 ,1, 王力1, 龙凤1, 昝林森1,2, 成功,1,2
,1, 王力1, 龙凤1, 昝林森1,2, 成功,1,2Codon Optimization of Human Lysozyme and High-Efficiency Expression in Bovine Mammary Cells
TIAN Yuan,1, WANG Li1, LONG Feng1, ZAN LinSen1,2, CHENG Gong,1,2通讯作者:
责任编辑: 林鉴非
收稿日期:2019-09-18接受日期:2020-05-29网络出版日期:2020-09-16
| 基金资助: |
Received:2019-09-18Accepted:2020-05-29Online:2020-09-16
作者简介 About authors
田媛,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2385KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
田媛, 王力, 龙凤, 昝林森, 成功. 人溶菌酶密码子优化及其在牛乳腺细胞中高效表达[J]. 中国农业科学, 2020, 53(18): 3805-3817 doi:10.3864/j.issn.0578-1752.2020.18.015
TIAN Yuan, WANG Li, LONG Feng, ZAN LinSen, CHENG Gong.
0 引言
【研究意义】抗生素滥用所引发的细菌耐药性、抗生素残留及食品安全等问题已被视为全球危害公众健康的问题,人们迫切需要开发新型广谱抗菌剂。溶菌酶又称胞壁质酶或N-乙酰胞壁质聚糖水解酶,在唾液、泪液、血清、人乳、牛乳和禽类蛋清中广泛分布,是一种能水解致病菌中黏多糖的碱性酶,具有广谱抗菌、消炎、抗病毒及免疫调节等多种功效,在畜牧、农产品、食品和医疗等行业广泛应用[1,2,3]。人溶菌酶和鸡蛋清溶菌酶氨基酸序列具有59%同源性,但抗菌活性远高于目前使用较广的鸡蛋清溶菌酶,具有更广阔的应用前景[4]。然而,通过传统方法从母乳、胎盘、唾液中提取分离或微生物表达系统生产重组人溶菌酶,获得量很少,稳定性低,无法满足研究和潜在市场应用的需求[5]。基因工程不断发展为大规模高效生产重组人溶菌酶提供了可能。因此,进一步优化表达系统,通过生物反应器实现重组人溶菌酶高效表达,将为今后重组人溶菌酶的工业化生产和应用奠定基础。【前人研究进展】2006年,MEGA首次利用山羊乳腺生物反应器生产了重组人溶菌酶[6],随后在牛[7,8,9]、猪[10]、山羊[11]等动物的研究中均有成功实现了重组人溶菌酶在乳腺中表达的报道。然而,不容忽视的是,异源重组蛋白表达量低是目前生物反应器中亟待解决的难题。研究发现,宿主细胞密码子使用偏好很大程度上影响了异源重组蛋白在宿主细胞的高效表达[12]。组成蛋白质的氨基酸有20种,而编码氨基酸的密码子有64种(简并密码子)。不同生物,甚至同种生物不同的蛋白质编码基因,对简并密码子使用频率并不相同,具有一定的偏爱性,其中使用频率高密码子称为最佳密码子,而那些很少被利用的密码子称为稀有或低频密码子。研究发现,少量的DNA密码子的改变,在很大程度上影响了蛋白翻译速率,即使替换单个简并密码子,其蛋白合成的速度会减缓到其正常速度的1/10甚至更慢[13],密码子使用偏好与蛋白翻译效率呈显著正相关[14]。通过蛋白质和mRNA表达数据库联合分析发现,密码子的选择决定了蛋白的表达量,生物体通过密码子的选择进而调控内源蛋白的表达丰度[15]。密码子的选择在生物反应器研究过程中十分重要,外源基因含有宿主细胞基因使用的低频密码子或者偏好密码子不一致情况下,往往会影响外源基因的mRNA的稳定性和翻译效率,造成重组蛋白表达量低[16]。前人研究结果比较发现,转基因山羊、牛乳腺中重组人乳铁蛋白表达量为1.5—30 g·L-1,而重组人溶菌酶的表达量只有0.025—0.27 g·L-1[17],表达量低造成提纯成本高,难以达到乳腺生物反应器工业化生产要求。【本研究切入点】因此,人溶菌酶基因密码子使用偏好性是否与牛主要乳蛋白密码子使用偏好一致?这种差异是否是导致人溶菌酶在乳腺生物反应器中表达量低的主要原因?密码子优化能否进一步提高重组人溶菌酶表达量?这些问题仍待进一步研究。【拟解决的关键问题】本试验通过对牛乳腺主要乳蛋白密码子使用偏好性进行分析,并根据牛乳蛋白高频密码子和低频密码子对人溶菌酶基因密码子进行翻译起始区优化和全局优化设计,通过荧光素酶、实时荧光定量PCR及Western-blot等方法从mRNA和蛋白水平对密码子优化效果进行分析比较,实现人溶菌酶基因的高效表达,为开展重组人溶菌酶的工业化生产及应用奠定基础。1 材料与方法
试验于2019年在西北农林科技大学国家肉牛改良中心实验室完成。1.1 材料
PGL3-Basic荧光素酶载体、海肾荧光素酶载体为实验室保存。E.coli DH 5a菌株和pMD19-T easy载体购自大连Takara公司。牛成纤维细胞通过耳组织块法分离获得,实验室冻存。牛乳腺上皮细胞系购自通派(上海)生物科技有限公司,小鼠乳腺上皮细胞系C127购自北京协和细胞库。主要试剂包括:高保真PCR酶、限制性内切酶、PrimeScript? RT reagent Kit with gDNA Eraser反转录试剂盒和SYBR Premix EX Taq定量PCR试剂盒购自宝生物工程(大连)有限公司;T4 DNA连接酶、Dual-Glo双荧光素酶检测试剂盒购自普洛麦格(北京)生物技术有限公司;Endo-free Plasmid Kit、胶回收试剂盒购自Omega Biotek股份有限公司;Lipo3000转染试剂及opti-MEM购自Invitrogen公司;Western-blot细胞裂解液、BCA蛋白浓度测定试剂盒及SDS-PAGE蛋白上样缓冲液购自上海碧云天生物技术有限公司;蛋白酶抑制剂购自罗氏公司。兔源溶菌酶多抗、兔源GAPDH单抗和HRP标记的羊抗兔二抗分别购自Santa Cruz(sc-292849),Abcam(ab181603)和生工生物工程(上海)股份有限公司(D110058)。1.2 牛主要乳蛋白基因和人溶菌酶基因密码子使用偏好分析
选取αs1、αs2、β、κ-酪蛋白,β-乳球蛋白,α-乳清白蛋白,乳铁蛋白等7种牛乳中主要乳蛋白基因CDS序列,用于密码子使用偏好性分析。具体乳蛋白基因名称、GenBank登录号、CDS序列长度见表1。Table 1
表1
表17种牛乳蛋白基因GenBank登录号及序列信息
Table 1
| 基因名称 Gene name | GenBank号 GenBank No. | 序列长度 Length of CDS |
|---|---|---|
| alpha-s1 casein (CSN1S1) | M33123 | 645 bp |
| alpha-s2 casein (CSN1S2) | BC114773 | 669 bp |
| beta casein (CSN2) | NM_181008 | 675 bp |
| kappa casein (CSN3) | BC102120 | 573 bp |
| beta-lactoglogbulin (LGB) | NM_173929 | 537 bp |
| alpha-lactalbumin (LALBA) | M18780 | 429 bp |
| lactotransferrin (LTF) | NM_180998 | 2127 bp |
新窗口打开|下载CSV
利用CodonW1.42和EMBOSS(http://imed.med. ucm.es/EMBOSS/)中CUSP、CHIPs等工具对牛主要乳蛋白基因密码子及人溶菌酶密码子使用特性进行分析。分析的特性参数主要包括:A3s,G3s,C3s,同义密码子在第3 位上相应碱基的出现频率(T3s)、基因的G+C含量(GC)、密码子第3位的G+C 含量(GC3s)、密码子适应性指数(CAI)、有效密码子数(ENc)等。同义密码子的相对使用度(RSCU)是对同义密码子的使用偏好性评估,该值等于同义密码子的实际观测值与同义密码子平均使用期望值的比值。如果密码子使用无偏好性,则RSCU值为1;如该密码子比其他同义密码子使用更频繁,则其RSCU值大于1,反之亦然。将7个牛乳蛋白基因和人溶菌酶基因各自作为一个对象,以密码子的RSCU 值作为变量,对应每个编码氨基酸密码子利用TBtools 0.665绘制热图并进行聚类分析。
1.3 溶菌酶密码子优化及基因克隆
根据牛主要乳蛋白基因密码子使用偏好性,对人溶菌酶基因局部(翻译起始区前22位密码子)和全局优化设计。利用mfold在线软件对优化后的人溶菌酶基因序列进行RNA二级结构分析(http://unafold.rna. albany.edu/)。采用Trizol 法提取人Hela细胞总RNA, 通过凝胶电泳和核酸定量检测RNA质量和浓度。根据Takara反转录试剂盒操作说明对RNA进行反转录获得cDNA。以cDNA为模板,利用P-pGL3-cw引物(表2)克隆获得人溶菌酶编码区序列(野生型溶菌酶序列,LYZcw)。反应条件为:98 ℃,30 s模板变性;98℃,5 s;57℃,15 s;72℃,15 s,30个循环。72℃,5 min延伸。以LYZcw cDNA为模板,利用P-pGL3-op22引物(表2)克隆获得翻译起始区密码子优化序列(LYZop22),PCR反应条件同上。对上述获得的LYZcw、LYZop22 PCR产物经过胶回收纯化、序列末端加A反应、T-A克隆并DH5α转化后进行测序验证(LYZcw-T、LYZop22-T)。全局密码子优化的溶菌酶序列(LYZop),经生工生物工程(上海)股份有限公司全基因合成并克隆到T载体中(LYZop-T)。
Table 2
表2
表2LYZcw、LYZop22、LYZop序列克隆引物信息及基因合成
Table 2
| 引物名称 Primer name | 基因名称 Gene name | 引物序列 Primer sequence | 产物长度 Product length | 酶切位点 Restriction site |
|---|---|---|---|---|
| P-pGL3-cw | LYZcw | F:CCATGGCCATGAAGGCTCTCATTG R:CCATGGTTACTCCACATCACTCCACAACCTTGAAC | 459bp | NcoI / Nco |
| P-pGL3-op22 | LYZop22 | F:CCATGGACCATGAAGGCCCTGATCGTGCTGGGAC TGGTGCTGCTGTCTGTGACCGTGCAGGGCAAGGT GTTTGAGAGGTGTGAGTTGGCCAGAACTCTG R:CCATGGTCACTCCACAACCTTGAAC | 459bp | NcoI / NcoI |
| —— | LYZop | 全基因合成 Whole gene synthesis | 459 bp | NcoI / NcoI |
| P-pcDNA-cw | LYZcw | F:AAGCTTCCATGAAGGCTCTCATTG R:CTCGAGCTCCACAACCTTGAAC | 456 bp | HindIII / XhoI |
| P-pcDNA-op22 | LYZop22 | F:CCATGGACCATGAAGGCCCTGATCG R:CTCGAGCTCCACAACCTTGAAC | 456bp | HindIII / XhoI |
| P-pcDNA-op | LYZop | F:CCATGGACCATGAAGGCCCTGATCG R:CTCGAG CACTCCACATCCCTGCACAT | 456 bp | HindIII / XhoI |
新窗口打开|下载CSV
1.4 pGL3-LYZcw/op22/op溶菌酶-荧光素酶融合表达构建
以Invitrogen公司pGL3-control载体为骨架载体,构建人溶菌酶基因与萤火虫荧光素酶基因融合表达载体。通过NcoI内切酶分别酶切LYZcw-T、LYZop22-T、LYZop- T和pGL3-control载体。1%琼脂糖凝胶电泳回收LYZcw、LYZop22、LYZop溶菌酶基因序列和线性化的pGL3-control载体。通过T4 DNA连接酶进行连接,分别构建pGL3-LYZcw/op22/op荧光素酶融合表达载体。测序筛选鉴定LYZcw/op22/op序列以正向插入并与萤火虫荧光素酶基因正确融合的质粒用于下一步研究。1.5 细胞转染及荧光素酶检测密码子优化效果
分别将实验室冻存的牛乳腺上皮细胞系(BMEC)、牛成纤维细胞(BFFC)及C127小鼠乳腺上皮细胞系复苏,用完全培养基(DMEM高糖+10% FBS +1% PS)重悬,接种到T75培养瓶中,置于37 ℃,5% CO2培养箱中培养。待细胞汇合度达到90%时进行传代接种于24孔培养板中(2×105/孔)。接种后的第二天,按照每孔0.8 μg质粒(PGL-LYZcw/ LYZop/LYZop22﹕pRL-tk = 10﹕1):2 μLlipo3000比例转染上述细胞,每组4个复孔,转染48 h后通过荧光素酶活性检测比较人溶菌酶密码子优化效果。1.6 pcDNA3.1-LYZcw/op22/op溶菌酶过表达载体构建
分别以pGL3-LYZcw/op22/op质粒为模板,利用P-pcDNA-cw、P-pcDNA-op22、P-pcDNA-op引物(表2)克隆LYZcw、LYZop22、LYZop序列(PCR反应条件参照1.3)并连接到T载体。使用HindIII、XhoI内切酶分别酶切pcDNA3.1(+)、LYZcw-T、LYZop22- T vector、LYZop- T,连接构建pcDNA-LYZcw/op22/op人溶菌酶过表达载体,测序做进一步鉴定。1.7 细胞转染及实时荧光定量PCR检测密码子优化效果
将培养的牛成纤维细胞(BFFC)、牛乳腺上皮细胞(BMEC)及C127细胞传代接种于12孔培养板中(2×105/孔)。接种后的第二天,按照lipo3000转染试剂说明书,每孔1.6 μg质粒(pcDNA空载、pcDNA-LYZcw/op22/op):4 μL lipo3000比例转染上述细胞,每组4个复孔。24 h后按照1.3中方法提取细胞总RNA并反转录获得cDNA,用于下一步定量PCR检测。对不同密码子优化方式对重组人溶菌酶mRNA水平表达量进行定量分析。反应条件为:预变性95 ℃,30 s。95 ℃,5 s;60 ℃,34 s,40个循环。循环结束后进行熔解曲线分析,确保扩增条带单一。
1.8 Western-blot检测密码子优化效果
将培养的牛乳腺细胞(BMEC)经胰酶消化后接种于6孔培养板中(4×105 / 孔)。接种后的第二天,按照lipo3000转染试剂说明书,每孔4.0 μg质粒(pcDNA空载、pcDNA-LYZcw/op22/op):10 μL lipo3000比例转染上述细胞,每组4个复孔。转染48 h后,收集蛋白用于Western-blot检测。蛋白电泳采用分离胶浓度为12%,蛋白上样量40 μg。电泳条件为浓缩胶80 V,分离胶120 V,待溴酚蓝跑出胶后终止电泳。蛋白转膜按照伯乐半干转膜仪操作说明,200 mA, 1h进行转膜。转膜结束后,PVDF膜经5%脱脂奶粉封闭——一抗4 ℃孵育过夜(人溶菌酶、GAPDH 1﹕1 000稀释)——TBST洗涤(3次,每次10 min)——二抗室温孵育2 h(1﹕3 000稀释)——TBST洗涤(3次,每次10 min)——显影——ChemiDoc XRS+仪器成像。1.9 数据分析
采用2-△△Ct 方法对real-time qPCR 分析计算目的基因相对表达量。GraphPad Prism 6.0进行One-way ANOVA分析和作图。数据均以平均数±标准误(Mean±SEM)表示。2 结果
2.1 牛乳蛋白及人溶菌酶基因密码子组成分析
通过CodonW软件对牛乳蛋白及人溶菌酶基因密码子组成进行分析,结果表明,牛乳蛋白基因密码子GC含量平均为0.485 ± 0.026,第三位碱基GC3s含量平均为0.537 ± 0.062。人溶菌酶基因密码子GC含量为0.473,第三位碱基GC3s含量为0.407。上述结果表明,人溶菌酶基因整体GC含量与牛乳蛋白基因GC含量相近,但第三位碱基GC含量低于牛乳蛋白基因GC含量。牛乳蛋白基因密码子偏好以GC结尾,而人溶菌酶基因密码子偏好以AT结尾(表3)。Table 3
表3
表3牛乳蛋白及人溶菌酶基因密码子组成分析
Table 3
| Bos taurus | A3s | T3s | C3s | G3s | GC3s | GC | CAI | ENc |
|---|---|---|---|---|---|---|---|---|
| αs1酪蛋白alpha-s1 casein | 0.311 | 0.461 | 0.290 | 0.258 | 0.408 | 0.445 | 0.227 | 52.300 |
| αs2酪蛋白alpha-s2 casein | 0.253 | 0.453 | 0.340 | 0.335 | 0.484 | 0.417 | 0.317 | 49.970 |
| β酪蛋白beta casein | 0.201 | 0.421 | 0.342 | 0.298 | 0.505 | 0.512 | 0.234 | 48.750 |
| κ-酪蛋白 kappa casein | 0.399 | 0.412 | 0.243 | 0.194 | 0.341 | 0.430 | 0.240 | 58.250 |
| 乳球蛋白lactoglogbulin | 0.079 | 0.147 | 0.574 | 0.519 | 0.825 | 0.586 | 0.349 | 36.270 |
| α乳清白蛋白alpha-lactalbumin | 0.283 | 0.349 | 0.395 | 0.337 | 0.526 | 0.441 | 0.281 | 55.510 |
| 乳铁蛋白lactotransferrin | 0.170 | 0.242 | 0.410 | 0.449 | 0.671 | 0.561 | 0.262 | 47.650 |
| Mean ± SEM | 0.242±0.039 | 0.355±0.045 | 0.371±0.040 | 0.341±0.042 | 0.537±0.062 | 0.485±0.026 | 0.273±0.017 | 49.814±2.668 |
| 人溶菌酶 human lysozyme | 0.3143 | 0.4032 | 0.2742 | 0.2323 | 0.407 | 0.473 | 0.193 | 50.270 |
新窗口打开|下载CSV
ENc值范围为20(每个氨基酸只使用一个密码子)到61(各个密码子被均衡使用),其值越低,偏好性越强。牛乳蛋白及人溶菌酶ENc值分别为49.814和50.27,表明牛乳蛋白和人溶菌酶基因密码子使用相对较平均。ENC vs GC3s线性回归分析结果表明,7种乳蛋白基因有效密码子数ENc值与第三位碱基GC3s呈显著负相关(R = - 0.8968,P<0.01)(图1),表明密码子使用偏好性越强(ENc值越小)的基因,其GC3s值越高,主要偏好以GC碱基结尾的密码子。因此,牛乳蛋白基因密码子的使用偏好性受GC3s影响。
图1
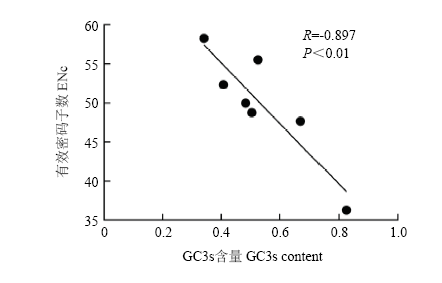 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1牛乳蛋白基因ENc vs GC3s相关性分析
圆点表示不同乳蛋白基因GC3s值对应的ENc值
Fig. 1Correlation analysis of ENc vs GC3s of bovine milk protein genes
The dot represents the ENc value corresponding to the GC3s value of different milk protein genes
2.2 牛乳蛋白基因密码子使用偏好性分析
对编码氨基酸且为简并密码子的7种牛乳蛋白基因使用的共59个密码子(不含Met、Trp及3个终止密码子)进行同义密码子的相对使用度(RSCU)分析。结果表明,牛乳蛋白基因对CTG(2.71)、AGG(2.32)、GCC(1.73)、GTG(1.68)、ATC(1.64)等5个密码子具有明显的偏好性(RSCU>1.5),为高频密码子。而TTA(0.12)、ATA(0.18)、TCG(0.19)、CTA(0.25)、CCG(0.26)、GCG(0.45)、ACG(0.48)等7个密码子在牛乳蛋白基因中使用较小(RSCU<0.5),为低频密码子(表4)。
Table 4
表4
表4牛乳蛋白基因密码子使用偏好性分析
Table 4
| 密码子 Codon | 氨基酸 Amino acid | 频率/1000 Frequency/1000 | 个数 Number | 同义密码子 相对使用度RSCU | 密码子 Codon | 氨基酸 Amino acid | 频率/1000 Frequency/1000 | 个数 Number | 同义密码子 相对使用度RSCU | |
|---|---|---|---|---|---|---|---|---|---|---|
| GCA | A | 13.793 | 26 | 0.73 | AAT | N | 19.629 | 37 | 0.96 | |
| GCC | A | 32.891 | 62 | 1.73 | CCA | P | 17.507 | 33 | 1.06 | |
| GCG | A | 8.488 | 16 | 0.45 | CCC | P | 19.629 | 37 | 1.19 | |
| GCT | A | 20.69 | 39 | 1.09 | CCG | P | 4.244 | 8 | 0.26 | |
| TGC | C | 14.854 | 28 | 0.98 | CCT | P | 24.403 | 46 | 1.48 | |
| TGT | C | 15.385 | 29 | 1.02 | CAA | Q | 16.976 | 32 | 0.57 | |
| GAC | D | 22.281 | 42 | 1.09 | CAG | Q | 42.44 | 80 | 1.43 | |
| GAT | D | 18.568 | 35 | 0.91 | AGA | R | 5.836 | 11 | 1.06 | |
| GAA | E | 33.952 | 64 | 0.90 | AGG | R | 12.732 | 24 | 2.32 | |
| GAG | E | 41.91 | 79 | 1.10 | CGA | R | 2.122 | 4 | 0.39 | |
| TTC | F | 21.22 | 40 | 1.13 | CGC | R | 3.183 | 6 | 0.58 | |
| TTT | F | 16.446 | 31 | 0.87 | CGG | R | 6.897 | 13 | 1.26 | |
| GGA | G | 11.671 | 22 | 1.07 | CGT | R | 2.122 | 4 | 0.39 | |
| GGC | G | 15.385 | 29 | 1.41 | AGC | S | 14.324 | 27 | 1.30 | |
| GGG | G | 9.019 | 17 | 0.83 | AGT | S | 13.263 | 25 | 1.20 | |
| GGT | G | 7.427 | 14 | 0.68 | TCA | S | 7.427 | 14 | 0.67 | |
| CAC | H | 11.141 | 21 | 1.35 | TCC | S | 14.854 | 28 | 1.34 | |
| CAT | H | 5.305 | 10 | 0.65 | TCG | S | 2.122 | 4 | 0.19 | |
| ATA | I | 2.653 | 5 | 0.18 | TCT | S | 14.324 | 27 | 1.30 | |
| ATC | I | 24.403 | 46 | 1.64 | ACA | T | 11.141 | 21 | 0.84 | |
| ATT | I | 17.507 | 33 | 1.18 | ACC | T | 18.037 | 34 | 1.36 | |
| AAA | K | 33.952 | 64 | 0.88 | ACG | T | 6.366 | 12 | 0.48 | |
| AAG | K | 42.971 | 81 | 1.12 | ACT | T | 17.507 | 33 | 1.32 | |
| CTA | L | 4.244 | 8 | 0.25 | GTA | V | 6.897 | 13 | 0.41 | |
| CTC | L | 19.629 | 37 | 1.14 | GTC | V | 15.915 | 30 | 0.95 | |
| CTG | L | 46.684 | 88 | 2.71 | GTG | V | 28.117 | 53 | 1.68 | |
| CTT | L | 18.037 | 34 | 1.05 | GTT | V | 15.915 | 30 | 0.95 | |
| TTA | L | 2.122 | 4 | 0.12 | TAC | Y | 15.385 | 29 | 0.91 | |
| TTG | L | 12.732 | 24 | 0.74 | TAT | Y | 18.568 | 35 | 1.09 | |
| AAC | N | 21.22 | 40 | 1.04 |
新窗口打开|下载CSV
2.3 基因密码子偏好性的聚类分析
以密码子的RSCU 值作为变量,对具有简并性的59个编码氨基酸的密码子进行聚类分析。依据RSCU 值可以将7种牛乳蛋白较好的分成为两大类:酪蛋白类(CSN2、CSN1S2、CSN3和CSN1S1)和乳清蛋白类(LTF、LGB和LALBA),而人溶菌酶基因密码子使用偏好与牛乳酪蛋白类更接近(图2)。结果表明,即使在同一乳腺组织内,编码牛乳酪蛋白类基因密码子和乳清蛋白类基因密码子在使用偏好性上可能也存在一定差异。图2
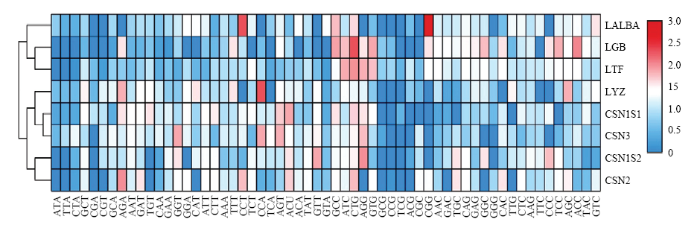 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2牛乳蛋白及人溶菌酶基因密码子使用RSCU值聚类分析
Fig. 2Clustering analysis of bovine milk protein and human lysozyme genes based on RSCU value
2.4 人溶菌酶基因密码子优化及分析
根据表3结果中牛乳蛋白基因高频密码子及低频密码子使用情况,对人溶菌酶密码子进行了优化。其中LYZop对溶菌酶全局密码子进行了优化,而LYZop22只对翻译起始区前22位密码子进行了优化(表5)。Table 5
表5
表5人溶菌酶基因密码子优化情况
Table 5
| LYZcw | LYZop22 | LYZop |
|---|---|---|
| 1 ATG AAG GCT CTC ATT GTT 19 CTG GGG CTT GTC CTC CTT 37 TCT GTT ACG GTC CAG GGC 55 AAG GTC TTT GAA AGG TGT 73 GAG TTG GCC AGA ACT CTG 91 AAA AGA TTG GGA ATG GAT 109 GGC TAC AGG GGA ATC AGC 127 CTA GCA AAC TGG ATG TGT 145 TTG GCC AAA TGG GAG AGT 163 GGT TAC AAC ACA CGA GCT 181 ACA AAC TAC AAT GCT GGA 199 GAC AGA AGC ACT GAT TAT 217 GGG ATA TTT CAG ATC AAT 235 AGC CGC TAC TGG TGT AAT 253 GAT GGC AAA ACC CCA GGA 271 GCA GTT AAT GCC TGT CAT 289 TTA TCC TGC AGT GCT TTG 307 CTG CAA GAT AAC ATC GCT 325 GAT GCT GTA GCT TGT GCA 343 AAG AGG GTT GTC CGT GAT 361 CCA CAA GGC ATT AGA GCA 379 TGG GTG GCA TGG AGA AAT 397 CGT TGT CAA AAC AGA GAT 415 GTC CGT CAG TAT GTT CAA 433 GGT TGT GGA GTG TAA | 1 ATG AAG GCC CTG ATC GTG 19 CTG GGA CTG GTG CTG CTG 37 TCT GTG ACC GTG CAG GGC 55 AAG GTG TTT GAG AGG TGT 73 GAG TTG GCC AGA ACT CTG 91 AAA AGA TTG GGA ATG GAT 109 GGC TAC AGG GGA ATC AGC 127 CTA GCA AAC TGG ATG TGT 145 TTG GCC AAA TGG GAG AGT 163 GGT TAC AAC ACA CGA GCT 181 ACA AAC TAC AAT GCT GGA 199 GAC AGA AGC ACT GAT TAT 217 GGG ATA TTT CAG ATC AAT 235 AGC CGC TAC TGG TGT AAT 253 GAT GGC AAA ACC CCA GGA 271 GCA GTT AAT GCC TGT CAT 289 TTA TCC TGC AGT GCT TTG 307 CTG CAA GAT AAC ATC GCT 325 GAT GCT GTA GCT TGT GCA 343 AAG AGG GTT GTC CGT GAT 361 CCA CAA GGC ATT AGA GCA 379 TGG GTG GCA TGG AGA AAT 397 CGT TGT CAA AAC AGA GAT 415 GTC CGT CAG TAT GTT CAA 433 GGT TGT GGA GTG TAA | 1 ATG AAG GCC CTG ATC GTG 19 CTG GGA CTG GTG CTG CTG 37 TCT GTG ACC GTG CAG GGC 55 AAG GTG TTT GAG AGG TGC 73 GAG CTG GCC AGG ACT CTG 91 AAG AGG CTG GGA ATG GAT 109 GGC TAT AGG GGA ATC AGC 127 CTG GCC AAC TGG ATG TGT 145 CTG GCC AAG TGG GAG TCT 163 GGC TAT AAC ACC AGG GCC 181 ACC AAC TAT AAC GCC GGA 199 GAC AGG AGC ACC GAC TAT 217 GGC ATC TTT CAG ATC AAC 235 TCT AGG TAT TGG TGT AAC 253 GAC GGC AAG ACC CCT GGC 271 GCC GTG AAT GCC TGT CAC 289 CTG TCT TGT AGC GCC CTG 307 CTG CAG GAC AAC ATC GCC 325 GAT GCT GTG GCC TGC GCC 343 AAG AGG GTG GTG AGG GAT 361 CCT CAG GGA ATC AGG GCT 379 TGG GTG GCC TGG AGG AAT 397 AGG TGC CAG AAC AGG GAC 415 GTG AGG CAG TAT GTG CAG 433 GGA TGT GGA GTG TAA |
新窗口打开|下载CSV
CodonW比较优化前后序列,结果表明,LYZcw、LYZop22、LYZop第三位碱基即GC3s含量分别为0.407、0.450、0.757。mRNA二级结构最小自由能MFE从-138.83 kcal/mol(LYZcw)分别降低到了-141.02 kcal/mol(LYZop22)和-178.12 kcal/mol(LYZop),mRNA二级结构稳定性明显提高。对mRNA翻译起始区100个碱基进行二级结构分析,发现相比于LYZcw(-29.7 kcal/mol),LYZop22(-26.4 kcal/mol)和LYZop(-31.0 kcal/mol)密码子优化并没有明显改变翻译起始区二级结构最小自由能,相反在5′端引入了更多的茎环结构(图3),有利于核糖体与mRNA的结合并启动翻译。
图3
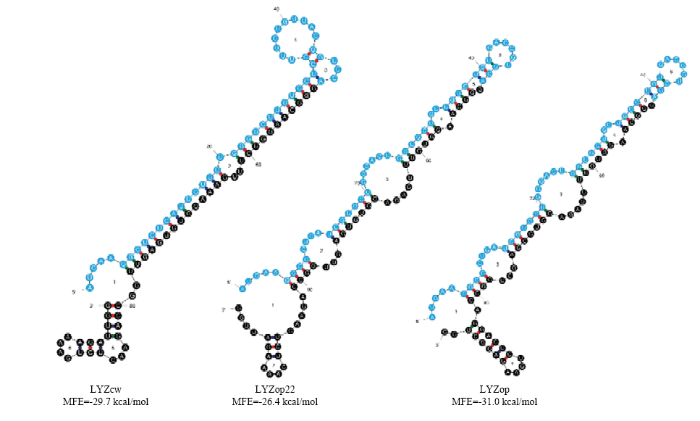 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3人溶菌酶密码子优化翻译起始区(1-100 bp)mRNA二级结构示意图
Fig. 3Schematic diagram of RNA second structure of codon optimized human lysozyme gene (1-100bp)
2.5 人溶菌酶-荧光素融合载体构建及溶菌酶表达分析
电泳和测序结果表明,成功克隆到了人溶菌酶野生型LYZcw、翻译起始区密码子优化的LYZop22 CDS区序列及全基因合成的全局密码子优化的LYZop CDS区序列,且与预期序列信息一致(图4)。Nco1酶切和测序结果表明,成功构建了pGL3-LYZcw/ op22/op 3个人溶菌酶-荧光素酶融合表达载体,且LYZcw、LYZop22、LYZop编码区与萤火虫荧光素酶基因正确融合,没有移码突变(图5)。图4
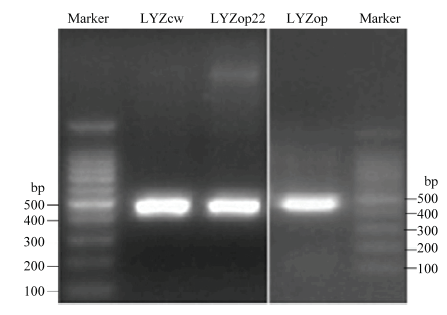 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4LYZcw、LYZop22和LYZop电泳检测
Fig. 4LYZcw, LYZop22 and LYZop electrophoresis detection
图5
 新窗口打开|下载原图ZIP|生成PPT
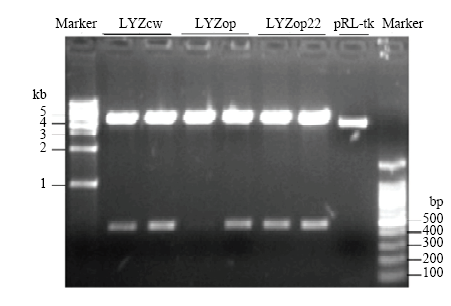
新窗口打开|下载原图ZIP|生成PPT图5pGL3-LYZcw/op22/op载体Nco1酶切鉴定
Fig. 5pGL3-LYZcw/op22/op vector identified by Nco1 enzyme digestion
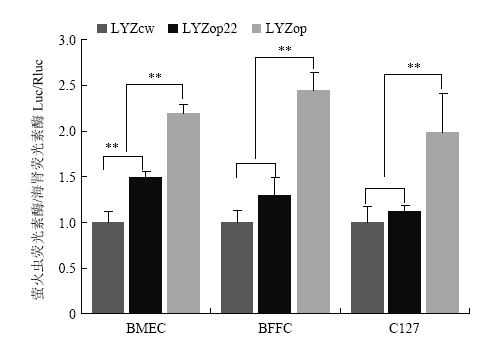
通过lipo3000 将pGL-LYZcw+pRL-tk、pGL-LYZop + pRL-tk、pGL-LYZop22 + pRL-tk分别转染BMEC、BFFC、C127细胞,48 h后检测荧光素酶表达量。荧光素酶检测结果表明,相比LYZcw,全局密码子优化的LYZop在BMEC和对照BFFC、C127细胞中分别提高了2.20倍(P<0.01)、2.44倍(P<0.01)和1.99倍(P<0.01)。翻译起始区密码子优化的LYZop22在BMEC细胞中相比野生型提高了1.48倍(P<0.01),而在对照BFFC、C127细胞中差异不显著(P>0.05)(图6)。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6荧光素酶检测人溶菌酶基因密码子优化效果
Fig. 6The effect of codon optimization analysis of human lysozyme gene by luciferase
2.6 mRNA、蛋白水平检测人溶菌酶基因密码子优化效果
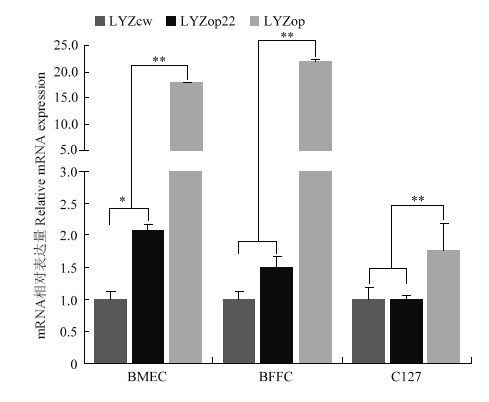
构建pcDNA-LYZcw/op22/op过表达载体,并分别转染BMEC、BFFC和C127细胞,检测密码子优化对人溶菌酶mRNA表达量的影响。实时荧光定量PCR结果表明,LYZop22相比LYZcw在BMEC和BFFC细胞中分别提高了2.08倍(P<0.05)和1.5倍(P>0.05)。而LYZop相比野生型LYZcw在上述两个细胞中分别提高了17.8倍(P<0.01)和22倍(P<0.01)(图7)。上述结果表明,根据牛乳腺主要蛋白进行密码子优化后的溶菌酶能显著提高其在牛乳腺上皮细胞和成纤维细胞转录水平表达量。图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7mRNA水平检测人溶菌酶基因密码子优化效果
Fig. 7The effect of codon optimization analysis on human mRNA level of human lysozyme gene
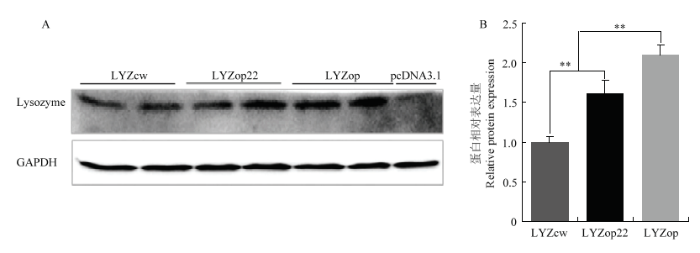
进一步对pcDNA-LYZcw/op22/op过表达载体转染的牛BMEC细胞进行人溶菌酶蛋白表达检测。相比野生型LYZcw,密码子优化的LYZop、LYZop22明显提高了人溶菌酶在牛BMEC细胞中的表达量,且全局密码子优化的LYZop溶菌酶表达量高于翻译起始区密码子优化的LYZop22(图8-A)。上述结果表明,密码子优化能明显提高人溶菌酶在牛乳腺上皮细胞中的表达。然而,经全局密码子优化的人溶菌酶基因在牛乳腺上皮细胞中蛋白表达水平提高倍数低于mRNA水平提高倍数(图8-B)。
图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8蛋白水平检测密码子优化对人溶菌酶表达量的影响
Fig. 8The effect of codon optimization analysis on protein level of human lysozyme
3 讨论
人溶菌酶作为非特异性免疫因子,对金黄色葡萄球菌、链球菌、大肠杆菌等均具有广谱的抗菌作用,在体内发挥着杀菌消炎、免疫调节、改善胃肠道菌群的作用。同时,在畜牧、农产品、食品、医疗和化妆品轻工等行业也具有广泛应用前景。利用现代基因工程和乳腺生物反应器生产重组人溶菌酶克服了传统提取方法蛋白稳定性差、成本高的不足,但如何进一步提高重组蛋白的表达量并用于下一步工业化生产仍是乳腺生物反应器首要考虑的问题。近年来研究发现,不同生物密码子的选择不仅对蛋白高水平表达发挥作用,同时对微量的调控蛋白表达也起到调节作用[13],这一发现对于提高外源重组蛋白的表达和生产具有重要的指导意义。研究发现,同一物种,不同组织甚至同一组织内不同蛋白其密码子使用偏好性均存在差异,正是这种差异可能进一步调节着蛋白的表达量[18]。本研究中根据牛乳中7种主要乳蛋白基因密码子偏好性进行聚类分析发现,牛乳中酪蛋白类(CSN2、CSN1S2、CSN3和CSN1S1)和乳清蛋白类(LTF、LGB和LALBA)可以明显的分为两类。表明,同一组织内牛乳酪蛋白和乳清蛋白基因密码子使用偏好存在一定的差异,而这种密码子使用偏好性可能在一定程度上影响着牛乳酪蛋白和乳清蛋白表达量。密码子使用偏好性分析发现,牛乳蛋白基因中发现5个高频密码子和7个低频密码子,且牛乳蛋白基因密码子第三位碱基偏好以GC结尾,而人溶菌酶偏好以AT结尾。而较高的GC含量,特别是简并碱基GC含量在哺乳动物细胞中与mRNA丰度和蛋白表达量呈正相关[19,20]。表明,人溶菌酶密码子使用偏好性不利于在牛乳中高效表达,这也可能是造成牛乳腺生物反应器中重组人溶菌酶表达量低的主要原因[17, 21]。
密码子使用偏好性在调节mRNA稳定性、蛋白翻译起始、延伸等方面发挥了重要的作用[22,23]。本研究中局部和全局密码子优化后的人溶菌酶mRNA二级结构自由能MFE分别由-138.83 kcal/mol降低到了-141.02 kcal/mol和-178.12 kcal/mol。前人研究发现,mRNA折叠强度与mRNA丰度和蛋白表达量呈正相关[24,25]。本研究发现,随着密码子优化后人溶菌酶mRNA二级结构稳定性的增加,其mRNA表达量也相应提高。在牛BMEC等3种细胞中,全局密码子优化的人溶菌酶mRNA水平显著高于局部密码子优化的人溶菌酶,这一结果也与优化后mRNA 稳定性呈正相关。表明,密码子优化可能通过增加mRNA二级结构稳定性和mRNA转录效率进而提高了人溶菌酶mRNA表达水平。
研究发现,mRNA 二级结构5′端的茎环结构有利于蛋白翻译起始和延伸,进而提高蛋白表达量[26,27]。密码子优化后的溶菌酶尽管整体mRNA MFE明显降低,但进一步对5′端翻译起始后100 bp mRNA碱基进行二级结构分析发现,密码子优化并没有明显改变人溶菌酶基因前100bp 二级结构自由能,反而在翻译起始区引入了更多的茎环结构,而5′端茎环结构的引入,有利于提高蛋白翻译起始效率[28]。酿酒酵母中研究发现,蛋白表达丰度与mRNA折叠强度存在正相关,而有效的mRNA翻译起始效率和较高的mRNA二级结构缩短了核糖体之间的距离,使mRNA的翻译效率提高,进而提高了蛋白的表达丰度[25, 29]。本研究中也发现,翻译区密码子优化后牛BMEC和BFFC细胞中人溶菌酶基因mRNA水平和蛋白水平相比野生型都有一定程度的提高。全局密码子优化的人溶菌酶基因,在mRNA翻译起始区引入了更多的茎环结构,同时,mRNA整体折叠也更稳定。因此,密码子优化后的人溶菌酶表达量在mRNA 水平和蛋白水平相比野生型均得到明显的提高。
密码子优化是提高外源重组蛋白表达的重要途径之一。尽管目前已开发许多软件可根据宿主密码子使用偏好对目的基因密码子进行优化和设计,然而关于密码子优化的“金标准”仍未统一,密码子优化的效果也不尽如人意,归根结底是由于目前关于密码子优化是如何影响外源蛋白表达仍存在很大的争议。密码子优化在mRNA转录效率[30]、稳定性[31],蛋白翻译起始[32]、延伸效率、蛋白折叠及蛋白稳定性[33]等多个环节均发挥了调节作用。ZHOU等研究发现,密码子优化提高了目的基因蛋白和RNA表达水平,但主要是体现在mRNA转录水平的提高[30]。PRESNYAK等[31]认为,密码子优化是决定mRNA稳定性的主要因素。本研究中发现,经全局密码子优化的人溶菌酶基因mRNA二级结构稳定性得到明显提高,其在牛乳腺上皮细胞中转录水平提高了17.8倍,然而其蛋白水平提高倍数却低于mRNA水平提高倍数。表明,密码子优化大幅提高了重组人溶菌酶mRNA的表达水平,但mRNA表达水平与蛋白表达水平并不一定呈强相关[34],而mRNA转录后的修饰可能进一步影响了蛋白的表达。因此,通过密码子优化提高mRNA表达水平的同时如何进一步提高蛋白表达水平及其内在分子机制仍有待于进一步研究。
4 结论
本研究通过生物信息学分析了牛乳腺主要乳蛋白基因密码子使用偏好,获得了牛乳蛋白基因密码子使用偏好及使用的高低频密码子;依据牛乳蛋白基因密码子使用偏好性对人溶菌酶全局密码子优化能显著提高重组人溶菌酶mRNA水平和蛋白水平的表达量,为今后利用生物反应器高效生产重组人溶菌酶奠定基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.ppat.1006512URLPMID:28934357 [本文引用: 1]

Lysozyme is a cornerstone of innate immunity. The canonical mechanism for bacterial killing by lysozyme occurs through the hydrolysis of cell wall peptidoglycan (PG). Conventional type (c-type) lysozymes are also highly cationic and can kill certain bacteria independently of PG hydrolytic activity. Reflecting the ongoing arms race between host and invading microorganisms, both gram-positive and gram-negative bacteria have evolved mechanisms to thwart killing by lysozyme. In addition to its direct antimicrobial role, more recent evidence has shown that lysozyme modulates the host immune response to infection. The degradation and lysis of bacteria by lysozyme enhance the release of bacterial products, including PG, that activate pattern recognition receptors in host cells. Yet paradoxically, lysozyme is important for the resolution of inflammation at mucosal sites. This review will highlight recent advances in our understanding of the diverse mechanisms that bacteria use to protect themselves against lysozyme, the intriguing immunomodulatory function of lysozyme, and the relationship between these features in the context of infection.
DOI:10.30918/MRIURL [本文引用: 1]
DOI:10.3109/07388551.2015.1084263URLPMID:26383819 [本文引用: 1]
Lysozyme is an antimicrobial peptide with a high enzymatic activity and positive charges. Therefore, it has applications in food and pharmaceutical industries as an antimicrobial agent. Lysozyme is ubiquitous in both animal and plant kingdoms. Currently, egg-white lysozyme is the most commercially available form of lysozyme. The main concerns of egg-white lysozyme are high recovery cost, low activity and most importantly the immunological problems to some people. Therefore, human lysozyme production has gained importance in recent years. Scientists have developed transgenic plants, animals and microorganisms that can produce human lysozyme. Out of these, microbial production has advantages for commercial productions, because high production levels are achievable in a relatively short time. It has been reported that fermentation parameters, such as pH, temperature, aeration, are key factors to increase the effectiveness of the human lysozyme production. Moreover, purification of the lysozyme from the fermentation broth needs to be optimized for the economical production. In conclusion, this review paper covers the mechanism of lysozyme, its sources, production methods and recovery of lysozyme.
DOI:10.3168/jds.S0022-0302(06)72114-2URLPMID:16428620 [本文引用: 1]
The potential for applying biotechnology to benefit animal agriculture and food production has long been speculated. The addition of human milk components with intrinsic antimicrobial activity and positive charge to livestock milk by genetic engineering has the potential to benefit animal health, as well as food safety and production. We generated one line of transgenic goats as a model for the dairy cow designed to express human lysozyme in the mammary gland. Here we report the characterization of the milk from 5 transgenic females of this line expressing human lysozyme in their milk at 270 microg/mL or 68% of the level found in human milk. Milk from transgenic animals had a lower somatic cell count, but the overall component composition of the milk and milk production were not different from controls. Milk from transgenic animals had a shorter rennet clotting time and increased curd strength. Milk of such nature may be of benefit to the producer by influencing udder health and milk processing.
DOI:10.1098/rspb.2013.3368URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1371/journal.pone.0017593URLPMID:21436886 [本文引用: 1]
BACKGROUND: There is great potential for using transgenic technology to improve the quality of cow milk and to produce biopharmaceuticals within the mammary gland. Lysozyme, a bactericidal protein that protects human infants from microbial infections, is highly expressed in human milk but is found in only trace amounts in cow milk. METHODOLOGY/PRINCIPAL FINDINGS: We have produced 17 healthy cloned cattle expressing recombinant human lysozyme using somatic cell nuclear transfer. In this study, we just focus on four transgenic cattle which were natural lactation. The expression level of the recombinant lysozyme was up to 25.96 mg/L, as measured by radioimmunoassay. Purified recombinant human lysozyme showed the same physicochemical properties, such as molecular mass and bacterial lysis, as its natural counterpart. Moreover, both recombinant and natural lysozyme had similar conditions for reactivity as well as for pH and temperature stability during in vitro simulations. The gross composition of transgenic and non-transgenic milk, including levels of lactose, total protein, total fat, and total solids were not found significant differences. CONCLUSIONS/SIGNIFICANCE: Thus, our study not only describes transgenic cattle whose milk offers the similar nutritional benefits as human milk but also reports techniques that could be further refined for production of active human lysozyme on a large scale.
DOI:10.1371/journal.pone.0123551URLPMID:25955256 [本文引用: 1]
Human lysozyme is a natural non-specific immune factor in human milk that plays an important role in the defense of breastfed infants against pathogen infection. Although lysozyme is abundant in human milk, there is only trace quantities in pig milk. Here, we successfully generated transgenic cloned pigs with the expression vector pBAC-hLF-hLZ-Neo and their first generation hybrids (F1). The highest concentration of recombinant human lysozyme (rhLZ) with in vitro bioactivity was 2759.6 +/- 265.0 mg/L in the milk of F0 sows. Compared with wild-type milk, rhLZ milk inhibited growth of Escherichia coli K88 during the exponential growth phase. Moreover, rhLZ in milk from transgenic sows was directly absorbed by the intestine of piglets with no observable anaphylactic reaction. Our strategy may provide a powerful tool for large-scale production of this important human protein in pigs to improve resistance to pathogen infection.
DOI:10.1016/j.jbiotec.2013.10.023URL [本文引用: 1]
Human lysozyme (hLZ), an essential protein against many types of microorganisms, has been expressed in transgenic livestock to improve their health status and milk quality. However, the large-scale production of hLZ in transgenic livestock is currently unavailable. Here we describe the generation of transgenic goats, by somatic cell-mediated transgenic cloning, that express large amounts of recombinant human lysozyme (rhLZ) in milk. Specifically, two optimized lysozyme expression cassettes (beta-casein/hLZ and beta-lactoglobulin/hLZ) were designed and introduced into goat somatic cells by cell transfection. Using transgenic cell colonies, which were screened by 0.8 mg/mL G418, as a nuclear donor, we obtained 10 transgenic cloned goats containing one copy of hLZ hybrid gene. An ELISA assay indicated that the transgenic goats secreted up to 6.2 g/L of rhLZ in their milk during the natural lactation period, which is approximately 5-10 times higher than human milk. The average rhLZ expression levels in beta-casein/hLZ and beta-lactoglobulin/hLZ transgenic goats were 2.3 g/L and 3.6 g/L, respectively. Therefore, both rhLZ expression cassettes could induce high levels of expression of the rhLZ in goat mammary glands. In addition, the rhLZ purified from goat milk has similar physicochemical properties as the natural human lysozyme, including the molecular mass, N-terminal sequence, lytic activity, and thermal and pH stability. An antibacterial analysis revealed that rhLZ and hLZ were equally effective in two bacterial inhibition experiments using Staphylococcus aureus and Escherichia coli. Taken together, our experiments not only underlined that the large-scale production of biologically active rhLZ in animal mammary gland is realistic, but also demonstrated that rhLZ purified from goat milk will be potentially useful in biopharmaceuticals. (C) 2013 Elsevier B.V.
DOI:10.1007/978-1-4939-9086-3_20URLPMID:30788833 [本文引用: 1]
Lung transplantation is the only option for patients with end-stage lung disease, but there is a shortage of available lung donors. Furthermore, efficiency of lung transplantation has been limited due to primary graft dysfunction. Recent mouse models mimicking lung disease in humans have allowed for deepening our understanding of disease pathomechanisms. Moreover, new techniques such as decellularization and recellularization have opened up new possibilities to contribute to our understanding of the regenerative mechanisms involved in the lung. Stripping the lung of its native cells allows for unprecedented analyses of extracellular matrix and sets a physiologic platform to study the regenerative potential of seeded cells. A comprehensive understanding of the molecular pathways involved for lung development and regeneration in mouse models can be translated to regeneration strategies in higher organisms, including humans. Here we describe and discuss several techniques used for murine lung de- and recellularization, methods for evaluation of efficacy including histology, protein/RNA isolation at the whole lung, as well as lung slices level.
DOI:10.1038/nature10965URL [本文引用: 2]
Protein synthesis by ribosomes takes place on a linear substrate but at non-uniform speeds. Transient pausing of ribosomes can affect a variety of co-translational processes, including protein targeting and folding(1). These pauses are influenced by the sequence of the messenger RNA(2). Thus, redundancy in the genetic code allows the same protein to be translated at different rates. However, our knowledge of both the position and the mechanism of translational pausing in vivo is highly limited. Here we present a genome-wide analysis of translational pausing in bacteria by ribosome profiling-deep sequencing of ribosome-protected mRNA fragments(3-5). This approach enables the high-resolution measurement of ribosome density profiles along most transcripts at unperturbed, endogenous expression levels. Unexpectedly, we found that codons decoded by rare transfer RNAs do not lead to slow translation under nutrient-rich conditions. Instead, Shine-Dalgarno-(SD) 6-like features within coding sequences cause pervasive translational pausing. Using an orthogonal ribosome(7,8) possessing an altered anti-SD sequence, we show that pausing is due to hybridization between the mRNA and 16S ribosomal RNA of the translating ribosome. In protein-coding sequences, internal SD sequences are disfavoured, which leads to biased usage, avoiding codons and codon pairs that resemble canonical SD sites. Our results indicate that internal SD-like sequences are a major determinant of translation rates and a global driving force for the coding of bacterial genomes.
DOI:10.1038/nrm.2017.91URLPMID:29018283 [本文引用: 1]
The advent of ribosome profiling and other tools to probe mRNA translation has revealed that codon bias - the uneven use of synonymous codons in the transcriptome - serves as a secondary genetic code: a code that guides the efficiency of protein production, the fidelity of translation and the metabolism of mRNAs. Recent advancements in our understanding of mRNA decay have revealed a tight coupling between ribosome dynamics and the stability of mRNA transcripts; this coupling integrates codon bias into the concept of codon optimality, or the effects that specific codons and tRNA concentrations have on the efficiency and fidelity of the translation machinery. In this Review, we first discuss the evidence for codon-dependent effects on translation, beginning with the basic mechanisms through which translation perturbation can affect translation efficiency, protein folding and transcript stability. We then discuss how codon effects are leveraged by the cell to tailor the proteome to maintain homeostasis, execute specific gene expression programmes of growth or differentiation and optimize the efficiency of protein production.
DOI:10.1093/nar/gkz033URLPMID:30698781 [本文引用: 1]
The codon stabilization coefficient (CSC) is derived from the correlation between each codon frequency in transcripts and mRNA half-life experimental data. In this work, we used this metric as a reference to compare previously published Saccharomyces cerevisiae mRNA half-life datasets and investigate how codon composition related to protein levels. We generated CSCs derived from nine studies. Four datasets produced similar CSCs, which also correlated with other independent parameters that reflected codon optimality, such as the tRNA abundance and ribosome residence time. By calculating the average CSC for each gene, we found that most mRNAs tended to have more non-optimal codons. Conversely, a high proportion of optimal codons was found for genes coding highly abundant proteins, including proteins that were only transiently overexpressed in response to stress conditions. We also used CSCs to identify and locate mRNA regions enriched in non-optimal codons. We found that these stretches were usually located close to the initiation codon and were sufficient to slow ribosome movement. However, in contrast to observations from reporter systems, we found no position-dependent effect on the mRNA half-life. These analyses underscore the value of CSCs in studies of mRNA stability and codon bias and their relationships with protein expression.
DOI:10.1016/j.tibtech.2004.04.006URL [本文引用: 1]
Abstract
The expression of functional proteins in heterologous hosts is a cornerstone of modern biotechnology. Unfortunately, proteins are often difficult to express outside their original context. They might contain codons that are rarely used in the desired host, come from organisms that use non-canonical code or contain expression-limiting regulatory elements within their coding sequence. Improvements in the speed and cost of gene synthesis have facilitated the complete redesign of entire gene sequences to maximize the likelihood of high protein expression. Redesign strategies are discussed here, including modification of translation initiation regions, alteration of mRNA structural elements and use of different codon biases.DOI:10.1007/s11248-015-9885-5URLPMID:26059245 [本文引用: 2]
Genetic engineering, which was first developed in the 1980s, allows for specific additions to animals' genomes that are not possible through conventional breeding. Using genetic engineering to improve agricultural animals was first suggested when the technology was in the early stages of development by Palmiter et al. (Nature 300:611-615, 1982). One of the first agricultural applications identified was generating transgenic dairy animals that could produce altered or novel proteins in their milk. Human milk contains high levels of antimicrobial proteins that are found in low concentrations in the milk of ruminants, including the antimicrobial proteins lactoferrin and lysozyme. Lactoferrin and lysozyme are both part of the innate immune system and are secreted in tears, mucus, and throughout the gastrointestinal (GI) tract. Due to their antimicrobial properties and abundance in human milk, multiple lines of transgenic dairy animals that produce either human lactoferrin or human lysozyme have been developed. The focus of this review is to catalogue the different lines of genetically engineered dairy animals that produce either recombinant lactoferrin or lysozyme that have been generated over the years as well as compare the wealth of research that has been done on the in vitro and in vivo effects of the milk they produce. While recent advances including the development of CRISPRs and TALENs have removed many of the technical barriers to predictable and efficient genetic engineering in agricultural species, there are still many political and regulatory hurdles before genetic engineering can be used in agriculture. It is important to consider the substantial amount of work that has been done thus far on well established lines of genetically engineered animals evaluating both the animals themselves and the products they yield to identify the most effective path forward for future research and acceptance of this technology.
DOI:10.1073/pnas.0404957101URLPMID:15314228 [本文引用: 1]
A diverse array of mechanisms regulate tissue-specific protein levels. Most research, however, has focused on the role of transcriptional regulation. Here we report systematic differences in synonymous codon usage between genes selectively expressed in six adult human tissues. Furthermore, we show that the codon usage of brain-specific genes has been selectively preserved throughout the evolution of human and mouse from their common ancestor. Our findings suggest that codon-mediated translational control may play an important role in the differentiation and regulation of tissue-specific gene products in humans.
DOI:10.1073/pnas.1518976113URL [本文引用: 1]
DOI:10.1371/journal.pbio.0040180URLPMID:16700628 [本文引用: 1]
Mammalian genes are highly heterogeneous with respect to their nucleotide composition, but the functional consequences of this heterogeneity are not clear. In the previous studies, weak positive or negative correlations have been found between the silent-site guanine and cytosine (GC) content and expression of mammalian genes. However, previous studies disregarded differences in the genomic context of genes, which could potentially obscure any correlation between GC content and expression. In the present work, we directly compared the expression of GC-rich and GC-poor genes placed in the context of identical promoters and UTR sequences. We performed transient and stable transfections of mammalian cells with GC-rich and GC-poor versions of Hsp70, green fluorescent protein, and IL2 genes. The GC-rich genes were expressed several-fold to over a 100-fold more efficiently than their GC-poor counterparts. This effect was not due to different translation rates of GC-rich and GC-poor mRNA. On the contrary, the efficient expression of GC-rich genes resulted from their increased steady-state mRNA levels. mRNA degradation rates were not correlated with GC content, suggesting that efficient transcription or mRNA processing is responsible for the high expression of GC-rich genes. We conclude that silent-site GC content correlates with gene expression efficiency in mammalian cells.
DOI:10.1038/nrg2899URLPMID:21102527 [本文引用: 1]
Despite their name, synonymous mutations have significant consequences for cellular processes in all taxa. As a result, an understanding of codon bias is central to fields as diverse as molecular evolution and biotechnology. Although recent advances in sequencing and synthetic biology have helped to resolve longstanding questions about codon bias, they have also uncovered striking patterns that suggest new hypotheses about protein synthesis. Ongoing work to quantify the dynamics of initiation and elongation is as important for understanding natural synonymous variation as it is for designing transgenes in applied contexts.
DOI:10.1038/nature16509URLPMID:26760206 [本文引用: 1]
Degeneracy in the genetic code, which enables a single protein to be encoded by a multitude of synonymous gene sequences, has an important role in regulating protein expression, but substantial uncertainty exists concerning the details of this phenomenon. Here we analyse the sequence features influencing protein expression levels in 6,348 experiments using bacteriophage T7 polymerase to synthesize messenger RNA in Escherichia coli. Logistic regression yields a new codon-influence metric that correlates only weakly with genomic codon-usage frequency, but strongly with global physiological protein concentrations and also mRNA concentrations and lifetimes in vivo. Overall, the codon content influences protein expression more strongly than mRNA-folding parameters, although the latter dominate in the initial ~16 codons. Genes redesigned based on our analyses are transcribed with unaltered efficiency but translated with higher efficiency in vitro. The less efficiently translated native sequences show greatly reduced mRNA levels in vivo. Our results suggest that codon content modulates a kinetic competition between protein elongation and mRNA degradation that is a central feature of the physiology and also possibly the regulation of translation in E. coli.
DOI:10.1038/nrm.2017.91URLPMID:29018283 [本文引用: 1]
The advent of ribosome profiling and other tools to probe mRNA translation has revealed that codon bias - the uneven use of synonymous codons in the transcriptome - serves as a secondary genetic code: a code that guides the efficiency of protein production, the fidelity of translation and the metabolism of mRNAs. Recent advancements in our understanding of mRNA decay have revealed a tight coupling between ribosome dynamics and the stability of mRNA transcripts; this coupling integrates codon bias into the concept of codon optimality, or the effects that specific codons and tRNA concentrations have on the efficiency and fidelity of the translation machinery. In this Review, we first discuss the evidence for codon-dependent effects on translation, beginning with the basic mechanisms through which translation perturbation can affect translation efficiency, protein folding and transcript stability. We then discuss how codon effects are leveraged by the cell to tailor the proteome to maintain homeostasis, execute specific gene expression programmes of growth or differentiation and optimize the efficiency of protein production.
[本文引用: 1]
DOI:10.1038/embor.2011.262URL [本文引用: 2]
One of the open questions in regulatory genomics is how the efficiency of gene translation is encoded in the coding sequence. Here we analyse recently generated measurements of folding energy in Saccharomyces cerevisiae, showing that genes with high protein abundance tend to have strong mRNA folding (mF; R = 0.68). mF strength also strongly correlates with ribosomal density and mRNA levels, suggesting that this relation at least partially pertains to the efficiency of translation elongation, presumably by preventing aggregation of mRNA molecules.
DOI:10.1016/j.molcel.2017.08.015URLPMID:28918899 [本文引用: 1]
Initiation is the rate-limiting step of translation, and in bacteria, mRNA secondary structure has been extensively reported as limiting the efficiency of translation by occluding the ribosome-binding site. In striking contrast with this inhibitory effect, we report here that stem-loop structures located within coding sequences instead activate translation initiation of the Escherichia coli fepA and bamA mRNAs involved in iron acquisition and outer membrane proteins assembly, respectively. Both structures promote ribosome binding in vitro, independently of their nucleotide sequence. Moreover, two small regulatory RNAs, OmrA and OmrB, base pair to and most likely disrupt the fepA stem-loop structure, thereby repressing FepA synthesis. By expanding our understanding of how mRNA cis-acting elements regulate translation, these data challenge the widespread view of mRNA secondary structures as translation inhibitors and show that translation-activating elements embedded in coding sequences can be targeted by small RNAs to inhibit gene expression.
DOI:10.1093/nar/gku1313URLPMID:25505165 [本文引用: 1]
The codon composition of the coding sequence's (ORF) 5' end first few dozen codons is known to be distinct to that of the rest of the ORF. Various explanations for the unusual codon distribution in this region have been proposed in recent years, and include, among others, novel regulatory mechanisms of translation initiation and elongation. However, due to the fact that many overlapping regulatory signals are suggested to be associated with this relatively short region, its research is challenging. Here, we review the currently known signals that appear in this region, the theories related to the way they regulate translation and affect the organismal fitness, and the debates they provoke.
DOI:10.1093/nar/gnh076URLPMID:15163763 [本文引用: 1]
The key step in bacterial translation is formation of the pre-initiation complex. This requires initial contacts between mRNA, fMet-tRNA and the 30S subunit of the ribosome, steps that limit the initiation of translation. Here we report a method for improving translational initiation, which allows expression of several previously non-expressible genes. This method has potential applications in heterologous protein synthesis and high-throughput expression systems. We introduced a synthetic RNA stem-loop (stem length, 7 bp; DeltaG(0) = -9.9 kcal/mol) in front of various gene sequences. In each case, the stem-loop was inserted 15 nt downstream from the start codon. Insertion of the stem-loop allowed in vitro expression of five previously non-expressible genes and enhanced the expression of all other genes investigated. Analysis of the RNA structure proved that the stem-loop was formed in vitro, and demonstrated that stabilization of the ribosome binding site is due to stem-loop introduction. By theoretical RNA structure analysis we showed that the inserted RNA stem-loop suppresses long-range interactions between the translation initiation domain and gene-specific mRNA sequences. Thus the inserted RNA stem-loop supports the formation of a separate translational initiation domain, which is more accessible to ribosome binding.
DOI:10.1093/nar/gku159URLPMID:24561808 [本文引用: 1]
Messenger RNA (mRNA) secondary structure decreases the elongation rate, as ribosomes must unwind every structure they encounter during translation. Therefore, the strength of mRNA secondary structure is assumed to be reduced in highly translated mRNAs. However, previous studies in vitro reported a positive correlation between mRNA folding strength and protein abundance. The counterintuitive finding suggests that mRNA secondary structure affects translation efficiency in an undetermined manner. Here, we analyzed the folding behavior of mRNA during translation and its effect on translation efficiency. We simulated translation process based on a novel computational model, taking into account the interactions among ribosomes, codon usage and mRNA secondary structures. We showed that mRNA secondary structure shortens ribosomal distance through the dynamics of folding strength. Notably, when adjacent ribosomes are close, mRNA secondary structures between them disappear, and codon usage determines the elongation rate. More importantly, our results showed that the combined effect of mRNA secondary structure and codon usage in highly translated mRNAs causes a short ribosomal distance in structural regions, which in turn eliminates the structures during translation, leading to a high elongation rate. Together, these findings reveal how the dynamics of mRNA secondary structure coupling with codon usage affect translation efficiency.
DOI:10.1073/pnas.1606724113URL [本文引用: 2]
DOI:10.1016/j.cell.2015.02.029URLPMID:25768907 [本文引用: 2]
mRNA degradation represents a critical regulated step in gene expression. Although the major pathways in turnover have been identified, accounting for disparate half-lives has been elusive. We show that codon optimality is one feature that contributes greatly to mRNA stability. Genome-wide RNA decay analysis revealed that stable mRNAs are enriched in codons designated optimal, whereas unstable mRNAs contain predominately non-optimal codons. Substitution of optimal codons with synonymous, non-optimal codons results in dramatic mRNA destabilization, whereas the converse substitution significantly increases stability. Further, we demonstrate that codon optimality impacts ribosome translocation, connecting the processes of translation elongation and decay through codon optimality. Finally, we show that optimal codon content accounts for the similar stabilities observed in mRNAs encoding proteins with coordinated physiological function. This work demonstrates that codon optimization exists as a mechanism to finely tune levels of mRNAs and, ultimately, proteins.
DOI:10.1016/j.jmb.2008.06.068URLPMID:18619977 [本文引用: 1]
Individual mRNAs are translated by multiple ribosomes that initiate translation with an interval of a few seconds. The ribosome speed is codon dependent, and ribosome queuing has been suggested to explain specific data for translation of some mRNAs in vivo. By modeling the stochastic translation process as a traffic problem, we here analyze conditions and consequences of collisions and queuing. The model allowed us to determine the on-rate (0.8 to 1.1 initiations/s) and the time (1 s) the preceding ribosome occludes initiation for Escherichia coli lacZ mRNA in vivo. We find that ribosome collisions and queues are inevitable consequences of a stochastic translation mechanism that reduce the translation efficiency substantially on natural mRNAs. The cells minimize collisions by having its mRNAs being unstable and by a highly selected codon usage in the start of the mRNA. The cost of mRNA breakdown is offset by the concomitant increase in translation efficiency.
DOI:10.1016/j.molcel.2015.07.018URLPMID:26321254 [本文引用: 1]
Codon usage bias is a universal feature of eukaryotic and prokaryotic genomes and has been proposed to regulate translation efficiency, accuracy, and protein folding based on the assumption that codon usage affects translation dynamics. The roles of codon usage in translation, however, are not clear and have been challenged by recent ribosome profiling studies. Here we used a Neurospora cell-free translation system to directly monitor the velocity of mRNA translation. We demonstrated that the preferred codons enhance the rate of translation elongation, whereas non-optimal codons slow elongation. Codon usage also controls ribosome traffic on mRNA. These conclusions were supported by ribosome profiling results in vitro and in vivo with template mRNAs designed to increase the signal-to-noise ratio. Finally, we demonstrate that codon usage regulates protein function by affecting co-translational protein folding. These results resolve a long-standing fundamental question and suggest the existence of a codon usage code for protein folding.
DOI:10.1038/srep10775URLPMID:26053859 [本文引用: 1]
Differential mRNA expression studies implicitly assume that changes in mRNA expression have biological meaning, most likely mediated by corresponding changes in protein levels. Yet studies into mRNA-protein correspondence have shown notoriously poor correlation between mRNA and protein expression levels, creating concern for inferences from only mRNA expression data. However, none of these studies have examined in particular differentially expressed mRNA. Here, we examined this question in an ovarian cancer xenograft model. We measured protein and mRNA expression for twenty-nine genes in four drug-treatment conditions and in untreated controls. We identified mRNAs differentially expressed between drug-treated xenografts and controls, then analysed mRNA-protein expression correlation across a five-point time-course within each of the four experimental conditions. We evaluated correlations between mRNAs and their protein products for mRNAs differentially expressed within an experimental condition compared to those that are not. We found that differentially expressed mRNAs correlate significantly better with their protein product than non-differentially expressed mRNAs. This result increases confidence for the use of differential mRNA expression for biological discovery in this system, as well as providing optimism for the usefulness of inferences from mRNA expression in general.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}