0 引言
【研究意义】三江黄牛原产于四川省阿坝藏族自治州汶川县,其中三江、白石、水磨和映秀等乡镇为主产区,在理县、茂汶等县市均有分布[1];三江黄牛具有躯干较长、役用性能良好、肉质好、抗病力和适应性强等特点,是经长期自然选择和人工选育而成,在遗传资源上是一个极为宝贵的基因库,但由于当地经济社会的发展和农业生产方式的改变,以及2008年汶川特大地震的发生导致三江黄牛产区功能的布局空间受限,使三江黄牛养殖规模、种群数量锐减,已濒临灭绝,因此,保护三江黄牛遗传资源显得尤为重要[2]。【前人研究进展】基因组包含了一个物种全部的遗传信息,全基因测序是解读基因组的核心技术,揭示基因组的多样性和信息的复杂性,最早的测序技术源于20世纪60年代中期[3-5],70年代后期第一代测序体系逐渐建立,主要有SANGER等[6]发明的双脱氧链末端终止法、MAXAM等[7]发明的化学降解法;随着生物技术的发展,二代测序技术逐渐走入大众视野,即大规模平行测序平台(massively parallel DNA sequeneing platform),主要包括:焦磷酸测序Roche/454 FLX、基于边合成边测序的Illumina/Solexa技术和边连接边测序的SOLID技术。测序技术不断发展,有助于对基因组进行更全面和更深入的解析,使得解析稀有物种的基因组,以及对转录组、表达谱、小RNA等大规模的功能基因组学的研究成为可能。人类基因组计划的完成,开启了不同物种全基因组测序的时代。牛基因组测序和HapMap计划的完成[8-9],鉴定出相当数量的遗传变异,其中单核苷酸多态性(SNP)是研究最为广泛的遗传变异类型,用于鉴定基因与牛表型变异相关的基因组区域,现已测序了多个牛种的全基因组[8,10-17]。利用Illumina HiSeq II平台,ECK等[8]在花斑牛上检测到240万SNPs,利用相同的测序平台,KAWAHARA-MIKI等[10]共鉴定了603万SNPs,采用SOLID技术,STOTHARD等[11]成功比对了黑安格斯和荷斯坦公牛的基因组变异,确定了约700万个SNPs和790拷贝数变异。同时,WGS-SNP位点衍生到全基因组关联研究,能够以更高的精度预测物种的重要经济性状,以及检测整个基因组的重要信息[18-19]。【本研究切入点】近年来对黄牛基因组层面的变异研究较多,但对三江黄牛全基因组研究尚未见报道,对三江黄牛品种的遗传资源研究相对匮乏。【拟解决的关键问题】从基因组层面揭示三江黄牛的变异情况,探讨三江黄牛遗传多样性,为进一步分析与经济性状相关的遗传学机制和保护三江黄牛品种遗传多样性提供基因组数据支持。1 材料与方法
1.1 供试材料
样本采集于2015年4月,地点是四川省阿坝州汶川县的三江乡和水磨镇,两乡镇是三江黄牛主要分布区域,选取毛色黄色、体型较大、特征明显的三江黄牛个体,采集其耳组织样50个,75%乙醇保存,带回实验室倒出保存液,-80℃保存备用。1.2 DNA文库的构建及测序
采用苯酚-氯仿法提取基因组DNA,1.5%琼脂糖凝胶电泳和A260/A280的比值检测DNA的纯度和浓度,将50个样本的基因组DNA等浓度等体积混合,利用CovarisS2进行随机打断,电泳回收所需长度的DNA片段(—500 bp),加上接头,进行cluster制备,最后应用Illumina HiSeq 2000测序仪,Paired-end法对插入片段进行测序,双端测序的长度为150 bp,最终得到测序数据。1.3 测序数据的质量控制和数据过滤
为保证数据的质量,测序原始数据要经过质量控制控和数据过滤,在信息分析前对数据进行质控,并通过数据过滤来减少数据的噪音。通过分析碱基的组成和质量值可控制原始数据的质量(图1)。由图1-a可以看出测序得到低质量(Q<20)碱基含量较低,图1-b看出A、T曲线重合,G、C曲线重合,说明碱基组成平衡,测序结果较好,可以进一步对数据进行处理分析。将得到的原始测序序列(raw reads)里有部分带接头或低质量的reads进行过滤,得到高质量的净数据(clean data),后续分析都基于净数据。数据过滤主要是去除带接头的成对reads;去除单端read中N碱基(N表示无法确定碱基信息)比例大于10%的成对reads;当单端测序read中含有低质量(质量值Q≤7)碱基数超过该条read碱基总数的30%时,去除此成对reads。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1三江黄牛数据碱基的组成和质量分布(a)碱基质量值;(b)碱基分布比
-->Fig. 1Composed bases and quality distribution of Sanjiang cattle data(a) The base quality values; (b) The base percentage distribution
-->
1.4 基因组数据组装和测序数据处理
利用BWA[20]软件将序列比对到参考基因组。应用工具包SAMtools、Picard-tools对比对结果进行统计、预处理(排序,去重复等),基因组分析工具包(genome analysis tool kit, GATK)[21]完成SNP/indel检测,即经比对在获得样本所有SNP信息的基础上,将检测到的基因型与参考序列之间存在多态性的位点进行过滤,得到高可信度的SNP/indel数据集,将所得到的SNPs和indels调用为VCF格式,比对到dbSNP数据库,鉴定新的SNPs及indels。Break Dancer[22]工具包分析结构变异(SV),最后应用Reseqtools[23]工具对变异结果进行注释统计作图等。2 结果
2.1 数据产出
2.1.1 净数据 测序共获得三江黄牛基因组77.8G净数据(Clean data),将所得到的Clean data进行统计(表1)。以普通牛基因组(UMD 3.1)(GCA_000003055.3)为参考,使用BWA软件[21]将clean reads比对到参考基因组(表2),测序深度为25.32×,覆盖率达99%以上,说明具有较高的单碱基正确性,比对到参考基因组reads和碱基的比率分别为86.55%和86.51%,说明测序样品同参考物种相似度高,亲缘关系较近。Table 1
表1
表1三江黄牛净数据
Table 1Clean data of Sanjiang cattle
| 样本 Sample | 所有reads All reads | 所有碱基 All bases | GC占总reads长度比 The percentage of GC accounted for the total reads length | 质量值大于20%的碱基比例 Bases quality value over 20% | 质量值大于30%的碱基比例 Bases quality value over 30% |
|---|---|---|---|---|---|
| SH | 778 403 444 | 77 840 344 400 | 42.41% | 96.23% | 93.59% |
新窗口打开
Table 2
表2
表2三江黄牛数据比对到参考基因组
Table 2Sanjiang cattle data aligned to the reference genome
| 样本 Sample | 测序深度 Sequencing depth | 覆盖率 Coverage | 所有比对上的reads All reads map | 成对比对上的reads Paired-end reads map | 所有比对上的碱基 All bases map | 成对比对上的碱基 Paired-end bases map |
|---|---|---|---|---|---|---|
| SH | 25.32× | 99.31% | 673 670 505 | 635 242 898 | 67 341 451 555 | 63 512 636 924 |
| 86.55% | 81.61% | 86.51% | 81.59% |
新窗口打开
2.1.2 染色体测序深度和覆盖度 对三江黄牛每条染色体测序深度和覆盖度统计。整个基因组测序深度为25.32×,深度最高为14号染色体26.55×,最低为X染色体21.54×。基因组的覆盖率为99.22%,其中X染色体覆盖率97.59%。由图2可知,覆盖上的reads和染色体长度成正比。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2reads覆盖各染色体区域的长度
-->Fig. 2Length of the regions that are covered by reads for each chromosome
-->
2.1.3 GC含量 GC含量对测序有一定的影响,高GC和低GC的区域会使测序的难度加大,导致部分序列无法准确测出,由图3可知,整个GC分布范围内覆盖深度较好(25×),GC含量结果无明显偏向性,说明建库与测序质量良好。
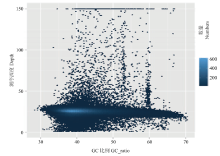 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3GC含量和测序深度
-->Fig. 3GC content and sequencing depth
-->
2.2 SNP检测
GATK的unifiedGenotyper完成对三江黄牛样品的SNP检测,共检测到20 477 130个SNPs位点,SNP密度确定为大约每131个碱基含有一个突变位点,突变分布能够定位各种与经济性状相关联的候选基因组区域。将SNP比对到dbSNP数据库(图4),数据库中共计90 045 399个SNPs,三江黄牛SNPs与数据库未匹配上的为2 147 988个,说明其为新发现的SNPs,占总SNPs的2.4%。纯合SNPs数为989 686(4.83%),杂合SNPs数为19 487 444(95.17%),纯合/杂合SNP比为1:19.7。基因间隔区的SNPs为15 009 500,占总SNPs的73.29%,大多数的SNPs集中在基因间隔区和内含子区,少部分在外显子、剪接位点和非编码区域(表4)。转换TS(transition)/颠换TV(transversion)是检测随机序列误差的重要指标,是对SNP的质量评估,经验值TS/TV>2.1[24],三江黄牛SNP转换数为14 800 438个,颠换为6 680 058个,转换/颠换(TS/TV)为2.215(图4),高于经验值,说明所识别大多数的SNP是准确的。在所有SNP中,由于SNP位点突变导致剪切位点突变和编码氨基酸密码子变化的SNP共1 462个,其中剪切位点突变SNP 727个,开始密码子变为非开始密码子SNP 117个,提前终止密码子SNP 530个,终止密码子变非终止密码子SNP 88个,在染色体上分布情况如图5。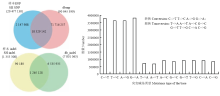 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4SNPs(indels)比对到dbSNP数据库和转换/颠换比
-->Fig. 4SNPs(indels)map to dbSNP database and transition/transversion ratio
-->
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5SNPs位点突变效应在每个染色体上的数量分布
-->Fig. 5Mutagenic effects of SNPs distributed on each chromosome
-->
人类和其他动物许多表型都与非同义SNP(non- synonymous SNP, nsSNP)相关[25],SNP注释是提供SNP位点与功能相关联的依据。本研究共检测到57 621个非同义突变,83 797个同义突变,非同义/同义SNP比率为0.69。ENSEMBL[26]数据库对非同义SNP注释得到9 017个基因(电子附表1,http:// pan.baidu.com/s/1qXN18dA)。DAVID[27]数据库对含nsSNP较多的108个基因进行功能基因富集分析(电子附表2,http://pan.baidu.com/s/1qXN18dA),基因功能注释可分为7类,主要集中在生化、代谢、免疫等过程(电子附表3,http://pan.baidu.com/s/1qXN18dA),其中免疫功能相关的GO:0006955涉及到免疫应答,GO:0019882涉及到抗原加工和呈递,对机体有重要作用,包括ULBP3、JSP.1、BOLA-DRB3等基因。同时还分析了nsSNP与肉质、产奶量、生长速度等重要经济性状的相关性,发现567个基因与已报道的重要经济性状相关[10,28-34],并对其基因进行注释(电子附表4,http://pan.baidu.com/s/1qXN18dA),471个与肉质相关基因,77个与抗病相关的基因,21个与产奶性状相关基因,10个与生长性状相关的基因,8个与生殖相关的基因,567个基因中还包括一些功能相重叠的基因,例如同时和肉质、生长性状相关的POMGNT2、SLC25A38、ADIPOR2、ACAD8基因,与肉质和抗病均相关的IL6R、GHRHR、NFKB1、LBP、ACAD8、SREBF1、CRHR1、PRLR、SEMA5A、IGF1R、FEZF2、MUT、FAS基因,产奶和抗病均相关的TMEM186、LBP、SEMA5、AFEZF2基因等。还有一些研究相对较多且机制较为清晰的基因,包括生长激素(GH),生长激素受体(GHR)和瘦肉素受体(LEPR)催乳素受体(PRLR)基因[29,32]等。
Table 3
表3
表3SNPs和indels的注释
Table 3SNPs and indels annotate
| 单核苷酸多态性 SNP | 数量 Number | 插入缺失标记 indel | 数量 Number | |
|---|---|---|---|---|
| 超级重叠群 Super-contig | 103943 | 超级重叠群 Super-contig | 3773 | |
| 假基因加工 Pro- pseudo-gene | 929 | 假基因加工 Pro-pseudo-gene | 18 | |
| 转录本 Transcript | 5404410 | 转录本 Transcript | 371744 | |
| 核糖核酸 RNA | 417 | 核糖核酸 RNA | 16 | |
| 假基因 Pseudo-gene | 9616 | 假基因 Pseudo-gene | 482 | |
| CDS区 CDS | 141412 | CDS区 CDS | 1545 | |
| 小核RNA snRNA | 2518 | 小核RNA snRNA | 110 | |
| 核糖体RNA rRNA | 822 | 核糖体RNA rRNA | 42 | |
| 核仁小RNA snoRNA | 1132 | 核仁小RNA snoRNA | 46 | |
| 3’ UTR Three prime UTR | 38925 | 3’ UTR Three prime UTR | 3606 | |
| 外显子 Exon | 166818 | 外显子 Exon | 4137 | |
| 5 ’UTR Five prime UTR | 4698 | 5’ UTR Five prime UTR | 240 | |
| MicroRNA miRNA | 978 | MicroRNA miRNA | 112 | |
| Mt基因 Mt gene | 12 | 内含子剪接 Intron splice | 368 | |
| 基因间隔区 NA(intergenic) | 15009500 | 基因间隔区 NA(intergenic) | 982443 |
新窗口打开
2.3 indel检测
最近研究中认为高于50 bp的缺失(deletion)和插入(insertion)为结构变异,而低于50 bp的deletion和insertion合称indel[35]。indel是基因组中除SNP数量最多的变异,三江黄牛共检测到1 355 308个indels,缺失和插入数量分别为693 180和662 148个,比例分别为51.15%、48.85%,比对到数据库共发现90 180个新indels,占总indels的6.7%(图4)。纯合indels为161 198(11.89%),杂合indels为1 194 110(88.11%)。indels注释情况(表3),基因间隔区的indels最多为982 443个,占总indels的72.49%。CDS区indels1 545个,外显子indels 4 137个,3′端非编码区和5′端非编码区indels数分别为3 606和240。SNPs和indels在每个染色体上数量分布(图6),除11、12、13号染色体上的SNP外,其余每条染色体上的SNP数均与染色体长度相关,随染色体长度的减小而降低。indels在每条染色体上的长度分布随染色体长度减小而降低。插入和缺失在CDS区和全基因组上的分布情况(图7),由图可知,缺失和插入的数量在全基因组上的分布随长度增加而减少,在CDS区未发现类似情况。但由两图都可看出插入和缺失数量集中在1—10 bp,其中1—3 bp最多。基于成对比对上reads的结果,检查插入的长度是否异常,针对缺失部分进行分析,共检测出1 906个结构变异。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图6SNPs和indels在每个染色体上数量分布
-->Fig. 6The number of SNPs and indels on each chromosome
-->
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图7Indels在全基因组上的长度分布
-->Fig. 7The size of indels distributed over the whole genome
-->
2.4 基因组的杂合度和群体核苷酸多样性指数
杂合度(H)和核苷酸多样性(Pi)是反映多态性高低的指标(图8)。将reads比对到参考基因组,识别三江黄牛19 487 444个杂合SNPs,其整个基因组的杂合度为7.6×10-3,说明三江黄牛品种的遗传多样性较高。三江黄牛全基因组Pi为0.0039,说明遗传多样性较为丰富。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图8indels在CDS上的长度分布
-->Fig. 8The size of indels distributed on CDS
-->
2.5 基因组Tajima'D和theta W指数
群体Tajima'D值是目标DNA序列在进化过程中是否遵循中性进化模型,导致D值为负可能是搭载效应[36]。三江黄牛群体Tajima'D为-0.06832(图9),说明群体中存在不平衡的选择。theta W是反映群体多态性的指标,是群体在核苷酸多态性水平上偏离中性进化且处于突变-漂移平衡的理想模型,三江黄牛基因组theta W为0.0040(图9),说明三江黄牛群体遗传多样性较为丰富。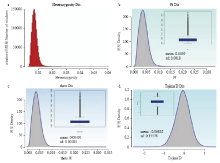 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图9(a)全基因组的杂合率H;(b)全基因组的多态性指标Pi;(c)全基因组的多态性指标theta W;(d)全基因组的Tajima'D
-->Fig. 9(a) Genome-wide heterozygosity rate H; (b) Polymorphic parameters Pi of the whole genome; (c) Polymorphism index theta W of the whole genome; (d) Genome-wide Tajima'D index
-->
3 讨论
在研究中,笔者使用Illumina 2000测序平台对濒危三江黄牛品种进行了全基因组测序,三江黄牛品种数量少,选择能够代表品种的个体进行测序尤为重要,为避免个体差异,在较低成本下聚集更多个体的信息来反映三江黄牛品种群体遗传多样性情况,因此将50个体采用混合DNA样本进行测序。测序获得三江黄牛基因组77.8G净数据,86.55%的reads、81.61%的成对reads、86.51%的碱基、81.59%的成对碱基比对到参考基因组,测序深度为25.32×,覆盖率达99%以上,具有较高的单碱基正确性,与先前对普通牛测序的结果相比[8,11],测序深度较高,检测到的变异充分可信[15]。通过GATK分析,在29条常染色体和X染色体上共发现20 477 130个SNPs位点和1 355 308个indels,三江黄牛种群数量较少,仅2 000多头,SNP数量较多,说明该品种遗传多样性较为丰富。总SNP中,杂合SNPs 19 487 444个(95.17%),纯合SNPs 989 686个(4.83%),与SHIN等[37]测序10个韩国公牛所得到2 234 514个(90.3%)杂合SNPs和239 370个(9.7%)的纯合SNPs结果相似。纯合/杂合SNPs的比为1:19.7,明显低于KAWAHARA-MIK等[10]研究的日本牛(1:1.2)和CHOI等[12]研究的韩牛(1:1.92)。从测序角度阐述纯和SNP是该混和样本中的所有样品在这个位点都是同一个碱基型且和参考基因组一致,杂合SNP是这个位点在所有混和样品里有多个碱基型,三江黄牛SNP纯合比率低,杂合度高,推测可能测序的50个体之间变异差异较大。还可能是三江黄牛选育程度低,近亲繁殖的概率低,与其他牛之间的基因交流较多,本身特异的功能基因正在丢失,保护三江黄牛品种显得尤为重要。测序所得indels大约占总变异(indels和SNPs)的5.3%,稍高于KAWAHARA-MIK等[10]和CHOI等[12]研究的结果。三江黄牛转换/颠换值为2.21,高于CHOI等[16]研究韩国牛牛种所得到2.1。将SNPs和indels比对到数据库发现2 147 988个新的SNPs和90 180个新的indels,分别占总SNPs的2.4%和总indels的6.7%,推测可能由于近年来随着基因组测序的发展,发现了较多新的SNPs及indels,数据库越来越完善,使比对上的比例明显增大,新发现的逐渐变少。大多数indels的长度较短,缺失的长度范围在1—29 kb,插入的范围在1—44 kb,缺失和插入的数量集中在1—10 bp,其中1—3 bp最多,从人类基因组数据上也观察到类似现象[38]。三江黄牛数据中,接近84.7%插入和79.6%缺失长度小于3 kb。29个常染色体上检测到的SNPs和indels与染色体长度成正比,结果符合预期,其中X染色体突变率最低,为4.33%,在小种群研究上,X染色体相比常染色体有较低的突变率[39]。
通过Ensemble数据库对NSSNP注释得到9 017个基因,与Eck等[8]报道的结果相一致,高于KAWAHARA-MIK等[10]研究的日本牛和LEE等[14]研究的韩国牛。非同义SNP注释发现567个与经济性状相关的基因,其中肉质、抗病、产奶、生长、生殖等相关的基因分别为471、77、21、10、8个。三江黄牛主要是供耕地役用,近年来逐渐向役肉兼用方向发展,研究中一些肉质相关基因中在其他黄牛品种上已有报道,例如脂肪酸结合蛋白4(FABP4)的突变与棕榈油酸肌肉内脂肪含量相关[40],加压素Ⅱ受体(UTS2R)突变与骨骼肌脂肪堆积相关[41],钙蛋白酶1(CAPN1)突变与阿伯丁安格斯牛肉嫩度有关[42],LEP和LEPR基因被发现与内洛尔、荷斯坦黑白花、安格斯、夏洛来、海福特、西门塔尔牛牛肉的脂肪含量有关[34,43],肌联蛋白基因(TNN)也被发现是影响肉质重要的候选基因[44],在本研究中TNN、ANKRD26、VCAN、NEB、COL4A3、TICAM1、ANKRD26、MYH4、NOTCH4等肉质相关基因上分布了较多nsSNP(>10)。还有一些与发育和疾病相关的基因,如NOD2和CARD15基因与荷斯坦牛育种值相关[45],蛋白激酶(PRKCD)基因与早期胚胎发育相关[46],Y连锁肽重复序列结构域(UTY)在公牛精子发生过程中发挥关键作用[47],性别决定区Y(SRY)是检测公牛精子质量和生育能力的重要候选标记[48]。PNPLA8、NRCAM和CTTNBP2是与瑞士褐牛韦弗综合征疾病相关的重要候选基因[49]。哺乳动物中的色素沉淀是由于毛发或皮肤缺乏或存在黑色素引起的,影响色素合成的主要基因有酪氨酸酶蛋白1(TYRP1)、多巴色素互变异构酶(DCT)、丝氨酸肽酶(CORIN)、黑皮素受体1(MC1R)、酪氨酸酶(TYP)、信号蛋白(ASIP)。在啮齿动物,黑色和黄色间的变化是由MC1R和拮抗剂ASIP所引起的。MITF基因能够调节TYP,TYRP1和DCT基因的表达。三江黄牛毛色以黄色为主,其次为黑色和草黑色,在其他牛种上很多与颜色相关的基因在三江黄牛上也发现了,如CORIN基因发现与韩牛黄毛色有关[37],推测可能也是影响三江黄牛黄色毛的重要基因。TYRP1、DCT、MLPH、KIT、KITLGD等基因发现能够导致头发毛囊表型变化[50]。
4 结论
本研究通过对三江黄牛全基因组测序得到77.8 Gb净数据,鉴定出大量的遗传变异,说明三江黄牛遗传多样性较为丰富,为进一步研究三江黄牛品种的遗传特性提供基因组数据支持。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}