Abstract: To elucidate the genetic structure and genetic diversity of Cynomorium songaricum, modern molecular biology techniques at the DNA level were used to study the genetic structure of 188 C. songaricum individuals from 18 wild populations in the Hexi Corridor Region of Gansu and Qinghai. After alignment, all amplified sequence lengths were 687 bp. The 687 bp ITS sequence detected 7 mutation sites in 188 individuals, defining 9 haplotypes. Processing these 9 haplotype sequences led to a data matrix for calculating the haplotype diversity (Hd=0.294 20) and nucleotide diversity (π=0.000 49). In the haplotype network map, H1 is located in the center and distributed in all populations, and is the ancient and core haplotype. AMOVA revealed that the variation in C. songaricum mainly occurs in populations. According to the genetic differentiation coefficient and Mantel test of ITS sequences, we found no significant relation between genetic and geographic distances, so the current distribution of C. songaricum represents the fragmentation product in recent time. Detection of historical expansion of populations showed that the Tajima’s D test rejected a neutral mutation evolution, and the population expansion history or gene locus is under negative selection pressure, so the null hypothesis cannot be ruled out. Our study provides molecular evidence for the classification system, identification and protection measures of C. songaricum.
表2 锁阳核糖体ITS序列的单倍型多态性位点 Table 2 Variable sites of ITS sequence haplotypes of Cynomorium songaricum
表3 Table 3 表3 表3 锁阳18个居群的单倍型遗传多样性组成 Table 3 Haplotype diversity and composition of Cynomorium songaricum from18 populations
Population code
Samples
Haptotypes
Hd
R1
11
H1, H2, H3, H4
0.25974
R2
17
H1, H5, H6
0.11586
R3
7
H1, H5
0.43956
R4
10
H1, H2, H5
0.42632
R5
11
H1, H6
0.24675
R6
10
H1, H7, H8
0.19474
R7
12
H1, H5
0.08333
R8
4
H1, H5, H9
0.60714
R9
10
H1, H8
0.10000
R10
11
H1, H5, H8
0.17749
R11
11
H1, H4, H6
0.17749
R12
10
H1
0
R13
11
H1
0
R14
7
H1, H6
0.14286
R15
12
H1, H9
0.08333
R16
12
H1, H9
0.08333
R17
11
H1, H8
0.09091
R18
11
H1
0
Total
188
0.29420
Hd: 单倍型多态性 Hd: Haplotype diversity
表3 锁阳18个居群的单倍型遗传多样性组成 Table 3 Haplotype diversity and composition of Cynomorium songaricum from18 populations
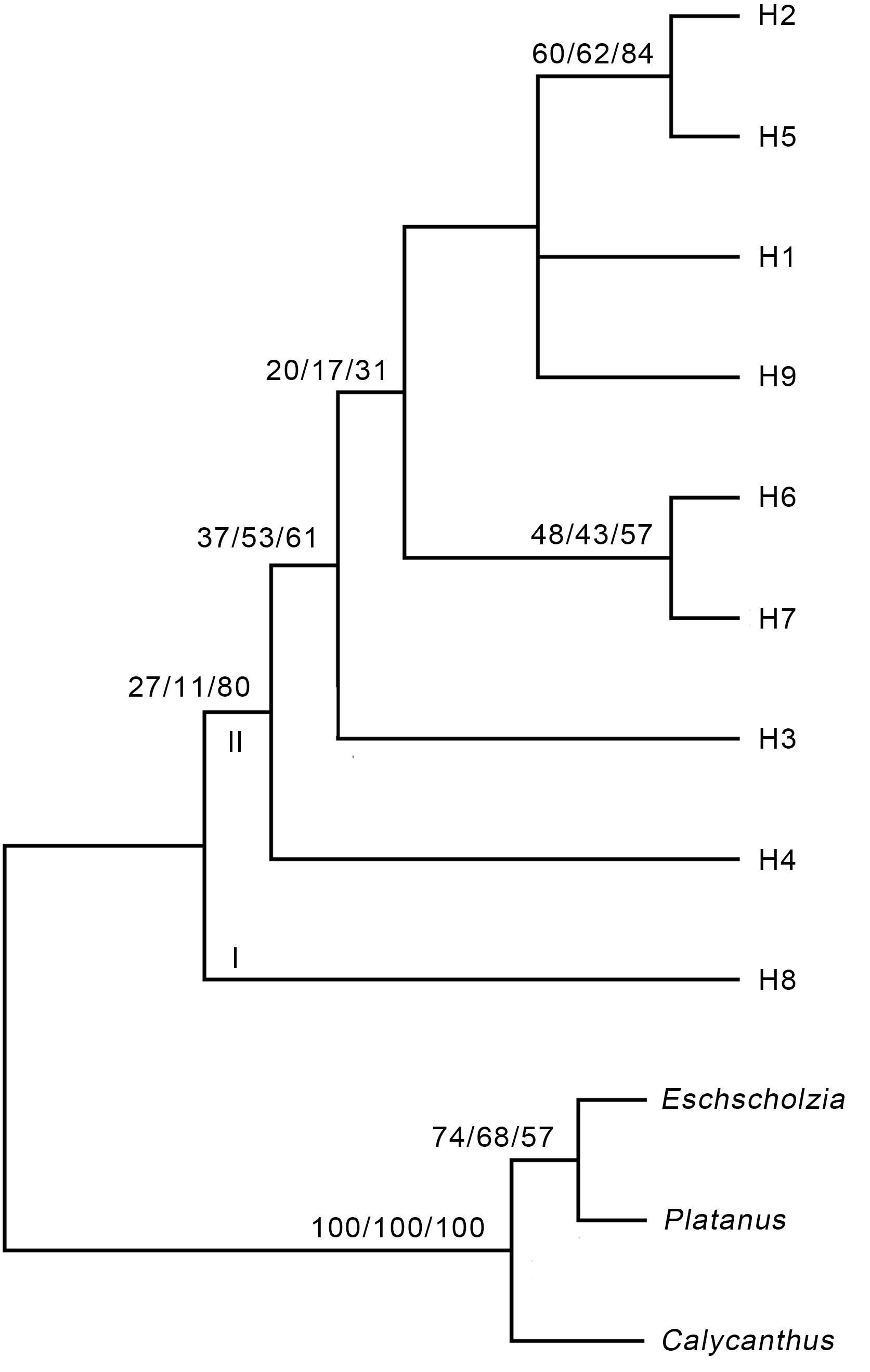
2.2 nrDNA单倍型之间的亲缘关系利用Paup 4.0中的最大简约法(MP)、最大似然法(ML)及贝叶斯(Bayes)法分别对单倍型进行分析, 以花菱草、美国蜡梅和一球悬铃木为外类群, Gap作为缺失状态, 使用启发式(heuristic)搜索建树, 利用自展法(bootstrap)进行重抽样检验, 自展重复次数设定为 1 000。结果显示, 3种方法构建的进化树基本一致。MP进化树中, 系统进化信息由一致性指数(consisten- cy index, CI)、保持性指数(retention index, RI)和恢复性指数(rescaled consistency index, RCI)组成。在进化树的构建过程中, 锁阳属的类群被划分为2支, 分支I仅包含单倍型H8; 分支II包含其它8种单倍型(图1)。 图1https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-3-313/img_1.png<b>图1</b> 基于锁阳ITS序列的严格一致性树<br/>步长=784, CI=0.949 0, RI=0.911 7, RCI=0.865 2。分支上的数字分别代表MP/ML/BI的支持率。<br/><b>Figure 1</b> Strict consensus tree based on the ITS sequence of <i>Cynomorium songaricum<br/></i>Length=784, CI=0.949 0, RI=0.911 7, RCI=0.865 2. The num- bers on the branch represent the support rate of MP/ML/BI, respectively. Figure 1https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-3-313/img_1.png<b>图1</b> 基于锁阳ITS序列的严格一致性树<br/>步长=784, CI=0.949 0, RI=0.911 7, RCI=0.865 2。分支上的数字分别代表MP/ML/BI的支持率。<br/><b>Figure 1</b> Strict consensus tree based on the ITS sequence of <i>Cynomorium songaricum<br/></i>Length=784, CI=0.949 0, RI=0.911 7, RCI=0.865 2. The num- bers on the branch represent the support rate of MP/ML/BI, respectively.
图1 基于锁阳ITS序列的严格一致性树 步长=784, CI=0.949 0, RI=0.911 7, RCI=0.865 2。分支上的数字分别代表MP/ML/BI的支持率。 Figure 1 Strict consensus tree based on the ITS sequence of Cynomorium songaricum Length=784, CI=0.949 0, RI=0.911 7, RCI=0.865 2. The num- bers on the branch represent the support rate of MP/ML/BI, respectively.
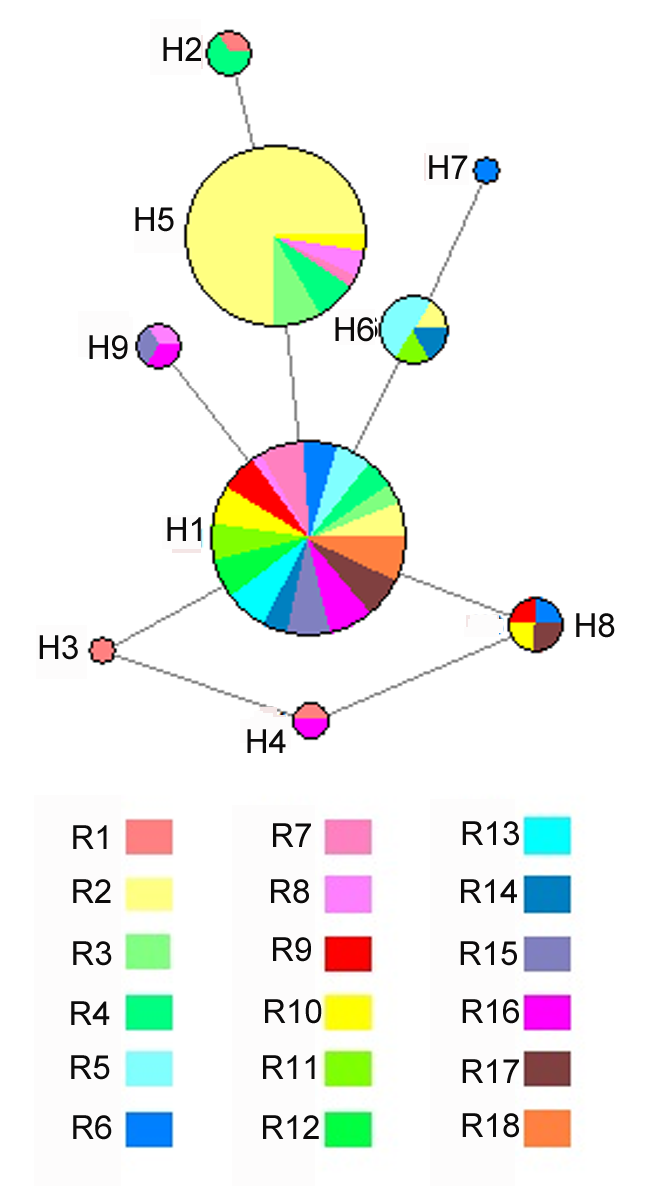
在18个居群中, ITS片段共出现7个变异位点, 9种单倍型。基于9种单倍型构建的不同地区网络分布图呈现星状结构(图2)。在不同地区, H1是共有单倍型。H7是R6特有单倍型, H2是R1和R4共有单倍型, H4是R1和R11共有单倍型。特有单倍型类群均来源于H1单倍型, 构成了西北地区特有的锁阳单倍型地理谱系结构。因此, 我们推测H1可能是锁阳属类群在中国西北地区的古老单倍型。总体来看, 单倍型分布的主要规律包括以下2种形式: (1) 所有种群共享某一种单倍型(H1); (2) 部分种群具有自己的特有单倍型。 图2https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-3-313/img_2.png<b>图2</b> 锁阳18个居群基于ITS序列的单倍型网络图<br/>圆圈的大小与单倍型的相对频率成正比; 不同颜色代表锁阳属不同种群。<br/><b>Figure 2</b> Haplotype network based on ITS sequence of <i>Cynomorium songaricum</i> from 18 populations<br/>The size of circles are proportional to the relative frequency of the haplotype; Different colors represent different populations of <i>Cynomorium songaricum.</i> Figure 2https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-3-313/img_2.png<b>图2</b> 锁阳18个居群基于ITS序列的单倍型网络图<br/>圆圈的大小与单倍型的相对频率成正比; 不同颜色代表锁阳属不同种群。<br/><b>Figure 2</b> Haplotype network based on ITS sequence of <i>Cynomorium songaricum</i> from 18 populations<br/>The size of circles are proportional to the relative frequency of the haplotype; Different colors represent different populations of <i>Cynomorium songaricum.</i>
图2 锁阳18个居群基于ITS序列的单倍型网络图 圆圈的大小与单倍型的相对频率成正比; 不同颜色代表锁阳属不同种群。 Figure 2 Haplotype network based on ITS sequence of Cynomorium songaricum from 18 populations The size of circles are proportional to the relative frequency of the haplotype; Different colors represent different populations of Cynomorium songaricum.
表5 锁阳核糖体基因单倍型分子方差分析 Table 5 Analysis of molecular variance for ribosome haplotypes of Cynomorium songaricum
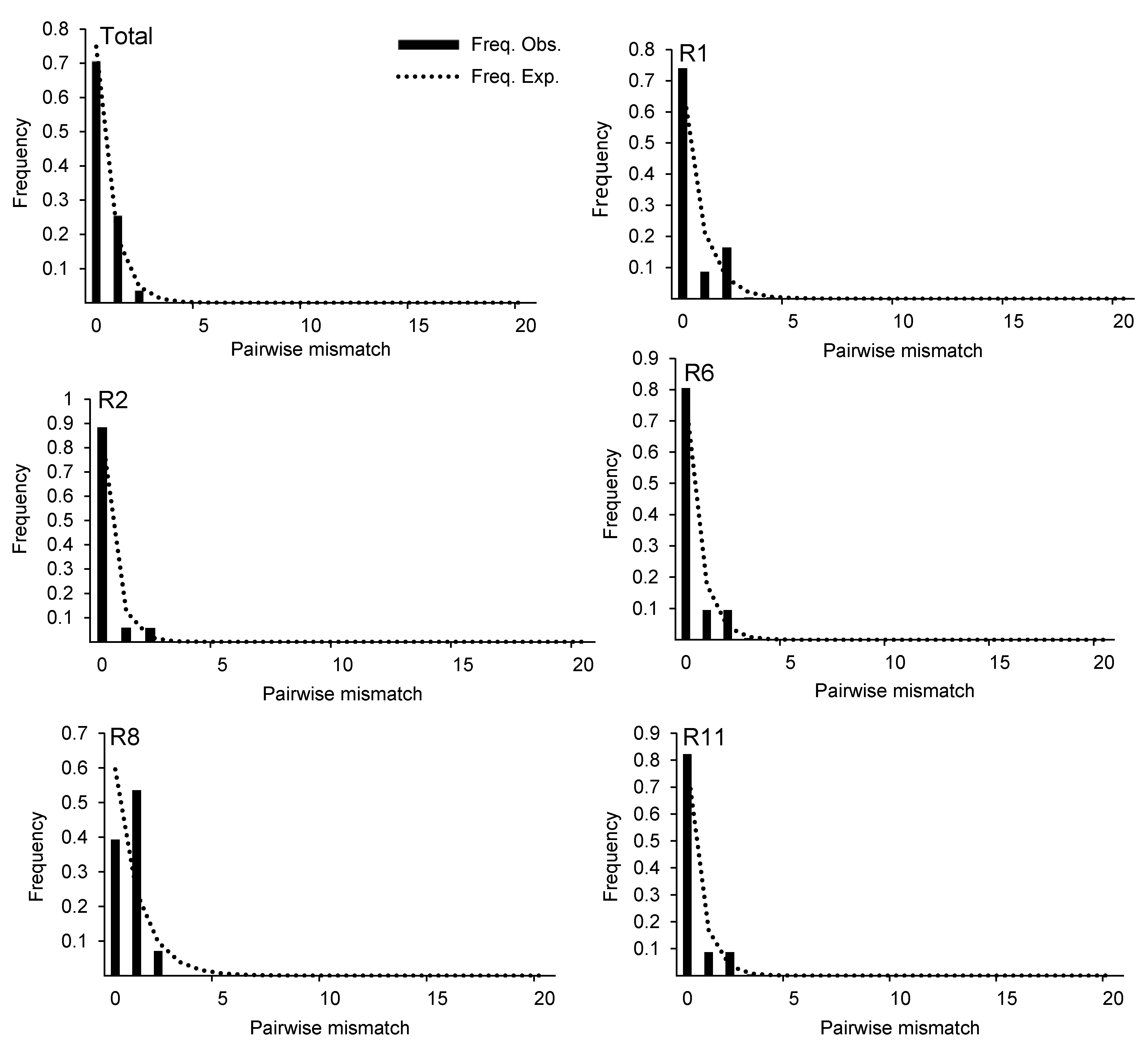
2.4 基于ITS序列的中性检验与失配分析为检验锁阳属整体分布区和不同种群是否发生扩张, 我们基于ITS序列对不同地区锁阳种群分布进行中性检测。结果表明, 整体数据显示Tajima’s D=-1.354 78 (不显著), Fu and Li’s D*和F*的中性检验值分别为0.079 80和-0.518 47 (不显著), 表明锁阳属拒绝中性进化, 群体历经扩张或者基因座位受到负选择作用, 其中性零假说不能被排除。同时对各种群进行中 性检验后发现, 除R1、R6和R11 (0.05<P<0.10)外, 其它种群的Tajima’s D值均不显著。利用DnaSP软件进行失配分析, 结果显示, 总体种群的失配分布曲线出现单峰, R1、R2、R6、R8和R11种群的失配分布曲线出现双峰(图3)。绝大多数种群的失配分布曲线呈现单峰, 但对这些种群进行中性检验, 均不显著。 图3https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-3-313/img_3.png<b>图3</b> 基于ITS序列的锁阳种群失配分析<br/>柱形代表变异位点在群体扩张模型下的预期分布; 虚线代表变异位点的实际分布。<br/><b>Figure 3</b> Mismatch distribution analysis for the populations of <i>Cynomorium songaricum</i> based on ITS sequence<br/>Cylindricality represents the expected distribution of variation sites under the population expansion model; dotted line represents the actual distribution of variation sites. Figure 3https://www.chinbullbotany.com/article/2018/1674-3466/1674-3466-53-3-313/img_3.png<b>图3</b> 基于ITS序列的锁阳种群失配分析<br/>柱形代表变异位点在群体扩张模型下的预期分布; 虚线代表变异位点的实际分布。<br/><b>Figure 3</b> Mismatch distribution analysis for the populations of <i>Cynomorium songaricum</i> based on ITS sequence<br/>Cylindricality represents the expected distribution of variation sites under the population expansion model; dotted line represents the actual distribution of variation sites.
图3 基于ITS序列的锁阳种群失配分析 柱形代表变异位点在群体扩张模型下的预期分布; 虚线代表变异位点的实际分布。 Figure 3 Mismatch distribution analysis for the populations of Cynomorium songaricum based on ITS sequence Cylindricality represents the expected distribution of variation sites under the population expansion model; dotted line represents the actual distribution of variation sites.
BarkmanTJ, McNealJR, LimSH, CoatG, CroomHB, YoungND, DepamphilisCW (2007). Mitochondrial DNA suggests at least 11 origins of parasitism in angiosperms and reveals genomic chimerism in parasitic plants.BMC Evol Biol 7, 248. [本文引用: 1]
[15]
Cibrián-JaramilloA, BaconCD, GarwoodNC, BatemanRM, ThomasMM, RussellS, BaileyCD, HahnWJ, BridgewaterSG, DeSalleR (2009). Population genetics of the understory fishtail palm Chamaedorea ernestiau- gusti in Belize: high genetic connectivity with local differen- tiation.BMC Genet 10, 65. [本文引用: 1]
[16]
CookeDEL, DuncanJM (1997). Phylogenetic analysis of Phytophthora species based on ITS1 and ITS2 sequences of the ribosomal RNA gene repeat.Mycol Res 101, 667-677. [本文引用: 1]
[17]
CosacovA, JohnsonLA, PaiaroV, CocucciAA, CórdobaFE, SérsicAN, CrisciJ (2013). Precipitation rather than temperature influenced the phylogeography of the endemic shrub Anarthrophyllum desideratum in the Patagonian steppe.J Biogeogra 40, 168-182. [本文引用: 1]
[18]
ExcoffierL (2004). Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model.Mol Ecol 13, 853-864. [本文引用: 2]
[19]
ExcoffierL, LavalG, SchneiderS (2007). Arlequin (version 3.0): an integrated software package for population genetics data analysis.Evol Bioinform 1, 47-50. [本文引用: 1]
[20]
ExcoffierL, SmousePE, QuattroJM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data.Genetics 131, 479-491. [本文引用: 1]
[21]
HallTA (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/ 98/NT. In: Information Retrieval Ltd., Nucleic Acids Symposium Series, vol. 41. London: IRL Press. pp. 95-98. [本文引用: 1]
[22]
HouDY, SongJY, ShiLC, MaXC, XinTY, HanJP, XiaoW, SunZY, ChengRY, YaoH (2013a). Stability and accuracy assessment of identification of traditional Chinese materia medica using DNA barcoding: a case study on Flos Lonicerae Japonicae.Biomed Res Int 2013, 549037. [本文引用: 1]
[23]
HouDY, SongJY, YaoH, HanJP, PangXH, ShiLC, WangXC, ChenSL (2013b). Molecular identification of corni fructus and its adulterants by ITS/ITS2 sequences.Chin J Nat Med 11, 121-127. [本文引用: 1]
LahayeR, Van der BankM, BogarinD, WarnerJ, PupulinF, GigotG, MaurinO, DuthoitS, BarracloughTG, SavolainenV (2008). DNA barcoding the floras of biodiversity hotspots.Proc Natl Acad Sci USA 105, 2923-2928. [本文引用: 2]
[26]
LibradoP, RozasJ (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data.Bioinformatics 25, 1451-1452. [本文引用: 1]
[27]
LiuGD, ChenGL, LiW, LiCX (2013). Genetic and phytochemical diversities of Cynomorium songaricum Rupr. in Northwest China indicated by ISSR markers and HPLC- fingerprinting.Biochem Syst Ecol 48, 34-41. [本文引用: 1]
[28]
NewmasterSG, FazekasAJ, SteevesRAD, JanovecJ (2008). Testing candidate plant barcode regions in the My- risticaceae.Mol Ecol Res 8, 480-490. [本文引用: 1]
[29]
NickrentDL, DerJP, AndersonFE (2005). Discovery of the photosynthetic relatives of the “Maltese mushroom” Cynomorium.BMC Evol Biol 5, 38. [本文引用: 1]
[30]
PonsO, PetitRJ (1996). Measwring and testing genetic differentiation with ordered Versus unordered alleles.Genetics 144, 1237-1245. [本文引用: 1]
[31]
RogersAR, HarpendingH (1992). Population growth makes waves in the distribution of pairwise genetic differences.Mol Biol Evol 9, 552-569. [本文引用: 1]
[32]
SlatkinM, HudsonRR (1991). Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations.Genetics 129, 555-562. [本文引用: 1]
[33]
SongJY, ShiLC, LiDZ, SunYZ, NiuYY, ChenZD, LuoHM, PangXH, SunZY, LiuC, LvAP, DengYP, Larson-RabinZ, WilkinsonM, ChenSL (2012). Extensive pyrosequencing reveals frequent intra-genomic variations of internal transcribed spacer regions of nuclear ribosomal DNA.PLoS One 7, e43971. [本文引用: 1]
[34]
SwaffordDL (2002). PAUP*: Phylogenetic Analysis Using Parsimony (* and Other Methods). Version 4.0 10b. Sunderland, MA:Sinauer Associates. [本文引用: 1]
[35]
ZhangZH, LiCQ, LiJH (2009). Phylogenetic placement of Cynomorium in Rosales inferred from sequences of the inverted repeat region of the chloroplast genome.J Syst Evol 47, 297-304. [本文引用: 1]
锁阳愈伤组织体系的建立及遗传多样性研究 1 2011
... 近年来, 国内外****已利用DNA分子标记等方法对锁阳的遗传多样性和系统发育关系进行了探讨.例如, 利用ISSR分子标记技术分析16个居群锁阳的遗传多样性(陈贵林等, 2011); 利用线粒体相关基因、核基因以及叶绿体相关基因等验证锁阳的系统发育地位(Nickrent et al., 2005; Barkman et al., 2007; Zhang et al., 2009).但鲜见利用核基因序列来探讨中国西北地区锁阳属种群谱系地理学的相关报道.本研究对来自18个居群的188个锁阳样本进行ITS序列分析, 揭示锁阳的遗传结构与遗传多样性, 进一步阐明不同地理区域对锁阳系统发育的影响, 以期为中国的锁阳谱系地理学研究提供更为丰富的资料, 同时为锁阳的资源保护和开发利用提供分子证据. ...
 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}