friedland et al., 2015 ; Kleinstiver et al., 2015a , 2015b ; Lee et al., 2016 ).Tsai等开发了用于GUIDE- seq测序结果分析的软件(https://github.com/aryee- lab/umi), 并公开供广大研究者使用. ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...图3 ) (Kuscu et al., 2014 ; Duan et al., 2014 ).这种方法能够覆盖全基因组, 但由于只检测基因组中与Cas9结合的位点, 没有考虑Cas9的切割活性, 因此其准确度较低, 容易过度估计脱靶效率.Cencic等(2014) 对43个通过ChIP-seq预测的脱靶位点进行验证, 发现只有1个目标位点和1个脱靶位点被切割, 其它41个预测位点均没有产生切割和突变.另据报道, 应用dCas9和免疫共沉淀方法预测12个sgRNA的潜在脱靶位点, 其中, 根据sgRNA的不同, 脱靶位点的数量由10到1 000不等.经过有活性的Cas9蛋白和相应sgRNA组合对基因组进行编辑, 结果显示大部分预测的脱靶位点都是可被切割的, 只是切割频率低于目标位点(Kuscu et al., 2014 ).因此, 用该方法预测的脱靶位点与实际的脱靶位点可能存在很大差异.其主要原因是由于使用了无活性的dCas9而不是有活性的Cas9, 因此切割活性无法体现. ...Kuscu et al., 2014 ).因此, 用该方法预测的脱靶位点与实际的脱靶位点可能存在很大差异.其主要原因是由于使用了无活性的dCas9而不是有活性的Cas9, 因此切割活性无法体现. ...图1 ).此方法灵敏度较高, 能检测到细胞中0.1%的突变.此外, 在定位切割位点的过程中, 可以通过测序得到的序列数目推测CRISPR/Cas9的切割活性.但由于细胞转染的局限, 使其只能应用于易转化的细胞.此后, 应用GUIDE-seq方法成功检测CRI- SPR/Cas9系统中的脱靶切割屡见报道, 且评价较好(Friedland et al., 2015 ; Kleinstiver et al., 2015a , 2015b ; Lee et al., 2016 ).Tsai等开发了用于GUIDE- seq测序结果分析的软件(https://github.com/aryee- lab/umi), 并公开供广大研究者使用. ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Gly- cine max )的成熟期育种过程中, 已通过确定成熟期相关基因和开发分子标记的方法加快育种进程(Li et al., 2017 ).分子标记技术在玉米(Zea mays )中也得到了广泛的应用, 目前已有55 K的SNP标记应用到玉米辅助育种(Xu et al., 2017 ).移除脱靶突变、保留目标位点的最佳选择便是定向回交, 结合分子标记辅助育种方法能够加速筛选, 缩短遗传背景纯化周期.一方面, 可以通过全基因组分子标记检测, 保留目标位点, 加速脱靶突变移除进程; 另一方面, 可以根据本文所述的脱靶检测方法推测脱靶位点, 开发相应的分子标记, 在回交或杂交的过程中移除脱靶位点.分子标记在育种中应用较为成熟, 通过分子标记移除脱靶位点与育种中纯化品种十分相似.因此分子标记技术在植物基因组定点编辑中将发挥助力作用并得到广泛应用. ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ... 设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ... 设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...trans -activating crRNA, tracrRNA).向导RNA和转运激活RNA可以组装成一个元件, 称为单一向导RNA (sgRNA).向导RNA的前20个碱基依靠碱基互补配对原则识别目标基因, Cas9蛋白和sgRNA复合体首先识别目标基因中的PAM序列, 从而完成与目标基因的结合.在整个目标基因的识别过程中, sgRNA和PAM发挥非常重要的作用.脱靶的产生也很大程度上来自对目标基因识别的准确性降低.sgRNA在与目标基因互补配对过程中允许1-5个碱基的错配, 从而导致目标以外的相似序列被编辑(Fu et al., 2013 ).sgRNA中的前20个负责互补配对的碱基中, 靠近PAM端的8-12个碱基被认为是核心序列, 在识别特异性上起重要作用(Patrick et al., 2013; Pattanayak et al., 2013 ).也有报道显示, 通过染色质免疫共沉淀测序(ChIP-seq)的方法检测到核心序列是靠近PAM端1-5个碱基(Wu et al., 2014 ).但ChIP-seq方法由于是利用没有切割功能的Cas9去识别并结合目标基因组中的序列, 只能反映识别和结合的情况, 无法推测切割的情况, 因此会高估脱靶位点的数量.PAM序列对CRISPR/Cas9识别和切割的准确性也产生较大影响.起初人们认为NGG是PAM序列(N代表A\T\G\C), 但经过研究发现Cas9不仅可以识别NGG, 还可以识别NRG (R代表A或G), 但NRG的切割频率仅为NGG的1/5 (Hsu et al., 2013 ).尽管识别NRG的频率远低于NGG, 但仍然存在脱靶的可能性.植物中关于这方面的研究很少, Meng等(2018) 在水稻中验证了野生型的spCas9能够识别NGG和NAG, 且经过对若干目标位点进行验证, 表明无论PAM序列是NGG还是NAG, 均能完成高效率的编辑.因此, 在选择目标位点时应考虑PAM序列.除sgRNA和PAM之外, Cas9的切割方式也影响该系统的精确度.原始的Cas9含有2个负责切割的元件, 即HNH区和RuvC区, 分别负责切割2条DNA链, 产生DNA双链断裂.整个切割过程由1个Cas9蛋白完成.研究表明, 若将1个Cas9蛋白中HNH区的氨基酸突变, 将另1个Cas9蛋白的RuvC区氨基酸突变, 然后将2个突变后的Cas9蛋白及2个目标位点相距较近的sgRNA共同转入细胞中, 从而产生DNA双链断裂, 能够降低脱靶频率(Cho et al., 2014 ). ...Cho et al., 2014 ).目前, 研究人员已开发出许多脱靶预测和sgRNA设计软件(表2 ).人们可以根据具体需求选择合适的软件设计sgRNA和预测脱靶可能性.另外, 使用较短(16-18个碱基)的间隔序列, 能够降低脱靶率(Fu et al., 2014 ).经过不断总结经验, 研究人员发现在设计sgRNA时遵循一定规律可以降低脱靶率, 如在设计sgRNA时要选择基因组中特异的序列或尽可能增加所选择的目标序列与基因组中其它序列的错配碱基数目; 靠近PAM序列的1-6个碱基最好是基因组中完全特异的; 另外也要考虑PAM序列, 在设计sgRNA时除比较sgRNA的序列是否特异外, 还要在基因组中查找是否存在同样的序列位于除NGG以外的其它PAM序列的下游, 如NAG和NGC等(Doench et al., 2016 ; Meng et al., 2018 ). ... 设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Osakabe et al., 2016 ).水稻耐寒基因编辑的脱靶研究表明, 通过BLASTN找到的潜在脱靶位点, 在突变体中均未检测到突变(Shen et al., 2017 ).综合其它植物基因编辑文献对脱靶的报道, 大多数研究都是通过软件预测潜在的脱靶位点, 然后定向检测预测所得的可能脱靶序列(Peterson et al., 2016 ; Kaya et al., 2016 ).这种方式无疑可以一定程度地检测脱靶效应, 但软件的选择对检测结果影响很大.因此, 广大科研工作者在今后的研究中, 可以用试验结合软件预测来选择潜在的脱靶位点(如FIND-seq、Circle-seq和SITE-seq), 从而提高脱靶检测的准确性.对于那些用于产品生产的突变体, 要结合全基因组深度测序进一步筛查脱靶编辑. 当然, 许多实验中没有检测到脱靶现象, 也与实验之前精心选择Cas9蛋白及目标位点有密切关系.本文介绍了一些在实验设计过程中减少产生脱靶的方法, 广大****可以根据实验需求参考借鉴. ...Gabriel et al., 2011 ).这一整合特性使其得以应用到基因组编辑的脱靶检测(图1 ) (Wang et al., 2015 ; Osborn et al., 2016 ).Wang等(2015) 研究表明IDLV的检测精度达1%, 并检测到CRISPR/Cas9的脱靶位点与目标位点之间的差异高达1-13个碱基.Osborn等(2016) 应用IDLV方法在人类T-细胞中检测出TALEN、CRISPR和Meg- Tal三种基因编辑工具的脱靶频率, 结果在TALEN和CRISPR的实验中所有IDLV检测到的脱靶位点均没有被切割.在Meg-Tal实验中确认了所有IDLV实验产生的脱靶位点均被切割产生变异.这2项研究均证明IDLV方法可以无偏见地检测全基因组中的脱靶位点, 但精确度只有1%. ...Osborn等(2016) 应用IDLV方法在人类T-细胞中检测出TALEN、CRISPR和Meg- Tal三种基因编辑工具的脱靶频率, 结果在TALEN和CRISPR的实验中所有IDLV检测到的脱靶位点均没有被切割.在Meg-Tal实验中确认了所有IDLV实验产生的脱靶位点均被切割产生变异.这2项研究均证明IDLV方法可以无偏见地检测全基因组中的脱靶位点, 但精确度只有1%. ...Pan et al., 2016 ; Tian et al., 2017 ; Zong et al., 2017 ; Chen et al., 2017 ).也有研究表明, CRISPR系统在植物基因组编辑中产生大量脱靶编辑, 需要通过使用Cas9突变体和其它方法才能降低脱靶频率(Zhang et al., 2017b ), 该研究是在拟南芥(Arabidopsis thaliana )中进行的, 而在拟南芥的其它报道中并未发现高脱靶频率现象, 这可能与选择的目标位点有关(Jiang et al., 2014 ).另有研究表明, 在应用Cas9突变体降低脱靶频率的同时, 若选择与目标序列完全一致的20个碱基作为gRNA, 则可以大大提高定点编辑的频率(Zhang et al., 2017a ).因此, 在选择目标位点时应尽量选择基因组中特异的序列以减小脱靶的可能性, 过多的脱靶将大大影响优良性状选育的商业化进程. ...Pan et al., 2016 ; Zhang et al., 2017a , 2017b ; Zong et al., 2017 ). ...trans -activating crRNA, tracrRNA).向导RNA和转运激活RNA可以组装成一个元件, 称为单一向导RNA (sgRNA).向导RNA的前20个碱基依靠碱基互补配对原则识别目标基因, Cas9蛋白和sgRNA复合体首先识别目标基因中的PAM序列, 从而完成与目标基因的结合.在整个目标基因的识别过程中, sgRNA和PAM发挥非常重要的作用.脱靶的产生也很大程度上来自对目标基因识别的准确性降低.sgRNA在与目标基因互补配对过程中允许1-5个碱基的错配, 从而导致目标以外的相似序列被编辑(Fu et al., 2013 ).sgRNA中的前20个负责互补配对的碱基中, 靠近PAM端的8-12个碱基被认为是核心序列, 在识别特异性上起重要作用(Patrick et al., 2013; Pattanayak et al., 2013 ).也有报道显示, 通过染色质免疫共沉淀测序(ChIP-seq)的方法检测到核心序列是靠近PAM端1-5个碱基(Wu et al., 2014 ).但ChIP-seq方法由于是利用没有切割功能的Cas9去识别并结合目标基因组中的序列, 只能反映识别和结合的情况, 无法推测切割的情况, 因此会高估脱靶位点的数量.PAM序列对CRISPR/Cas9识别和切割的准确性也产生较大影响.起初人们认为NGG是PAM序列(N代表A\T\G\C), 但经过研究发现Cas9不仅可以识别NGG, 还可以识别NRG (R代表A或G), 但NRG的切割频率仅为NGG的1/5 (Hsu et al., 2013 ).尽管识别NRG的频率远低于NGG, 但仍然存在脱靶的可能性.植物中关于这方面的研究很少, Meng等(2018) 在水稻中验证了野生型的spCas9能够识别NGG和NAG, 且经过对若干目标位点进行验证, 表明无论PAM序列是NGG还是NAG, 均能完成高效率的编辑.因此, 在选择目标位点时应考虑PAM序列.除sgRNA和PAM之外, Cas9的切割方式也影响该系统的精确度.原始的Cas9含有2个负责切割的元件, 即HNH区和RuvC区, 分别负责切割2条DNA链, 产生DNA双链断裂.整个切割过程由1个Cas9蛋白完成.研究表明, 若将1个Cas9蛋白中HNH区的氨基酸突变, 将另1个Cas9蛋白的RuvC区氨基酸突变, 然后将2个突变后的Cas9蛋白及2个目标位点相距较近的sgRNA共同转入细胞中, 从而产生DNA双链断裂, 能够降低脱靶频率(Cho et al., 2014 ). ...Osakabe et al., 2016 ).水稻耐寒基因编辑的脱靶研究表明, 通过BLASTN找到的潜在脱靶位点, 在突变体中均未检测到突变(Shen et al., 2017 ).综合其它植物基因编辑文献对脱靶的报道, 大多数研究都是通过软件预测潜在的脱靶位点, 然后定向检测预测所得的可能脱靶序列(Peterson et al., 2016 ; Kaya et al., 2016 ).这种方式无疑可以一定程度地检测脱靶效应, 但软件的选择对检测结果影响很大.因此, 广大科研工作者在今后的研究中, 可以用试验结合软件预测来选择潜在的脱靶位点(如FIND-seq、Circle-seq和SITE-seq), 从而提高脱靶检测的准确性.对于那些用于产品生产的突变体, 要结合全基因组深度测序进一步筛查脱靶编辑. 当然, 许多实验中没有检测到脱靶现象, 也与实验之前精心选择Cas9蛋白及目标位点有密切关系.本文介绍了一些在实验设计过程中减少产生脱靶的方法, 广大****可以根据实验需求参考借鉴. ... 设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ... 设计软件 网址 软件功能 Cas-OFFinder http://www.rgenome.net/ 针对CRISPR/Cas9系统, 通过使用者提供的目标位点序列, 推测所选择的目标基因组中潜在的脱靶位点.可选择不同的PAM、错配数量和是否允许错位配对(Bae et al., 2014 ) CHOPCHOP https://chopchop.rc.fas.harvard. edu/ 针对CRISPR/Cas9和TALEN系统, 根据用户给出的目标基因序列, 在目标基因组或染色体中查找潜在的脱靶位点.可查找2个以内碱基错配的脱靶序列(Montague et al., 2014 ) CRISPR Design http://crispr.mit.edu/ 在所给基因序列中设计sgRNA, 能够预测该sgRNA在基因组中的脱靶情况, 并标出最特异的sgRNA CRISPR/Cas9 gRNA Finder http://spot.colorado.edu/~slin/cas9.html 在所给出的基因序列中, 查找可能的目标切割位点, 给出合适的sgRNA序列, 并推测其二级结构(Mali et al., 2013 ) CRISPRfinder Christine http://crispr.u-psud.fr/Server/ 在公开的微生物基因组中定位CRISPR重复序列位置, 并能报告间隔序列(Pourcel and Drevet, 2013 ) E-CRISP http://www.e-crisp.org/E-CRISP/ 设计并评估CRISPR目标位点, 输入基因ID、FASTA序列进行搜索.可针对不同的CRISPR系统进行设计(Hsu et al., 2014 ) CRISPR-Plant http://www.genome.arizona.edu/crispr/ 此软件针对一系列植物基因组设计CRISPR目标位点, 在所给出的基因序列或染色体序列中查找合适的目标位点(Lozano-Juste and Cutler, 2014 ) CRISPR MultiTargeter http://www.multicrispr.net 可用于设计同时靶向几个基因或1个基因中的多个位点的sgRNA (Prykhozhij et al., 2015 ) sgRNA Designer http://www.broadinstitute.org/rnai/public/ 适用于人类和小鼠基因组中sgRNA的设计.能够推荐最高特异性的sgRNA, 但不会给出可能的脱靶位点(Doench et al., 2016 ) sgRNA Scorer https://crispr.med.harvard.edu/sgRNAScorer/ 可以设计sgRNA并评估sgRNA的体内切割活性(Chari et al., 2015 )
3.2 CRISPR系统的优化 在SpCas9蛋白的结构解析基础上, 张峰实验室通过突变HNH与RuvC两个切割元件中间区域的单个氨基酸, 使SpCas9的脱靶率大幅度降低, 由野生型的10%降低到0.5%.通过同时突变几个效果较好的氨基酸, 进一步将脱靶频率降低且保持与野生型SpCas9相同的切割频率(Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Osakabe et al., 2016 ).水稻耐寒基因编辑的脱靶研究表明, 通过BLASTN找到的潜在脱靶位点, 在突变体中均未检测到突变(Shen et al., 2017 ).综合其它植物基因编辑文献对脱靶的报道, 大多数研究都是通过软件预测潜在的脱靶位点, 然后定向检测预测所得的可能脱靶序列(Peterson et al., 2016 ; Kaya et al., 2016 ).这种方式无疑可以一定程度地检测脱靶效应, 但软件的选择对检测结果影响很大.因此, 广大科研工作者在今后的研究中, 可以用试验结合软件预测来选择潜在的脱靶位点(如FIND-seq、Circle-seq和SITE-seq), 从而提高脱靶检测的准确性.对于那些用于产品生产的突变体, 要结合全基因组深度测序进一步筛查脱靶编辑. 当然, 许多实验中没有检测到脱靶现象, 也与实验之前精心选择Cas9蛋白及目标位点有密切关系.本文介绍了一些在实验设计过程中减少产生脱靶的方法, 广大****可以根据实验需求参考借鉴. ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Veres et al., 2014 ; Smith et al., 2014 ).全基因组测序方法的关键是选择合适的对照基因组, 将自然的突变背景尽量去除, 然后通过数据分析, 寻找含有PAM基序的突变位点, 最后再通过PCR和Sanger测序进行验证.如此得到的可能脱靶序列数量较大, 还需利用辅助软件在其中找到那些与目标序列相似的序列进行PCR和Sanger测序验证, 因此会存在遗漏现象.由于测序的精确度问题, 该方法只能用于检测高频率的脱靶现象, 而对于低频率事件则无法检出. ...Pan et al., 2016 ; Tian et al., 2017 ; Zong et al., 2017 ; Chen et al., 2017 ).也有研究表明, CRISPR系统在植物基因组编辑中产生大量脱靶编辑, 需要通过使用Cas9突变体和其它方法才能降低脱靶频率(Zhang et al., 2017b ), 该研究是在拟南芥(Arabidopsis thaliana )中进行的, 而在拟南芥的其它报道中并未发现高脱靶频率现象, 这可能与选择的目标位点有关(Jiang et al., 2014 ).另有研究表明, 在应用Cas9突变体降低脱靶频率的同时, 若选择与目标序列完全一致的20个碱基作为gRNA, 则可以大大提高定点编辑的频率(Zhang et al., 2017a ).因此, 在选择目标位点时应尽量选择基因组中特异的序列以减小脱靶的可能性, 过多的脱靶将大大影响优良性状选育的商业化进程. ...Joung and Tsai, 2017b ; Tsai et al., 2017 ).该方法通过对剪切后的基因组片段进行环化, 然后用Cas9切割环化后的基因组片段, 所有能够被切割的环均被切开, 为线型, 再对线性DNA片段进行二代测序, 从而得到可能的脱靶位点(图4 ).与其它检测方法相比, 此方法能够检测到更多低频率的可能脱靶位点.例如, 在测定人的HBB 基因中相同的目标序列时, Circle-seq方法比Digenome-seq方法多检测出156个潜在脱靶位点, 经确认其中有29个位点是真实的脱靶位点.为了方便Circle-seq方法的应用, 研究人员还开发了公开的数据分析软件(python package circleseq), 使用者可直接得到1个含有脱靶位点的表格及比对后的潜在脱靶序列. ...Veres et al., 2014 ; Smith et al., 2014 ).全基因组测序方法的关键是选择合适的对照基因组, 将自然的突变背景尽量去除, 然后通过数据分析, 寻找含有PAM基序的突变位点, 最后再通过PCR和Sanger测序进行验证.如此得到的可能脱靶序列数量较大, 还需利用辅助软件在其中找到那些与目标序列相似的序列进行PCR和Sanger测序验证, 因此会存在遗漏现象.由于测序的精确度问题, 该方法只能用于检测高频率的脱靶现象, 而对于低频率事件则无法检出. ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...Gabriel et al., 2011 ).这一整合特性使其得以应用到基因组编辑的脱靶检测(图1 ) (Wang et al., 2015 ; Osborn et al., 2016 ).Wang等(2015) 研究表明IDLV的检测精度达1%, 并检测到CRISPR/Cas9的脱靶位点与目标位点之间的差异高达1-13个碱基.Osborn等(2016) 应用IDLV方法在人类T-细胞中检测出TALEN、CRISPR和Meg- Tal三种基因编辑工具的脱靶频率, 结果在TALEN和CRISPR的实验中所有IDLV检测到的脱靶位点均没有被切割.在Meg-Tal实验中确认了所有IDLV实验产生的脱靶位点均被切割产生变异.这2项研究均证明IDLV方法可以无偏见地检测全基因组中的脱靶位点, 但精确度只有1%. ...Wang等(2015) 研究表明IDLV的检测精度达1%, 并检测到CRISPR/Cas9的脱靶位点与目标位点之间的差异高达1-13个碱基.Osborn等(2016) 应用IDLV方法在人类T-细胞中检测出TALEN、CRISPR和Meg- Tal三种基因编辑工具的脱靶频率, 结果在TALEN和CRISPR的实验中所有IDLV检测到的脱靶位点均没有被切割.在Meg-Tal实验中确认了所有IDLV实验产生的脱靶位点均被切割产生变异.这2项研究均证明IDLV方法可以无偏见地检测全基因组中的脱靶位点, 但精确度只有1%. ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...trans -activating crRNA, tracrRNA).向导RNA和转运激活RNA可以组装成一个元件, 称为单一向导RNA (sgRNA).向导RNA的前20个碱基依靠碱基互补配对原则识别目标基因, Cas9蛋白和sgRNA复合体首先识别目标基因中的PAM序列, 从而完成与目标基因的结合.在整个目标基因的识别过程中, sgRNA和PAM发挥非常重要的作用.脱靶的产生也很大程度上来自对目标基因识别的准确性降低.sgRNA在与目标基因互补配对过程中允许1-5个碱基的错配, 从而导致目标以外的相似序列被编辑(Fu et al., 2013 ).sgRNA中的前20个负责互补配对的碱基中, 靠近PAM端的8-12个碱基被认为是核心序列, 在识别特异性上起重要作用(Patrick et al., 2013; Pattanayak et al., 2013 ).也有报道显示, 通过染色质免疫共沉淀测序(ChIP-seq)的方法检测到核心序列是靠近PAM端1-5个碱基(Wu et al., 2014 ).但ChIP-seq方法由于是利用没有切割功能的Cas9去识别并结合目标基因组中的序列, 只能反映识别和结合的情况, 无法推测切割的情况, 因此会高估脱靶位点的数量.PAM序列对CRISPR/Cas9识别和切割的准确性也产生较大影响.起初人们认为NGG是PAM序列(N代表A\T\G\C), 但经过研究发现Cas9不仅可以识别NGG, 还可以识别NRG (R代表A或G), 但NRG的切割频率仅为NGG的1/5 (Hsu et al., 2013 ).尽管识别NRG的频率远低于NGG, 但仍然存在脱靶的可能性.植物中关于这方面的研究很少, Meng等(2018) 在水稻中验证了野生型的spCas9能够识别NGG和NAG, 且经过对若干目标位点进行验证, 表明无论PAM序列是NGG还是NAG, 均能完成高效率的编辑.因此, 在选择目标位点时应考虑PAM序列.除sgRNA和PAM之外, Cas9的切割方式也影响该系统的精确度.原始的Cas9含有2个负责切割的元件, 即HNH区和RuvC区, 分别负责切割2条DNA链, 产生DNA双链断裂.整个切割过程由1个Cas9蛋白完成.研究表明, 若将1个Cas9蛋白中HNH区的氨基酸突变, 将另1个Cas9蛋白的RuvC区氨基酸突变, 然后将2个突变后的Cas9蛋白及2个目标位点相距较近的sgRNA共同转入细胞中, 从而产生DNA双链断裂, 能够降低脱靶频率(Cho et al., 2014 ). ...Gly- cine max )的成熟期育种过程中, 已通过确定成熟期相关基因和开发分子标记的方法加快育种进程(Li et al., 2017 ).分子标记技术在玉米(Zea mays )中也得到了广泛的应用, 目前已有55 K的SNP标记应用到玉米辅助育种(Xu et al., 2017 ).移除脱靶突变、保留目标位点的最佳选择便是定向回交, 结合分子标记辅助育种方法能够加速筛选, 缩短遗传背景纯化周期.一方面, 可以通过全基因组分子标记检测, 保留目标位点, 加速脱靶突变移除进程; 另一方面, 可以根据本文所述的脱靶检测方法推测脱靶位点, 开发相应的分子标记, 在回交或杂交的过程中移除脱靶位点.分子标记在育种中应用较为成熟, 通过分子标记移除脱靶位点与育种中纯化品种十分相似.因此分子标记技术在植物基因组定点编辑中将发挥助力作用并得到广泛应用. ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...Slaymaker et al., 2016 ).根据SpCas9的结构改造SpCas9蛋白, 从而提高编辑的准确度并不局限于这一种方式.Kleinstiver等(2015a) 通过改变几个负责与目标DNA结合的氨基酸, 提高了编辑的准确度, 使脱靶率降低至接近背景值的水平.该团队还在人类细胞中证实了经过改造的Cas9突变体不仅减少了脱靶, 还能保持原有的编辑效率(Kleinstiver et al., 2015b , 2016 ).Cas9突变体可以降低脱靶率, 但很多突变体在降低脱靶率的同时定点编辑的频率也降低了, 因此人们希望能快速找到既能降低脱靶率又能保持原有编辑效率的Cas9突变体.Lee等(2017) 运用大肠杆菌作为筛选Cas9突变体的工具, 成功地从100多个Cas9突变体中找到2个可以降低脱靶率且保持高效定点编辑频率的Cas9蛋白.Zhang等(2107a) 在植物细胞中测试了2个含有与DNA 结合的氨基酸突变的spCas9 (espCAs9和spCas9- HF1), 发现gRNA序列如果含有20个与目标基因序列完全相同的碱基, 可以大幅度提高espCAs9和spCas9-HF1的定点编辑频率, 且保持较低的脱靶率.另外, 应用1对Cas9切口酶或使用dCas9突变体连接FokI的方法, 均可降低脱靶率(Shen et al., 2014 ; Havlicek et al., 2017 ).一方面, 通过对Cas9蛋白进行改造来提高识别目标序列的准确性; 另一方面, 也可以通过利用Cas9蛋白的抑制酶或基因沉默的方式来控制Cas9蛋白的表达, 使其只在特定的时间段表达, 从而降低脱靶率(Shin et al., 2017 ; Harrington et al., 2017 ). ...Pan et al., 2016 ; Tian et al., 2017 ; Zong et al., 2017 ; Chen et al., 2017 ).也有研究表明, CRISPR系统在植物基因组编辑中产生大量脱靶编辑, 需要通过使用Cas9突变体和其它方法才能降低脱靶频率(Zhang et al., 2017b ), 该研究是在拟南芥(Arabidopsis thaliana )中进行的, 而在拟南芥的其它报道中并未发现高脱靶频率现象, 这可能与选择的目标位点有关(Jiang et al., 2014 ).另有研究表明, 在应用Cas9突变体降低脱靶频率的同时, 若选择与目标序列完全一致的20个碱基作为gRNA, 则可以大大提高定点编辑的频率(Zhang et al., 2017a ).因此, 在选择目标位点时应尽量选择基因组中特异的序列以减小脱靶的可能性, 过多的脱靶将大大影响优良性状选育的商业化进程. ...Pan et al., 2016 ; Zhang et al., 2017a , 2017b ; Zong et al., 2017 ). ...Homo sapiens )细胞中应用广泛(Jiang et al., 2013 ; Li et al., 2013 ; Wang et al., 2014 , 2017 ).该技术在基因定点突变、目标基因敲除、目标基因沉默和激活、基因定点整合以及单碱基替换等方面的研究中发挥了巨大的推进作用.然而, CRISPR系统作为细菌中的获得性免疫系统(Deveau et al., 2010 ; Sampson and Weiss, 2013 ), 依赖间隔区序列与目标基因序列的碱基互补配对识别外源入侵DNA, 对目标DNA的匹配程度具有一定的容忍度, 允许个别碱基的错配.在基因编辑过程中, CRISPR的这一特点导致基因组中与目标DNA只有较少碱基差别的其它DNA也会被切割, 称为脱靶现象.脱靶现象的存在很大程度上阻碍了CRISPR技术在生产实践中的应用.自CRISPR技术应用于真核生物以来, 人们对脱靶现象进行了大量研究, 早期用测序等方法来检测脱靶现象的发生情况.从最初的体外检测到体内检测, 从复杂的测序检测到各种简便测定方法的建立, 从了解脱靶现状到降低脱靶率的方法研究, 都取得了长足的进展.Jinek等(2012) 在体外实验中发现与目标基因存在1个碱基差异的crRNA (CRISPR RNA)能够切割目标基因, 但切割频率低于与目标基因完全匹配的crRNA.2013年, 继张锋实验室首次发表了CRISPR/Cas9在人类细胞中定点切割的应用, J Keith团队即在人类细胞中验证了CRISPR/Cas9的脱靶现象(Fu et al., 2013 ).他们发现CRISPR/Cas9系统在人类细胞中具有很高的脱靶切割频率, 与crRNA存在5个碱基差异的非靶标基因仍然有可能被切割, 并发生突变.这一发现迅速引起了科研人员的关注.Patrick等随后也系统地验证了CRISPR/Cas9在人 类细胞中的脱靶率(Hsu et al., 2013 ).他们在人EMX1 基因中选择了4个目标位点, 在每个位点设计了57个不同的sgRNA (single-guide RNA), 这些sgRNA包含了所有与目标位点中靠近PAM (protospacer adjacent motif)序列的1-19个碱基存在1个碱基差异的可能性.结果显示, CRISPR/Cas9对于PAM远端序列的单碱基差异容忍度高于对PAM近端的8-12个核心序列.同时, 他们还设计了一系列不同的sgRNA, 这些sgRNA与目标位点存在2-5个碱基差异, 尤其是核心序列中2-3个碱基的差异, 可以大大降低脱靶切割的发生, 而存在5个碱基的差异则几乎检测不到脱靶切割.关于植物细胞中CRISPR/Cas9脱靶现象的研究较少.现有文献显示, 植物中的脱靶切割频率较低.例如, Belhaj等(2013) 在烟草(Nicoti- ana tabacum )的瞬时表达实验中, 设计了18个含有与目标位点存在3-6个碱基差异的sgRNA, 用以测试CRISPR/Cas9的脱靶切割, 没有发现CRISPR的脱靶切割.朱健康实验室在水稻(Oryza sativa )中检测到13个潜在的脱靶位点, 这些位点的序列与目标位点存在1-7个碱基的差异.结果显示, 只有与目标位点存在1个碱基差异的位点发生了脱靶切割(Zhang et al., 2014 ).同年, 另一研究团队在水稻中检测到82-90株通过CRISPR/Cas9产生的突变体, 其中3个与目标位点存在1-3个碱基差异的潜在脱靶位点, 均没有检测到突变(Xu et al., 2014 ).本文阐述了CRISPR脱靶现象的产生机理和检测方法, 以及如何减少脱靶编辑, 以期让广大****更深入地了解CRISPR脱靶现象, 并在设计和选择目标位点时能够尽量减少脱靶产生的可能性. ...Pan et al., 2016 ; Tian et al., 2017 ; Zong et al., 2017 ; Chen et al., 2017 ).也有研究表明, CRISPR系统在植物基因组编辑中产生大量脱靶编辑, 需要通过使用Cas9突变体和其它方法才能降低脱靶频率(Zhang et al., 2017b ), 该研究是在拟南芥(Arabidopsis thaliana )中进行的, 而在拟南芥的其它报道中并未发现高脱靶频率现象, 这可能与选择的目标位点有关(Jiang et al., 2014 ).另有研究表明, 在应用Cas9突变体降低脱靶频率的同时, 若选择与目标序列完全一致的20个碱基作为gRNA, 则可以大大提高定点编辑的频率(Zhang et al., 2017a ).因此, 在选择目标位点时应尽量选择基因组中特异的序列以减小脱靶的可能性, 过多的脱靶将大大影响优良性状选育的商业化进程. ...Pan et al., 2016 ; Zhang et al., 2017a , 2017b ; Zong et al., 2017 ). ...Pan et al., 2016 ; Tian et al., 2017 ; Zong et al., 2017 ; Chen et al., 2017 ).也有研究表明, CRISPR系统在植物基因组编辑中产生大量脱靶编辑, 需要通过使用Cas9突变体和其它方法才能降低脱靶频率(Zhang et al., 2017b ), 该研究是在拟南芥(Arabidopsis thaliana )中进行的, 而在拟南芥的其它报道中并未发现高脱靶频率现象, 这可能与选择的目标位点有关(Jiang et al., 2014 ).另有研究表明, 在应用Cas9突变体降低脱靶频率的同时, 若选择与目标序列完全一致的20个碱基作为gRNA, 则可以大大提高定点编辑的频率(Zhang et al., 2017a ).因此, 在选择目标位点时应尽量选择基因组中特异的序列以减小脱靶的可能性, 过多的脱靶将大大影响优良性状选育的商业化进程. ...Pan et al., 2016 ; Zhang et al., 2017a , 2017b ; Zong et al., 2017 ). ...  ,
, 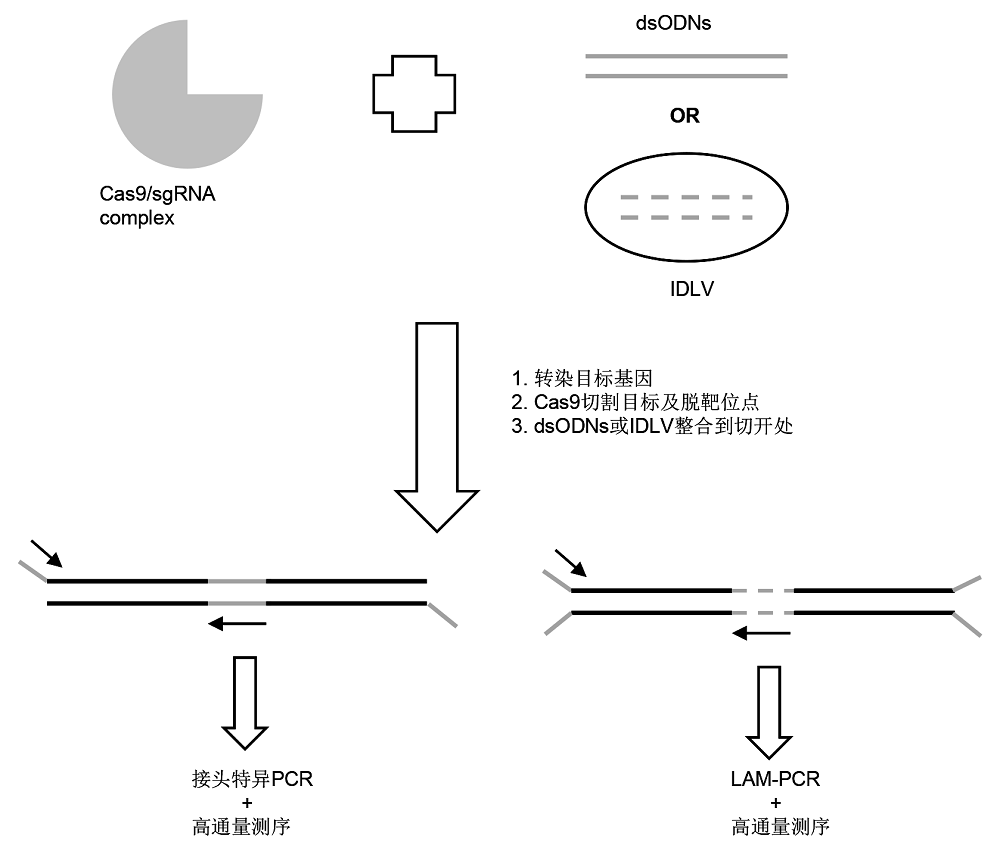
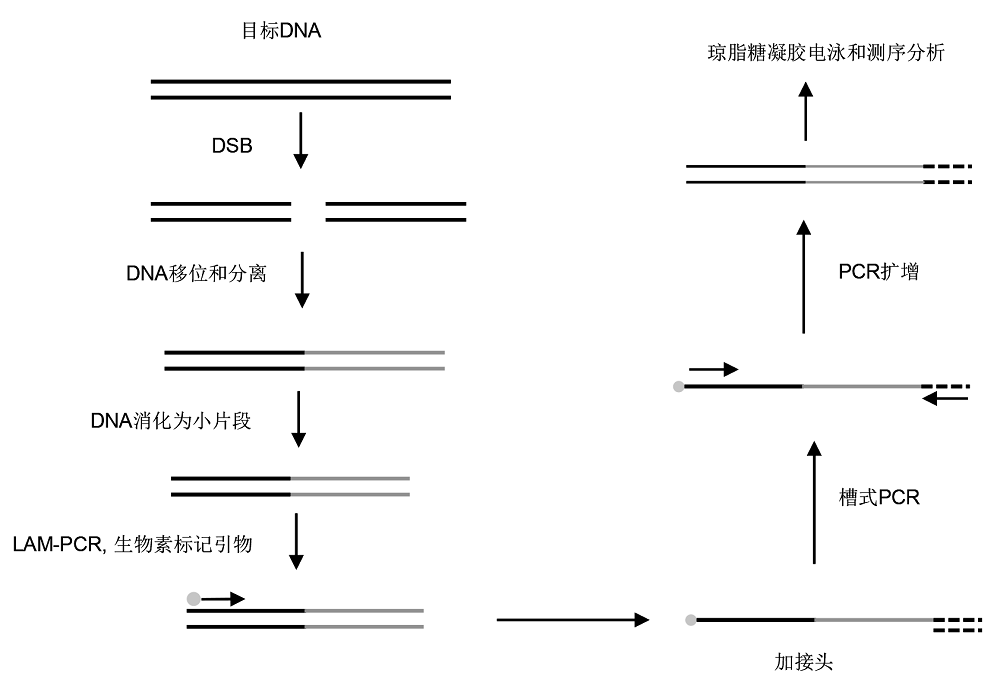
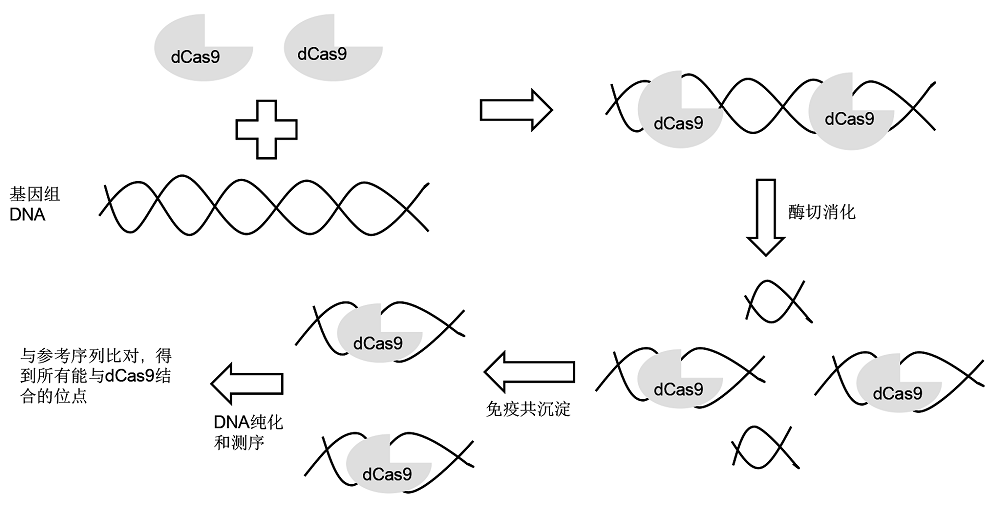
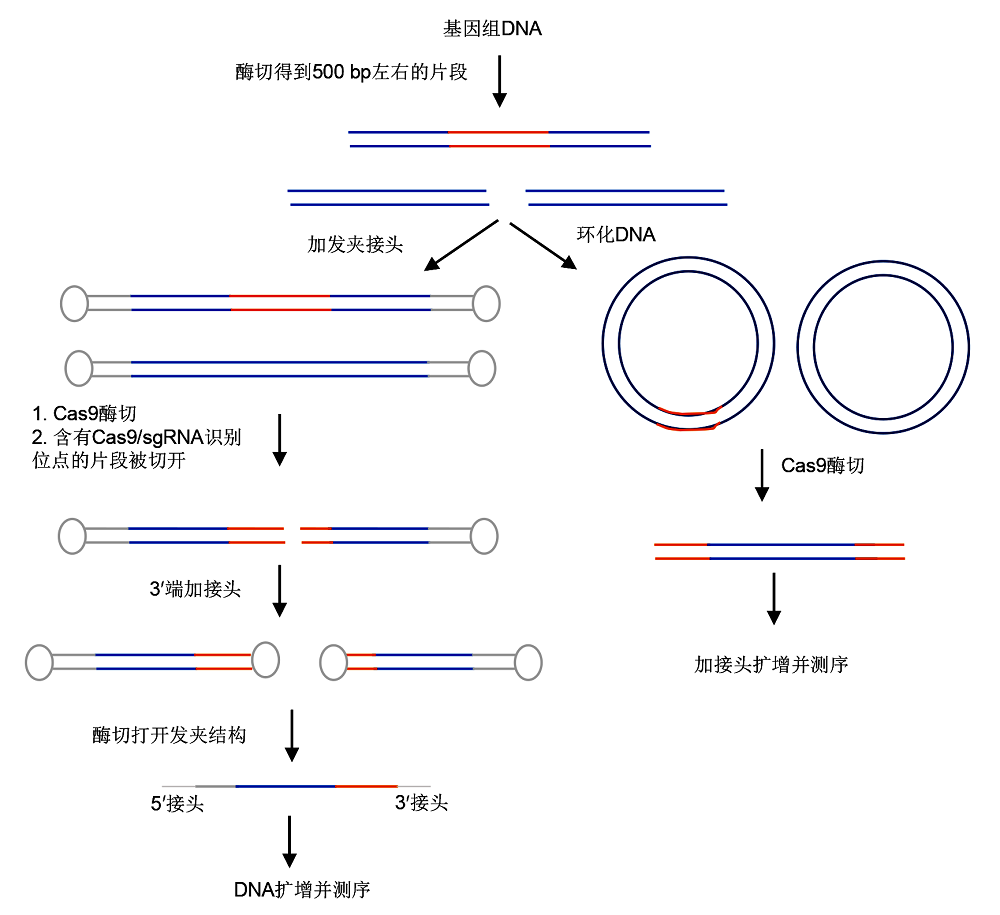

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}