 ,1, 江东,2, 李永平3, 彭磊,4, 李冬云5, 朱延松2, 杨云光1
,1, 江东,2, 李永平3, 彭磊,4, 李冬云5, 朱延松2, 杨云光1Identification of Bud Sport Mutation of Satsuma Mandarin by Target SSR-seq Technology
HU DongMei,1, JIANG Dong,2, LI YongPing3, PENG Lei,4, LI DongYun5, ZHU YanSong2, YANG YunGuang1通讯作者:
责任编辑: 赵伶俐
收稿日期:2021-02-22接受日期:2021-05-20
| 基金资助: |
Received:2021-02-22Accepted:2021-05-20
作者简介 About authors
胡冬梅,Tel:15008875127;E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1628KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
胡冬梅, 江东, 李永平, 彭磊, 李冬云, 朱延松, 杨云光. 利用Target SSR-seq技术鉴定温州蜜柑芽变材料. 中国农业科学, 2021, 54(23): 5083-5096 doi:10.3864/j.issn.0578-1752.2021.23.013
HU DongMei, JIANG Dong, LI YongPing, PENG Lei, LI DongYun, ZHU YanSong, YANG YunGuang.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】柑橘极易发生芽变,从芽变和实生选种中获得的柑橘新品种占所有品种的比例高达77%[1,2],尤其是一些无核柑橘品种,如冰糖橙、温州蜜柑等多数来源于芽变。通常,根据枝、叶、果等植物形态学特征就能够对芽变材料予以区分,但环境、气候、栽培条件的改变会造成植物形态学特征的较大差异,很难通过形态特征就将一些品种区分开[3],所以仅靠形态标记很难准确用于亲子关系的区分和鉴定,因此利用分子标记开展品种辨别已成为资源鉴定、品种选育和品种权保护的一项重要工作,其关键又在于能够发掘出区分芽变材料差异的分子标记。【前人研究进展】目前,许多分子标记技术如RAPD[4,5,6]、ISSR[7]、SRAP[8]等都被用于芽变材料的区分和柑橘遗传背景的研究。随着深度测序技术的发展,芽变材料的基因组重测序结果表明,芽变材料间存在大量的重复序列变异[9],这为芽变材料的分子标记开发提供了重要方向。实际上,在柑橘基因组上广泛分布着简单重复序列SSR,又称作微卫星DNA,它是以1—6个核苷酸为重复单元构成的串联重复,在不同个体或群体之间由于串联数目的不同而呈现出高度的多态性和变异性。由于SSR在染色体上分布广泛,基因组覆盖率较高、数量丰富,呈共显性遗传、并由于其高可靠性和稳定性,而被广泛应用于柑橘的遗传多样性研究、遗传图谱构建[10]、种质资源鉴定[11,12,13]和分子标记辅助选择[14]等研究中[1,4-6,15]。由于柑橘芽变材料间的遗传背景高度相似[11],前人利用ISSR标记仅能鉴别部分芽变品种[11],而温州蜜柑、甜橙、椪柑、柠檬、葡萄柚中的芽变材料鉴定则相当困难。目前SSR的检测方法常采用聚丙烯酰胺凝胶电泳或毛细管荧光电泳等进行区分,由于凝胶分辨率以及制胶、电泳等问题,常造成读带困难、自动化程度较低、耗时费力,一次只能对少数SSR位点进行基因分型。随着大量基因组数据的公布,对基因组的比较研究发现在染色体个别位置上存在变异热区[16,17],利用这些变异热区开发SSR标记,将有助于材料的区分和鉴别。同时利用深度测序技术如AmpSeq-SSR、Target-SSR等技术可一次性对大量SSR位点进行基因分型,突破了毛细管电泳检测对SSR位点检测数量的限制[18],且通过测序数据与已知的参考基因组比对,能够发现一些新的转录本和新基因[19],将有助于个体识别以及亲缘关系的鉴定。目前靶位点SSR测序技术已经在水稻[20]、黄瓜[21]、大戟[22]以及法医物证鉴定[23]中得到广泛应用,但该技术在柑橘芽变材料的鉴定中还没有相关报道。【本研究切入点】为提高柑橘芽变品种的分子鉴别能力,本研究首先从变异热点区域筛选多态性较高的SSR位点,利用这些SSR位点的高多态性来显著提高芽变材料的鉴别区分能力。由于多数情况下单个SSR并不能区分芽变材料,因此通过SSR位点的组合能够提高芽变材料的检测能力,为增加SSR位点的检测数量、通量和准确性,并利于自动化检测,本研究采用了Target-SSR测序技术,利用多路复用靶位点PCR扩增建立测序文库,并运用Miseq进行测序和基因分型。可一次性获得多个SSR位点基因分型数据,不仅降低了分型成本,而且分型的结果更加准确可靠,能直接确定突变的基因位点,对后续研究基因变异对性状影响意义重大。在本研究中综合应用了高多态性SSR的数字挖掘和Target-SSR深度测序两种策略,提高了柑橘芽变品种的鉴别能力。【拟解决的关键问题】玉溪是云南省第一大柑橘产地,2019年,全市生产柑橘48.2万t,占云南省柑橘总产量的44.40%。近年来通过大力开展芽变资源选优,发掘了一批有潜力的极早熟、早熟、优质的柑橘芽变资源。为高效鉴定这些芽变材料,本研究通过生物信息学方法发掘高多态性SSR位点,多路复用SSR靶位点扩增构建测序文库,并利用Miseq高通量测序平台验证了发掘出的SSR位点在区分芽变材料中的有效性。同时通过这些SSR对22份柑橘材料进行遗传多态性的分析。本研究建立了柑橘芽变材料区分和鉴定的高效方法,为今后柑橘种质资源的保存、利用和研究奠定了重要基础。1 材料与方法
1.1 植物材料与DNA提取
试验于2019年6月至2020年12月在西南大学柑桔研究所国家柑橘资源圃实验室进行。研究共选用22份柑橘资源,这些材料来源于云南省玉溪市华宁县、新平县在柑橘芽变选优活动中获得的疑似芽变材料及其母本(表1),样本分两次采集,第二次由于未采集到13号标本,故未纳入后续的聚类分析。这些材料包括一些地方优良品种。取每份材料的幼嫩叶片,利用CTAB法[24]提取DNA进行遗传多样性研究和Target-SSR的PCR扩增建库。
Table 1
表1
表122份柑橘种质资源
Table 1
| 序号 Code | 种质号 Germplasm number | 采集地 Collection site | 对应母本 Corresponding maternal parent | 序号 Code | 种质号 Germplasm number | 采集地 Collection site | 对应母本 Corresponding maternal parent | |
|---|---|---|---|---|---|---|---|---|
| 1 | 芽变1号 Bud sports No1. | 华宁县 Huaning county | 4 | 12 | 芽变5号 Bud sports No5. | 华宁县 Huaning county | 11 | |
| 2 | 芽变2号 Bud sport No2. | 华宁县 Huaning county | 4 | 13 | 大分1号 Oita No1. | 华宁县 Huaning county | ||
| 3 | 芽变3号 Bud sport No3. | 华宁县 Huaning county | 9 | 14 | HX10 | 华宁县 Huaning county | ||
| 4 | 冰糖橙 Bingtang Orange | 华宁县 Huaning county | 15 | MK11 | 华宁县 Huaning county | |||
| 5 | BJ2 | 华宁县 Huaning county | 16 | 芽变6号 Bud sport No6. | 新平县 Xinping county | 4 | ||
| 6 | SC3 | 华宁县 Huaning county | 17 | 芽变7号 Bud sport No7. | 新平县 Xinping county | 4 | ||
| 7 | 芽变4号 Bud sport No4. | 华宁县 Huaning county | 不详(温州蜜柑) Unknown (Satsuma) | 18 | 芽变8号 Bud sport No8. | 新平县 Xinping county | 4 | |
| 8 | 宫本 Miyanoto Wase | 华宁县 Huaning county | 19 | 芽变9号 Bud sport No9. | 新平县 Xinping county | 4 | ||
| 9 | 大浦 Ooura Wase | 华宁县 Huaning county | 20 | 芽变10号 Bud Sport No10. | 新平县 Xinping county | 4 | ||
| 10 | 兴津 Okitsu Wase | 华宁县 Huaning county | 21 | 芽变11号 Bud sport No11. | 新平县 Xinping county | 4 | ||
| 11 | 日南1号 Nichihan No1. | 华宁县 Huaning county | 22 | HG12 | 新平县 Xinping county |
新窗口打开|下载CSV
1.2 利用柑橘基因组数据发掘SSR位点
通过全基因组扫描方式发掘SSR,这些SSR位点将用于靶位点的扩增和文库构建。利用GMATA软件[25]对克里曼丁红橘参考基因组(1.3 对22份柑橘资源的多态性检测
根据获得的SSR侧翼序列设计引物,引物设计采用Primer3软件[26]进行。为验证设计引物在芽变材料上的区分能力,首先对22 份柑橘材料进行检测。试验共设计引物53 对,引物长度为18—22 bp,引物Tm值为60—65℃,从中筛选出4对多态性较高的SSR引物用于22份柑橘材料的遗传多样性研究(表2)。PCR反应体系为20 μL,其中包括100 ng DNA模板、10 μL 2×TSINGKE Mastermix酶和1 μL的SSR引物,PCR条件为:94℃ 5 min;94℃ 30 s,60℃ 30 s,72℃ 30 s,35个循环;72℃延伸5 min。反应产物利用聚丙烯凝胶电泳后,银染显带进行观测。Table 2
表2
表2本试验筛选出的4对多态性引物
Table 2
| 引物编号 Primer number | 上游Forward (5′-3′) | 下游Reward(5′-3′) |
|---|---|---|
| MK7 | CTGGGGCTCACATAAATCGT | GATTTGTCCGCCAATCAAGT |
| MK9 | GCATTGCAGCACTTTTGTCAT | GCATTGCAGCACTTTTGTCAT |
| MK19 | GGGTTGGGAATGTGAATGAA | CCTAGGGGTGGGCATATTTT |
| MK17 | CCCACCTCGTTGAATCTCTC | ACCAGTACCACTGCTTACTCTTTT |
新窗口打开|下载CSV
1.4 数据统计与分析
对22份柑橘资源开展遗传多样性分析,利用聚丙烯酰凝胶电泳法对SSR位点进行基因分型,根据SSR电泳条带的有无记录供试材料基因型。1表示有条带(包括弱带),0表示无条带,构建(0,1)矩阵。利用Cervus 3.07软件[27]对4对引物的观测等位基因数、观测杂合度、期望杂合度、PIC值等进行计算,通过GenAlEx 6.51[28]软件对22份材料进行PCoA分析。1.5 测序验证
为验证SSR差异条带在区分品种上的可靠性和稳定性,对有差异的PCR条带采用割胶回收纯化,回收PCR产物,将PAGE胶捣碎后加入100 μL ddH2O;0℃金属浴20 min;以水浴后溶液为模板做PCR扩增;对PCR产物进行琼脂糖凝胶电泳后使用微量琼脂糖凝胶DNA回收试剂盒回收;因回收量过低,对回收产物TA克隆后送深圳华大基因公司进行检测。1.6 Target SSR-Seq文库构建
为一次性对多个SSR靶位点进行基因分型,本研究在采用多路复用扩增的方式来构建Target SSR-seq文库。其步骤包括:第一步使用在线工具Multiple Primer Analyzer对引物互补结构分析并设计合适的引物组合,共设计18个引物组合包含77对引物,使用多路复用PCR对2份温州蜜柑芽变材料DNA样品进行靶位点SSR的扩增。多重PCR反应在20 μL反应体系中进行,其中包括100 ng DNA模板、10 μL 2×Hieff Canace® Gold PCR Master Mix高保真酶预混液和3—6 μL的多重SSR引物。PCR条件为:98℃ 3 min;98℃ 30 s,60℃ 30 s,72℃ 30 s,35个循环;72℃ 5 min延伸。用PCR产物小量回收试剂盒回收。第二步,使用MaxUp II DNA Library Prep Kit for Illumina建库试剂盒对PCR产物建库,主要步骤包括PCR产物3′端添加A尾的末端修复反应、为产物添加用于Illumina Miseq平台的测序接头,DNA clean beads纯化、为每个样品添加特异标签的Index Primer PCR反应,DNA clean beads纯化。此后,将Target SSR-seq文库在Illumina MiSeq测序平台上进行测序。1.7 Target SSR-Seq测序数据的分析
对测序后的fastq原始数据利用fastqc(2 结果
2.1 柑橘基因组中SSR的发掘
利用GMATA软件在克里曼丁红橘参考基因组中获得69 101个SSR,其中2基序SSR占69.5%,3基序SSR占25.7%,4基序SSR占3.8%,5基序SSR占0.7%,6基序SSR占0.3%。在2基序SSR中,以AT/TA的基序数量最多,占17.5%;在3基序SSR中,以AAT基序最多(3.9%)(图1)。图1
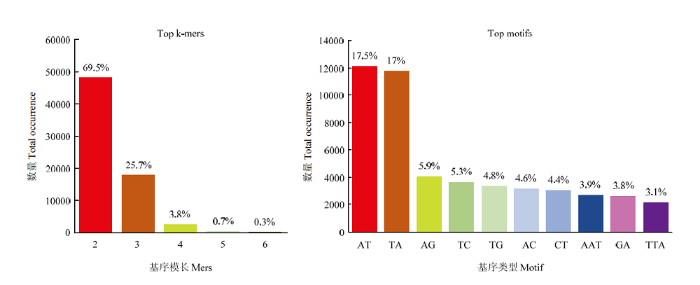 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1克里曼丁红橘全基因组中鉴定的SSR基序及其比例
Fig. 1The SSR motifs and its proportion in citrus reference genome
在温州蜜柑全基因组中获得的80 193个SSR,其中2基序SSR占69.8%,3基序SSR占25.6%,4基序SSR占3.7%,5基序SSR占0.7%,6基序SSR占0.3%。在2基序SSR中,以AT/TA基序最多,占17.8%;3基序中以AAT最多,占3.9%(图2)。从两个柑橘品种的全基因组序列中,鉴定到的SSR类型及比例几乎一致,均是以2基序的AT/TA重复所占比例最多。
图2
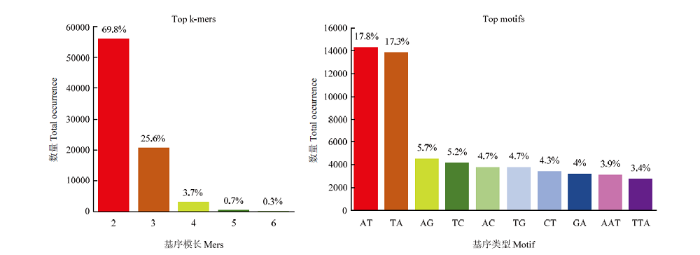 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2温州蜜柑全基因组中鉴定的SSR基序及其比例
Fig. 2The SSR motifs and its proportion of SSR in satsuma genome
为寻找能够鉴定柑橘芽变的SSR标记,对温州蜜柑和克里曼丁红橘的SSR序列去除重复后,再利用BLASTn比对从中发掘两个品种之间存在差异的SSR位点,这些存在于品种间的差异SSR在理论上属于胚性突变。进一步对温州蜜柑基因组数据库进行SSR的搜索和比对。发现其中一些SSR序列比如BDQV01000095.1_33999 具有284个匹配项(hit matches)、BDQV01000126.1_38180具有277个匹配项、BDQV01000074.1_30393具有170个匹配项,表明这些序列在温州蜜柑上的同源序列较多,具有较高的多态性,且在基因组上分布较广,暗示这些序列有可能来源于突变热点区域,能够反映体细胞突变的差异。而且这些序列中的一些往往具有反向重复特性且含有SSR,暗示其具有一定的生物学功能。利用Perl脚本取得其同源序列两端各150 bp的序列,根据获得的序列利用Clustal W进行序列对齐后,发现其中有SSR的插入和缺失的变异,如图3所示。利用这些体细胞突变序列来区分柑橘芽变材料时,可能会提供更多的多态信息,从而有助于芽变材料的鉴定。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3同源性较高的序列中的一些SSR变异
Fig. 3SSR variation presented in some homologous sequences
为此,针对这些在温州蜜柑全基因组上存在较多同源序列的位点进行了PCR引物设计,不仅在全基因组上能够同时对多个位点进行靶向扩增,且SSR的覆盖程度更高,SSR的多态性更高,这样使得测序文库的构建效率更加高效。
2.2 22份柑橘材料SSR扩增结果
从53对SSR引物中筛选出4对扩增效果及重复性较好、条带清晰、多态性较高的引物用于22份柑橘材料的分析。4个引物共获得46个等位条带,每个位点的平均等位基因为1.913,平均期望杂合率为0.2056,平均PIC值为0.1718。图4为9号引物的扩增效果图。图4
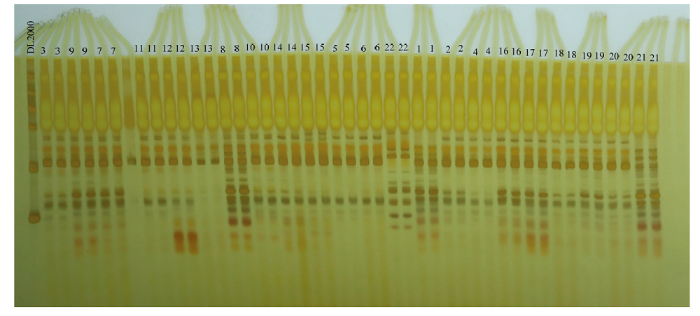 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图49号引物在22份柑橘种质上的扩增效果图
Fig. 4The amplification band profiles by No.9 primer on 22 citrus accessions
从丙烯酰胺凝胶电泳结果来看,4份引物区分芽变材料的能力均较强,获得的带型较多,可用于芽变材料的基因分型。在22份材料中共产生多态性条带84条,平均每对引物检测到6.11个多态性位点。对比9#母本和3#变异的扩增效果图发现,9号引物、30号引物和53号引物均能扩增出差异性条带,在图谱中不仅有较多新条带的出现,也发现了部分亲本条带的缺失。
根据22份柑橘材料的基因型,采用GenAlEx 6.51软件进行 PCoA主坐标轴分析,可明显将一些早熟芽变材料3、9、7、8、10、11、12号等区分开(图5),这些材料位于第一坐标轴的右侧,与中晚熟材料能够明显地区分开。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图522份材料SSR标记的PCoA分析
3、9、7、8、10、11、12号为早熟材料,其余为中晚熟材料;图中13表示14号样品,以此类推
Fig. 5The principal coordinates analysis graph of 22 citrus accessions based on SSR genotype
3, 9, 7, 8, 10, 11, and 12 on behalf early ripening accessions, and the others are medium and late ripening accessions; Diagram 13 said 14 samples, and so on
2.3 验证
为验证SSR引物扩增结果的稳定性和可靠性,取差异条带进行割胶回收后进行Sanger测序。9号引物的9#1、9#9、9#17等多个克隆的测序结果相同,均为同一基因位点。对基因进行注释,证实为编码柑橘双功能核黄素生物合成蛋白RIBA 1的基因,NCBI参考序列为XM_006492553.2,GenBank编号为NW_006257091.1。根据基因序列重新设计正义引物(5′-GGGATTGCCTGTTGACTC-3′),反义引物(5′- GTTTGAATTGCTGCTTATGT-3′)再次进行扩增,扩增得到单一条带,没有扩增出条带的为变异株(图6),结果与9号引物的扩增结果吻合。图6
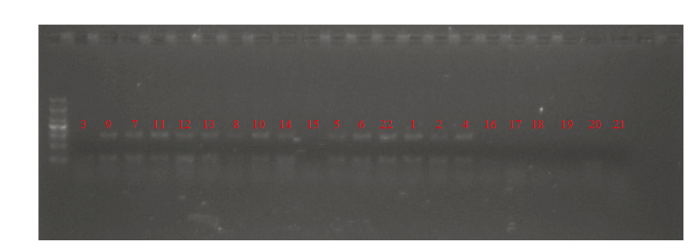 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6目标条带9#1扩增结果
Fig. 6The amplification bands by No.9 (9#1) primer checked with agarose gel electrophoresis
2.4 深度测序数据分析
为验证从温州蜜柑基因组上挖掘到的SSR位点在区分柑橘芽变材料上的有效性,选择2个温州蜜柑的芽变材料3号和7号,利用77对SSR引物进行多路复用靶向扩增来构建测序文库,之后在Illumina Miseq测序平台上进行测序,测序数据经过质量控制并去除接头后,分别获得40.725 M和38.559 M的短读序列。利用NextGENe_v2.4.3与克里曼丁红橘参考基因组进行对比后,3号共鉴定到1 208个与克里曼丁红橘基因组存在差异的变异位点,平均测序深度为273.12,最高测序深度为7 955。7号材料共鉴定到1 202个变异位点,平均测序深度为269.57,最高测序深度为6 366。利用两份材料的vcf文件,采用vcftools提取两个文库中各个位点的测序深度DP、等位基因频率AF、等位基因覆盖度SGACOV以及插入及重复序列,通过数据库中等位基因频率AF和测序深度的比对,可寻找到能够区分芽变材料的重要分子标记。足够的测序深度使SSR和SNP的鉴定更为准确可靠。在测序深度较高的序列中或者附近往往含有SSR位点,并且两个芽变材料的最高测序深度均对应在相同的SCAFFOLD位置上,比如在测序深度较高的SCAFFOLD_2上的9 464 527—9 464 586位置含有GAA重复,SCAFFOLD_6上23 400 572—23 400 596含有TTA重复,表明从短读序列cluster的测序深度的分布上,能够确定扩增产物的主带及其位置。从而通过测序深度,可以获得多个位置SSR的基因型,实现了同时对多位点SSR的基因分型。对相同位点上SSR等位基因的统计,可发现在一个位点上存在多个等位基因的现象,这为芽变品种的鉴定提供了更多的可选择标记。
通过数据库中测序深度DP、等位基因频率AF、等位基因覆盖度SGACOV以及插入及重复序列的对比,发现在SCAFFOLD_6和SCAFFOLD_2两个位置上存在显著的SSR差异(表3),进一步用IGV审视后发现在SCAFFOLD_6的23 400 572—23 400 596上和SCAFFOLD_2位置9 464 550—9 464 651上有重复单元的插入或缺失差异,另外在SSR位点的侧翼也存在SNP变异(图7、图8)。通过深度测序,不仅验证了Target-SSR测序技术在区分芽变材料上的有效性,同时能够对SSR进行精准的基因分型和定位,尤其是对SSR中插入或缺失的重复单元差异数目进行精确的鉴定。
Table 3
表3
表32个柑橘芽变材料的SSR和SNP基因型差异比较
Table 3
| 3.CHROM | 3.POS | 3.REF | 3.ALT | 7.ALT | 3.DP | 7.DP | 3.AF | 7.AF | 3.SGACOV | 7.SGACOV |
|---|---|---|---|---|---|---|---|---|---|---|
| SCAFFOLD_2 | 9464551 | A | C | 7955 | .212 | 1688 | ||||
| SCAFFOLD_2 | 9464547 | GAGAAGAAGAAGAAGA | GAGAAGAAGAAGA | GAGAAGAAGAAGA | 7954 | 6366 | .342 | .397 | 2718 | 2528 |
| SCAFFOLD_2 | 9464536 | GAAA | GA | GA | 7919 | 6328 | .347 | .409 | 2750 | 2590 |
| SCAFFOLD_2 | 9464527 | G | C | C | 7914 | 6328 | .366 | .427 | 2894 | 2703 |
| SCAFFOLD_2 | 9464521 | A | G | 7912 | .213 | 1689 | ||||
| SCAFFOLD_6 | 23400577 | GTATTATTATTATTATT | GTATTATTATTATTATTATTATT | GTATTATTATTATTATTATTATT | 5603 | 5709 | .606 | .373 | 3396 | 2131 |
| SCAFFOLD_6 | 23400577 | GTATTATTATTATTATT | GTATTATTATTATTATTATTATT | T | 5603 | 5709 | .606 | .227 | 3396 | 1298 |
新窗口打开|下载CSV
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7两个温州蜜柑芽变品种在Scaffold_6 上的SSR等位变异
Fig. 7SSR alleles variation of two satsuma bud sports aligned on scaffold_6
图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8两个温州蜜柑芽变品种在Scaffold_2 上的SSR等位变异
Fig. 8SSR allel variation of two satsuma bud sports aligned on scaffold_2
通过对两个温州蜜柑芽变VCF变异数据的比对发现,测序深度在600以上的差异位点在2个芽变品种中仅有3个,其中一个位于SCAFFOLD_6的23 400 572 —23 400 596上,另外一个位于SCAFFOLD_2位置9 464 550—9 464 651上和5 693 245—5 693 275上。而减少测序深度的筛选条件,则可以得到更多的差异位点,但在结果的可靠性上存疑。这进一步证实了利用常规SSR技术来检测芽变的效率确实不高。在2个芽变品种中,通过Target-SSR测序获得的变异位点并不多,为进一步提高芽变品种的分辨力,可以利用更多的SSR引物来进行靶位点的扩增建库。
3 讨论
芽变选种是柑橘品种选育的重要途径之一,植物形态学上的差异往往是芽变鉴定的重要标记。但在区分芽变材料时,由于外界环境条件的改变或者受表观遗传因素的影响,常导致植物形态特征出现较大差异,以形态特征差异来区分芽变,其可靠性和准确性不高,需要进一步开展遗传特异性、稳定性和一致性观测,来确定变异的可靠性,而这样往往会花费较长时间。分子标记技术作为芽变材料鉴定的辅助手段,在芽变选种中已得到广泛应用。其中SSR标记由于稳定性高、重复性好、数量丰富、呈共显性遗传、高多态性、易于自动化检测,在芽变材料鉴定、品种区分、品种权保护、遗传多态性研究、遗传图谱构建等多个方面得到广泛应用。SSR在用于芽变材料检测时,缺点是SSR所能提供的遗传多态性水平不高,往往需要对大量的SSR引物进行筛选,用常规的聚丙烯酰胺凝胶电泳有时难以找到可区分标记,且由于聚丙烯酰胺凝胶电泳的分辨率限制,不能准确区分SSR扩增产物碱基个数的微小差异,偶尔也会产生假阳性或假阴性结果。而以往采用区分柑橘芽变材料的AFLP技术,又存在读带困难、自动化程度低、耗时费力等问题,尤其区分遗传背景高度相似的柑橘品种,如区分脐橙[3,11]、温州蜜柑[11]、冰糖橙等芽变材料时,也存在多态性低而不能有效区分芽变材料的相同问题。比如,李益等[11]利用SSR开展柑橘品种鉴定,从362对引物中筛选出21对SSR核心引物,尽管通过增加引物对组合可提高品种的区分能力,但仍有279个品种无法单独一一区分,虽可划分为87个小组,但组内品种无法区分。罗静等[6]利用RAPD标记对25份柑橘资源及其芽变系进行鉴定和多样性分析,RAPD的多态性检出率只有50.7%。在其他果树上也存在类似问题。在桃芽变株系的鉴定中,通过50对SSR检测,只有对照C2在CP-PCT022和BPPCT023标记上出现了与供试样品不同的等位基因位点[30];在梨的芽变检测中,10对EST-SSR引物在4组材料中没有扩增出差异条带,不能鉴别芽变材料[31],说明利用常规手段采用多态性水平较低的SSR标记来区分鉴定芽变材料,其效率并不高。要克服这个问题,一个办法是选用基因组上变异热点区域中多态性较高的SSR进行相关区域的扩增,另一个办法就是增加SSR的检测位点数量。利用下一代测序技术开展SSR基因分型是目前新兴的技术,按照文库构建方法的差异可分为Target SSR-seq[21]和AmpSeq-SSR[32]两类。Target SSR-seq是在构建文库前对已有的SSR进行筛选,确定目标SSR后建库测序。YANG等[21]利用122个SSR通过Target SSR-seq技术对382个黄瓜品种进行基因分型,获得16个核心引物用于黄瓜品种鉴定。AmpSeq-SSR是一种大规模SSR-seq方法,它通过设计可多重扩增的AmpliSeq引物,使用专用的AmpliTaq酶扩增,理论上可同时扩增上百万个位点,但其引物设计要求高、价格贵。LI等[32]利用3 105个SSR通过AmpSeq-SSR技术构建水稻的指纹图谱。Target SSR-seq相对于AmpliSeq在小规模SSR基因分型应用上成本更低,但其操作更加繁琐,需对每个SSR位点单独扩增。本研究所使用Target SSR-seq技术在引物设计上结合了Target SSR-seq和AmpSeq-SSR两种方法的优点,首先通过对芽变近似材料基因组的扫描和同源序列比对,来获得基因序列中同源相似度高、匹配数量多、多态性高的SSR位点。这些SSR位点在基因组上的匹配度高,一方面说明其在基因组上的分布广,有可能存在高度重复,因而变异的可能性就更大,相应的SSR多态性指数也就提高了。利用其侧翼序列来设计引物,不仅能大幅减少引物的设计数量,同时又能保证SSR的多态性水平;另一方面,若这些变异的SSR来自于同一个位点,且多态性水平高,表明存在复等位基因,在同一材料中存在这种复等位变异,就证明了有部分细胞产生了变异,从而为芽变形态的发生奠定了基础。
一般情况下,SSR在整个基因组上均有分布,但近年来利用基因组序列开展SSR鉴定时,发现在不同染色体上的SSR数量并不是均匀分布[33,34,35]。在禾本科不同种类植物中,SSR在基因组上的分布密度在种间存在较大差异[34],着丝粒附近的密度往往小于染色体臂上的密度,非编码区(UTRs)SSR的覆盖率比编码区、外显子区和内含子区更高,SSR的丰度与重组率呈显著的线形正相关[34]。为提高SSR区分芽变品种的能力,往往需要筛选多态性高的SSR用于靶向测序,本研究通过同源序列的比较,以发现匹配数量多且多态性水平高的SSR位点,进而设计引物。这与以往通过基因组序列来鉴定SSR位点有本质区别,以往SSR的生物信息学挖掘只是对SSR位点和模式的鉴定,缺少同源序列的比较,因此不能提供SSR多态性方面的信息。本研究利用BLASTn发现了一些在基因组上同源匹配数量很高的SSR序列,表明这些SSR位点来自于序列重复区域或者是变异热点区域,这些位点除了与基因组上的多次复制事件有关外,还可能与染色体的多次重排事件有关,因此这些SSR能够提供更多的遗传变异信息,因此有助于芽变材料的鉴定。MA等[33]采用对4个物种基因组开展SSR引物的e-PCR比较,来评价不同物种之间SSR的多态性和SSR产物在物种之间的特异性,显著提高了SSR引物区分不同物种的能力。
本研究利用温州蜜柑和克里曼丁橘全基因组序列发掘SSR位点,发现在基因组上AT/TA二单元是最多的串联重复,而3基序中以AAT最多,这与PAVAN KUMAR等[36]在叶子花中的结果相似。利用同源序列比对的方式来获得多态性SSR位点,采用在全基因组序列中同源比对匹配度高、数量多的SSR位点的侧翼序列来设计引物,共设计了53对引物,筛选到能够区分芽变材料的4对引物,其效率比其他方式利用SSR来区分柑橘芽变材料的效率更高。4对引物就可对22份柑橘种质材料进行有效区分,并且对有差异性的条带进行回收和Sanger测序,进一步证实了这些引物在区分柑橘品种上的可靠性、稳定性及分辨能力,同样这些引物还能够用于柑橘其他种类及品种的区分和鉴定。本研究中Target-SSR Miseq的测序结果也证实,在测序深度较高的扩增位点中,往往存在多个SSR等位基因,这些等位基因有可能来自于基因组上的不同位点,代表着基因的复制或者跳跃事件,有一定的生物学功能;也有可能来自于基因组上的同一位点,若是存在于同一位点上的复等位基因,则暗示有个别细胞产生了变异从而形成混合的细胞亚群,因此根据不同SSR等位基因的比例差异,能够实现柑橘芽变品种的区分。
另外,增加SSR的检测位点数量,也能有效提高鉴别区分芽变材料的能力。本研究采用靶位点的多路复用扩增来构建测序文库,通过18个组合,对2个温州蜜柑芽变材料采用77对SSR进行了有效扩增,构建了测序文库,并采用Illumina Miseq的深度测序一次性获得了1 202—1 208个变异位点,平均每对引物获得了15.6个变异位点,显著高于凝胶电泳的检测能力。并且能对变异位点获得了准确的基因分型,同时还可获得SSR侧翼的SNP基因型,进一步提高信息量和芽变材料的鉴定能力。该方法还克服了凝胶电泳存在的读带困难,可组合的SSR基因座的个数受荧光种类限制,检测的变异位点少,一次难以进行多个SSR位点的基因分型的问题。YANG等[21]利用Target-SSR测序开展黄瓜品种的SSR基因分型,表明该技术不仅经济高效,测序深度较高(~1 000×),同时基因分型的成功率高达100%。利用群体材料进行SSR基因分型时,发现SSR位点上的等位基因数量与观察杂和度、PIC值呈显著正相关。在进行SSR基因分型开展黄瓜遗传群体的划分时,作者采用了遗传多样性高而等位基因数最少的SSR位点用于基因分型。而本研究中Target-SSR测序技术主要用于2个芽变材料的区分,因此采用遗传多态性高而等位基因数较多的SSR,能够显著提高芽变材料的鉴别能力。
本研究中Target-SSR深度测序的结果表明,2份温州蜜柑芽变材料间的SSR基因型至多在3个等位基因上出现了差异,有差异的SSR位点数并不多。这一方面印证了SSR技术区分柑橘芽变的能力有限,另一方面也表明通过PCR扩增建库,由于引物的特异性强,建库较为成功。对于同一位点SSR基因型中3等位基因的出现,还需要进一步区分是否存在基因组上的多位点扩增。只有通过测序深度、等位基因覆盖度、等位基因频率等多个指标的结合,才能更精准地发现芽变导致的SSR位点差异。由于这些有差异的SSR位点往往来源于体细胞变异而非胚性变异,因此存在变异细胞数量少,在测序深度低、PCR扩增建库存在偏向性时,这种一个位点上存在多等位基因变异的现象,往往难以被检测到。本研究为提高柑橘芽变品种的区分能力,在进行SSR靶位点的深度测序前,使用了较多数量的SSR引物进行多路复用靶位点PCR扩增,增加了检测位点数量,同时选用覆盖度高、同源序列多、多态性高的SSR位点进行引物设计,使文库构建更加高效,也更加容易发现芽变材料中的突变位点。深度测序结果不仅准确探明了芽变材料的SSR基因型,同时也定位了SSR在基因组上的位置,利用SSR的测序深度和等位基因频率两个参数结合,发现了在芽变材料间具有差异的SNP和SSR位点,从而实现了芽变材料的准确区分,也为芽变材料目标性状变异的遗传机理研究提供了更多有用信息。鉴于Target-SSR靶位点的深度测序技术能够在24 h内对多个样本中的数百个目标SSR位点进行快速基因分型,不仅成本更低,而且发掘变异的能力更强,未来还可通过进一步增加SSR扩增靶位点的数量来提高芽变材料的鉴别能力。该技术除了用于芽变材料的鉴定外,一次能够获得覆盖全基因组的大量位点的SSR基因型,更有利于柑橘资源的遗传演化及多样性研究。
4 结论
本研究采用基因组数据库挖掘和比对,发掘突变热点位置的SSR,利用优选出的靶位点SSR设计引物进行多路扩增构建测序文库,采用Miseq二代测序技术,成功鉴定了温州蜜柑的芽变材料及其亲本,同时对玉溪地区的22份优异柑橘材料进行了多样性研究。本研究综合了芽变检测的两种技术,选用来源于变异热点的多态性较高的SSR位点进行扩增构建测序文库,增加SSR的检测位点数量,并基于深度测序技术获得了精准的SSR靶位点及其基因型,为今后的柑橘芽变品种鉴定、资源遗传多样性研究奠定了重要基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11033-019-04884-7URL [本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 6]
[本文引用: 6]
[本文引用: 1]
[本文引用: 1]
DOI:10.1270/jsbbs.53.35URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.cell.2012.11.019URL [本文引用: 1]
[本文引用: 1]
DOI:10.1093/nar/gkx093URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3389/fpls.2019.00531URL [本文引用: 4]
DOI:10.1016/j.ejbt.2020.11.004URL [本文引用: 1]
DOI:10.2144/000113721URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/nar/gks596URL [本文引用: 1]
DOI:10.1111/j.1365-294X.2007.03089.xURL [本文引用: 1]
DOI:10.1111/men.2006.6.issue-1URL [本文引用: 1]
DOI:10.1093/bib/bbs017URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 2]
DOI:10.1016/j.ejbt.2020.01.004URL [本文引用: 2]
DOI:10.1186/s12863-015-0178-zURL [本文引用: 3]
DOI:10.18632/aging.v12i5URL [本文引用: 1]
DOI:10.1016/j.gene.2020.144794URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}