 ,中国农业科学院作物科学研究所,北京100081
,中国农业科学院作物科学研究所,北京100081Genome-Wide Detection of Selection Signal in Temperate and Tropical Maize Populations with Use of FST and XP-EHH
YANG YuXin, ZOU Cheng,Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing 100081通讯作者:
收稿日期:2018-10-30接受日期:2018-12-9网络出版日期:2019-02-16
| 基金资助: |
Received:2018-10-30Accepted:2018-12-9Online:2019-02-16
作者简介 About authors
杨宇昕,

摘要
关键词:
Abstract
Keywords:
PDF (2348KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
杨宇昕, 邹枨. 基于温带和热带玉米群体全基因组FST和XP-EHH的选择信号检测[J]. 中国农业科学, 2019, 52(4): 579-590 doi:10.3864/j.issn.0578-1752.2019.04.001
YANG YuXin, ZOU Cheng.
0 引言
【研究意义】玉米是非常重要的粮食和饲料作物,考古学和遗传学研究证明现代玉米起源于约9 000年前的墨西哥西南部,其野生祖先是大刍草[1,2,3]。玉米的起源中心位于低纬度短日照的热带环境,经过不断地人工选择和驯化,逐渐降低了光周期敏感性,并且被广泛种植于58°N到40°S的温带长日照地区[4,5]。根据玉米对地理环境的适应性,其可以划分为适应长日照环境的温带玉米和适应短日照环境的热带玉米2种类型[6]。热带玉米不仅具有优良的农艺性状(例如根系发达、抗病虫性强[7]等特点),而且具有丰富的遗传变异[8],是进行群体改良的优良材料。不同的玉米群体在人工选择和驯化的过程中,逐渐适应当地的生长环境,导致染色体区段上控制目标性状基因的优势等位基因频率逐渐增加,多态性发生改变,根据中性遗传假说[9],这将会导致与目的基因紧密连锁的染色体序列多态性也会随之发生改变,这一个过程称之为搭车效应[10]。由于人工选择而导致的DNA结构变化称之为即选择信号(selection signal),其特点是染色体区段上出现较长距离的单倍型纯合、连锁不平衡值改变以及优势等位基因频率的增加。因此,利用选择信号法探究温热带玉米群体的基因组变化,可以鉴定在热带玉米适应性改良过程中受到选择的基因区段,并且深入的挖掘开花基因,对于揭示玉米的驯化改良的遗传机制具有重要意义。【前人研究进展】选择信号检测方法根据其分析原理可以分为以下3种类型:(1)基于等位基因频率谱的方法,代表的计算方法包括Tajima's D[11]和CLR(composite likelihood ratio)[12]等;(2)基于连锁不平衡的方法,主要包括EHH(extended haplotype homozygosity)[13]、iHS(integrated haplotype score)[14]和XP-EHH[15]等;(3)基于群体分化进行群体遗传分化的方法,主要包括FST法[16,17,18]。目前,应用较为广泛的选择信号检测方法包括群体分化指数法(FST)和群体间扩展单倍型纯合度法(XP-EHH)。FST统计理论最早由WRIGHT[16]提出,经过对其分析方法和计算理论的不断改进,如今使用较多的是由WEIR等[17]提出的无偏估计的FST。FST选择信号法可以扫描全基因组范围内的SNP位点,计算每个SNP的FST值,其取值范围为0—1,0代表群体间所有位点都没有出现分化,1代表群体间已经完全分化。XP-EHH选择信号是基于基因的连锁不平衡原理,群体经过人工选择和改良,将会出现较大范围的染色体重组,由于连锁作用的存在,导致突变基因附近的中性位点也会逐代的传递,因此,在染色体上形成较长范围的单倍型纯合。XP-EHH的计算方法是EHH和iHS统计原理的扩展,相比于FST方法只能计算出发生分化的位点,XP-EHH统计量可以得到选择作用所在的群体,即XP-EHH统计值为正数时,表明选择发生在试验群体,反之则发生在参考群体。随着越来越多物种的参考基因组构建完成,利用FST和XP-EHH选择信号法,已经在遗传进化、基因挖掘等生物学领域取得了很大的进展。AXELSSON等[19]对狼和狗采取全基因组重测序得到3.8亿个SNP遗传变异标记,通过进行FST选择信号检测,在淀粉消化和脂肪代谢中起到重要作用的10个调控基因表现出明显的选择信号,这些调控基因可以促进狗的祖先在以淀粉为主要食用能量的人类社会中生存,相比于肉食性习性动物的狼,可以得到更多食物,这种可以消化淀粉的适应性演化是狼驯化为狗的关键步骤。LIU等[20]利用温带、热带和亚热带玉米中代表性的260个玉米自交系,结合FST的计算方法,发现热带玉米相比于温带玉米具有更高的遗传多样性和更多的等位基因位点。通过研究地方品种和自交系的位点间差异,表明现代玉米低于80%的位点来源于地方品种,为利用玉米地方品种进行改良提供了有力的试验支持。HE等[21]利用XP-EHH选择信号的方法,在温带玉米改良进程中一共鉴定到超过1 100个候选基因区段,通过基因富集分析,发现这些基因主要参与蔗糖合成和油分含量调控等。【本研究切入点】随着高通量测序技术在农业领域的广泛应用,高质量的玉米参考基因组图谱[22,23]和单倍型图谱[24,25]相继构建完成,极大地促进了玉米功能基因组学的发展,同时这些公共数据也为广大科研人员进行其他的生物学问题研究提供了宝贵的数据资源。热带玉米在经历人工驯化和改良过程中逐渐适应温带环境,因此,有必要利用选择信号法揭示在改良过程中发生选择的位点,挖掘候选驯化基因,进一步从基因层面上探究玉米的群体改良。【拟解决的关键问题】本研究选择具有代表性的30个温带玉米自交系和21个热带玉米自交系作为材料,基于全基因组单倍型数据,分析温热带玉米基因组多态性,并且结合FST和XP-EHH 2种方法进行群体间选择信号的检测,挖掘发生选择的驯化基因, 为玉米驯化的遗传机理解析提供理论依据。1 材料与方法
1.1 温、热带玉米自交系基因型的获取
选取具有代表性的30个温带玉米自交系和21个热带玉米自交系,其基因型数据来自于玉米单倍型图谱第三版[25],选择单倍型图谱中测序深度较高的重测序数据,以确保其准确率。原始基因型数据以VCF文件存储。考虑到原始基因型数据等位基因频率偏低可能影响后续分析[26],因此,利用VCFtools软件将缺失率较高和最小等位基因频率低于0.05的SNP剔除,相关参数为--maf 0.05。首先进行温带玉米群体和热带玉米群体的SNP筛选,分别得到温带和热带的基因型数据,这两个基因型数据只进行SNP的功能注释。使用的选择信号计算方法要求温带和热带玉米群体具有相同的SNP数目,因此,将温带玉米和热带玉米一起进行SNP的筛选,得到共存于温带和热带玉米群体的高质量SNP标记,该基因型用于FST和XP-EHH选择信号的计算。为了评估温热带玉米群体全基因组水平上的变异情况,利用SnpEff[27](版本号4.3p)软件对温热带玉米群体的变异信息进行功能注释。SNP密度分布利用R软件包CMplot绘制。玉米参考基因组下载于Ensemble数据库。1.2 主成分分析
主成分分析选择Tassel(版本号5)软件。首先将过滤得到的温热带玉米群体的基因型文件导入到Tassel软件,按照5个成分进行分析,选择前2个主成分进行绘图展示。1.3 FST选择信号检测
采用群体分化指数(FST)法进行温热带玉米间选择信号的检测,其计算原理是依据染色体等位基因频率变化。群体间选择信号的检测可以揭示不同群体驯化过程中经历的自然选择。按照WEIR等[17]的统计方法进行FST值的计算,考虑到基于单位点SNP扫描的方法容易受到遗传漂变等因素的影响,因此,为降低假阳性,选择滑动窗口的计算方法来增加选择信号的灵敏度[28]。利用VCFtools软件计算滑动窗口内的FST值,相关参数设置为--fst-window-size 200 kb、--fst-window-step 100 kb。利用R包qqman展示全基因组水平上的FST值。为了鉴定FST值的受选择位点,选择FST值的top 1%作为显著阈值线[29],高于阈值线的SNP位点定义为“受选择位点”,FST的计算公式参考WEIR等[17]报道。1.4 XP-EHH选择信号计算
同时利用基于群体间扩展单倍型纯合度(XP- EHH)的方法检测群体间存在分化的基因区段。XP-EHH选择信号的计算使用selscan[30]软件(版本号v1.1.0),将温带玉米群体作为试验群体,热带玉米群体作为参考群体。当基因组某一区段的XP-EHH的值为正,代表在试验群体中发生了选择,反之则表示参考群体的基因组片段发生选择。基于XP-EHH选择信号得到的统计值近似符合正态分布[15],对XP-EHH的值进行标准化正态分布处理,使用selscan的norm参数对原始的XP-EHH值进行标准化处理。将标准正态化处理后的XP-EHH值从大到小排序,取其top 1%[29]作为显著的阈值线用以判断温、热带群体间是否发生选择,并且筛选出受选择基因区段,XP-EHH的计算原理参考SABETI等[15]的报道。1.5 候选基因的鉴定
FST法是按照滑动窗口计算得到的受选择位点,因此,将滑动窗口的起始和终止位点各向上游和下游扩增50 kb作为受选择选择区段,使用Bedtools[31](版本号v2.26)软件将受选择位点附近的候选驯化基因与玉米参考基因组进行基因比对,编码基因若是落在滑动窗口内则定义为候选基因。针对XP-EHH法得到的受选择位点,以显著SNP位点向上下游各扩增50 kb作为受选择的区域,同样利用Bedtools软件筛选出候选基因。由于篇幅所限,利用maizeGDB(https://www. maizegdb.org/)对选择信号值较高的基因进行基因功能注释。1.6 候选基因的富集分析
利用在线工具agriGO进行候选基因的富集分析[32]。分析方法采用奇异富集分析(singular enrichment analysis,SEA),参考数据库选择Zea mays ssp. v5a,显著性GO条目的检验使用Fisher精确校验,显著性阈值为0.05。分析内容包括细胞组分、生物过程和分子功能等。2 结果
2.1 温、热带玉米群体的SNP功能注释
利用SnpEff软件评估温、热带玉米群体在基因组水平上的多态性情况(图1),基因组注释信息来源于B73自交系。结果表明,热带玉米染色体的SNP数目显著的高于温带群体。热带玉米群体一共鉴定到14 123 408个SNP,染色体上平均每145个碱基存在一个变异位点;在温带玉米群体中共鉴定到个8 791 673个SNP,平均每234个碱基存在一个变异位点。温带和热带玉米群体一起进行SNP筛选,最终得到了204 752个符合过滤标准的SNP位点。此外,还分析了温带玉米和热带玉米群体中SNP的分布区域(表1),结果表明,温带和热带玉米群体中的SNP变异主要都发生在基因区间(Intergentic),此外依次是下游区间(Downstream)、上游区间(Upstream)、内含子区间(Intron)、外显子区间(Exon)等。图1
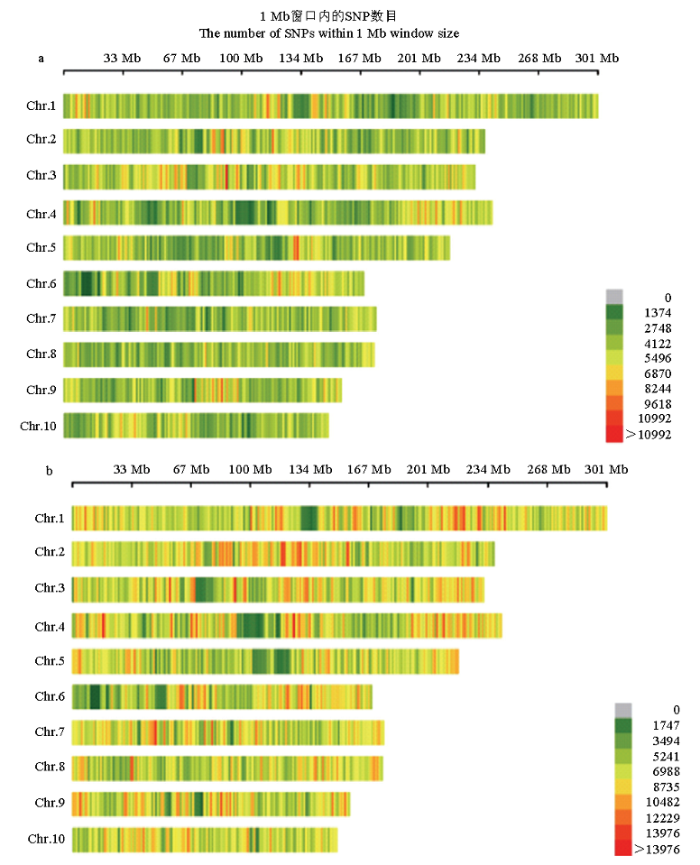 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1温、热带玉米群体SNP在基因组上的分布
a:温带玉米群体SNP分布;b:热带玉米群体SNP分布。横轴代表染色体的物理位置,窗口大小为1 Mb区间。深绿色代表SNP密度小的区域,红色代表SNP密度高的区域
Fig. 1The SNP distribution in the temperate and tropical maize genome
a: Represents the SNP distribution in temperate chromosomes; b: Represents the SNP distribution in tropical chromosomes. The abscissa represents the physical position of chromosome, and window size is 1 Mb. The darkgreen represents the SNP-poor regions, and red color indicates SNP-rich regions
Table 1
表1
表1温带和热带玉米群体的SNP在基因组区间的分布比例
Table 1
| 类型 Type | 温带 Temperate | 热带 Tropical | ||
|---|---|---|---|---|
| 数目 Count | 百分比 Percent (%) | 数目 Count | 百分比 Percent (%) | |
| Downstream | 2722265 | 18.14 | 4507749 | 18.54 |
| Exon | 326417 | 2.18 | 463543 | 1.91 |
| Intergenic | 7686519 | 51.22 | 12389340 | 50.97 |
| Intron | 1257112 | 8.38 | 2046559 | 8.42 |
| Splice_Site_Acceptor | 647 | 0.00 | 1005 | 0.00 |
| Splice_Site_Donor | 602 | 0.00 | 977 | 0.00 |
| Spice_Site_Region | 28482 | 0.19 | 45454 | 0.19 |
| Upstream | 2667996 | 17.78 | 4376510 | 18.00 |
| UTR_3_Prime | 192736 | 1.28 | 297085 | 1.22 |
| UTR_5_Prime | 124412 | 0.83 | 180849 | 0.74 |
新窗口打开|下载CSV
2.2 温、热带玉米群体的主成分分析
温带玉米和热带玉米由于对光周期敏感性的不同,其表型存在较大差异(图2),例如株高、叶片数、开花期等。温带材料可以在温带环境正常散粉开花,热带材料生殖生长期较长,未能在温带环境散粉开花。此外,热带材料的株高也显著的高于温带材料。利用温带玉米和热带玉米基因组信息,借助主成分分析策略来揭示温带和热带玉米群体之间的遗传关系(图3),按照PC1和PC2 2个维度可以将使用的玉米自交系分为温带和热带2个类群。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2温带(a)和热带(b)玉米自交系表型
Fig. 2The phenotype of the temperate (a) and tropical (b) maize inbred line
图3
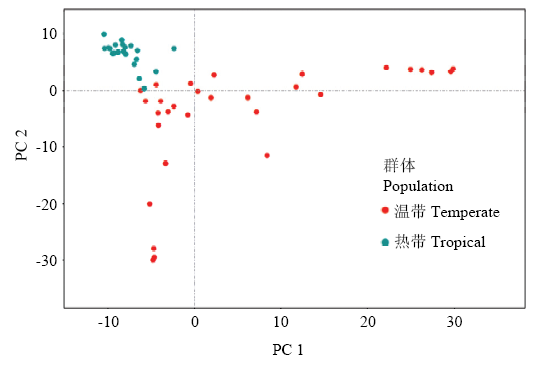 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3温、热带群体的主成分分析
Fig. 3The principle component analysis of temperate and tropical maize
2.3 FST值分析结果
温带玉米光周期敏感性低,可以在长日照的温带环境下正常生长结实;而热带玉米则具有较强的光周期敏感性,主要栽培于热带环境。利用2个群体共有的204 752个SNP标记计算得到每个滑动窗口内的FST值,全基因组水平上FST的top 1%是0.3593(图4),高于阈值线的受选择位点一共是1 908个,占总变异位点数的0.9%。其中第4染色体受到选择的显著性位点最多,是414个;第7染色体最少,是61个。第1染色体66 460 001—66 510 000区间内含有最高的FST值,其值为0.77。在第1、2、3、4、5、6、8、9、10染色体上都有较强的选择信号。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4全基因组水平上的FST分布
阈值线代表FST值的top 1%
Fig. 4Genome-wide FST values between temperate and tropical maize populations
The cutoff line represent top 1% of FST
2.4 XP-EHH的分析
XP-EHH选择信号检测的原理是根据原有的突变区段由于连锁效应较难被打破,而产生新的突变需要较长的选择周期才能达到很高的基因频率。因此,当某个单倍型区段在群体中出现频率较高,代表该基因区段经历了选择,可以利用XP-EHH选择信号的方法鉴定出来。利用selscan软件计算全基因组的上每个SNP位点的XP-EHH值,使用温带群体作为试验群体,热带群体作为参考群体,当XP-EHH值为正值时,代表在温带群体发生了选择,其值为负值时,表明驯化选择发生在热带群体。由于主要关注热带玉米向温带玉米的驯化,因此,只分析了XP-EHH值大于0的情况,即温带玉米群体中发生选择的染色体区段(图5),结果表明,在第2、3、7、8、10染色体存在较多的选择区段,其top 1%为3.32,高于阈值线的SNP位点数目是39 664个,XP-EHH值最高的位点是位于Chr.2:69.324 Mb附近,其值为7.3298。以受选择SNP位点为核心向上下游各扩增50 kb得到100 kb的候选基因区段,利用Bedtools软件将其与玉米B73参考基因组进行比对,共得到1 913个候选基因,其中包含2个与玉米适应性改良关系密切相关的基因,分别是ZmCCT9[33]和COL1[34]。ZmCCT9(Chr.9:115 786 897—115 789 787),其FST值为3.58,最新的一项研究表明ZmCCT9是玉米开花控通路中的一个重要调节因子[33],在长日照环境下,ZmCCT9抑制开花促进因子ZCN8[35,36]的表达,导致玉米晚花,通过CRISPR/Cas9技术介导的ZmCCT9敲除,发现可以促进玉米提早开花。鉴定到ZmCCT9在温带群体受到选择,因而加速热带玉米群体对温带环境的适应,这与前人报道一致[33]。COL1(Chr.9:108 447 974—108 449 794)是玉米光周期调控通路中的重要基因,该基因编码蛋白可以和开花促进因子FT蛋白互作,加速玉米开花以适应长日照环境,是一个重要的开花调控基因。图5
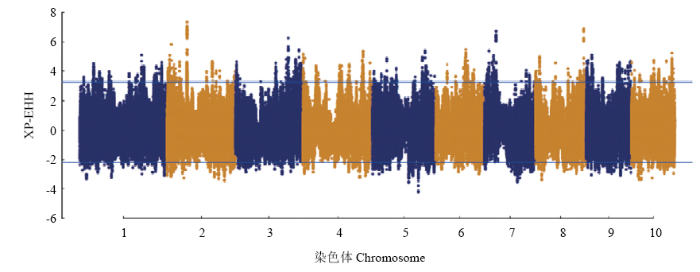 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5温、热带玉米全基因组水平上的XP-EHH选择信号分布
阈值线代表选择信号值的top 1%
Fig. 5Genome-wide distribution of XP-EHH signatures between temperate and tropical maize
The cutoff lines represent the top 1% of the XP-EHH selection signal
2.5 候选基因的富集分析和功能注释
利用FST鉴定得到557个候选基因和XP-EHH得到的1 913个候选基因。基因富集分析表明FST法得到的候选基因富集在生物学过程(biological process,P),共19个显著的GO条目(表2)。这些GO条目主要涉及到肌动蛋白发育调控、微丝的发育调控和细胞骨架构成等。利用XP-EHH得到的1 913个基因没有富集得到显著的GO条目,推测是因为热带玉米驯化温带玉米涉及到众多农艺性状的改变,并且许多农艺性状是由一系列微效数量位点控制,因此未能得到显著的GO条目。尽管如此,XP-EHH的方法仍然可以得到较多的选择基因,并且这些基因中鉴定得到了与玉米花期调控相关的基因,例如ZmCCT9和COL1。因此,通过选择信号的方法可以为解析玉米驯化的遗传机理、鉴定候选驯化基因提供实验依据。除了这些已经报道过的调控基因,还统计了选择信号值高于top 1%和top 0.1%的候选基因(表3)。另外,通过maizeGDB网站进行基因功能注释,发现这些基因均是玉米生长发育过程中的重要调控因子,例如GRMZM2G387528是一个光敏色素调节因子,参与玉米开花通路调控;GRMZM2G474769参与下胚轴的发育调控。Table 2
表2
表2FST选择信号得到的基因进行GO富集分析的结果
Table 2
| GO条目 GO term | 通路 Ontology | 描述 Description | P值 P-value | 错误发现率 FDR |
|---|---|---|---|---|
| GO:0008064 | P | 肌动蛋白聚合或解聚的调节 Regulation of actin polymerization or depolymerization | 1.30E-06 | 0.00011 |
| GO:0032271 | P | 蛋白质聚合的调节Regulation of protein polymerization | 1.30E-06 | 0.00011 |
| GO:0030832 | P | 微丝长度的调节Regulation of actin filament length | 1.30E-06 | 0.00011 |
| GO:0030833 | P | 微丝聚合反应的调节Regulation of actin filament polymerization | 1.30E-06 | 0.00011 |
| GO:0032970 | P | 肌动蛋白丝的调控过程Regulation of actin filament-based process | 1.30E-06 | 0.00011 |
| GO:0044087 | P | 细胞组分生物合成的调节Regulation of cellular component biogenesis | 1.30E-06 | 0.00011 |
| GO:0030041 | P | 肌动蛋白丝的聚合Actin filament polymerization | 2.20E-06 | 0.00014 |
| GO:0008154 | P | 激动蛋白丝的聚合或者解聚Actin polymerization or depolymerization | 2.20E-06 | 0.00014 |
| GO:0007015 | P | 肌动蛋白丝组织Actin filament organization | 2.20E-06 | 0.00014 |
| GO:0032535 | P | 细胞组分大小的调节Regulation of cellular component size | 3.10E-06 | 0.00016 |
| GO:0090066 | P | 解剖学结构大小的调节Regulation of anatomical structure size | 3.10E-06 | 0.00016 |
| GO:0033043 | P | 细胞器组织的调节Regulation of organelle organization | 4.20E-06 | 0.00021 |
| GO:0051128 | P | 细胞成分组织的调节Regulation of cellular component organization | 1.20E-05 | 0.00057 |
| GO:0030036 | P | 肌动蛋白细胞骨架组织Actin cytoskeleton organization | 0.00071 | 0.029 |
| GO:0030029 | P | 肌动蛋白丝基过程Actin filament-based process | 0.00071 | 0.029 |
| GO:0006996 | P | 细胞器组织Organelle organization | 0.0012 | 0.046 |
新窗口打开|下载CSV
Table 3
表3
表3选择信号鉴定得到的候选基因功能注释
Table 3
| 基因 Gene | 染色体 Chromosome | 物理位置 Position (Mb) | FST值a FST value | XP-EHH值b XP-EHH value | 注释c Annotation |
|---|---|---|---|---|---|
| GRMZM2G366434d | 5 | 17.336—17.338 | 0.3688** | 4.6263** | AP2-ErEBP转录因子206,ereb206 AP2-EREBP-transcription factor 206, ereb206 |
| GRMZM2G145579d | 4 | 47.618—47.620 | 0.5071** | 3.8724* | bHLH转录因子165,bhlh165 bHLH-transcription factor 165, bhlh165 |
| GRMZM2G023325 | 1 | 66.506—66.507 | 0.7845** | 氧氮杂萘酮合成12,bx12 Benzoxazinone synthesis12,bx12 | |
| GRMZM2G074094 | 1 | 199.519—199.525 | 0.4380** | SET结构域,sdg103 SET domain group 103, sdg103 | |
| GRMZM2G092091 | 1 | 220.057—220.061 | 0.4136** | bHLH转录因子27,bhlh27 bHLH-transcription factor 27,bhlh27 | |
| GRMZM2G478417 | 1 | 272.109—272.113 | 0.3706** | bZIP转录因子49,bzip49 bZIP-transcription factor 49,bzip49 | |
| GRMZM2G174834 | 4 | 159.692—159.694 | 0.3952** | WRI1转录因子,wri2 WRI1 transcription factor2, wri2 | |
| GRMZM2G126505 | 4 | 185.907—185.912 | 0.3879** | 脱落酸8'-羟化酶2,abh12 Abscisic acid 8'-hydroxylase2, abh12 | |
| GRMZM2G070523 | 5 | 20.219—20.221 | 0.3693** | MYB转录因子129,myb129 MYB-transcription factor 129, myb129 | |
| GRMZM2G004334 | 6 | 109.244—109.247 | 0.5006** | 同源框转录因子10,hb10 Homeobox-transcription factor 10, hb10 | |
| GRMZM2G170148 | 9 | 110.859—110.870 | 0.3954** | MYB相关转录因子86,mybr86 MYB-related-transcription factor 86, mybr86 | |
| GRMZM2G156013 | 10 | 141.014—141.018 | 0.4835** | 丝氨酸-苏氨酸激酶4,stk4 Serine-threonine kinase4, stk4 | |
| GRMZM2G143602 | 2 | 8.289—8.295 | 4.1470* | CK2蛋白激酶α1,cka1 CK2 protein kinase alpha 1, cka1 | |
| GRMZM2G001289 | 2 | 15.033—15.035 | 3.7898* | 同源框转录因子75,hb75 Homeobox-transcription factor 75, hb75 | |
| GRMZM2G102059 | 2 | 236.711—236.716 | 3.6779* | ABI3-VP1转录因子12,abi12 ABI3-VP1-transcription factor 12, abi12 | |
| GRMZM2G089638 | 3 | 1.464—1.469 | 4.1762* | TCP转录因子31,tcptf31 TCP-transcription factor 31, tcptf31 | |
| GRMZM2G087804 | 3 | 140.859—140.861 | 3.9809* | 金色植物2,g2 Golden plant2, g2 | |
| GRMZM2G015875 | 4 | 47.618—47.620 | 4.5721* | NMCP/CRWN同源因子1,nch1 NMCP/CRWN-Homologous1, nch1 | |
| GRMZM2G145579 | 4 | 161.101—161.108 | 3.8724* | bHLH转录因子165,bhlh165 bHLH-transcription factor 165, bhlh165 | |
| GRMZM5G880069 | 5 | 184.337—184.342 | 3.8136* | WRKY转录因子109,wrky109 WRKY-transcription factor 109, wrky109 | |
| GRMZM2G129783 | 6 | 106.637—106.640 | 3.7924* | 五肽重复蛋白346,ppr346 Pentatricopeptide repeat protein346, ppr346 | |
| GRMZM2G387528 | 8 | 0.935—0.938 | 4.2271* | 光敏色素作用因子3,pif3 Phytochrome interacting factor3, pif3 | |
| GRMZM2G028054 | 8 | 170.725—170.728 | 3.7593* | MYB转录因子74,myb74 MYB-transcription factor 74, myb74 | |
| GRMZM2G009808 | 9 | 107.641—107.645 | 5.3817** | 乌头酸梅3,aco30 Aconitase3, aco30 | |
| GRMZM2G139082 | 9 | 116.292—116.294 | 4.3570* | 热冲击互补因子1,hscf1 Heat shock complementing factor1, hscf1 | |
| GRMZM2G013671 | 9 | 152.030—152.038 | 3.7187* | β扩增蛋白5 Beta expansin5, expb5 | |
| GRMZM2G474769 | 10 | 78.160—78.161 | 4.8528** | 下胚轴伸长蛋白1,lhy1 Late hypocotyl elongation protein ortholog1, lhy1 | |
| GRMZM2G180168 | 10 | 78.333—78.337 | 4.6254** | ABI3-VP1转录因子23,abi23 ABI3-VP1-transcription factor 23, abi23 |
新窗口打开|下载CSV
3 讨论
3.1 温热带玉米基因组变异分析
玉米起源于热带环境的墨西哥西南部,经过对其进行适应性改良,玉米逐渐的扩散到世界各地。根据光周期敏感性的不同,玉米可以被划分为温带玉米和热带玉米[6]。热带玉米靠近玉米起源中心,很少经历人工选择和改良,保留着大量的有利变异,是进行群体遗传改良的重要材料[8]。本研究选择30份温带玉米自交系和21份热带玉米作为研究材料进行重测序。通过SnpEff软件对变异文件进行基因组的功能的注释和预测,发现热带玉米基因组具有更多的SNP(图1),无论是SNP的总数还是平均分布密度均显著高于温带玉米。该结果证明了热带玉米具有更高的遗传多样性,这与前人研究即热带玉米具有更高的遗传多样性一致[8]。主成分分析发现温带玉米群存在更大的变异幅度(图2),推测是因为温带玉米群体包含较多的亚群,因此其变异幅度更广,该结果和前人的主成分分析一致[6]。这也表明本研究所使用的基因型数据以及群体划分的准确性,为后续的选择信号分析奠定了数据基础。玉米作为一个广适性的作物,其在基因组水平存在多态性变化,进而导致相关表型变化,是使其成为有重要影响力作物的一个重要条件。研究揭示了温热带玉米基因组水平SNP变异情况,可为后续的玉米功能基因组学研究提供理论基础。3.2 FST和XP-EHH的计算原理
随着海量生物学数据的产出,使用高密度的SNP标记进行群体间选择信号的分析在很多物种中都已成功进行,包括人类[37]、绵羊[38]、水稻[39]等。选择信号是一种重要的群体遗传学研究手段,利用合适的选择信号可以显著的揭示玉米适应性进化的遗传机理,并且可以深入挖掘与农艺性状相关的候选基因[21]。本研究利用FST和XP-EHH的方法进行选择信号的鉴定。FST可以利用等位基因的频率变化来筛选基因组受到驯化选择的基因区段[16],这是因为同一个物种由于驯化的目的、种植环境、改良时间不同将会导致基因组上的等位基因频率发生变化,虽然它可以鉴定出受到选择的基因,但是不能确定选择的方向[40],因此,本研究进一步引入XP-EHH选择信号方法。XP-EHH是一种基于基因组上单倍型纯合度的方法,鉴定不同群体间发生选择作用的区段,XP-EHH统计分析方法引入了参考群体和试验群体的概念,因此可以根据统计值判断基因组发生选择的方向,这为深入筛选候选驯化基因提供了更有力的数据支撑[41]。3.3 选择信号检测结果的评价
传统的选择信号检测方法主要利用RFLP、AFLP和SSR等分子标记。有研究利用玉米和大刍草的99个SSR标记作为选择信号进行分析[2],揭示了现代玉米起源于约9 000年前的墨西哥西南部。然而RFLP、AFLP和SSR等分子标记开发程序繁琐,并且定位精度不高,如今已经很少应用于选择信号的研究之中。随着新一代测序技术的发展,基于高通量测序技术得到的SNP标记由于其精度高、密度大以及定位更加准确等优点,逐渐取代了传统的分子标记,成为进行选择信号检测的主要方法[19,20,21]。本研究选择具有代表性的温带和热带玉米群体的重测序数据,进行全基因组水平的FST和XP-EHH的2种选择信号的鉴定,分别筛选到了557和1 913个候选基因。利用FST鉴定得到的基因进行富集分析,结果表明这些基因富集在一些重要的生物学通路上,主要参与肌动蛋白的发育调节(GO:0008064,GO:0032956)、微丝的发育调节(GO:0030832)、细胞骨架发育调控(GO:0030036)等。本研究利用FST得到基因进行富集分析,得到的显著GO条目较少,此外XP-EHH选择信号法鉴定到的候选基因未能得到显著的GO条目,不能准确地反映群体分化的结果。推测是目前所用的参考数据库信息不够全面,因此未能特异的鉴定出富有生物学意义的GO条目。本研究通过对具有较高选择信号的基因进行功能注释,发现这些基因是在热带玉米改良为温带玉米过程中受到了极大的选择,同时也是玉米生长发育过程中重要的调控因子(表3)。因此,结合相关的表型性状和选择信号法可以鉴定玉米适应改良过程中发生变化的基因。其中通过XP-EHH选择信号法,鉴定到温热带玉米适应性改良相关的调控基因ZmCCT9[33]和玉米开花调控相关的基因COL1[34]。尽管前人曾经利用过相关的选择信号鉴定了玉米在驯化过程中的选择基因[21],然而本研究着重挖掘了与开花相关的调控基因,开花是玉米生育期的一个重要农艺性状,关系到植株从营养生长到生殖生长的转变。其中包括已经报道过的ZmCCT9和COL1。此外在候选基因集中鉴定到了一些开花调控候选基因,例如GRMZM2G387528,基因功能注释表明该基因是一个光敏色素互作因子(表3),与光周期基因ZmphyB1互作[42]。因此,本研究鉴定得到的候选基因可以为后续的开花遗传机理解析提供理论基础。这些研究表明结合FST和XP-EHH选择信号法是进行群体遗传分化、功能富集分析和驯化基因挖掘的一个重要手段。4 结论
基于对温带和热带玉米的基因型数据分析,发现热带玉米具有更高的遗传多样性,表明其是玉米分子育种的重要资源。鉴定到玉米驯化改良过程中受到选择的基因,且部分受选择的基因参与玉米开花发育调控途径。(责任编辑 李莉)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1073/pnas.0812525106URLPMID:19307570 [本文引用: 1]

Questions that still surround the origin and early dispersals of maize (Zea mays L.) result in large part from the absence of information on its early history from the Balsas River Valley of tropical southwestern Mexico, where its wild ancestor is native. We report starch grain and phytolith data from the Xihuatoxtla shelter, located in the Central Balsas Valley, that indicate that maize was present by 8,700 calendrical years ago (cal. B.P.). Phytolith data also indicate an early preceramic presence of a domesticated species of squash, possibly Cucurbita argyrosperma. The starch and phytolith data also allow an evaluation of current hypotheses about how early maize was used, and provide evidence as to the tempo and timing of human selection pressure on 2 major domestication genes in Zea and Cucurbita. Our data confirm an early Holocene chronology for maize domestication that has been previously indicated by archaeological and paleoecological phytolith, starch grain, and pollen data from south of Mexico, and reshift the focus back to an origin in the seasonal tropical forest rather than in the semiarid highlands.
DOI:10.1073/pnas.052125199URLPMID:11983901 [本文引用: 2]
There exists extraordinary morphological and genetic diversity among the maize landraces that have been developed by pre-Columbian cultivators. To explain this high level of diversity in maize, several authors have proposed that maize landraces were the products of multiple independent domestications from their wild relative (teosinte). We present phylogenetic analyses based on 264 individual plants, each genotyped at 99 microsatellites, that challenge the multiple-origins hypothesis. Instead, our results indicate that all maize arose from a single domestication in southern Mexico about 9,000 years ago. Our analyses also indicate that the oldest surviving maize types are those of the Mexican highlands with maize spreading from this region over the Americas along two major paths. Our phylogenetic work is consistent with a model based on the archaeological record suggesting that maize diversified in the highlands of Mexico before spreading to the lowlands. We also found only modest evidence for postdomestication gene flow from teosinte into maize.
DOI:10.1073/pnas.1013011108URLPMID:21189301 [本文引用: 1]
The last two decades have seen important advances in our knowledge of maize domestication, thanks in part to the contributions of genetic data. Genetic studies have provided firm evidence that maize was domesticated from Balsas teosinte (Zea mays subspecies parviglumis), a wild relative that is endemic to the mid- to lowland regions of southwestern Mexico. An interesting paradox remains, however: Maize cultivars that are most closely related to Balsas teosinte are found mainly in the Mexican highlands where subspecies parviglumis does not grow. Genetic data thus point to primary diffusion of domesticated maize from the highlands rather than from the region of initial domestication. Recent archeological evidence for early lowland cultivation has been consistent with the genetics of domestication, leaving the issue of the ancestral position of highland maize unresolved. We used a new SNP dataset scored in a large number of accessions of both teosinte and maize to take a second look at the geography of the earliest cultivated maize. We found that gene flow between maize and its wild relatives meaningfully impacts our inference of geographic origins. By analyzing differentiation from inferred ancestral gene frequencies, we obtained results that are fully consistent with current ecological, archeological, and genetic data concerning the geography of early maize cultivation.
DOI:10.1126/science.1174276URL [本文引用: 1]
DOI:10.1126/science.aam9425URLPMID:28774930 [本文引用: 1]
By 4000 years ago, people had introduced maize to the southwestern United States; full agriculture was established quickly in the lowland deserts but delayed in the temperate highlands for 2000 ye ...
DOI:10.1007/s00122-009-1162-7URLPMID:19823800 [本文引用: 3]
Characterization of genetic diversity is of great value to assist breeders in parental line selection and breeding system design. We screened 770 maize inbred lines with 1,034 single nucleotide polymorphism (SNP) markers and identified 449 high-quality markers with no germplasm-specific biasing effects. Pairwise comparisons across three distinct sets of germplasm, CIMMYT (394), China (282), and Brazil (94), showed that the elite lines from these diverse breeding pools have been developed with only limited utilization of genetic diversity existing in the center of origin. Temperate and tropical/subtropical germplasm clearly clustered into two separate groups. The temperate germplasm could be further divided into six groups consistent with known heterotic patterns. The greatest genetic divergence was observed between temperate and tropical/subtropical lines, followed by the divergence between yellow and white kernel lines, whereas the least divergence was observed between dent and flint lines. Long-term selection for hybrid performance has contributed to significant allele differentiation between heterotic groups at 20% of the SNP loci. There appeared to be substantial levels of genetic variation between different breeding pools as revealed by missing and unique alleles. Two SNPs developed from the same candidate gene were associated with the divergence between two opposite Chinese heterotic groups. Associated allele frequency change at two SNPs and their allele missing in Brazilian germplasm indicated a linkage disequilibrium block of 142 kb. These results confirm the power of SNP markers for diversity analysis and provide a feasible approach to unique allele discovery and use in maize breeding programs.
DOI:10.1007/s001220051039URL [本文引用: 1]
Commercial maize ( Zea mays L.) in the USA has a restricted genetic base as newer hybrids are largely produced from crosses among elite inbred lines representing a small sample (predominantly about 6- to 8-base inbreds) of the Stiff stalk and Lancaster genetic backgrounds. Thus, expansion of genetic diversity in maize has been a continuous challenge to breeders. Tropical germplasm has been viewed as a useable source of diversity, although the integration of tropical germplasm into existing inbred line and hybrid development is laborious. The present study is an evaluation of the potential of tropical germplasm for temperate maize improvement. All possible single-, three-way-, and double-cross hybrids among three largely temperate and three temperate-adapted, all-tropical inbred lines were evaluated in yield-trial tests. Single-cross hybrids containing as much as 50–60% tropical germplasm produced 8.061t65ha -1 of grain yield, equivalent to the mean yield of the commercial check hybrids. On the other hand, three-way and double-cross hybrids with the highest mean yield contained lower amounts of tropical germplasm, 10–19% and 34–44%, respectively. Overall, hybrids containing 10–60% tropical germplasm yielded within the range of the commercial hybrid checks. Hybrids with more than 60% tropical germplasm had significantly lower yields, and 100% tropical hybrids yielded the least among all hybrids evaluated. The results indicate that inbred lines containing tropical germplasm are not only a useful source to expand the genetic diversity of commercial maize hybrids, but they, also are competitive in crosses with temperate materials, producing high-yielding hybrids. These experimental hybrids exhibited good standability (comparable to the commercial check hybrids) but contained 1–2% higher grain moisture, leading to delayed maturity. Recurrent selection procedures are being conducted on derivatives of these materials to extract lines with superior yield, good standability, and reduced grain moisture which can be used for commercial exploitation.
DOI:10.1007/s10681-013-1017-9URL [本文引用: 3]
Maize ( Zea mays L.) is one of few crops that can offer significant genetic gains with the utilization of genetic diversity. Genetically broad-based germplasm has the potential to contribute useful and unique alleles to U.S. Corn Belt breeding programs not present in current U.S. genome sequences (e.g. B73, NAM, etc.). Our objectives were to determine if unique tropical genetic materials have been effectively adapted to temperate environments and how their agronomic performance was relative to adapted populations. An important long-term objective of the Iowa and North Dakota maize breeding programs has been, in addition to the typical elite by elite line pedigree selection cultivar development process, to adapt exotic and unique germplasm, maximize their genetic improvement, and develop unique products for breeding and commercial uses. Stratified mass selection methodology for earliness has been utilized for the adaptation of tropical and temperate populations to Iowa and North Dakota environments. This method has allowed screening of up to 25,000 genotypes per population cycle at a rate of one cycle per year. In addition, the estimated cost per year our programs had for the adaptation of each population was less than $2,000 which could successfully be applied in any breeding program across the globe. This cost has been less than 1 % of the total cost for finding minor genes on the same trait. Our results showed the successful adaptation of exotic populations was independent from genetic background. We can speculate there are a few major genes responsible for most of flowering date expression. We encourage the use of technology to target traits according to their genetic complexity. Stratified mass selection at the phenotypic level has been successful. Each of the populations with either 25 of 100 % tropical germplasm are available for anyone who may desire to expand the germplasm base of their breeding programs with tropical germplasm adapted to temperate mid- and short-season U.S. Corn Belt environments.
DOI:10.1038/217624a0 [本文引用: 1]
DOI:10.1017/S0016672300014634URLPMID:4407212 [本文引用: 1]
When a selectively favourable gene substitution occurs in a population, changes in gene frequencies will occur at closely linked loci. In the case of a neutral polymorphism, average heterozygosity will be reduced to an extent which varies with distance from the substituted locus. The aggregate effect of substitution on neutral polymorphism is estimated; in populations of total size 106or more (and perhaps of 104or more), this effect will be more important than that of random fixation. This may explain why the extent of polymorphism in natural populations does not vary as much as one would expect from a consideration of the equilibrium between mutation and random fixation in populations of different sizes. For a selectively maintained polymorphism at a linked locus, this process will only be important in the long run if it leads to complete fixation. If the selective coefficients at the linked locus are small compared to those at the substituted locus, it is shown that the probability of complete fixation at the linked locus is approximately exp (c), wherecis the recombinant fraction andNthe population size. It follows that in a large population a selective substitution can occur in a cistron without eliminating a selectively maintained polymorphism in the same cistron.
DOI:10.1101/gad.3.11.1801URLPMID:2513255 [本文引用: 1]
Abstract The relationship between the two estimates of genetic variation at the DNA level, namely the number of segregating sites and the average number of nucleotide differences estimated from pairwise comparison, is investigated. It is found that the correlation between these two estimates is large when the sample size is small, and decreases slowly as the sample size increases. Using the relationship obtained, a statistical method for testing the neutral mutation hypothesis is developed. This method needs only the data of DNA polymorphism, namely the genetic variation within population at the DNA level. A simple method of computer simulation, that was used in order to obtain the distribution of a new statistic developed, is also presented. Applying this statistical method to the five regions of DNA sequences in Drosophila melanogaster, it is found that large insertion/deletion (greater than 100 bp) is deleterious. It is suggested that the natural selection against large insertion/deletion is so weak that a large amount of variation is maintained in a population.
DOI:10.1101/gr.4252305URLPMID:16251466 [本文引用: 1]
Detecting selective sweeps from genomic SNP data is complicated by the intricate ascertainment schemes used to discover SNPs, and by the confounding influence of the underlying complex demographics and varying mutation and recombination rates. Current methods for detecting selective sweeps have little or no robustness to the demographic assumptions and varying recombination rates, and provide no method for correcting for ascertainment biases. Here, we present several new tests aimed at detecting selective sweeps from genomic SNP data. Using extensive simulations, we show that a new parametric test, based on composite likelihood, has a high power to detect selective sweeps and is surprisingly robust to assumptions regarding recombination rates and demography (i.e., has low Type I error). Our new test also provides estimates of the location of the selective sweep(s) and the magnitude of the selection coefficient. To illustrate the method, we apply our approach to data from the Seattle SNP project and to Chromosome 2 data from the HapMap project. In Chromosome 2, the most extreme signal is found in the lactase gene, which previously has been shown to be undergoing positive selection. Evidence for selective sweeps is also found in many other regions, including genes known to be associated with disease risk such as DPP10 and COL4A3.
DOI:10.1038/nature01140URLPMID:12397357 [本文引用: 1]
Abstract The ability to detect recent natural selection in the human population would have profound implications for the study of human history and for medicine. Here, we introduce a framework for detecting the genetic imprint of recent positive selection by analysing long-range haplotypes in human populations. We first identify haplotypes at a locus of interest (core haplotypes). We then assess the age of each core haplotype by the decay of its association to alleles at various distances from the locus, as measured by extended haplotype homozygosity (EHH). Core haplotypes that have unusually high EHH and a high population frequency indicate the presence of a mutation that rose to prominence in the human gene pool faster than expected under neutral evolution. We applied this approach to investigate selection at two genes carrying common variants implicated in resistance to malaria: G6PD and CD40 ligand. At both loci, the core haplotypes carrying the proposed protective mutation stand out and show significant evidence of selection. More generally, the method could be used to scan the entire genome for evidence of recent positive selection.
DOI:10.1371/journal.pbio.0040072URLPMID:1892825 [本文引用: 1]
The identification of signals of very recent positive selection provides information about the adaptation of modern humans to local conditions. We report here on a genome-wide scan for signals of very recent positive selection in favor of variants that have not yet reached fixation. We describe a new analytical method for scanning single nucleotide polymorphism (SNP) data for signals of recent selection, and apply this to data from the International HapMap Project. In all three continental groups we find widespread signals of recent positive selection. Most signals are region-specific, though a significant excess are shared across groups. Contrary to some earlier low resolution studies that suggested a paucity of recent selection in sub-Saharan Africans, we find that by some measures our strongest signals of selection are from the Yoruba population. Finally, since these signals indicate the existence of genetic variants that have substantially different fitnesses, they must indicate loci that are the source of significant phenotypic variation. Though the relevant phenotypes are generally not known, such loci should be of particular interest in mapping studies of complex traits. For this purpose we have developed a set of SNPs that can be used to tag the strongest 鈭250 signals of recent selection in each population. Applying their newly developed method, the authors search International HapMap Project data representing three populations for signals of recent selection across the human genome.
DOI:10.1038/nature06250URL [本文引用: 3]
DOI:10.1007/978-3-642-88415-3URL [本文引用: 3]
The French edition of Professor Jacquard's book, published in 1970, has already been reviewed [see ABA 39, 2681]. This translation was undertaken with the aim of reaching a wider audience, in view of the inability or unwillingness of many English speaking scientists to read French texts. The translation is by Drs Deborah and Brian Charlesworth of Liverpool University, UK, who are to be commended both on its accuracy and readability. The new edition is some 50% longer than the original, with new material added and some sections replaced or brought up to date, both by the author (particularly on genetic distance and on French population studies) and by the translators (on overlapping generations). Some formulations have also been simplified.The book is concerned primarily with the mathematical theory of population genetics, and specifically its extension and application to human populations. Proofs are derived in some detail which, though useful for the student, result in concepts being buried in a mass of mathematics. Whilst the mathematics is complicated, it does not require advanced knowledge on the part of the reader. Much of the material can be found in other texts, sometimes more clearly, but this book is useful in that it provides alternative approaches and derivations.The overall plan of the book is to outline the conditions of Hardy-Weinberg equilibrium and then devote large chapters to removal of each of the assumptions in turn. The combined effects of removal of several assumptions, i.e. the effects of several evolutionary forces acting together, are dealt with rather briefly and less adequately.Strong points of the text are the analyses of genetic structure: relationships among individuals, inbreeding and non-random mating (with or without finite population size), where the contributions of Malecot and his students receive proper attention, and the theory of overlapping generations. In addition to the theory, many interesting examples of human population studies by French workers are used for illustration. Weaker parts of the text are the attempts to explain the necessary basic genetics in a few introductory pages, the discussion of inheritance of quantitative traits and the omission of problems of selection at more than one locus.Outside of the mathematics, Professor Jacquard discusses many of the problems of analysing human populations, but makes little contribution and expresses no views on some of the more contentious problems in population genetics such as neutral versus selective polymorphisms.In summary, "The genetic structure of populations" is a worthwhile addition to the few books available on theoretical population genetics.W.G. Hill
DOI:10.1111/j.1558-5646.1984.tb05657.xURLPMID:28563791 [本文引用: 4]
Formulae are given for estimators for the parameters F, 胃, f (FIT, FST, FIS) of population structure. As with all such estimators, ratios are used so that their properties are not known exactly, but they have been found to perform satisfactorily in simulations. Unlike the estimators in general use, the formulae do not make assumptions concerning numbers of populations, sample sizes, or heterozygote frequencies. As such, they are suited to small data sets and will aid the comparisons of results of different investigators. A simple weighting procedure is suggested for combining information over alleles and loci, and sample variances may be estimated by a jackknife procedure.
DOI:10.1017/S0016672310000121URLPMID:20515517 [本文引用: 1]
A two-step procedure is presented for analysis of θ (FST) statistics obtained for a battery of loci, which eventually leads to a clustered structure of values. The first step uses a simple Bayesian model for drawing samples from posterior distributions of θ-parameters, but without constructing Markov chains. This step assigns a weakly informative prior to allelic frequencies and does not make any assumptions about evolutionary models. The second step regards samples from these posterior distributions as ‘data’ and fits a sequence of finite mixture models, with the aim of identifying clusters of θ-statistics. Hopefully, these would reflect different types of processes and would assist in interpreting results. Procedures are illustrated with hypothetical data, and with published allelic frequency data for type II diabetes in three human populations, and for 12 isozyme loci in 12 populations of the argan tree in Morocco.
DOI:10.1038/nature11837URL [本文引用: 2]
DOI:10.1017/S001667230006426URLPMID:14704191 [本文引用: 2]
Two hundred and sixty maize inbred lines, representative of the genetic diversity among essentially all public lines of importance to temperate breeding and many important tropical and subtropical lines, were assayed for polymorphism at 94 microsatellite loci. The 2039 alleles identified served as raw data for estimating genetic structure and diversity. A model-based clustering analysis placed the inbred lines in five clusters that correspond to major breeding groups plus a set of lines showing evidence of mixed origins. A hylogenetic tree was constructed to further assess the genetic structure of maize inbreds, showing good agreement with the pedigree information and the cluster analysis. Tropical and subtropical inbreds possess a greater number of alleles and greater gene diversity than their temperate counterparts. The temperate Stiff Stalk lines are on average the most divergent from all other inbred groups. Comparison of diversity in equivalent samples of inbreds and open-pollinated landraces revealed that maize inbreds capture <80% of the alleles in the landraces, suggesting that landraces can provide additional genetic diversity for maize breeding. The contributions of four different segments of the landrace gene pool to each inbred group's gene pool were estimated using a novel likelihood-based model. The estimates are largely consistent with known histories of the inbreds and indicate that tropical highland germplasm is poorly represented in maize inbreds. Core sets of inbreds that capture maximal allelic richness were defined. These or similar core sets can be used for a variety of genetic applications in maize.
DOI:10.1371/journal.pone.0169806URLPMID:5242465 [本文引用: 4]
The extensive genetic variation present in maize (Zea mays) germplasm makes it possible to detect signatures of positive artificial selection that occurred during temperate and tropical maize improvement. Here we report an analysis of 532,815 polymorphisms from a maize association panel consisting of 368 diverse temperate and tropical inbred lines. We developed a gene-oriented approach adapting exonic polymorphisms to identify recently selected alleles by comparing haplotypes across the maize genome. This analysis revealed evidence of selection for more than 1100 genomic regions during recent improvement, and included regulatory genes and key genes with visible mutant phenotypes. We find that selected candidate target genes in temperate maize are enriched in biosynthetic processes, and further examination of these candidates highlights two cases, sucrose flux and oil storage, in which multiple genes in a common pathway can be cooperatively selected. Finally, based on available parallel gene expression data, we hypothesize that some genes were selected for regulatory variations, resulting in altered gene expression.
DOI:10.1126/science.1178534URL [本文引用: 1]
DOI:10.1038/nature22971URLPMID:28605751 [本文引用: 1]
Abstract Complete and accurate reference genomes and annotations provide fundamental tools for characterization of genetic and functional variation. These resources facilitate the determination of biological processes and support translation of research findings into improved and sustainable agricultural technologies. Many reference genomes for crop plants have been generated over the past decade, but these genomes are often fragmented and missing complex repeat regions. Here we report the assembly and annotation of a reference genome of maize, a genetic and agricultural model species, using single-molecule real-time sequencing and high-resolution optical mapping. Relative to the previous reference genome, our assembly features a 52-fold increase in contig length and notable improvements in the assembly of intergenic spaces and centromeres. Characterization of the repetitive portion of the genome revealed more than 130,000 intact transposable elements, allowing us to identify transposable element lineage expansions that are unique to maize. Gene annotations were updated using 111,000 full-length transcripts obtained by single-molecule real-time sequencing. In addition, comparative optical mapping of two other inbred maize lines revealed a prevalence of deletions in regions of low gene density and maize lineage-specific genes.
DOI:10.1038/ng.2313URL [本文引用: 1]
DOI:10.1093/gigascience/gix134URLPMID:29300887 [本文引用: 2]
Abstract Background: Characterization of genetic variations in maize has been challenging, mainly due to deterioration of collinearity between individual genomes in the species. An international consortium of maize research groups combined resources to develop the maize haplotype version 3 (HapMap 3), built from whole genome sequencing data from 1,218 maize lines, covering pre-domestication and domesticated Zea mays varieties across the world. Results: A new computational pipeline was set up to process over 12 trillion bp of sequencing data and a set of population genetics filters were applied to identify over 83 million variant sites. Conclusions: We identified polymorphisms in regions where collinearity is largely preserved in the maize species. However, the fact that the B73 genome used as the reference only represents a fraction of all haplotypes is still an important limiting factor.
DOI:10.1093/bioinformatics/btr330URLPMID:21653522 [本文引用: 1]
The variant call format (VCF) is a generic format for storing DNA polymorphism data such as SNPs, insertions, deletions and structural variants, together with rich annotations. VCF is usually stored in a compressed manner and can be indexed for fast data retrieval of variants from a range of positions on the reference genome. The format was developed for the 1000 Genomes Project, and has also been adopted by other projects such as UK10K, dbSNP and the NHLBI Exome Project. VCFtools is a software suite that implements various utilities for processing VCF files, including validation, merging, comparing and also provides a general Perl API.http://vcftools.sourceforge.net
DOI:10.4161/fly.19695URL [本文引用: 1]
DOI:10.1038/hdy.2015.42URLPMID:4611237 [本文引用: 1]
Identifying signatures of recent or ongoing selection is of high relevance in livestock population genomics. From a statistical perspective, determining a proper testing procedure and combining various test statistics is challenging. On the basis of extensive simulations in this study, we discuss the statistical properties of eight different established selection signature statistics. In the considered scenario, we show that a reasonable power to detect selection signatures is achieved with high marker density (>1 SNP/kb) as obtained from sequencing, while rather small sample sizes (~15 diploid individuals) appear to be sufficient. Most selection signature statistics such as composite likelihood ratio and cross population extended haplotype homozogysity have the highest power when fixation of the selected allele is reached, while integrated haplotype score has the highest power when selection is ongoing. We suggest a novel strategy, called de-correlated composite of multiple signals (DCMS) to combine different statistics for detecting selection signatures while accounting for the correlation between the different selection signature statistics. When examined with simulated data, DCMS consistently has a higher power than most of the single statistics and shows a reliable positional resolution. We illustrate the new statistic to the established selective sweep around the lactase gene in human HapMap data providing further evidence of the reliability of this new statistic. Then, we apply it to scan selection signatures in two chicken samples with diverse skin color. Our analysis suggests that a set of well-known genes such as BCO2, MC1R, ASIP and TYR were involved in the divergent selection for this trait.
[本文引用: 2]
DOI:10.1093/molbev/msu211URLPMID:4166924 [本文引用: 1]
Haplotype-based scans to detect natural selection are useful to identify recent or ongoing positive selection in genomes. As both real and simulated genomic datasets grow larger, spanning thousands of samples and millions of markers, there is a need for a fast and efficient implementation of these scans for general use. Here we present selscan, an efficient multi-threaded application that implements Extended Haplotype Homozygosity (EHH), Integrated Haplotype Score (iHS), and Cross-population Extended Haplotype Homozygosity (XPEHH). selscan accepts phased genotypes in multiple formats, including TPED, and performs extremely well on both simulated and real data and over an order of magnitude faster than existing available implementations. It calculates iHS on chromosome 22 (22,147 loci) across 204 CEU haplotypes in 353s on one thread (33s on 16 threads) and calculates XPEHH for the same data relative to 210 YRI haplotypes in 578s on one thread (52s on 16 threads). Source code and binaries (Windows, OSX and Linux) are available at https://github.com/szpiech/selscan .
DOI:10.1093/bioinformatics/btq033URLPMID:20110278 [本文引用: 1]
Motivation: Testing for correlations between different sets of genomic features is a fundamental task in genomics research. However, searching for overlaps between features with existing web-based methods is complicated by the massive datasets that are routinely produced with current sequencing technologies. Fast and flexible tools are therefore required to ask complex questions of these data in an efficient manner.Results: This article introduces a new software suite for the comparison, manipulation and annotation of genomic features in Browser Extensible Data (BED) and General Feature Format (GFF) format. BEDTools also supports the comparison of sequence alignments in BAM format to both BED and GFF features. The tools are extremely efficient and allow the user to compare large datasets (e.g. next-generation sequencing data) with both public and custom genome annotation tracks. BEDTools can be combined with one another as well as with standard UNIX commands, thus facilitating routine genomics tasks as well as pipelines that can quickly answer intricate questions of large genomic datasets.Availability and implementation: BEDTools was written in C++. Source code and a comprehensive user manual are freely available at http://code.google.com/p/bedtoolsContact:aaronquinlan@gmail.com; imh4y@virginia.eduSupplementary information:Supplementary data are available at Bioinformatics online.
DOI:10.1093/nar/gkx382URLPMID:28472432 [本文引用: 1]
The agriGO platform, which has been serving the scientific community for >10 years, specifically focuses on gene ontology (GO) enrichment analyses of plant and agricultural species. We continuously maintain and update the databases and accommodate the various requests of our global users. Here, we present our updated agriGO that has a largely expanded number of supporting species (394) and datatypes (865). In addition, a larger number of species have been classified into groups covering crops, vegetables, fish, birds and insects closely related to the agricultural community. We further improved the computational efficiency, including the batch analysis andP-value distribution (PVD), and the user-friendliness of the web pages. More visualization features were added to the platform, including SEACOMPARE (cross comparison of singular enrichment analysis), direct acyclic graph (DAG) and Scatter Plots, which can be merged by choosing any significant GO term. The updated platform agriGO v2.0 is now publicly accessible athttp://systemsbiology.cau.edu.cn/agriGOv2/.
DOI:10.1073/pnas.1718058115URLPMID:29279404 [本文引用: 4]
Flowering time is a critical determinant of crop adaptation to local environments. As a result of natural and artificial selection, maize has evolved a reduced photoperiod sensitivity to adapt to regions over 90掳 of latitude in the Americas. Here we show that a distant Harbinger-like transposon acts as acis-regulatory element to repressZmCCT9expression to promote flowering under the long days of higher latitudes. The transposon atZmCCT9and another functional transposon at a second flowering-time gene,ZmCCT10, arose sequentially following domestication and were targeted by selection as maize spread from the tropics to higher latitudes. Our results demonstrate that new functional variation created by transposon insertions helped maize to spread over a broad range of latitudes rapidly. From its tropical origin in southwestern Mexico, maize spread over a wide latitudinal cline in the Americas. This feat defies the rule that crops are inhibited from spreading easily across latitudes. How the widespread latitudinal adaptation of maize was accomplished is largely unknown. Through positional cloning and association mapping, we resolved a flowering-time quantitative trait locus to a Harbinger-like transposable element positioned 57 kb upstream of a CCT transcription factor (ZmCCT9). The Harbinger-like element acts incisto repressZmCCT9expression to promote flowering under long days. Knockout ofZmCCT9by CRISPR/Cas9 causes early flowering under long days.ZmCCT9is diurnally regulated and negatively regulates the expression of the florigenZCN8, thereby resulting in late flowering under long days. Population genetics analyses revealed that the Harbinger-like transposon insertion atZmCCT9and the CACTA-like transposon insertion at another CCT paralog,ZmCCT10, arose sequentially following domestication and were targeted by selection for maize adaptation to higher latitudes. Our findings help explain how the dynamic maize genome with abundant transposon activity enabled maize to adapt over 90掳 of latitude during the pre-Columbian era.
DOI:10.1186/1471-2229-10-126URLPMID:20576144 [本文引用: 2]
pAbstract/p pBackground/p pThe plant circadian clock orchestrates 24-hour rhythms in internal physiological processes to coordinate these activities with daily and seasonal changes in the environment. The circadian clock has a profound impact on many aspects of plant growth and development, including biomass accumulation and flowering time. Despite recent advances in understanding the circadian system of the model plant itArabidopsis thaliana/it, the contribution of the circadian oscillator to important agronomic traits in itZea mays /itand other cereals remains poorly defined. To address this deficit, this study investigated the transcriptional landscape of the maize circadian system./p pResults/p pSince transcriptional regulation is a fundamental aspect of circadian systems, genes exhibiting circadian expression were identified in the sequenced maize inbred B73. Of the over 13,000 transcripts examined, approximately 10 percent displayed circadian expression patterns. The majority of cycling genes had peak expression at subjective dawn and dusk, similar to other plant circadian systems. The maize circadian clock organized co-regulation of genes participating in fundamental physiological processes, including photosynthesis, carbohydrate metabolism, cell wall biogenesis, and phytohormone biosynthesis pathways./p pConclusions/p pCircadian regulation of the maize genome was widespread and key genes in several major metabolic pathways had circadian expression waveforms. The maize circadian clock coordinated transcription to be coincident with oncoming day or night, which was consistent with the circadian oscillator acting to prepare the plant for these major recurring environmental changes. These findings highlighted the multiple processes in maize plants under circadian regulation and, as a result, provided insight into the important contribution this regulatory system makes to agronomic traits in maize and potentially other C4 plant species./p
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/ncomms5800URLPMID:25226282 [本文引用: 1]
Abstract Facial recognition plays a key role in human interactions, and there has been great interest in understanding the evolution of human abilities for individual recognition and tracking social relationships. Individual recognition requires sufficient cognitive abilities and phenotypic diversity within a population for discrimination to be possible. Despite the importance of facial recognition in humans, the evolution of facial identity has received little attention. Here we demonstrate that faces evolved to signal individual identity under negative frequency-dependent selection. Faces show elevated phenotypic variation and lower between-trait correlations compared with other traits. Regions surrounding face-associated single nucleotide polymorphisms show elevated diversity consistent with frequency-dependent selection. Genetic variation maintained by identity signalling tends to be shared across populations and, for some loci, predates the origin of Homo sapiens. Studies of human social evolution tend to emphasize cognitive adaptations, but we show that social evolution has shaped patterns of human phenotypic and genetic diversity as well.
DOI:10.11843/j.issn.0366-6964.2013.12.005URLMagsci [本文引用: 1]
<p><span >本研究基于苏尼特羊(</span><em ><span lang="EN-US">n</span></em><span lang="EN-US">=66</span><span >)、德国肉用美利奴羊</span><span lang="EN-US">(<em >n</em>=159)</span><span >和杜泊羊</span><span lang="EN-US">(<em >n</em>=93)3</span><span >个群体的</span><span lang="EN-US">Illumina Ovine SNP50 </span><span >芯片分型数据,借助群体分化指数</span><em ><span lang="EN-US">F<sub>ST</sub></span></em><span >法进行群体间选择信号的检测分析,应用生物信息学方法分析寻找选择信号区域内的重要基因。结果,共找到</span><span lang="EN-US">343</span><span >个“离群”位点,通过基因注释找到这些位点对应的</span><span lang="EN-US">365</span><span >个候选基因。其中部分基因与绵羊生长、繁殖及肉质性状相关,如</span><em ><span lang="EN-US">IGF</span></em><span lang="EN-US">2PB3</span><span >、</span><em ><span lang="EN-US">IGF</span></em><span lang="EN-US">1R</span><span >、</span><em ><span lang="EN-US">BMP</span></em><span lang="EN-US">2</span><span >、</span><em ><span lang="EN-US">BMPR</span></em><span lang="EN-US">1B</span><span >、</span><em ><span lang="EN-US">CAPN</span></em><span lang="EN-US">3</span><span >等,本研究结果也进一步验证了前期利用</span><span lang="EN-US">CNV</span><span >和</span><span lang="EN-US">GWAS</span><span >研究检测到的基因结果。通过</span><span lang="EN-US">GO</span><span >分析发现,这些基因主要富集在新陈代谢和去甲基化等条目。通过群体分化指数</span><em ><span lang="EN-US">F<sub>ST</sub></span></em><span >能有效地检测到具有选择信号的基因,其中包括绵羊部分重要经济性状的候选基因,研究结果能为绵羊的育种提供参考。</span></p>
DOI:10.11843/j.issn.0366-6964.2013.12.005URLMagsci [本文引用: 1]
<p><span >本研究基于苏尼特羊(</span><em ><span lang="EN-US">n</span></em><span lang="EN-US">=66</span><span >)、德国肉用美利奴羊</span><span lang="EN-US">(<em >n</em>=159)</span><span >和杜泊羊</span><span lang="EN-US">(<em >n</em>=93)3</span><span >个群体的</span><span lang="EN-US">Illumina Ovine SNP50 </span><span >芯片分型数据,借助群体分化指数</span><em ><span lang="EN-US">F<sub>ST</sub></span></em><span >法进行群体间选择信号的检测分析,应用生物信息学方法分析寻找选择信号区域内的重要基因。结果,共找到</span><span lang="EN-US">343</span><span >个“离群”位点,通过基因注释找到这些位点对应的</span><span lang="EN-US">365</span><span >个候选基因。其中部分基因与绵羊生长、繁殖及肉质性状相关,如</span><em ><span lang="EN-US">IGF</span></em><span lang="EN-US">2PB3</span><span >、</span><em ><span lang="EN-US">IGF</span></em><span lang="EN-US">1R</span><span >、</span><em ><span lang="EN-US">BMP</span></em><span lang="EN-US">2</span><span >、</span><em ><span lang="EN-US">BMPR</span></em><span lang="EN-US">1B</span><span >、</span><em ><span lang="EN-US">CAPN</span></em><span lang="EN-US">3</span><span >等,本研究结果也进一步验证了前期利用</span><span lang="EN-US">CNV</span><span >和</span><span lang="EN-US">GWAS</span><span >研究检测到的基因结果。通过</span><span lang="EN-US">GO</span><span >分析发现,这些基因主要富集在新陈代谢和去甲基化等条目。通过群体分化指数</span><em ><span lang="EN-US">F<sub>ST</sub></span></em><span >能有效地检测到具有选择信号的基因,其中包括绵羊部分重要经济性状的候选基因,研究结果能为绵羊的育种提供参考。</span></p>
DOI:10.1038/ng.695URLPMID:20972439 [本文引用: 1]
Uncovering the genetic basis of agronomic traits in crop landraces that have adapted to various agro-climatic conditions is important to world food security. Here we have identified 65 3.6 million SNPs by sequencing 517 landraces and constructed a high-density haplotype map of the genome using a novel data-imputation method. We performed genome-wide association studies (GWAS) for 14 agronomic traits in the population of subspecies. The loci identified through GWAS explained 65 36% of the phenotypic variance, on average. The peak signals at six loci were tied closely to previously identified genes. This study provides a fundamental resource for genetics research and breeding, and demonstrates that an approach integrating second-generation genome sequencing and GWAS can be used as a powerful complementary strategy to classical biparental cross-mapping for dissecting complex traits in .
DOI:10.1371/journal.pgen.1000471URLPMID:19424416 [本文引用: 1]
Abstract Selection acting on genomic functional elements can be detected by its indirect effects on population diversity at linked neutral sites. To illuminate the selective forces that shaped hominid evolution, we analyzed the genomic distributions of human polymorphisms and sequence differences among five primate species relative to the locations of conserved sequence features. Neutral sequence diversity in human and ancestral hominid populations is substantially reduced near such features, resulting in a surprisingly large genome average diversity reduction due to selection of 19-26% on the autosomes and 12-40% on the X chromosome. The overall trends are broadly consistent with "background selection" or hitchhiking in ancestral populations acting to remove deleterious variants. Average selection is much stronger on exonic (both protein-coding and untranslated) conserved features than non-exonic features. Long term selection, rather than complex speciation scenarios, explains the large intragenomic variation in human/chimpanzee divergence. Our analyses reveal a dominant role for selection in shaping genomic diversity and divergence patterns, clarify hominid evolution, and provide a baseline for investigating specific selective events.
URL [本文引用: 1]
畜禽选择信号的研究随着基因组大数据的爆发式增长,已经成为目前畜禽群体遗传学研究与重要经济性状基因定位的一个重要手段。使用恰当的选择信号方法,了解选择信号检测中的统计学问题,对于准确解析畜禽适应性进化的潜在遗传机制,精细定位重要经济性状的候选基因具有重要的意义。围绕畜禽基因组选择信号研究概况,本文对选择信号的概念、检验统计量类别、畜禽选择信号检测的影响因素及相关统计学问题进行了综述,以期为进一步拓展选择信号的研究思路提供参考。
URL [本文引用: 1]
畜禽选择信号的研究随着基因组大数据的爆发式增长,已经成为目前畜禽群体遗传学研究与重要经济性状基因定位的一个重要手段。使用恰当的选择信号方法,了解选择信号检测中的统计学问题,对于准确解析畜禽适应性进化的潜在遗传机制,精细定位重要经济性状的候选基因具有重要的意义。围绕畜禽基因组选择信号研究概况,本文对选择信号的概念、检验统计量类别、畜禽选择信号检测的影响因素及相关统计学问题进行了综述,以期为进一步拓展选择信号的研究思路提供参考。
DOI:10.1093/jxb/erw217URLPMID:27262126 [本文引用: 1]
phyB and PIF3 share a common protein-protein signaling mechanism in maize and Arabidopsis. However, phyB1 and phyB2 in maize do not share this mechanism, despite being closely related, indicating recent evolutionary divergence. Two maize phytochrome-interacting factor (PIF) basic helix-loop-helix (bHLH) family members, ZmPIF3.1 and ZmPIF3.2, were identified, cloned and expressedin vitroto investigate light-signaling interactions. A phylogenetic analysis of sequences of the maize bHLH transcription factor gene family revealed the extent of the PIF family, and a total of seven predicted PIF-encoding genes were identified from genes encoding bHLH family VIIa/b proteins in the maize genome. To investigate the role of maize PIFs in phytochrome signaling, full-length cDNAs for phytochromesPhyA2,PhyB1,PhyB2andPhyC1from maize were cloned and expressedin vitroas chromophorylated holophytochromes. We showed that ZmPIF3.1 and ZmPIF3.2 interact specifically with the Pfr form of maize holophytochrome B1 (ZmphyB1), showing no detectable affinity for the Pr form. Maize holophytochrome B2 (ZmphyB2) showed no detectable binding affinity for PIFs in either Pr or Pfr forms, but phyB Pfr from Arabidopsis interacted with ZmPIF3.1 similarly to ZmphyB1 Pfr. We conclude that subfunctionalization at the protein-protein interaction level has altered the role of phyB2 relative to that of phyB1 in maize. Since thephyB2mutant shows photomorphogenic defects, we conclude that maize phyB2 is an active photoreceptor, without the binding of PIF3 seen in other phyB family proteins.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}