 ,1, 陈宏伟2, 廖芳丽3, 李莉2, 刘昌燕2, 刘良军2, 万正煌,2, 沙爱华,1
,1, 陈宏伟2, 廖芳丽3, 李莉2, 刘昌燕2, 刘良军2, 万正煌,2, 沙爱华,1Analysis of F-Box Gene Family Based on Salt-Stressed Transcriptome Sequencing in Vicia faba L.
HAO ShuLin,1, CHEN HongWei2, LIAO FangLi3, LI Li2, LIU ChangYan2, LIU LiangJun2, WAN ZhengHuang,2, SHA AiHua,1通讯作者:
责任编辑: 李莉
收稿日期:2019-12-19接受日期:2020-03-5网络出版日期:2020-09-01
| 基金资助: |
Received:2019-12-19Accepted:2020-03-5Online:2020-09-01
作者简介 About authors
郝树琳,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (6356KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郝树琳, 陈宏伟, 廖芳丽, 李莉, 刘昌燕, 刘良军, 万正煌, 沙爱华. 基于盐胁迫转录组信息的蚕豆F-box基因家族分析[J]. 中国农业科学, 2020, 53(17): 3443-3454 doi:10.3864/j.issn.0578-1752.2020.17.003
HAO ShuLin, CHEN HongWei, LIAO FangLi, LI Li, LIU ChangYan, LIU LiangJun, WAN ZhengHuang, SHA AiHua.
0 引言
【研究意义】蚕豆(Vicia faba L.)是巢菜属一年生或越年生草本植物,有生物固氮之王的美誉,具有较高经济价值[1]。在蚕豆生长发育过程中,生物和非生物胁迫等因素对蚕豆产生影响,最终导致其产量和品质下降,造成经济损失。盐胁迫是其中一种主要的非生物胁迫,严重危害蚕豆的生长发育。F-box基因广泛参与植物生长发育及响应非生物胁迫途径,如细胞蛋白质降解、激素感知和信号传导等[2,3,4]。因此,研究蚕豆F-box基因功能和培育蚕豆耐盐品种具有重要意义。【前人研究进展】自然条件下,植物的生长会受到内部或外部的刺激,为了应对产生的刺激,植物已经发展出多种调节机制来协调它们的生命活动。其中,泛素26s蛋白酶体系统(ubiquitin-26s proteasome system,UPS)降解蛋白是一种重要的翻译后调控机制,并在真核生物中高度保守[5]。UPS催化多种泛素与靶底物的共价连接,使底物被26S蛋白酶体识别并进行蛋白水解。泛素化主要由泛素激活酶(ubiquitin-activating enzymes,E1s)、泛素偶联酶(ubiquitin-conjugating enzymes,E2s)和泛素蛋白连接酶(ubiquitin protein ligases,E3s)3种酶来完成。其中,E3s起到关键的底物识别功能[6]。F-box蛋白作为E3s的核心成分,即SCF(Skp1-Cul1-F-box)复合物,介导不同底物的特异性降解[7]。F-box基因广泛存在于各种植物,是植物中最大、进化最快的基因家族之一,越来越多的真核F-box基因被发现[8,9,10,11]。F-box基因在不同物种间的数量相差较大。动物体内的F-box基因数量相对较少,秀丽线虫是目前发现含有F-box基因最多的动物,具有326个,而人体内仅含有38个,果蝇只含有22个[12];植物中F-box基因家族成员个数远多于动物,模式植物苜蓿和拟南芥分别含有972和694个[13,14],农作物水稻、大豆、玉米和鹰嘴豆分别含有687、509、359和285个[15,16],经济作物苹果和梨分别含有517和226个[17,18]。F-box结构域一般位于蛋白的N端,含有40—50个氨基酸,与SKP1相互作用形成SCF复合物,而C端通常是介导底物识别特异性的二级结构[14]。在哺乳动物中,根据C末端二级结构的不同,F-box蛋白可分为3类。具有LRR结构域的FBXL,具有WD40重复结构域的FBXW和具有其他结构域的FBXO[19]。在植物中,通过对拟南芥和水稻F-box蛋白的全基因组分析,发现了大量结构域,如WD40(Tryptophan-aspartic acid 40)、kelch重复序列、LRR(Leucine-rich repeat)、FBD(F-Box and BRCT domain)、环指结构、PAS/PAC(Per Arnt Sim)、PPR(pentatricopeptide repeat)和TUB(tubby)等[15]。因此,F-box蛋白可以识别多种类型的底物,在植物生长发育、生物和非生物胁迫、激素信号转导等生物学过程中发挥重要作用。UFO是植物中发现的第一个F-box蛋白,在花分生组织鉴定和花器官发育中具有重要作用[20]。随着深入研究,越来越多的F-box蛋白被发现,并且这些蛋白的功能变得多样化。在拟南芥中,许多F-box蛋白参与植物激素反应。例如,含有F-box结构域的TIR1是拟南芥的生长素受体[21]。AFB 1-3是一种生长素信号转导的F-box蛋白,可以结合类似TIR1的Aux/IAA蛋白,促进生长素的应答[22]。F-box蛋白也可调控赤霉素信号通路,例如拟南芥的SLEEPY1(SLY1)和SNEEZY(SNE),水稻中的赤霉素不敏感矮杆2(GID2)[23]。在乙烯信号转导途径中,乙烯不敏感3(EIN3)通过结合F-box1/2(EBF1/2)进行降解[24]。COI1也属F-box蛋白,它是茉莉酸(JA)信号反应的一个重要因子,也是拟南芥JA依赖性反应必需的[25]。此外,F-box蛋白LKP1/ZTL、LKP2/FKL和FKF1参与光形态发生、昼夜节律和开花时间[26]。DOR是拟南芥中与ASK14和CUL1特异性相互作用的F-box蛋白,在耐旱条件下起负调控作用[27]。水稻F-box基因OsFbx352与葡萄糖延迟种子萌发有关[28]。此外,早期研究中也发现了一些植物的S位点F-box基因,证明其参与花粉管伸长,甚至参与自交不亲和[29]。【本研究切入点】目前,对于蚕豆的研究多集中在生理层面,在蚕豆中对F-box基因家族的鉴定和系统分析鲜见报道。【拟解决的关键问题】本研究利用生物信息学的方法对蚕豆F-box基因家族成员进行鉴定,并对其基因结构、亚细胞定位、蛋白保守结构域及系统进化等进行分析,同时利用qRT-PCR技术对F-box家族成员响应盐胁迫的差异表达模式进行分析,为后续研究蚕豆F-box基因的功能提供一定的理论依据。1 材料与方法
1.1 材料
蚕豆(Vicia faba L.)种子由湖北省农业科学院杂粮研究工程中心提供。于2018年9月,选取饱满、大小均等的2个不同品种(Y134耐盐和Y078不耐盐)的蚕豆种子进行发芽试验,用0.8% NaCl处理,分别于16和24 h收集具有胚胎1/4部分子叶的材料,每个处理设置3个生物重复,材料收集后液氮速冻,-80℃保存备用。试验于2018年在长江大学农学院耐渍油料种质资源湖北省重点实验室进行。
1.2 蚕豆总RNA的提取、文库构建及转录组测序
植物总RNA提取采用TRNzol试剂法(TRNzol Universal Reagent,天根生化科技有限公司,北京,中国)分别提取蚕豆芽期胚的总RNA,并用TGem spectrophotometer Plus(天根生化科技有限公司,北京,中国)微量分光光度计检测RNA纯度(OD260/280比值)以及RNA浓度,最后用1%琼脂糖凝胶电泳检测。检测合格的样品送到广州赛哲生物科技股份有限公司进行转录组测序,并通过De Novo方法[30]组装得到116 093条Unigene,后续分析基于此测序结果进行。1.3 蚕豆和拟南芥F-box基因的筛选
以盐胁迫转录组数据为基础,在NR、Swiss-prot和Pfam 3个数据库中进行注释,初步注释到451个F-box相关基因,并用TBtools软件[31]批量提取相应的蛋白序列。将这些蛋白序列于NCBI进行BLAST搜索比对,同时利用CDD(conserved domain database)在线工具(1.4 蚕豆F-box基因家族保守域及其进化关系分析
利用序列分析软件Clustal W对161个蚕豆F-box蛋白进行多序列比对,去除保守域之外的区域,通过在线软件Web Logo 3(1.5 蚕豆F-box基因家族亚细胞定位预测、基因结构及Motif分析
利用在线软件Prot Comp 9.0(1.6 蚕豆F-box基因的表达模式分析与验证
蚕豆转录组测序文库中各基因片段表达量的计算采用FPKM(fragments per kilobase million)法[33]。将数据标准化后,利用TBtools软件对F-box基因在2个不同处理时间和2个不同品种间的表达绘制热图。以蚕豆2个不同处理时间的2个品种为材料,选取了5个在处理16 h上、下调明显的F-box基因,进行实时荧光定量PCR。各样品RNA采用第一链cDNA合成试剂盒(Promega,Madison,USA)反转录cDNA,选用蚕豆VfNADHD4作内参[34],采用Primer Premier 5.0设计荧光定量引物(表1),反应体系为模板cDNA 1 μL、正/反向引物各0.5 μL(10 μmol·L-1)、2×RealUniversal PreMix(SYBR Green, blue,FP201,TIANGEN,北京,中国)10 μL和ddH2O 8 μL。PCR反应程序为95℃ 15 min;95℃ 10 s,57℃ 20 s,72℃ 30 s,40个循环。每个样品设置3次技术重复。所用仪器为美国伯乐(BIO-RAD,CA,USA)CFX Connect实时荧光定量PCR仪,采用2-△△CT方法计算相对表达量的[35]。Table 1
表1
表1实时荧光定量PCR所用的引物
Table 1
| 基因Gene | 正向引物序列Forward primer sequence (5′-3′) | 反向引物序列Reverse primer sequence (5′-3′) |
|---|---|---|
| Vf056266.1 | CACGCCCAAAATCTCAAGGT | AAGTTGCTAGGCCTTCCAGT |
| Vf060904.1 | TGAACGAAACTGGCTTGA | CGATGGTGCGATAGAGGA |
| Vf062764.1 | TTACCAATAACCCTTCCAC | TCAATACGATTAGCACCCT |
| Vf024236.1 | CTGCTGCTGTTGTGAAGA | AACCAAACGGGAGAAGAT |
| Vf045761.1 | CCGTCTCATACTATCGTCTGTT | GGGTCGGCTTGGGAGGAAT |
| VfNADHD4 | AGGGTTAGTGAGCACCATGC | ATAGCCAAAGGGAATACGCC |
新窗口打开|下载CSV
2 结果
2.1 蚕豆F-box基因家族的鉴定及分类
基于盐胁迫转录组测序数据,在NR、Swiss-prot和Pfam 3个数据库中进行注释,初步注释到451个F-box相关基因,通过NCBI BLAST预测,去除重复序列和冗余转录本后,最终得到161个蚕豆F-box基因。利用SMART和CDD在线工具对所得到的161条F-box蛋白序列进行预测,并根据WANG等[18]对梨F-box基因家族的分类方法,按照每个F-box基因所包含的C端保守结构域类型,将其分为FBX、FBXFBA、FBXLRR、FBXPP2、FBXKelch、FBXTUB、FBXFBD、FBXDUF、FBXACTIN、FBXWD40和FBO 11类。其中FBX类基因64个,FBXFBA类基因27个,FBXLRR类基因19个,FBXPP2类基因8个,FBXKelch类基因16个,FBXTUB类基因7个,FBXFBD类基因10个,FBXDUF类基因2个,FBXACTIN类基因2个,FBXWD40类基因1个,FBO类基因5个。
2.2 蚕豆F-box蛋白的保守基序分析
利用序列分析软件Clustal W对161个蚕豆F-box蛋白进行多序列比对,去除保守域之外的不保守区域,对其保守结构域进行基序分析。通常F-box蛋白在N端含有40—50个氨基酸的F-box基序[18]。由图1蚕豆F-box蛋白的保守基序图可知,其约含49个氨基酸残基,包括1个极度保守的色氨酸残基(W)。此外,在F-box蛋白的保守基序中还存在其他保守氨基酸残基,如第4位的亮氨酸残基(L)、第5位的脯氨酸残基(P)、第12位的苏氨酸残基(T)、第30位的缬氨酸残基(V)。这些保守的氨基酸残基可能与色氨酸残基共同维持蚕豆F-box基序的α螺旋结构[36],进而发挥其特定的功能。图1
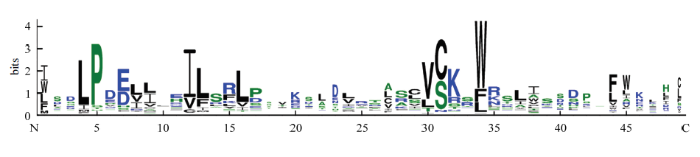 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1蚕豆F-box蛋白保守基序
Fig. 1Conserved Motif of Vicia faba L.F-box protein
2.3 蚕豆与拟南芥F-box基因的进化关系与功能预测分析
利用MEGA-X软件构建蚕豆与拟南芥F-box蛋白家族的系统进化树,根据进化树(图2)的拓扑结构将2个物种的F-box家族蛋白进行聚类分析。研究表明,对植物的F-box蛋白进行系统发育分析时,具有相同C端结构域的F-box蛋白倾向于聚集在系统发育树的同一类群中[11]。因此,根据不同的特异性区域将F-box基因家族成员分为11个不同的亚群。FBX亚族(64)是由只含有F-box结构域的基因组成,不包含其他任何特定结构域,其成员数量多于其他子群。其他10个亚族由包含一个不可缺少的F-box结构域和其他可变结构域的基因组成,如LRR(命名为FBXLRR)、Tub(FBXTUB)、Kelch(FBXKelch)、FBA (FBXFBA)、FBD(FBXFBD)等。例如,FBXFBA和FBXFBD亚群分别表示含有FBA结构域或FBD结构域而没有其他可变域的基因。为了验证上述分类,利用蚕豆F-box全长序列与部分功能明确的拟南芥全长序列共同构建了一个系统发育树,事实上,观察到一些分组成员聚集在一起,例如FBXFBA、FBXFBD、FBXTUB和FBXPP2,这在一定程度上支持了基于C端结构域分类的方法。图2
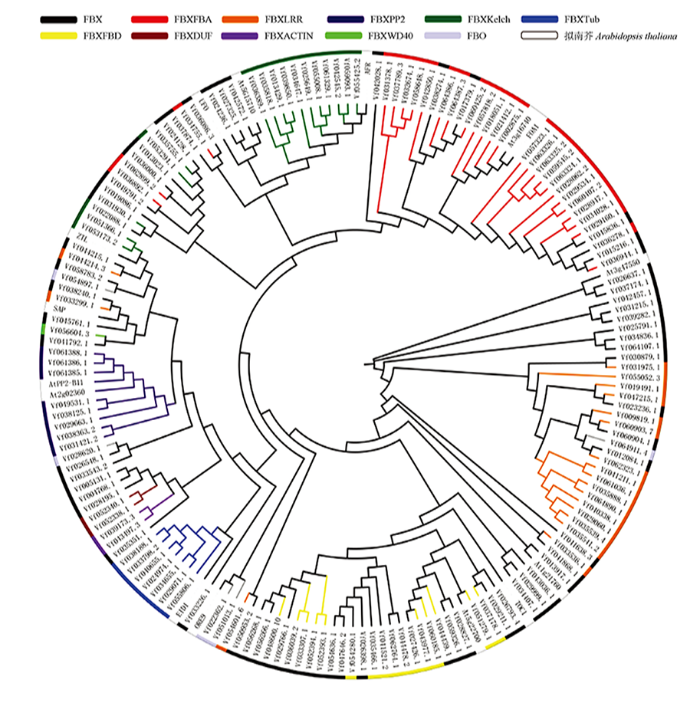 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2蚕豆与拟南芥F-box蛋白家族的进化分析
Fig. 2Phylogenetic analysis of F-box protein in Vicia faba L. and Arabidopsis thaliana
由图2可知,一些F-box亚族与拟南芥F-box蛋白进化关系较近。例如,FBXFBA亚族中Vf057818.2、Vf018051.2、Vf021412.1、Vf002875.1与拟南芥At3g16740、FOA1位于同一进化亚枝上;FBX亚族中Vf026637.1、Vf037174.1与拟南芥At3g47550w位于同一进化亚枝上,Vf026793.4、Vf029211.1与拟南芥TOC1进化关系较近;FBXFBD亚族中Vf051279.1与拟南芥At5g22700位于同一进化亚枝上;FBXTub亚族与拟南芥EID1位于同一进化亚枝上;FBXPP2亚族与拟南芥AtPP2-B11、At2g02360位于同一进化亚枝上;FBXKelch亚族与拟南芥AFR位于同一进化亚枝,表明它们可能具有相似的功能。
2.4 蚕豆F-box基因的亚细胞定位预测
通过在线软件Prot Comp 9.0(http://linux.softberry. com)对161个蚕豆F-box个成员的氨基酸序列进行亚细胞定位预测(图3),结果显示,共有124个候选F-box基因位于细胞外,其中FBX亚族多达59个,占候选基因总数的47.58%,说明这些F-box蛋白可能参与胞外某些特定的生物学过程;37个位于细胞核中,其中FBXPP2亚族多达8个,占候选基因总数的21.62%,表明其可能通过调控细胞核基因的转录和翻译来发挥特定功能。图3
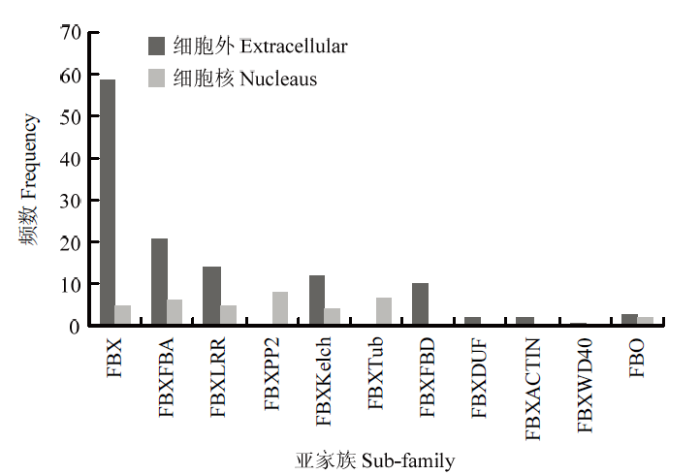 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3蚕豆F-box基因家族的亚细胞定位预测
Fig. 3Sub-cellular localization of F-box gene family in Vicia faba L.
2.5 蚕豆F-box基因的基因结构及Motif分析
根据基因组注释信息,利用TBtools软件对F-box基因的基因结构进行可视化分析(图4),发现F-box基因家族成员无内含子结构,且每一个成员都含有UTR区和CDS区。根据F-box家族成员的进化关系可知,同一亚族的编码区长度大致相同。因此,基因结构分析在一定程度上也支持了基于C端结构域分类的方法。利用在线软件MEME(http://meme-suite.org/)分析蚕豆F-box基因家族成员基序类型和排列顺序。如图4所示,F-box基因家族预测含有20个Motif,F-box基因家族成员之间所包含的Motif数目及种类存在一定的差异。除了Vf036806.3和Vf033536.1不含Motif1外,其他均含有Motif1,相比Motif1,Motif2在蚕豆F-box基因家族中有60个成员不含该基序,Motif3在蚕豆F-box基因家族中有131个成员不含该基序。Motif4出现29次,Motif5出现17次,Motif6出现21次,Motif7出现35次,Motif8出现5次,Motif9出现11次,Motif10出现7次,Motif11出现51次,Motif12出现40次,Motif13出现39次,Motif14出现40次,Motif15出现42次,Motif16出现6次,Motif17出现6次,Motif18出现22次,Motif19出现25次,Motif20出现7次。结果表明,出现频率较高的Motif为F-box基因家族中非常重要的保守基序。对于出现频率较低的Motif,通过Smart在线分析,其功能的相关信息未知,有待日后进一步深入研究。
图4
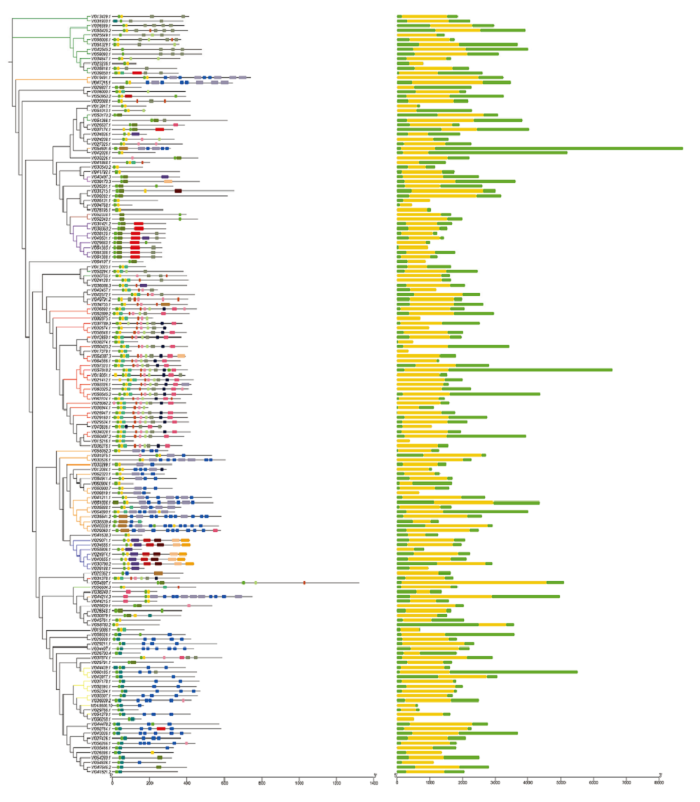 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4蚕豆F-box基因家族结构和Motif分析
Fig. 4Family structure and Motif analysis of F-box gene in Vicia faba L.
2.6 蚕豆F-box基因在盐处理不同时间的表达模式分析
基因的表达模式可以为基因功能提供重要的依据,从蚕豆盐胁迫转录组测序结果的基因表达数据库中,分别提取161个F-box家族基因在不同品种(Y134 T2-T1为以Y078(不耐盐)品种16 h盐处理表达量为参照,Y134(耐盐)品种16 h盐处理与之相比较的差异表达量;CK2-CK1为以Y078(不耐盐)品种16 h对照的表达量为参照,Y134(耐盐)品种16 h对照与之相比较的差异表达量,同理T4-T3为以Y078(不耐盐)品种24 h盐处理表达量为参照,Y134(耐盐)品种24 h盐处理与之相比较的差异表达量;CK4-CK3为以Y078(不耐盐)品种24 h盐处理表达量为参照,Y134(耐盐)品种24 h盐处理与之相比较的差异表达量。由图5可知,随着盐处理时间的增加,蚕豆F-box基因的表达模式出现差异化。通过横向比对每个家族成员的表达差异,发现大部分成员在不同的时间节点均有高表达和低表达,一部分成员在16 h处理表达量明显升高,还有一些成员在24 h处理表达量明显降低。说明这些家族成员可能在盐胁迫前16 h中参与主要作用。为了进一步验证上述结果,并确定F-box基因在蚕豆盐胁迫下的参与情况,采用荧光定量PCR,在蚕豆2个品种和2个处理时间中进行差异表达分析(图6)。选取5个差异表达基因(在盐胁迫的2个时间点,以至少以一个时间节点的FDR<0.05,且差异倍数2倍及2倍以上的基因为差异表达基因),其中3个上调基因,2个下调基因。结果表明,5个蚕豆F-box基因的表达变化各不相同,其中Vf056266.1在盐处理16和24 h的表达量上调;Vf060904.1在盐处理16 h的表达量下调非常明显,盐处理24 h表达量无显著差异;Vf062764.1在盐处理16 h上调非常明显,在盐处理24 h不明显,后者与转录组数据不符;Vf024236.1在盐处理16 h表达量上调,盐处理24 h表达量上调不明显;Vf045761.1在盐处理16 h下调非常明显,盐处理24 h表达量上调,这与转录组数据不符。盐处理16 h,qRT-PCR检测到的5个基因,有4个基因相对表达量与转录组数据基本一致。图5
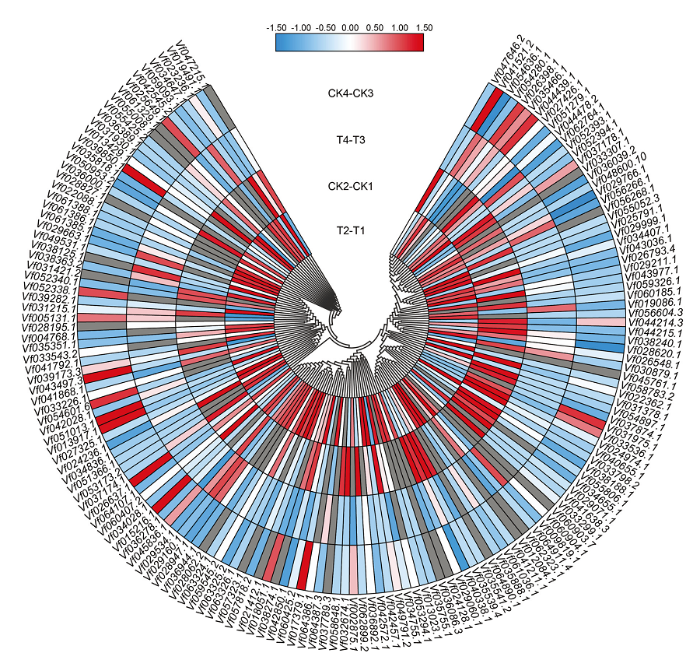 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5蚕豆F-box基因在盐胁迫下的表达模式
此图基于蚕豆盐胁迫转录组测序数据的计算得出,每种颜色代表相应表达量的数值,数值越大,表达量越高
Fig. 5The pattern of expression of F-box family in salt stress of Vicia faba L.
The heat map was generated based on the calculation of sequencing data of salt stress of Vicia faba L.. Each kind of color represents the value of the corresponding relative expression. The higher the value, the higher the expression
图6
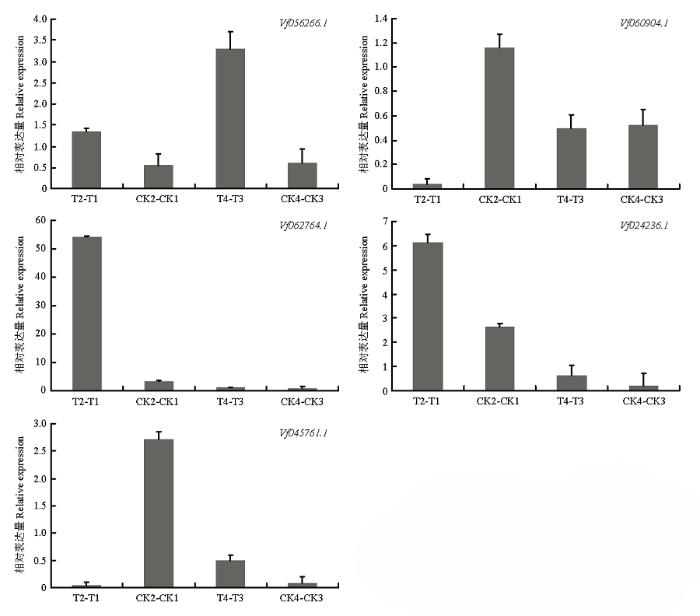 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6蚕豆F-box基因的实时荧光定量PCR
Fig. 6F-box genes of Vicia faba L.for salt stress based on qRT-PCR
3 讨论
在植物中,F-box作为一个较大的基因家族,它们参与了多种生物途径,在整个植物生命周期的一系列生物学过程中发挥重要作用。然而,大多数植物F-box基因的功能仍然未知,特别是在蚕豆中。由于蚕豆的全基因组序列还未公布,在很大程度上限制了其重要性状基因的研究。本研究基于蚕豆盐胁迫转录组数据,对蚕豆F-box基因家族进行系统分析。根据基因结构、保守基序、系统发育关系将其划分为不同亚族。在不同品种的不同盐胁迫下,对其表达模式进行了分析,并通过qRT-PCR进行了验证。通过对蚕豆F-box蛋白的保守结构域进行基序分析,得出F-box基序中包含一个极度保守的色氨酸残基,这与其他植物相类似。因此,蚕豆也有可能普遍形成SCF复合物。此外,大多数已知的蚕豆C端结构域也存在于其他植物中,表明进化基因的保守性以及F-box蛋白在不同植物中的相似性。根据C端结构域的不同,蚕豆F-box蛋白可分为11个亚家族,其中最丰富的F-box蛋白属于FBX亚家族,含有未知的C端结构域。Motif分析结果表明,大多数基序也存在于其他植物中,而少数基序是豆科或蚕豆特有的。与其他已知的C端保守结构域一样,这些推测的新基序也可能在F-box蛋白与其底物的相互作用中发挥作用。那些含有豆科或蚕豆特异性基序的蛋白可能具有与豆科特异性细胞过程相关的功能,但具体功能有待深入研究。
本文使用基于C端底物相互作用域的F-box基因分类标准,再次肯定了之前基于N端F-box基序的分类,说明N端F-box基序可能与C端基序共同进化,这一点与其他植物相似[13]。此外,F-box基序生成的系统发育树与基因结构分析的结果是一致的,说明系统发育树准确地代表了蚕豆F-box基因家族的系统发育关系。然而,蚕豆F-box家族基因结构中无内含子,无内含子F-box基因在拟南芥、水稻、玉米、鹰嘴豆和胡杨中所占比例较高[16]。由亚细胞定位预测结果可知,蚕豆F-box蛋白主要存在于细胞外和细胞核中,少数在细胞质、细胞质膜和液泡中。大量的豆类F-box蛋白被预测有多个亚细胞定位。据报道,在拟南芥中检测到17个F-box蛋白大部分定位于胞腔内,单个F-box蛋白可以形成多个SCF复合物[37]。这些都说明了F-box蛋白在调控植物的生物学过程中具有多种作用。
基因表达模式与基因功能关系十分密切,通过研究蚕豆盐胁迫过程中基因表达的变化,可发现参与盐胁迫的相关功能基因。F-box基因被证实参与非生物胁迫反应,ZHAO等[3]发现过表达的小麦TaFBA1的转基因烟草各项生理指标均高于野生型。JIA等[38]发现拟南芥F-box基因AtPP2-B11通过抑制ROS积累和影响体内Na+平衡来提高植株的耐盐性;AN等[39]研究发现MdMAX2超表达能够提高植株的耐盐和耐旱性。本文基于蚕豆盐胁迫转录组数据,分析F-box基因在蚕豆盐胁迫下的差异表达模式,筛选出5个表达差异显著的基因,用于进一步qRT-PCR分析。qRT-PCR验证发现,有3个表达上调的差异基因和2个表达下调的差异基因,说明它们可能在蚕豆遭受盐胁迫过程中起着正向或负向调控作用。为研究蚕豆中参与盐胁迫过程中的相关F-box基因的功能和机制提供基础。
拟南芥F-box家族蛋白功能的研究报道很多,UFO参与调节花器官发育[40],AtPP2-B11对耐旱性起负调控作用,提高盐胁迫抗性,并参与ABA信号转导过程[38],AtTR1参与盐胁迫反应[41]。可根据进化树中位于同一进化亚枝中的拟南芥蛋白功能,预测研究物种蛋白的功能,严莉等[42]通过构建拟南芥与黑果枸杞的MYB转录因子的系统进化树,将位于相邻位置的黑果枸杞和拟南芥MYB转录因子列为具有相同或相似结构的转录因子,从而推测其功能的一致性。本研究所得的161个蚕豆F-box蛋白与15个功能明确的拟南芥F-box蛋白作系统进化树,根据15个拟南芥F-box蛋白进化关系的远近推测蚕豆F-box蛋白的功能。如Vf002875.1与At3g16740相邻,根据拟南芥At3g16740的功能推测Vf002875.1可能参与影响种子萌发的过程[43];推测Vf061385.1可能与拟南芥AtPP2-B11功能一致,参与盐胁迫反应[38];Vf026793.4与拟南芥TOC1进化关系较近,可能参与生物钟调节[44]。通过对拟南芥F-box蛋白保守结构域的搜索发现,含有同一C端结构域的蛋白进化到同一亚枝或进化关系较近,证明相邻或较近进化关系上的蛋白有可能具有相同或相似的功能,为研究F-box蛋白的功能提供了一种思路。此外,在后续研究中,将对F-box基因家族成员进行转基因试验,分析其在盐胁迫及其他非生物胁迫中的具体表型影响。
4 结论
基于蚕豆盐胁迫转录组测序数据,筛选注释了161个蚕豆F-box基因,且均含有F-box保守结构域。根据C末端氨基酸序列不同使家族成员多样化,分为11个亚家族,家族成员均无内含子结构,且家族成员在胞外和细胞核中均存在。Vf056266.1、Vf062764.1和Vf024236.1正调控盐胁迫,Vf060904.1和Vf045761.1负调控盐胁迫,且盐胁迫下的F-box家族成员在不同处理时间下呈现出不同的响应。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1073/pnas.0812043106URLPMID:19126682 [本文引用: 2]

F-box proteins are substrate-recognition components of the Skp1-Rbx1-Cul1-F-box protein (SCF) ubiquitin ligases. In plants, F-box genes form one of the largest multigene superfamilies and control many important biological functions. However, it is unclear how and why plants have acquired a large number of F-box genes. Here we identified 692, 337, and 779 F-box genes in Arabidopsis, poplar and rice, respectively, and studied their phylogenetic relationships and evolutionary patterns. We found that the plant F-box superfamily can be divided into 42 families, each of which has a distinct domain organization. We also estimated the number of ancestral genes for each family and identified highly conservative versus divergent families. In conservative families, there has been little or no change in the number of genes since the divergence between eudicots and monocots approximately 145 million years ago. In divergent families, however, the numbers have increased dramatically during the same period. In two cases, the numbers of genes in extant species are >100 times greater than that in the most recent common ancestor (MRCA) of the three species. Proteins encoded by highly conservative genes always have the same domain organization, suggesting that they interact with the same or similar substrates. In contrast, proteins of rapidly duplicating genes sometimes have quite different domain structures, mainly caused by unusually frequent shifts of exon-intron boundaries and/or frameshift mutations. Our results indicate that different F-box families, or different clusters of the same family, have experienced dramatically different modes of sequence divergence, apparently having resulted in adaptive changes in function.
[本文引用: 1]
DOI:10.1007/s10142-015-0438-zURLPMID:25877816 [本文引用: 2]
F-box protein is a subunit of Skp1-Rbx1-Cul1-F-box protein (SCF) complex with typically conserved F-box motifs of approximately 40 amino acids and is one of the largest protein families in eukaryotes. F-box proteins play critical roles in selective and specific protein degradation through the 26S proteasome. In this study, we bioinformatically identified 972 putative F-box proteins from Medicago truncatula genome. Our analysis showed that in addition to the conserved motif, the F-box proteins have several other functional domains in their C-terminal regions (e.g., LRRs, Kelch, FBA, and PP2), some of which were found to be M. truncatula species-specific. By phylogenetic analysis of the F-box motifs, these proteins can be classified into three major families, and each family can be further grouped into more subgroups. Analysis of the genomic distribution of F-box genes on M. truncatula chromosomes revealed that the evolutional expansion of F-box genes in M. truncatula was probably due to localized gene duplications. To investigate the possible response of the F-box genes to abiotic stresses, both publicly available and customer-prepared microarrays were analyzed. Most of the F-box protein genes can be responding to salt and heavy metal stresses. Real-time PCR analysis confirmed that some of the F-box protein genes containing heat, drought, salicylic acid, and abscisic acid responsive cis-elements were able to respond to the abiotic stresses.
DOI:10.3390/ijms18040818URL [本文引用: 2]
URLPMID:17293439 [本文引用: 2]
DOI:10.1007/s00438-015-1004-zURLPMID:25855485 [本文引用: 2]
The F-box protein family is a large family that is characterized by conserved F-box domains of approximately 40-50 amino acids in the N-terminus. F-box proteins participate in diverse cellular processes, such as development of floral organs, signal transduction and response to stress, primarily as a component of the Skp1-cullin-F-box (SCF) complex. In this study, using a global search of the apple genome, 517 F-box protein-encoding genes (F-box genes for short) were identified and further subdivided into 12 groups according to the characterization of known functional domains, which suggests the different potential functions or processes that they were involved in. Among these domains, the galactose oxidase domain was analyzed for the first time in plants, and this domain was present with or without the Kelch domain. The F-box genes were distributed in all 17 apple chromosomes with various densities and tended to form gene clusters. Spatial expression profile analysis revealed that F-box genes have organ-specific expression and are widely expressed in all organs. Proteins that contained the galactose oxidase domain were highly expressed in leaves, flowers and seeds. From a fruit ripening expression profile, 166 F-box genes were identified. The expressions of most of these genes changed little during maturation, but five of them increased significantly. Using qRT-PCR to examine the expression of F-box genes encoding proteins with domains related to stress, the results revealed that F-box proteins were up- or down-regulated, which suggests that F-box genes were involved in abiotic stress. The results of this study helped to elucidate the functions of F-box proteins, especially in Rosaceae plants.
DOI:10.1186/1471-2164-16-1URL [本文引用: 1]
DOI:10.1016/j.plantsci.2016.09.009URLPMID:27968985 [本文引用: 3]
F-box gene family, as one of the largest gene families in plants, plays crucial roles in regulating plant development, reproduction, cellular protein degradation and responses to biotic and abiotic stresses. However, comprehensive analysis of the F-box gene family in pear (Pyrus bretschneideri Rehd.) and other Rosaceae species has not been reported yet. Herein, we identified a total of 226 full-length F-box genes in pear for the first time. And these genes were further divided into various subgroups based on specific domains and phylogenetic analysis. Intriguingly, we observed that whole-genome duplication and dispersed duplication have a major contribution to F-box family expansion. Furthermore, the dynamic evolution for different modes of gene duplication was dissected. Interestingly, we found that dispersed and tandem duplicate have been evolving at a high rate. In addition, we found that F-box genes exhibited functional specificity based on GO analysis, and most of the F-box genes were significantly enriched in the protein binding (GO: 0005515) term, supporting that F-box genes might play a critical role for gene regulation in pear. Transcriptome and digital expression profiles revealed that F-box genes are involved in the development of multiple pear tissues. Overall, these results will set stage for elaborating the biological role of F-box genes in pear and other plants.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00425-005-0138-3URLPMID:16244866 [本文引用: 1]
The UNUSUAL FLORAL ORGANS (UFO) gene of Arabidopsis encodes an F-box protein required for the determination of floral-organ and floral-meristem identity. Mutation of UFO leads to dramatic changes in floral-organ type which are well-characterized whereas inflorescence defects are more subtle and less understood. These defects include an increase in the number of secondary inflorescences, nodes that alternate between forming flowers and secondary inflorescences, and nodes in which a single flower is subtended by a bract. Here, we show how inflorescence defects correlate with the abnormal development of floral primordia and establish a temporal requirement for UFO in this process. At the inflorescence apex of ufo mutants, newly formed primordia are initially bract-like. Expression of the floral-meristem identity genes LFY and AP1 are confined to a relatively small adaxial region of these primordia with expression of the bract-identity marker FIL observed in cells that comprise the balance of the primordia. Proliferation of cells in the adaxial region of these early primordia is delayed by several nodes such that primordia appear
DOI:10.1038/nature03542URLPMID:15917798 [本文引用: 1]
Despite 100 years of evidence showing a pivotal role for indole-3-acetic acid (IAA or auxin) in plant development, the mechanism of auxin perception has remained elusive. Central to auxin response are changes in gene expression, brought about by auxin-induced interaction between the Aux/IAA transcriptional repressor proteins and the ubiquitin-ligase complex SCF(TIR1), thus targeting for them proteolysis. Regulated SCF-mediated protein degradation is a widely occurring signal transduction mechanism. Target specificity is conferred by the F-box protein subunit of the SCF (TIR1 in the case of Aux/IAAs) and there are multiple F-box protein genes in all eukaryotic genomes examined so far. Although SCF-target interaction is usually regulated by signal-induced modification of the target, we have previously shown that auxin signalling involves the modification of SCF(TIR1). Here we show that this modification involves the direct binding of auxin to TIR1 and thus that TIR1 is an auxin receptor mediating transcriptional responses to auxin.
URL [本文引用: 1]
【Objective】The purpose of this research is to analyze the genetic diversity of global faba bean germplasm, to explore their genetic similarity and population structure, and to provide essential information for germplasm evaluation and effective utilization of faba bean genetic resources. 【Method】The genetic similarity of 383 faba bean accessions from 35 countries was analyzed by using ISSR marker. 【Result】Eleven ISSR primers generated 229 unambiguous bands, of which 212 were polymorphic, and the rate of polymorphic bands was 0.93. Gene diversity index (H) and allelic richness (NA) of different geographic groups of germplasm ranged from 0.16 to 0.28 and from 104 to 193, respectively. Chinese spring-seeding area group showed the highest genetic diversity (H = 0.28, NA = 193), and America group showed the lowest (H = 0.16, NA = 104). The spring faba bean germplasm was clearly separated from winter faba bean germplasm of China in UPGMA clustering analysis based on ISSR molecular marker data. Germplasm from China is quite distinct to that from exotic accessions. The accessions from Europe had a closer genetic similarity with that from North Africa. While the germplasm resources from Asia, Europe and Africa are closely related to their geographical distribution.【Conclusion】Accessions from spring-seeding area of China were most diverse. Germplasm from America showed lowest diversity. The results indicated that the genetic similarity and diversity of faba bean germplasm are closely associated with their growth habit, their geographical origin and ecological distribution.
Key words: faba bean (Vcia faba L.); germplasm resourses; ISSR marker; genetic diversity; genetic similarity
URL [本文引用: 1]
【Objective】The purpose of this research is to analyze the genetic diversity of global faba bean germplasm, to explore their genetic similarity and population structure, and to provide essential information for germplasm evaluation and effective utilization of faba bean genetic resources. 【Method】The genetic similarity of 383 faba bean accessions from 35 countries was analyzed by using ISSR marker. 【Result】Eleven ISSR primers generated 229 unambiguous bands, of which 212 were polymorphic, and the rate of polymorphic bands was 0.93. Gene diversity index (H) and allelic richness (NA) of different geographic groups of germplasm ranged from 0.16 to 0.28 and from 104 to 193, respectively. Chinese spring-seeding area group showed the highest genetic diversity (H = 0.28, NA = 193), and America group showed the lowest (H = 0.16, NA = 104). The spring faba bean germplasm was clearly separated from winter faba bean germplasm of China in UPGMA clustering analysis based on ISSR molecular marker data. Germplasm from China is quite distinct to that from exotic accessions. The accessions from Europe had a closer genetic similarity with that from North Africa. While the germplasm resources from Asia, Europe and Africa are closely related to their geographical distribution.【Conclusion】Accessions from spring-seeding area of China were most diverse. Germplasm from America showed lowest diversity. The results indicated that the genetic similarity and diversity of faba bean germplasm are closely associated with their growth habit, their geographical origin and ecological distribution.
Key words: faba bean (Vcia faba L.); germplasm resourses; ISSR marker; genetic diversity; genetic similarity
DOI:10.1104/pp.106.085969URLPMID:16920877 [本文引用: 1]
Although a wide range of structurally diverse small molecules can act as auxins, it is unclear whether all of these compounds act via the same mechanisms that have been characterized for 2,4-dichlorophenoxyacetic acid (2,4-D) and indole-3-acetic acid (IAA). To address this question, we used a novel member of the picolinate class of synthetic auxins that is structurally distinct from 2,4-D to screen for Arabidopsis (Arabidopsis thaliana) mutants that show chemically selective auxin resistance. We identified seven alleles at two distinct genetic loci that conferred significant resistance to picolinate auxins such as picloram, yet had minimal cross-resistance to 2,4-D or IAA. Double mutants had the same level and selectivity of resistance as single mutants. The sites of the mutations were identified by positional mapping as At4g11260 and At5g49980. At5g49980 is previously uncharacterized and encodes auxin signaling F-box protein 5, one of five homologs of TIR1 in the Arabidopsis genome. TIR1 is the recognition component of the Skp1-cullin-F-box complex associated with the ubiquitin-proteasome pathway involved in auxin signaling and has recently been shown to be a receptor for IAA and 2,4-D. At4g11260 encodes the tetratricopeptide protein SGT1b that has also been associated with Skp1-cullin-F-box-mediated ubiquitination in auxin signaling and other pathways. Complementation of mutant lines with their corresponding wild-type genes restored picolinate auxin sensitivity. These results show that chemical specificity in auxin signaling can be conferred by upstream components of the auxin response pathway. They also demonstrate the utility of genetic screens using structurally diverse chemistries to uncover novel pathway components.
DOI:10.1111/j.1365-313x.2003.01990.xURLPMID:14756772 [本文引用: 1]
The phytohormone gibberellin (GA) controls growth and development in plants. Previously, we identified a rice F-box protein, gibberellin-insensitive dwarf2 (GID2), which is essential for GA-mediated DELLA protein degradation. In this study, we analyzed the biological and molecular biological properties of GID2. Expression of GID2 preferentially occurred in rice organs actively synthesizing GA. Domain analysis of GID2 revealed that the C-terminal regions were essential for the GID2 function, but not the N-terminal region. Yeast two-hybrid assay and immunoprecipitation experiments demonstrated that GID2 is a component of the SCF complex through an interaction with a rice ASK1 homolog, OsSkp15. Furthermore, an in vitro pull-down assay revealed that GID2 specifically interacted with the phosphorylated Slender Rice 1 (SLR1). Taken these results together, we conclude that the phosphorylated SLR1 is caught by the SCFGID2 complex through an interacting affinity between GID2 and phosphorylated SLR1, triggering the ubiquitin-mediated degradation of SLR1.
DOI:10.1073/pnas.0401698101URLPMID:15090654 [本文引用: 1]
Ubiquitination of various intracellular proteins by ubiquitin-protein ligases (or E3s) plays an essential role in eukaryotic cell regulation primarily through its ability to selectively target proteins for degradation by the 26S proteasome. Skp1, Cullin, F-box (SCF) complexes are one influential E3 class that use F-box proteins to deliver targets to a core ligase activity provided by the Skp1, Cullin, and Rbx1 subunits. Almost 700 F-box proteins can be found in Arabidopsis, indicating that SCF E3s likely play a pervasive role in plant physiology and development. Here, we describe the reverse genetic analysis of two F-box proteins, EBF1 and -2, that work coordinately in SCF complexes to repress ethylene action. Mutations in either gene cause hypersensitivity to exogenous ethylene and its precursor 1-aminocyclopropane-1-carboxylic acid. EBF1 and -2 interact directly with ethylene insensitive 3 (EIN3), a transcriptional regulator important for ethylene signaling. Levels of EIN3 are increased in mutants affecting either EBF1 or -2, suggesting that the corresponding SCF complexes work together in EIN3 breakdown. Surprisingly, double ebf1 ebf2 mutants display a substantial arrest of seedling growth and have elevated EIN3 levels, even in the absence of exogenous ethylene. Collectively, our results show that the SCF(EBF1/EBF2)-dependent ubiquitination and subsequent removal of EIN3 is critical not only for proper ethylene signaling but also for growth in plants.
DOI:10.1038/nature09430URLPMID:20927106 [本文引用: 1]
Jasmonates are a family of plant hormones that regulate plant growth, development and responses to stress. The F-box protein CORONATINE INSENSITIVE 1 (COI1) mediates jasmonate signalling by promoting hormone-dependent ubiquitylation and degradation of transcriptional repressor JAZ proteins. Despite its importance, the mechanism of jasmonate perception remains unclear. Here we present structural and pharmacological data to show that the true Arabidopsis jasmonate receptor is a complex of both COI1 and JAZ. COI1 contains an open pocket that recognizes the bioactive hormone (3R,7S)-jasmonoyl-l-isoleucine (JA-Ile) with high specificity. High-affinity hormone binding requires a bipartite JAZ degron sequence consisting of a conserved alpha-helix for COI1 docking and a loop region to trap the hormone in its binding pocket. In addition, we identify a third critical component of the jasmonate co-receptor complex, inositol pentakisphosphate, which interacts with both COI1 and JAZ adjacent to the ligand. Our results unravel the mechanism of jasmonate perception and highlight the ability of F-box proteins to evolve as multi-component signalling hubs.
DOI:10.1126/science.1110586URLPMID:16002617 [本文引用: 1]
The temporal control of CONSTANS (CO) expression and activity is a key mechanism in photoperiodic flowering in Arabidopsis. FLAVIN-BINDING, KELCH REPEAT, F-BOX 1 (FKF1) protein regulates CO transcription, although the molecular mechanism is unknown. We demonstrate here that FKF1 controls the stability of a Dof transcription factor, CYCLING DOF FACTOR 1 (CDF1). FKF1 physically interacts with CDF1, and CDF1 protein is more stable in fkf1 mutants. Plants with elevated levels of CDF1 flower late and have reduced expression of CO. CDF1 and CO are expressed in the same tissues, and CDF1 binds to the CO promoter. Thus, FKF1 controls daily CO expression in part by degrading CDF1, a repressor of CO transcription.
DOI:10.1104/pp.108.126912URLPMID:18835996 [本文引用: 1]
Guard cells, which form stoma in leaf epidermis, sense and integrate environmental signals to modulate stomatal aperture in response to diverse conditions. Under drought stress, plants synthesize abscisic acid (ABA), which in turn induces a rapid closing of stoma, to prevent water loss by transpiration. However, many aspects of the molecular mechanism for ABA-mediated stomatal closure are still not understood. Here, we report a novel negative regulator of guard cell ABA signaling, DOR, in Arabidopsis (Arabidopsis thaliana). The DOR gene encodes a putative F-box protein, a member of the S-locus F-box-like family related to AhSLF-S(2) and specifically interacting with ASK14 and CUL1. A null mutation in DOR resulted in a hypersensitive ABA response of stomatal closing and a substantial increase of drought tolerance; in contrast, the transgenic plants overexpressing DOR were more susceptible to the drought stress. DOR is strongly expressed in guard cells and suppressed by ABA treatment, suggesting a negative feedback loop of DOR in ABA responses. Double-mutant analyses of dor with ABA-insensitive mutant abi1-1 showed that abi1-1 is epistatic to dor, but no apparent change of phospholipase Dalpha1 was detected between the wild type and dor. Affymetrix GeneChip analysis showed that DOR likely regulates ABA biosynthesis under drought stress. Taken together, our results demonstrate that DOR acts independent of phospholipase Dalpha1 in an ABA signaling pathway to inhibit the ABA-induced stomatal closure under drought stress.
URLPMID:22859682 [本文引用: 1]
DOI:10.1105/tpc.104.026963URLPMID:15598801 [本文引用: 1]
Recently, an S haplotype-specific F-box (SFB) gene has been proposed as a candidate for the pollen-S specificity gene of RNase-mediated gametophytic self-incompatibility in Prunus (Rosaceae). We have examined two pollen-part mutant haplotypes of sweet cherry (Prunus avium). Both were found to retain the S-RNase, which determines stylar specificity, but one (S3' in JI 2434) has a deletion including the haplotype-specific SFB gene, and the other (S4' in JI 2420) has a frame-shift mutation of the haplotype-specific SFB gene, causing amino acid substitutions and premature termination of the protein. The loss or significant alteration of this highly polymorphic gene and the concomitant loss of pollen self-incompatibility function provides compelling evidence that the SFB gene encodes the pollen specificity component of self-incompatibility in Prunus. These loss-of-function mutations are inconsistent with SFB being the inactivator of non-self S-RNases and indicate the presence of a general inactivation mechanism, with SFB conferring specificity by protecting self S-RNases from inactivation.
DOI:10.1038/nbt.1883URLPMID:21572440 [本文引用: 1]
Massively parallel sequencing of cDNA has enabled deep and efficient probing of transcriptomes. Current approaches for transcript reconstruction from such data often rely on aligning reads to a reference genome, and are thus unsuitable for samples with a partial or missing reference genome. Here we present the Trinity method for de novo assembly of full-length transcripts and evaluate it on samples from fission yeast, mouse and whitefly, whose reference genome is not yet available. By efficiently constructing and analyzing sets of de Bruijn graphs, Trinity fully reconstructs a large fraction of transcripts, including alternatively spliced isoforms and transcripts from recently duplicated genes. Compared with other de novo transcriptome assemblers, Trinity recovers more full-length transcripts across a broad range of expression levels, with a sensitivity similar to methods that rely on genome alignments. Our approach provides a unified solution for transcriptome reconstruction in any sample, especially in the absence of a reference genome.
[本文引用: 1]
DOI:10.1016/j.plantsci.2016.09.009URLPMID:27968985 [本文引用: 1]
F-box gene family, as one of the largest gene families in plants, plays crucial roles in regulating plant development, reproduction, cellular protein degradation and responses to biotic and abiotic stresses. However, comprehensive analysis of the F-box gene family in pear (Pyrus bretschneideri Rehd.) and other Rosaceae species has not been reported yet. Herein, we identified a total of 226 full-length F-box genes in pear for the first time. And these genes were further divided into various subgroups based on specific domains and phylogenetic analysis. Intriguingly, we observed that whole-genome duplication and dispersed duplication have a major contribution to F-box family expansion. Furthermore, the dynamic evolution for different modes of gene duplication was dissected. Interestingly, we found that dispersed and tandem duplicate have been evolving at a high rate. In addition, we found that F-box genes exhibited functional specificity based on GO analysis, and most of the F-box genes were significantly enriched in the protein binding (GO: 0005515) term, supporting that F-box genes might play a critical role for gene regulation in pear. Transcriptome and digital expression profiles revealed that F-box genes are involved in the development of multiple pear tissues. Overall, these results will set stage for elaborating the biological role of F-box genes in pear and other plants.
DOI:10.1007/s13277-014-2832-xURLPMID:25412953 [本文引用: 1]
We aimed to identify the potential microRNA (miRNA) targets for colorectal cancer (CRC). Small RNA-seq and RNA-seq data of GSE46622 were downloaded from Gene Expression Omnibus (GEO) database, including samples of tumor tissue, metastasis tissue, and normal tissue from eight CRC patients. Data comparison of small RNA-seq and RNA-seq was performed through Bowtie and TopHat softwares, respectively. Then the expressed values of each sample were calculated by Cufflinks and Cuffdiff based on fragments per kilobase of exon per millionfragments mapped (FPKM) methods. The differentially expressed miRNAs and core miRNAs were identified by paired t-test. Besides, miRNA target genes were integrated through miRanda, MirTarget2, PicTar, PITI, TargetScan, and miRecords databases, followed by functional analysis of specific miRNA. The average comparison rate of sequence reads in miRNA, noncoding RNA, and other areas of the genome is 49.75, 2.90, and 47.35 %, respectively. A total of 49 miRNAs was differentially expressed. Compared with normal controls, 3 miRNAs were upregulated and 13 miRNAs were downregulated in tumor samples as well as 48 miRNAs were upregulated and 20 miRNAs were downregulated in the metastasis samples. Among them, 18 metastasis-specific expressed miRNAs, 2 tumor-specific expressed miRNAs, and 11 normal expressed miRNA were found. miRNA-1, miRNA-338-5p, miRNA-326, and miR-490-5p were selected as important miRNAs. Besides, miRNA-338-5p target gene RAB6B, FAP, and CTGF were identified as oncogenes. The miRNAs, such as miRNA-1, miRNA-338-5p, and miRNA-326 may be used as potential targets for CRC diagnosis and treatment.
DOI:10.3389/fpls.2017.02021URLPMID:29234338 [本文引用: 1]
Transfer cells (TCs) support high rates of membrane transport of nutrients conferred by a plasma membrane area amplified by lining a wall labyrinth comprised of an uniform wall layer (UWL) upon which intricate wall ingrowth (WI) papillae are deposited. A signal cascade of auxin, ethylene, extracellular hydrogen peroxide (H2O2) and cytosolic Ca(2+) regulates wall labyrinth assembly. To identify gene cohorts regulated by each signal, a RNA- sequencing study was undertaken using Vicia faba cotyledons. When cotyledons are placed in culture, their adaxial epidermal cells spontaneously undergo trans-differentiation to epidermal TCs (ETCs). Expressed genes encoding proteins central to wall labyrinth formation (signaling, intracellular organization, cell wall) and TC function of nutrient transport were assembled. Transcriptional profiles identified 9,742 annotated ETC-specific differentially expressed genes (DEGs; Log2fold change > 1; FDR p
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1016/s0092-8674(00)80098-7URLPMID:8706131 [本文引用: 1]
We have identified the yeast and human homologs of the SKP1 gene as a suppressor of cdc4 mutants and as a cyclin F-binding protein. Skp1p indirectly binds cyclin A/Cdk2 through Skp2p, and directly binds Skp2p, cyclin F, and Cdc4p through a novel structural motif called the F-box. SKP1 is required for ubiquitin-mediated proteolysis of Cin2p, Clb5p, and the Cdk inhibitor Sic1p, and provides a link between these molecules and the proteolysis machinery. A large number of proteins contain the F-box motif and are thereby implicated in the ubiquitin pathway. Different skp1 mutants arrest cells in either G1 or G2, suggesting a connection between regulation of proteolysis in different stages of the cycle.
DOI:10.1371/journal.pone.0050009URLPMID:23166809 [本文引用: 1]
F-Box (FBX) proteins are encoded by a multigene family present in major lineages of eukaryotes. A number of FBX proteins are shown to be subunits of SCF complex, a type of E3 ligases composed of SKP1, CULLIN, FBX and RBX1 proteins. The Arabidopsis SKP-LIKE (ASK) proteins are also members of a family and some of them interact with FBX proteins directly. To clarify how FBX and ASK proteins combine, we carried out a large-scale interaction analysis between FBX and ASK proteins using yeast two-hybrid assay (Y2H) in Arabidopsis thaliana. FBX proteins randomly chosen from those proteins that interacted with more than one ASK protein were further analyzed for their subcellular localization and in vivo interaction with ASK proteins. Furthermore, the expression profiles of FBX and ASK genes were compared. This work reveals that FBX proteins had a preference for interacting with ASK proteins depending on the domains they contain such as the FBX-associated (FBA) domain, the Kelch domain and leucine rich repeat (LRR). In addition, it was found that a single FBX protein could form multiple SCF complexes by interacting with several ASK proteins in many cases. Furthermore, it was suggested that the variation of SCF complexes were especially abundant in tissues related to male gametophyte and seed development. More than half of the FBX proteins studied did not interact with any of the ASK proteins, implying the necessity for certain regulations for their interaction in vivo and/or distinct roles from subunits of the SCF complex.
DOI:10.1093/jxb/erv245URLPMID:26041321 [本文引用: 3]
Skp1-Cullin-F-box (SCF) E3 ligases are essential to the post-translational regulation of many important factors involved in cellular signal transduction. In this study, we identified an F-box protein from Arabidopsis thaliana, AtPP2-B11, which was remarkably induced with increased duration of salt treatment in terms of both transcript and protein levels. Transgenic Arabidopsis plants overexpressing AtPP2-B11 exhibited obvious tolerance to high salinity, whereas the RNA interference line was more sensitive to salt stress than wild-type plants. Isobaric tag for relative and absolute quantification analysis revealed that 4311 differentially expressed proteins were regulated by AtPP2-B11 under salt stress. AtPP2-B11 could upregulate the expression of annexin1 (AnnAt1) and function as a molecular link between salt stress and reactive oxygen species accumulation in Arabidopsis. Moreover, AtPP2-B11 influenced the expression of Na(+) homeostasis genes under salt stress, and the AtPP2-B11 overexpressing lines exhibited lower Na(+) accumulation. These results suggest that AtPP2-B11 functions as a positive regulator in response to salt stress in Arabidopsis.
[本文引用: 1]
DOI:10.1007/s00425-005-0138-3URLPMID:16244866 [本文引用: 1]
The UNUSUAL FLORAL ORGANS (UFO) gene of Arabidopsis encodes an F-box protein required for the determination of floral-organ and floral-meristem identity. Mutation of UFO leads to dramatic changes in floral-organ type which are well-characterized whereas inflorescence defects are more subtle and less understood. These defects include an increase in the number of secondary inflorescences, nodes that alternate between forming flowers and secondary inflorescences, and nodes in which a single flower is subtended by a bract. Here, we show how inflorescence defects correlate with the abnormal development of floral primordia and establish a temporal requirement for UFO in this process. At the inflorescence apex of ufo mutants, newly formed primordia are initially bract-like. Expression of the floral-meristem identity genes LFY and AP1 are confined to a relatively small adaxial region of these primordia with expression of the bract-identity marker FIL observed in cells that comprise the balance of the primordia. Proliferation of cells in the adaxial region of these early primordia is delayed by several nodes such that primordia appear
[本文引用: 1]
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2017.20.013URL [本文引用: 1]
【Objective】MYB is one of the most common transcription factor families in plants. It is widely involved in plant growth, and metabolic regulations. So far, there is no systematic analysis of the MYB transcription factor family of tree crops. Analysis of MYB family based on the transcriptome data in Lycium ruthenicum Murr. was conducted in this study, which laid a foundation for the research on biological function, and mechanism of metabolic regulations of MYB genes. 【Method】Based on the transcriptome sequencing (RNA-Seq) data, the NR, NT, Swiss-Prot, PFAM and NCBI sites were used at the same time to screen and classify the MYB genes of L. ruthenicum Murr. The Web Logo3, Prot Comp 9.0, and MEGA5.0 were also applied to conservative structure prediction, subcellular localization, and phylogenetic analysis. The expression pattern of MYB genes related to fruit development was obtained and Real-time fluorescence quantitative PCR was used to detect the specific expression of those genes. 【Result】Based on the transcriptome sequencing (RNA-Seq) data, 83 transcription factors of MYB family were annotated, selected, and divided into four categories (R2R3-MYB, 1R-MYB, 3R-MYB and 4R-MYB) according to their structural characteristics. The R2 MYB motif of the R2R3-MYB transcription factor contains three highly conserved tryptophan residues, and the first tryptophan residue in the R3 MYB motif is replaced by some hydrophobic amino acids. The phylogenetic trees of MYB family of L. ruthenicum Murr. and Arabidopsis thaliana were constructed, which showed that the MYB family of L. ruthenicum Murr. contained three major branches, and six evolutionary branches. The result of the subcellular localization demonstrated that 44 MYB transcription factors were located in the cytoplasm, and 37 MYB transcription factors were located in the nucleus. The analysis of differential expression of MYB genes of L. ruthenicum Murr. based on transcriptome sequencing (RNA-Seq) showed that MYB genes might be involved in the regulation of anthocyanin in three fruit development periods. Additionally, differential expression data based on fluorescence quantitative PCR confirmed that some MYB transcription factors might play a role in the regulation of anthocyanin synthesis in different fruit development periods ofL. ruthenicum Murr.. 【Conclusion】83 transcription factors of MYB family were annotated of L. ruthenicum Murr. The findings have laid a foundation for further studies of the structures and biological functions of MYB family.
DOI:10.3864/j.issn.0578-1752.2017.20.013URL [本文引用: 1]
【Objective】MYB is one of the most common transcription factor families in plants. It is widely involved in plant growth, and metabolic regulations. So far, there is no systematic analysis of the MYB transcription factor family of tree crops. Analysis of MYB family based on the transcriptome data in Lycium ruthenicum Murr. was conducted in this study, which laid a foundation for the research on biological function, and mechanism of metabolic regulations of MYB genes. 【Method】Based on the transcriptome sequencing (RNA-Seq) data, the NR, NT, Swiss-Prot, PFAM and NCBI sites were used at the same time to screen and classify the MYB genes of L. ruthenicum Murr. The Web Logo3, Prot Comp 9.0, and MEGA5.0 were also applied to conservative structure prediction, subcellular localization, and phylogenetic analysis. The expression pattern of MYB genes related to fruit development was obtained and Real-time fluorescence quantitative PCR was used to detect the specific expression of those genes. 【Result】Based on the transcriptome sequencing (RNA-Seq) data, 83 transcription factors of MYB family were annotated, selected, and divided into four categories (R2R3-MYB, 1R-MYB, 3R-MYB and 4R-MYB) according to their structural characteristics. The R2 MYB motif of the R2R3-MYB transcription factor contains three highly conserved tryptophan residues, and the first tryptophan residue in the R3 MYB motif is replaced by some hydrophobic amino acids. The phylogenetic trees of MYB family of L. ruthenicum Murr. and Arabidopsis thaliana were constructed, which showed that the MYB family of L. ruthenicum Murr. contained three major branches, and six evolutionary branches. The result of the subcellular localization demonstrated that 44 MYB transcription factors were located in the cytoplasm, and 37 MYB transcription factors were located in the nucleus. The analysis of differential expression of MYB genes of L. ruthenicum Murr. based on transcriptome sequencing (RNA-Seq) showed that MYB genes might be involved in the regulation of anthocyanin in three fruit development periods. Additionally, differential expression data based on fluorescence quantitative PCR confirmed that some MYB transcription factors might play a role in the regulation of anthocyanin synthesis in different fruit development periods ofL. ruthenicum Murr.. 【Conclusion】83 transcription factors of MYB family were annotated of L. ruthenicum Murr. The findings have laid a foundation for further studies of the structures and biological functions of MYB family.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nature02163URLPMID:14654842 [本文引用: 1]
The underlying mechanism of circadian rhythmicity appears to be conserved among organisms, and is based on negative transcriptional feedback loops forming a cellular oscillator (or 'clock'). Circadian changes in protein stability, phosphorylation and subcellular localization also contribute to the generation and maintenance of this clock. In plants, several genes have been shown to be closely associated with the circadian system. However, the molecular mechanisms proposed to regulate the plant clock are mostly based on regulation at the transcriptional level. Here we provide genetic and molecular evidence for a role of ZEITLUPE (ZTL) in the targeted degradation of TIMING OF CAB EXPRESSION 1 (TOC1) in Arabidopsis thaliana (thale cress). The physical interaction of TOC1 with ZTL is abolished by the ztl-1 mutation, resulting in constitutive levels of TOC1 protein expression. The dark-dependent degradation of TOC1 protein requires functional ZTL, and is prevented by inhibiting the proteosome pathway. Our results show that the TOC1-ZTL interaction is important in the control of TOC1 protein stability, and is probably responsible for the regulation of circadian period by the clock.
DOI:10.1093/pcp/pcx035URLPMID:28340173 [本文引用: 1]
F-box proteins are part of one of the largest families of regulatory proteins that play important roles in protein degradation. In plants, F-box proteins are functionally very diverse, and only a small subset has been characterized in detail. Here, we identified a novel F-box protein FBX92 as a repressor of leaf growth in Arabidopsis. Overexpression of AtFBX92 resulted in plants with smaller leaves than the wild type, whereas plants with reduced levels of AtFBX92 showed, in contrast, increased leaf growth by stimulating cell proliferation. Detailed cellular analysis suggested that AtFBX92 specifically affects the rate of cell division during early leaf development. This is supported by the increased expression levels of several cell cycle genes in plants with reduced AtFBX92 levels. Surprisingly, overexpression of the maize homologous gene ZmFBX92 in maize had no effect on plant growth, whereas ectopic expression in Arabidopsis increased leaf growth. Expression of a truncated form of AtFBX92 showed that the contrasting effects of ZmFBX92 and AtFBX92 gain of function in Arabidopsis are due to the absence of the F-box-associated domain in the ZmFBX92 gene. Our work reveals an additional player in the complex network that determines leaf size and lays the foundation for identifying putative substrates.
DOI:10.1016/j.plantsci.2017.03.010URLPMID:28483055 [本文引用: 2]
F-box protein is a major subunit of the Skp1-Cullin-F-box (SCF) complex. We previously isolated an F-box gene from wheat, TaFBA1, and here we show that overexpression of TaFBA1 in transgenic plants under salt stress increases germination rate, root elongation, and biomass accumulation compared with WT plants. Improvements in the photosynthetic rate and its corresponding parameters were also found in the transgenic plants. These results suggest that overexpression of TaFBA1 improves salt stress tolerance in transgenic tobacco. Further, the transgenic plants displayed less membrane damage, higher antioxidant enzyme activity, and less accumulation of ROS under salt stress. The transgenic plants also had lower Na(+) content and higher K(+) content than WT plants in leaves and roots. The activity of H(+)-ATPase on the plasma membrane in the transgenic plants was higher than in WT plants, and was accompanied by a net Na(+) efflux. In the tonoplast, the activity levels of V-ATPase and PPase were also higher in the transgenic plants, thus helping to maximize intracellular Na(+) compartmentalization. The expression of some stress-related genes was upregulated by salt stress. This suggests that the enhancement of plant salt stress tolerance may be associated with an improvement in antioxidative competition and Na(+)/K(+) ion regionalization.
URLPMID:27716048 [本文引用: 1]
DOI:10.1146/annurev.arplant.55.031903.141801URLPMID:15377232 [本文引用: 1]
Much of plant physiology, growth, and development is controlled by the selective removal of short-lived regulatory proteins. One important proteolytic pathway involves the small protein ubiquitin (Ub) and the 26S proteasome, a 2-MDa protease complex. In this pathway, Ub is attached to proteins destined for degradation; the resulting Ub-protein conjugates are then recognized and catabolized by the 26S proteasome. This review describes our current understanding of the pathway in plants at the biochemical, genomic, and genetic levels, using Arabidopsis thaliana as the model. Collectively, these analyses show that the Ub/26S proteasome pathway is one of the most elaborate regulatory mechanisms in plants. The genome of Arabidopsis encodes more than 1400 (or >5% of the proteome) pathway components that can be connected to almost all aspects of its biology. Most pathway components participate in the Ub-ligation reactions that choose with exquisite specificity which proteins should be ubiquitinated. What remains to be determined is the identity of the targets, which may number in the thousands in plants.
DOI:10.1111/j.1469-8137.2012.04266.xURLPMID:22897362 [本文引用: 1]
Ubiquitin is well established as a major modifier of signalling in eukaryotes. However, the extent to which plants rely on ubiquitin for regulating their lifecycle is only recently becoming apparent. This is underlined by the over-representation of genes encoding ubiquitin-metabolizing enzymes in Arabidopsis when compared with other model eukaryotes. The main characteristic of ubiquitination is the conjugation of ubiquitin onto lysine residues of acceptor proteins. In most cases the targeted protein is rapidly degraded by the 26S proteasome, the major proteolysis machinery in eukaryotic cells. The ubiquitin-proteasome system is responsible for removing most abnormal peptides and short-lived cellular regulators, which, in turn, control many processes. This allows cells to respond rapidly to intracellular signals and changing environmental conditions. This review maps out the roles of the components of the ubiquitin-proteasome system with emphasis on areas where future research is urgently needed. We provide a flavour of the diverse aspects of plant lifecycle where the ubiquitin-proteasome system is implicated. We aim to highlight common themes using key examples that reiterate the importance of the ubiquitin-proteasome system to plants. The future challenge in plant biology is to define the targets for ubiquitination, their interactors and their molecular function within the regulatory context.
DOI:10.1007/s00018-008-7592-6URLPMID:18344020 [本文引用: 1]
A signature feature of all living organisms is their utilization of proteins to construct molecular machineries that undertake the complex network of cellular activities. The abundance of a protein element is temporally and spatially regulated in two opposing aspects: de novo synthesis to manufacture the required amount of the protein, and destruction of the protein when it is in excess or no longer needed. One major route of protein destruction is coordinated by a set of conserved molecules, the F-box proteins, which promote ubiquitination in the ubiquitin-proteasome pathway. Here we discuss the functions of F-box proteins in several cellular scenarios including cell cycle progression, synapse formation, plant hormone responses, and the circadian clock. We particularly emphasize the mechanisms whereby F-box proteins recruit specific substrates and regulate their abundance in the context of SCF E3 ligases. For some exceptions, we also review how F-box proteins function through non-SCF mechanisms.
DOI:10.1016/j.tplants.2009.01.003URLPMID:19285909 [本文引用: 1]
The importance of regulated proteolysis in the physiology and development of plants is highlighted by the large number of genes dedicated to proteasome-dependent protein degradation. Within the SCF class of E3 ubiquitin ligases are more than 700 F-box proteins that act as recognition modules to specifically target their dedicated substrates for ubiquitylation. This review focuses on very recent studies indicating that some F-box proteins function as phytohormone or light receptors, which directly perceive signals and facilitate specific target-protein degradation to regulate downstream pathways. If this new connection between ligand-regulated proteolysis and signaling proves to be more extensive, an entirely new way of understanding the control of signal transduction is in the offing.
DOI:10.1371/journal.pone.0016219URLPMID:21297981 [本文引用: 1]
The emergence of multigene families has been hypothesized as a major contributor to the evolution of complex traits and speciation. To help understand how such multigene families arose and diverged during plant evolution, we examined the phylogenetic relationships of F-Box (FBX) genes, one of the largest and most polymorphic superfamilies known in the plant kingdom. FBX proteins comprise the target recognition subunit of SCF-type ubiquitin-protein ligases, where they individually recruit specific substrates for ubiquitylation. Through the extensive analysis of 10,811 FBX loci from 18 plant species, ranging from the alga Chlamydomonas reinhardtii to numerous monocots and eudicots, we discovered strikingly diverse evolutionary histories. The number of FBX loci varies widely and appears independent of the growth habit and life cycle of land plants, with a little as 198 predicted for Carica papaya to as many as 1350 predicted for Arabidopsis lyrata. This number differs substantially even among closely related species, with evidence for extensive gains/losses. Despite this extraordinary inter-species variation, one subset of FBX genes was conserved among most species examined. Together with evidence of strong purifying selection and expression, the ligases synthesized from these conserved loci likely direct essential ubiquitylation events. Another subset was much more lineage specific, showed more relaxed purifying selection, and was enriched in loci with little or no evidence of expression, suggesting that they either control more limited, species-specific processes or arose from genomic drift and thus may provide reservoirs for evolutionary innovation. Numerous FBX loci were also predicted to be pseudogenes with their numbers tightly correlated with the total number of FBX genes in each species. Taken together, it appears that the FBX superfamily has independently undergone substantial birth/death in many plant lineages, with its size and rapid evolution potentially reflecting a central role for ubiquitylation in driving plant fitness.
DOI:10.1371/journal.pone.0068672URLPMID:23904908 [本文引用: 1]
F-box proteins (FBPs) represent one of the largest and fastest evolving gene/protein families in the plant kingdom. The FBP superfamily can be divided in several subfamilies characterized by different C-terminal protein-protein interaction domains that recruit targets for proteasomal degradation. Hence, a clear picture of their phylogeny and molecular evolution is of special interest for the general understanding of evolutionary histories of multi-domain and/or large protein families in plants. In an effort to further understand the molecular evolution of F-box family proteins, we asked whether the largest subfamily in Arabidopsis thaliana, which carries a C-terminal F-box associated domain (FBA proteins) shares evolutionary patterns and signatures of selection with other FBPs. To address this question, we applied phylogenetic and molecular evolution analyses in combination with the evaluation of transcriptional profiles. Based on the 2219 FBA proteins we de novo identified in 34 completely sequenced plant genomes, we compared their evolutionary patterns to a previously analyzed large subfamily carrying C-terminal kelch repeats. We found that these two large FBP subfamilies generally tend to evolve by massive waves of duplication, followed by sequence conservation of the F-box domain and sequence diversification of the target recruiting domain. We conclude that the earlier in evolutionary time a major wave of expansion occurred, the more pronounced these selection signatures are. As a consequence, when performing cross species comparisons among FBP subfamilies, significant differences will be observed in the selective signatures of protein-protein interaction domains. Depending on the species, the investigated subfamilies comprise up to 45% of the complete superfamily, indicating that other subfamilies possibly follow similar modes of evolution.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}