Comparison of Genomic Prediction Accuracy for Meat Type Chicken Carcass Traits Based on GBLUP and BayesB Method
ZHU Mo,1,2, ZHENG MaiQing2, CUI HuanXian2, ZHAO GuiPing,2, LIU Yang,11Department of Animal Genetics, Breeding and Reproduction, College of Animal Science and Technology, Nanjing Agricultural University, Nanjing 210095 2State Key Laboratory of Animal Nutrition/Institute of Animal Sciences, Chinese Academy of Agricultural Sciences, Beijing 100193
Received:2020-11-10Accepted:2021-04-7 作者简介 About authors 朱墨,E-mail: 2018105017@njau.edu.cn。
摘要 【目的】育种值估计是畜禽育种的核心内容,准确的育种值估计是提高选种准确性的重要手段。屠宰性状是肉鸡重要的经济性状,且无法活体测量,利用基因组信息进行育种值估计的基因组选择是十分有力的工具。文章通过比较GBLUP和BayesB方法对肉鸡屠宰性状的基因组预测的准确性,为肉鸡育种值估计的策略提供依据。【方法】用本课题组前期收集的3 362只白羽肉鸡的表型记录,性状包括胸肌率(BrR)、胸肌重(BrW)、屠体重(CW)、腿肌率(ThR)和腿肌重(ThW)。利用中国农业科学院自主研发的“京芯一号”鸡 55 K SNP芯片对所有个体进行了基因分型,采用PLINK软件对芯片的基因型数据进行质量控制,基因组选择的GBLUP和BayesB方法分别由基于R语言的ASReml包和hibayes包实施,并采用世代验证法评估两种方法的基因组育种值的预测准确性。【结果】研究发现基因组选择的准确性与性状的遗传力大致呈正相关。结果显示,在使用GBLUP方法时,预测准确性最高的性状为胸肌率。胸肌率、胸肌重、屠体重、腿肌率和腿肌重的基因组预测的准确性分别为0.3262、0.2871、0.2780、0.2153、0.2126;在使用BayesB方法时,预测准确性最高的性状为胸肌率。5个性状的基因组预测的准确性分别为0.3765、0.2257、0.1376、0.2525、0.2844。结果表明,对于白羽肉鸡屠宰性状的基因组选择研究,BayesB方法的预测准确性略高于GBLUP方法。对于3000的样本量和55 K的标记密度,GBLUP方法的计算时间大约为1h,BayesB方法的计算时间大约为7h。【结论】对于胸肌率、腿肌率和腿肌重,BayesB方法的预测准确性高于GBLUP方法;对于屠体重和胸肌重,GBLUP方法的预测准确性高于BayesB。畜禽的育种工作注重实效性。在实际的育种工作中,需要综合考虑育种值估计的准确性和育种的时效性来决定用何种方式估计基因组育种值。 关键词:鸡;基因组选择;世代验证;准确性
Abstract 【Objective】 Predicting the breeding value is the core content of livestock breeding and the accurate predicting of breeding value is an important approach to improve the selection accuracy of breeding. Carcass traits are economic important traits for broilers. However, carcass traits can only be measured postmortem. Genomic selection may be a powerful tool because of its accurate prediction of breeding values of animals without own phenotypic information. At present, there are few reports on genomic selection on carcass traits for broilers. The purpose of this study was to compare the accuracy of genomic prediction of broiler carcass traits by using GBLUP and BayesB method. 【Method】 This study investigated the efficiency of genomic prediction in a white-feathered broiler population, which was collected from 3 362 white-feathered broilers. Five carcass traits, including breast muscle rate (BrR), breast muscle weight (BrW), carcass weight (CW), thigh muscle rate (ThR) and thigh muscle weight (ThW) were taken into account. All the individuals were genotyped with “IASCHICK” chicken 55 K SNP array. PLINK software was used for the quality control of genotype data. GBLUP and BayesB method were implemented with ASReml and hibayes package in R software, respectively. Generation validation were utilized to evaluate the accuracy of genomic prediction for twenty replicates for each trait.【Result】The results showed that the accuracy of genomic selection was almost positively correlated with the heritability of each trait. The validation results indicated that the prediction accuracy of BrR was the highest with GBLUP method, and the accuracies of BrR, BrW, CW, ThR and ThW were 0.3262, 0.2871, 0.2780, 0.2153, and 0.2126, respectively. Meanwhile, the accuracy of BrR was the highest with BayesB method, and the accuracies of BrR, BrW, CW, ThR and ThW were 0.3765, 0.2257, 0.1376, 0.2525, and 0.2844, respectively. The results showed that the prediction accuracy of BayesB method was slightly higher than that of GBLUP method. It took about 1 h and 7 h for GBLUP and BayesB method, respectively, to carry out the calculating procedure for one trait. 【Conclusion】 In conclusion, the prediction accuracy of BrR, ThR and ThW with BayesB method was higher than that with GBLUP method, while the prediction accuracy of BrW and CW with GBLUP method was higher than that with BayesB method. However, the calculating time for BayesB method was longer than that for GBLUP method. In breeding practice, the balance of prediction accuracy and computational efficiency should be comprehensively considered to predict the genomic estimated breeding value. Keywords:chicken;genomic selection;generation validation;accuracy
PDF (429KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 朱墨, 郑麦青, 崔焕先, 赵桂苹, 刘杨. 基于GBLUP和BayesB方法对肉鸡屠宰性状基因组预测准确性的比较. 中国农业科学, 2021, 54(23): 5125-5131 doi:10.3864/j.issn.0578-1752.2021.23.016 ZHU Mo, ZHENG MaiQing, CUI HuanXian, ZHAO GuiPing, LIU Yang. Comparison of Genomic Prediction Accuracy for Meat Type Chicken Carcass Traits Based on GBLUP and BayesB Method. Scientia Acricultura Sinica, 2021, 54(23): 5125-5131 doi:10.3864/j.issn.0578-1752.2021.23.016
胸肌率、胸肌重、屠体重、腿肌率和腿肌重的描述性统计量汇总于表1。分别使用基于系谱构建的亲缘关系A矩阵和基于全基因组SNP信息构建的亲缘关系G矩阵,采用平均信息约束最大似然算法(average information restricted maximum likelihood, AIREML)估计加性遗传方差和残差方差,本研究中的方差组分使用ASReml 4.1.0 软件进行估计[21]。基于系谱构建的A阵估计BrR、BrW、CW、ThR和ThW的遗传力均高于基于基因组SNP信息构建的G阵估计BrR、BrW、CW、ThR和ThW的遗传力。各性状的遗传力估计结果见表2。
Table 2 表2 表2各屠宰性状的遗传力估计 Table 2Results of heritability for each carcass trait
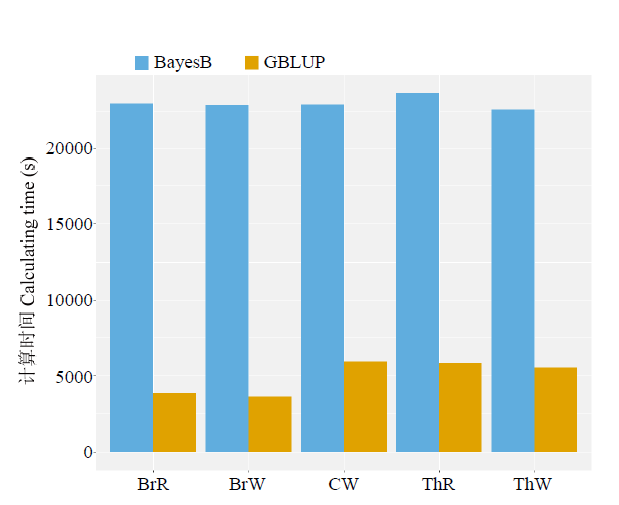
畜禽的育种工作注重时效性,计算效率是基因组选择在实际生产中应用时需要考虑的一个重要因素。贝叶斯模型的参数求解过程通过蒙特卡洛马尔科夫链(Markov chain monte carlo, MCMC)过程,采用高强度的吉布斯(Gibbs)抽样,往往需要经过上万次迭代,且无法并行计算,这常常限制了贝叶斯类的方法在育种中的应用,使得基于混合线性模型的方法(GBLUP为代表)在基因组选择中的应用最为广泛。但是,基于混合线性模型的方法的预测准确性要低于贝叶斯类模型[9]。在一些研究中,将最显著的SNP或验证过的QTL加入混合模型作为协变量来提高预测准确性,然而这些SNP或QTL解释的表型方差有限,可能会导致假阳性的结果[28,29]。还有研究者通过对SNP进行加权,构建性状特异性的亲缘关系矩阵,也能在一定程度上提高混合线性模型基因组预测的准确性[30,31,32]。然而,这些SNP的效应很容易被干扰。本研究中,BayesB方法的计算时长远远大于GBLUP方法(图 1),但是BayesB方法的预测准确性显著高于GBLUP方法。而且,对于3 000的样本量和55 K 的标记密度,BayesB的计算时间大约为7 h,尚在可接受的范围。在实际的育种中,使用低密度芯片数据,在样本和标记数量都不是十分庞大的情况下,为了提高基因组育种值预测的准确性,使用贝叶斯方法进行基因组育种值的估计是可行的方案。
HENDERSON CR. Best linear unbiased estimation and prediction under a selection model Biometrics, 1975, 31(2):423-447. DOI:10.2307/2529430URL [本文引用: 1]
LANDER, THOMPSONR. Efficiency of marker-assisted selection in the improvement of quantitative traits Genetics, 1990, 124(3):743. DOI:10.1093/genetics/124.3.743URL [本文引用: 1]
GODDARD ME, HAYES BJ. Mapping genes for complex traits in domestic animals and their use in breeding programmes Nature Reviews Genetics, 2009, 10(6):381-391. DOI:10.1038/nrg2575URL [本文引用: 1]
MEUWISSEN TH, HAYES BJ, GODDARD ME. Prediction of total genetic value using genome-wide dense marker maps Genetics, 2001, 157(4):1819-1829. DOI:10.1093/genetics/157.4.1819URL [本文引用: 5]
GODDARD ME, HAYES BJ, MEUWISSENT H E. Using the genomic relationship matrix to predict the accuracy of genomic selection Journal of animal breeding and genetics = Zeitschrift fur Tierzuchtung und Zuchtungsbiologie, 2011, 128(6):409-421. [本文引用: 1]
YANGJ, BENYAMINB, MCEVOY BP, GORDONS, HENDERS AK, NYHOLT DR, MADDEN PA, HEATH AC, MARTIN NG, MONTGOMERY GW, GODDARD ME, VISSCHER PM. Common SNPs explain a large proportion of the heritability for human height Nature Genetics, 2010, 42(7):565-569. DOI:10.1038/ng.608URL [本文引用: 1]
VANRADEN PM. Efficient methods to compute genomic predictions Journal of Dairy Science, 2008, 91(11):4414-4423. DOI:10.3168/jds.2007-0980URL [本文引用: 2]
AGUILARI, MISZTALI, JOHNSOND, LEGARRAA, TSURUTAS, LAWLORT. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score Journal of Dairy Science, 2010, 93(2):743-752. DOI:10.3168/jds.2009-2730URL [本文引用: 1]
HABIERD, FERNANDO RL, KIZILKAYAK, GARRICK DJ. Extension of the bayesian alphabet for genomic selection BMC Bioinformatics, 2011, 12:186. DOI:10.1186/1471-2105-12-186URL [本文引用: 4]
International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution Nature, 2004, 432(7018):695-716. DOI:10.1038/nature03154URL [本文引用: 1]
KRANISA, GHEYAS AA, BOSCHIEROC, TURNERF, YUL, SMITHS, TALBOTR, PIRANIA, BREWF, KAISERP, HOCKING PM, FIFEM, SALMONN, FULTONJ, STROM TM, HABERERG, WEIGENDS, PREISINGERR, GHOLAMIM, QANBARIS, SIMIANERH, WATSON KA, WOOLLIAMS JA, BURT DW. Development of a high density 600K SNP genotyping array for chicken BMC Genomics, 2013, 14:59. DOI:10.1186/1471-2164-14-59URL [本文引用: 1]
LIUR, XINGS, WANGJ, ZHENGM, CUIH, CROOIJMANSR P M A, LIQ, ZHAOG, WENJ. A new chicken 55K SNP genotyping array BMC Genomics, 2019, 20(1):410. DOI:10.1186/s12864-019-5736-8URL [本文引用: 2]
ZHANGZ, XU ZQ, LUO YY, ZHANG HB, GAON, HE JL, JI CL, ZHANG DX, LI JQ, ZHANG XQ. Whole genomic prediction of growth and carcass traits in a Chinese quality chicken population Journal of Animal Science, 2017, 95(1):72-80. [本文引用: 2]
LIUT, QUH, LUOC, SHUD, WANGJ, LUND MS, SUG. Accuracy of genomic prediction for growth and carcass traits in Chinese triple-yellow chickens BMC Genetics, 2014, 15:110. DOI:10.1186/s12863-014-0110-yURL [本文引用: 2]
WOLCA, DROBIK-CZWARNOW, JANKOWSKIT, ARANGOJ, SETTARP, FULTON JE, FERNANDO RL, GARRICK DJ, DEKKERSJ C M. Accuracy of genomic prediction of shell quality in a White Leghorn line Poultry Science, 2020, 99(6):2833-2840. DOI:10.1016/j.psj.2020.01.019URL [本文引用: 1]
CHEN CY, MISZTALI, AGUILARI, TSURUTAS, MEUWISSENT H E, AGGREYS E, WINGT, MUIRW M. Genome-wide marker- assisted selection combining all pedigree phenotypic information with genotypic data in one step: An example using broiler chickens Journal of Animal Science, 2011, 89(1):23-28. DOI:10.2527/jas.2010-3071URL [本文引用: 1]
GONZáLEZ-RECIOO, GIANOLAD, ROSA GJ, WEIGEL KA, KRANISA. Genome-assisted prediction of a quantitative trait measured in parents and progeny: Application to food conversion rate in chickens Genetics, Selection, Evolution: GSE, 2009, 41:3. DOI:10.1186/1297-9686-41-3URL [本文引用: 1]
LIUT, LUOC, WANGJ, MAJ, SHUD, LUND MS, SUG, QUH. Assessment of the genomic prediction accuracy for feed efficiency traits in meat-type chickens PLoS ONE, 2017, 12(3):e0173620. DOI:10.1371/journal.pone.0173620URL [本文引用: 1]
PURCELLS, NEALEB, TODD-BROWNK, THOMASL, FERREIRAM A R, BENDERD, MALLERJ, SKLARP, DEBAKKER P I W, DALYM J, SHAMP C. PLINK: A tool set for whole-genome association and population-based linkage analyses American Journal of Human Genetics, 2007, 81(3):559-575. DOI:10.1086/519795URL [本文引用: 1]
LIJ, WANGJ, KANG HM, LIU RR, LIH, ZHAO GP. The difference of genetic parameters for carcass and meat quality traits by BLUP and GBLUP methods in Beijing You chicken Acta Veterinaria et Zootechnica Sinica, 2020, 51(1):35-42. (in Chinese) [本文引用: 1]
ØDEGåRDJ, MEUWISSEN T HE. Estimation of heritability from limited family data using genome-wide identity-by-descent sharing Genetics, Selection, Evolution : GSE, 2012, 44:16. DOI:10.1186/1297-9686-44-16URL [本文引用: 1]
LEE SH, GODDARD ME, VISSCHER PM, VANDER WERF J H. Using the realized relationship matrix to disentangle confounding factors for the estimation of genetic variance components of complex traits Genetics, Selection, Evolution : GSE, 2010, 42:22. DOI:10.1186/1297-9686-42-22URL [本文引用: 1]
VEERKAMP RF, MULDER HA, THOMPSONR, CALUSM P L. Genomic and pedigree-based genetic parameters for scarcely recorded traits when some animals are genotyped Journal of Dairy Science, 2011, 94(8):4189-4197. DOI:10.3168/jds.2011-4223URL [本文引用: 1]
ZENGJ, PSZCZOLAM, WOLCA, STRABELT, FERNANDO RL, GARRICK DJ, DEKKERSJ C M. Genomic breeding value prediction and QTL mapping of QTLMAS2011 data using Bayesian and GBLUP methods BMC Proceedings, 2012, 6(2):S7. [本文引用: 1]
TENGJ, GAON, ZHANGH, LIX, LIJ, ZHANGH, ZHANGX, ZHANGZ. Performance of whole genome prediction for growth traits in a crossbred chicken population Poultry Science, 2019, 98(5):1968-1975. DOI:10.3382/ps/pey604URL [本文引用: 1]
LOPES MS, BOVENHUISH, VAN SONM, NORDBøØ, GRINDFLEK EH, KNOL EF, BASTIAANSENJ W M. Using markers with large effect in genetic and genomic predictions Journal of Animal Science, 2017, 95(1):59-71. [本文引用: 1]
MOORE JK, MANMATHAN HK, ANDERSON VA, POLAND JA, MORRIS CF, HALEY SD. Improving genomic prediction for pre-harvest sprouting tolerance in wheat by weighting large-effect quantitative trait loci Crop Science, 2017, 57(3):1315-1324. DOI:10.2135/cropsci2016.06.0453URL [本文引用: 1]
TIEZZIF, MALTECCAC. Accounting for trait architecture in genomic predictions of US Holstein cattle using a weighted realized relationship matrix Genetics, Selection, Evolution: GSE, 2015, 47:24. DOI:10.1186/s12711-015-0100-1URL [本文引用: 1]
ZHANGZ, ERBEM, HEJ, OBERU, GAON, ZHANGH, SIMIANERH, LIJ. Accuracy of whole-genome prediction using a genetic architecture-enhanced variance-covariance matrix G3 (Bethesda, Md.), 2015, 5(4):615-627. [本文引用: 1]
ZHANGZ, OBERU, ERBEM, ZHANGH, GAON, HEJ, LIJ, SIMIANERH. Improving the accuracy of whole genome prediction for complex traits using the results of genome wide association studies PLoS ONE, 2014, 9(3):e93017. DOI:10.1371/journal.pone.0093017URL [本文引用: 1]
 ,1,2, 郑麦青2, 崔焕先2, 赵桂苹
,1,2, 郑麦青2, 崔焕先2, 赵桂苹

 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}