0 引言
【研究意义】弧菌属(Vibrio)为革兰氏阴性菌,嗜盐,广泛存在于海水中,是引起鱼、虾等水产品疾病的重要病原菌[1-2]。其中,副溶血弧菌、溶藻弧菌、霍乱弧菌和创伤弧菌同时也是重要的食源性病原弧菌[3-4],对人类具有较强的致病性,例如霍乱弧菌能引起人腹泻、呕吐等病症,曾经在世界上多次爆发;溶藻弧菌也早在1979年就有报道称其对人类的致病性,能够引起中耳炎等疾病[5],而这些弧菌常常存在于人类食用的海产品中,对海产品中这些弧菌的检测、监测具有重要意义。【前人研究进展】弧菌的检测主要包括生理生化鉴定、PCR法等,生理生化鉴定方法周期长,不能满足快速检测的要求;而以PCR为基础的方法由于其高特异性、高灵敏度、快速等特点得到了广泛的应用。近年来,国内外用于弧菌快速检测的方法有PCR[6-7]、巢氏PCR[8-9]、多重PCR[10-11]、荧光定量PCR[12-13]、多重荧光定量PCR[14-15]等。多重PCR具有高通量的特点,但由于引物之间的竞争,其检测通量受到了限制,而且结果需要进一步凝胶电泳分析,费时费力;巢氏PCR拥有高灵敏度的特点,但在其检测通量方面无法与多重PCR相比;荧光定量PCR因无需凝胶电泳,得到了最为广泛的应用,但多重荧光定量PCR由于引物探针之间的竞争和仪器荧光检测通道的限制,难以实现高通量检测。因此,需要建立一种基于荧光定量PCR的高通量、高灵敏、具备定量能力的检测方法。【本研究切入点】本研究以4种重要食源性病原弧菌(副溶血弧菌、霍乱弧菌、创伤弧菌、溶藻弧菌)为靶目标,提出一种多重富集定量PCR(multiplex enrichment quantitative PCR, ME-qPCR)方法,改变现有方法存在的缺陷,真正实现高通量、高灵敏度、定量检测。【拟解决的关键问题】本研究拟针对常见的4种食源性病原弧菌——副溶血弧菌的collagenase、溶藻弧菌的gyrB、霍乱弧菌的ompW和创伤弧菌的vvhA,分别设计特异性内、外引物,并针对细菌16S rRNA保守区设计通用引物作为扩增内标,建立同时能检测4种食源性病原弧菌的ME-qPCR体系,同时能够指示PCR反应的假阴性,结果无需凝胶电泳,真正达到定量、高通量、快速、准确的检测要求。1 材料与方法
试验于2015年9月至2016年3月在汕头出入境检验检疫局检验检疫技术中心进行。1.1 材料
河流弧菌、拟态弧菌、鳗弧菌、哈维氏弧菌、梅氏弧菌保存于汕头出入境检验检疫局检验检疫技术中心动物检疫实验室。副溶血弧菌(ATCC17802)、溶藻弧菌(ATCC17749)、霍乱弧菌(ATCC14035)、创伤弧菌(ATCC27562)、大肠杆菌(ATCC8739)、单增李斯特菌(ATCC19111)、金黄色葡萄球菌(ATCC43300)、枯草芽孢杆菌(ATCC6633)、伤寒沙门氏菌(ATCC14028)保存于暨南大学食品科学与工程系。1.2 细菌培养及基因组DNA的提取
将弧菌单菌落接种于含3% NaCl的胰蛋白胨大豆肉汤(tryptic soy broth,TSB)培养基中,将非弧菌单菌落接种于脑心浸液(brian heart infusion,BHI)液体培养基中,37℃下振荡培养过夜。采用试剂盒法(TIANamp Bacteria DNA Kit,TIANGEN)提取细菌基因组DNA,提取后取1 μL用微量紫外分光光度计(ND-1000,NanoDrop)测定提取的基因组DNA的浓度和纯度,基因组DNA保存于-20℃待用。1.3 引物的设计与合成
根据副溶血弧菌collagenase(GenBank ID:AF326572.1)、溶藻弧菌gyrB(GenBank ID:AF007288)、霍乱弧菌ompW(GenBank ID:X51948.1)和创伤弧菌vvhA(GenBank ID:AF376027.1)序列,以及细菌16S rRNA的保守序列,利用Primer Premier V5.0和Oligo V6.22软件进行引物的设计和筛选,并保证内、外引物产物大小符合荧光定量PCR的要求(小于200 bp)。在参考了大量文献已报道的引物基础上筛选出特异性外引物,在外引物产物片段序列内设计内引物,副溶血弧菌、溶藻弧菌和创伤弧菌的内引物设计较困难,故保留外引物中特异性较强的那一条,仅设计一条内引物,形成3对半巢式引物组,16S rRNA基因作为IAC的靶基因引物参考了已报道的文献,但内引物设计细菌通用引物难度较大,内外引物均用同一对引物。上述所有 引物由生工生物工程(上海)股份有限公司合成 (表1)。Table 1
表1
表1各基因的内外引物序列
Table 1Outer and inner primers sequence of each gene
| 目标 Target | 基因 Gene | 引物 Primers | 正向引物(5′-3′) Forward primer | 反向引物(5′-3′) Reverse primer | 产物大小 Product size (bp) |
|---|---|---|---|---|---|
| 创伤弧菌 V. vulnificus | vvhA | 外引物[16] Outer primers | TTCCAACTTCAAACCGAACTATGA | TGAGAAGAGTGCTGAAGGGATTA | 136 |
| 内引物[16] Inner primers | GTAACGGATTTCGAGATGGG | TGAGAAGAGTGCTGAAGGGATTA | 78 | ||
| 副溶血弧菌 V. parahaemolyticus | collagenase | 外引物[17] Outer primers | ACGACCACAAACAGCAACGACT | TGGTCAGAATCAAACGCCG | 116 |
| 内引物[17] Inner primers | AAGTTGCATCAACGAGCTGTTC | TGGTCAGAATCAAACGCCG | 66 | ||
| 溶藻弧菌 V. alginolyticus | gyrB | 外引物[18] Outer primers | GCCGTAACCGTAAGAACCAAGC | CCTAGTGCGGTGATCAGTGTTG | 136 |
| 内引物[18] Inner primers | GAAAAAGCACGTTTCGACAAGA | CCTAGTGCGGTGATCAGTGTTG | 63 | ||
| 霍乱弧菌 V. cholerae | ompW | 外引物[19] Outer primers | GCTAATTCGACTTTCCGTCCATA | CGTTAGCAGCAAGTCCCCAT | 139 |
| 内引物 Inner primers | TATGTTGGTGCGGGTTTGA | CCCATGAGTCGTCCAGTTTT | 103 | ||
| 扩增内标 IAC | 16S rRNA | 内外引物[20] Outer and inner primers | CCTGGTAGTCCACGCCGTAA | CGAATTAAACCACATGCTCCA | 168 |
新窗口打开
1.4 ME-qPCR体系
多重富集定量PCR(Multiplex multiplex enrichment quantitative PCR,ME-qPCR)方法针对每一个靶基因序列均设计内、外两对引物,内外引物产物大小符合基于SYBR Green I的荧光定量PCR的要求。首先,将所有内、外引物全部混合,进行一个循环数较少(10—20 cycles)的高通量多重富集PCR,然后将PCR产物用ddH2O稀释作为模板,运用内引物进行第二轮巢式荧光定量PCR,结果根据扩增曲线和熔融曲线分析。在第一轮高通量多重PCR中,每个靶基因均有4种可能的产物类型,这4种产物类型均能作为第二轮巢式荧光定量PCR的模板,这意味着4条引物对应的4种组合只要有1种能够成功扩增,整个反应就能进行,这相当于将靶基因从模板中富集(enrichment)出来,这增加了本方法的成功率和稳定性。同时,由于第一轮循环数较少,引物之间的竞争较弱,各个基因均能得到均匀扩增(图1)。具体操作步骤如下:第一轮多重富集PCR采用多重PCR反应试剂盒(Multiplex PCR Assay Kit,TaKaRa),反应体系为:Mix2溶液25 μL,Mix1溶液0.25 μL,各基因的内、外引物混合物(各个引物终浓度均为0.1 μmol·L-1)和DNA模板各2 μL,用ddH2O调整体积至50 μL。反应条件为:94℃ 1 min;94℃ 30 s,60℃ 60 s,72℃ 60 s,10—20个循环;72℃ 5 min(Mastercycler ep,Eppendorf)。
将第一轮多重富集PCR产物用ddH2O稀释100倍作为第二轮巢式荧光定量PCR的模板,使用SYBR荧光定量PCR反应试剂盒(SYBR Premix Ex Taq,TaKaRa),每孔体系为:SYBR溶液12.5 μL、第一轮PCR产物稀释物1 μL、各基因的内引物(引物终浓度均为0.4 μmol·L-1),用ddH2O调整体积至25 μL。反应条件为95℃ 30 s;95℃ 5 s,60℃ 10 s,72℃ 10 s,35个循环,每个循环在72℃结束后检测荧光信号;反应结束后进行熔融曲线分析:65—95℃,每0.5℃扫描荧光一次(CFX96,Bio-rad)。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1ME-qPCR的原理
-->Fig. 1The principle of ME-qPCR
-->
1.5 ME-qPCR体系特异性的评价
利用1.1中的14株菌株,按照1.2的的方法提取基因组DNA,并按照1.4步骤进行检测,对结果进行分析,评价ME-qPCR体系的特异性。1.6 ME-qPCR体系的定量能力和灵敏度评价
按照1.2的方法分别提取溶藻弧菌、副溶血弧菌、霍乱弧菌和创伤弧菌的基因组DNA,采用10倍梯度稀释,分别制备成浓度分别为100、10、1、0.1、0.01和0.001 ng·μL-1的样品,对上述样品分别采用ME-qPCR和普通荧光定量PCR进行检测,普通荧光定量PCR直接采用内引物组,对比两种方法的定量能力和灵敏度,评价ME-qPCR的定量能力和灵敏度。1.7 ME-qPCR体系检测实际样品
采用本课题组前期分离的69株疑似弧菌菌株[20],利用已建立的ME-qPCR体系对上述弧菌菌落进行鉴定,同时对这些疑似菌落进行生理生化鉴定,对比两种方法。2 结果
2.1 ME-qPCR第一轮反应循环数的优化
第一轮多重富集PCR的循环数是ME-qPCR的关键参数。将4种弧菌的基因组DNA混合作为模板,进行第一轮多重富集循环数分别为10、15和20的ME-qPCR,并直接运用内引物对上述模板进行普通荧光定量PCR,对比在不同第一轮反应循环数下ME-qPCR与普通荧光定量PCR的差别。结果如表2所示,当第一轮反应循环数为10时,其Ct值大于普通荧光定量PCR,说明其灵敏度低于普通荧光定量PCR;当第一轮反应循环数为15和20时,其Ct值均小于普通荧光定量PCR。但理论上第一轮反应循环数越小,引物之间的竞争越少,PCR反应偏好性越弱。综合考虑,确定第一轮反应循环数为15。Table 2
表2
表2ME-qPCR第一轮循环数的优化
Table 2The optimization of first round reaction cycle number of ME-qPCR
| 目标菌 Target bacteria | Ct值 Ct values | |||
|---|---|---|---|---|
| ME-qPCR (10 cycles) | ME-qPCR (15 cycles) | ME-qPCR (20 cycles) | RT-qPCR | |
| 溶藻弧菌 V. alginolyticus | 21.35±0.27 | 16.05±0.13 | 11.94±0.23 | 20.10±0.36 |
| 副溶血弧菌 V. parahaemolyticus | 20.43±0.08 | 15.14±0.04 | 10.43±0.07 | 19.00±0.04 |
| 创伤弧菌 V. vulnificus | 22.12±0.19 | 16.54±0.08 | 11.57±0.36 | 21.75±0.34 |
| 霍乱弧菌 V. cholerae | 22.76±0.02 | 15.51±0.28 | 12.02±0.74 | 21.68±0.16 |
| 扩增内标 IAC | 18.47±0.13 | 13.74±0.15 | 9.01±0.12 | 17.46±0.23 |
新窗口打开
2.2 ME-qPCR体系的建立
在确定了ME-qPCR第一轮反应的循环数后,混合溶藻弧菌、副溶血弧菌、创伤弧菌和霍乱弧菌的基因组DNA为模板,建立了ME-qPCR体系用于4种食源性病原弧菌的检测,同时在体系中添加IAC能指示PCR反应的假阴性。结果如图2所示,5个基因均得到扩增,每个基因3个复孔的重复性好,熔融曲线结果表明产物单一,没有非特异性扩增产物的出现。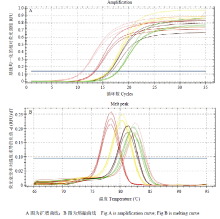 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2ME-qPCR同时检测4种食源性病原弧菌
-->Fig. 2The detection of four Vibiro spp. by ME-qPCR simultaneously
-->
2.3 ME-qPCR体系特异性评价
利用14株菌对ME-qPCR体系的特异性进行了评价。结果如图3所示,14种菌的IAC均表现为阳性扩增,说明反应不存在假阴性,4种目标菌顺利检出,其余非目标菌均为阴性结果,表明建立的ME-qPCR体系特异性强。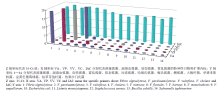 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3ME-qPCR体系特异性评价
-->Fig. 3The specificity test of ME-qPCR
-->
2.4 ME-qPCR体系定量能力及灵敏度的评价
混合4种食源性病原弧菌的基因组DNA并稀释成一系列浓度梯度,同时进行ME-qPCR和普通荧光定量PCR,对比两种方法的定量能力和灵敏度。结果如表3所示,ME-qPCR的灵敏度高于普通荧光定量PCR约1个数量级,2种方法的Ct值线性关系好,R2均大于0.99,PCR扩增效率符合荧光定量PCR的要求,这说明ME-qPCR拥有和普通荧光定量PCR相同的定量能力。Table 3
表3
表3多重富集定量PCR(ME-qPCR)与实时定量PCR(RT-qPCR)灵敏度及定量能力对比
Table 3Sensitivity and quantitation between ME-qPCR and RT-qPCR
| 模板 Template | 溶藻弧菌 V. alginolyticus | 副溶血弧菌 V. parahaemolyticus | 霍乱弧菌 V. cholerae | 创伤弧菌 V. vulnificus | ||||
|---|---|---|---|---|---|---|---|---|
| ME-qPCR | RT-qPCR | ME-qPCR | RT-qPCR | ME-qPCR | RT-qPCR | ME-qPCR | RT-qPCR | |
| 100 ng | 13.56±0.06 | 19.67±0.04 | 14.27±0.03 | 19.41±0.15 | 14.19±0.09 | 18.86±0.06 | 14.15±0.10 | 18.87±0.05 |
| 10 ng | 17.13±0.05 | 23.03±0.08 | 17.39±0.04 | 22.35±0.11 | 17.28±0.16 | 22.21±0.04 | 17.24±0.19 | 21.97±0.14 |
| 1 ng | 20.15±0.03 | 26.42±0.04 | 20.54±0.06 | 25.58±0.05 | 20.62±0.25 | 25.81±0.21 | 20.75±0.25 | 25.63±0.30 |
| 0.1 ng | 23.39±0.04 | 30.33±0.12 | 24.34±0.08 | 29.25±0.08 | 23.89±0.16 | 29.42±0.19 | 23.85±0.21 | 28.69±0.11 |
| 0.01 ng | 27.22±0.19 | 33.76±0.18 | 27.58±0.21 | 33.58±0.15 | 27.58±0.39 | 33.11±0.28 | 27.57±0.09 | 32.88±0.27 |
| 0.001 ng | 30.29±0.21 | — | 30.79±0.31 | — | 31.29±0.23 | — | 31.18±0.11 | — |
| R2 | 0.9991 | 0.9993 | 0.9993 | 0.994 | 0.9988 | 0.9997 | 0.999 | 0.9973 |
| 扩增效率 Amplification efficiency | 99% | 91% | 99% | 92% | 96% | 91% | 97% | 94% |
新窗口打开
2.5 ME-qPCR体系检测实际样品
利用建立好的ME-qPCR体系对课题组前期分离的69个疑似弧菌菌落进行PCR鉴定,结果与前期的API生理生化试验结果一致[20]。3 讨论
引物之间的竞争一直是以PCR为基础的高通量检测技术的瓶颈,传统多重PCR通过设计引物时降低理论上引物之间形成二级结构的可能性来提高多重PCR的成功率,而且同时要考虑不同靶基因的产物片段大小,这两方面加在一起大大增加了引物的设计难度,因此有****开发了多重PCR引物设计系统[21],这一定程度上提高了构建多重PCR体系的成功率。另外,还有****将多重PCR与毛细管电泳技术[22],或者改进引物的设计方法,例如DPO(dual priming oligonucleotide)引物[23]、通用引物多重PCR(universal primer multiplex PCR,UP-M-PCR)[24]等,从而提高了多重PCR的检测通量。但多重PCR的检测通量依然受到多方面限制,而且多重PCR需要凝胶电泳或毛细管电泳分析,增加了检测所需的时间。本研究建立的多重富集定量PCR(multiplex enrichment quantitative PCR,ME-qPCR)体系分2步反应,首先进行一轮高通量的多重富集PCR,然后将产物稀释作为模板分别进行第二轮荧光定量PCR。由于第一轮反应的循环数较少,引物之间的竞争减少,各个基因能得到均匀扩增,这与多重串联式PCR(multiplex tandem PCR,MT-PCR)的原理一致[25]。另外,将内、外引物同时添加进第一轮反应中,会产生4种可能的产物,这4种可能的产物均能够作为第二轮巢式荧光定量PCR的模板,这相当于增加了模板被富集出来的概率,从而提高了反应的成功率,这与扩增子拯救多重PCR(amplicon rescue multiplex PCR,Arm-PCR)的原理一致[26]。本研究建立的ME-qPCR体系既提高了检测的灵敏度,又能保证整个体系的定量能力,同时提高了检测通量,结果无需凝胶电泳,适合实验室对这4种常见食源性病原菌的检测。灵敏度是基于PCR检测方法的一个重要方面,荧光定量PCR的灵敏度一般高于普通PCR,而巢氏荧光定量PCR灵敏度又高于荧光定量PCR。COSTA等[27]建立了一种单管巢氏荧光定量PCR,高于普通荧光定量PCR法1个数量级。本研究建立的ME-qPCR方法,灵敏度高达0.001 ng,同样高于普通荧光定量PCR法1个数量级,这是由于ME-qPCR的第一轮进行了一次高通量多重富集PCR,然后将产物作为第二轮巢氏荧光定量PCR,这与巢氏PCR的效果相当,因而提高了该方法的灵敏度。
在实际检测过程中,样品中存在PCR抑制因子,或者仪器的故障等因素,都会导致PCR结果假阴性的出现[28],影响结果的准确性。目前,在PCR检测体系中添加扩增内标(internal amplification control,IAC)是指示是否存在PCR反应假阴性的重要方法[29]。NORDSTROM等[30]针对副溶血弧菌tdh、trh和tlh这3类溶血素基因,建立了含扩增内标的多重荧光定量PCR,在检测的同时能指示PCR反应的假阴性,提高了检测的准确性。魏霜等[20]针对溶藻弧菌、副溶血弧菌、创伤弧菌和霍乱弧菌,建立了添加扩增内标的多重PCR。本研究建立的ME-qPCR体系内添加了以16S rRNA为靶基因的扩增内标,能有效指示PCR反应的假阴性,提高了检测的准确性。
4 结论
本研究建立了一套多重富集定量PCR用于同时检测溶藻弧菌、副溶血弧菌、创伤弧菌和霍乱弧菌,并能有效指示PCR反应的假阴性,该方法特异性强,比普通荧光定量PCR灵敏度高1个数量级,而且拥有与普通荧光定量PCR相同的定量能力,结果无需凝胶电泳分析,适用于食品中4种常见病原弧菌的快速筛检。由于该方法同时兼顾了高通量、高灵敏度、定量等方面的优势,为某些痕量样品的分析如石蜡包埋样品各基因表达量的分析、深加工食品转基因成分检测等提供了一种新方法;同时,由于本方法是基于染料法的荧光定量PCR,也可将该方法与高分辨率溶解曲线(high resolution melting,HRM)技术相结合,用于SNP位点的检测,适合于某些大量样品、多位点的分析。(责任编辑 赵伶俐)
The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}