 ,西南大学农学与生物科技学院,重庆400716
,西南大学农学与生物科技学院,重庆400716Genome-Wide Association Analysis of Tribenuron-Methyl Tolerance Related Traits in Brassica napus L. Under Germination
ZHOU QingYuan, WANG Qian, YE Sang, CUI MinSheng, LEI Wei, GAO HuanHuan, ZHAO YuFeng, XU XinFu, TANG ZhangLin, LI JiaNa, CUI Cui,College of Agronomy and Biotechnology, Southwest University, Chongqing 400716通讯作者:
收稿日期:2018-09-17接受日期:2018-11-17网络出版日期:2019-02-13
| 基金资助: |
Received:2018-09-17Accepted:2018-11-17Online:2019-02-13
作者简介 About authors
周清元,Tel:13883388890;E-mail:
王倩,Tel:13251395292;E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2008KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
周清元, 王倩, 叶桑, 崔明圣, 雷维, 郜欢欢, 赵愉风, 徐新福, 唐章林, 李加纳, 崔翠. 苯磺隆胁迫下油菜萌发期相关性状的全基因组关联分析[J]. 中国农业科学, 2019, 52(3): 399-413 doi:10.3864/j.issn.0578-1752.2019.03.002
ZHOU QingYuan, WANG Qian, YE Sang, CUI MinSheng, LEI Wei, GAO HuanHuan, ZHAO YuFeng, XU XinFu, TANG ZhangLin, LI JiaNa, CUI Cui.
0 引言
【研究意义】苯磺隆作为一种磺酰脲类除草剂,可用于防除禾谷类和其他油料作物田中多种阔叶杂草[1,2]。虽然在推荐剂量下的使用较为安全[3],但仍存在短期土壤残留现象[4,5],容易导致对除草剂敏感的作物死亡[6]。近年来,随着油菜田阔叶杂草防除问题逐渐突出[7],采用阔叶除草剂和抗耐性油菜品种相配合是解决杂草问题最为经济有效的方式[8]。种子萌发期是作物生长的关键阶段,其发芽质量的好坏直接影响作物的出苗率、作物的生长发育以及经济效益[9]。研究表明,不同品种(系)油菜萌发期对苯磺隆的耐性具有显著性差异,利用苯磺隆对油菜种子的发芽性状进行分析,筛选出萌发期对苯磺隆耐性极强的品种(系)是可行的[10]。因此,针对油菜萌发期耐苯磺隆进行相关遗传因子研究和筛选耐性基因,对了解油菜苯磺隆响应的分子机理、培育耐苯磺隆油菜品种具有重要意义。【前人研究进展】全基因组关联分析(genome-wide association analysis,GWAS)是研究数量性状的重要方法之一,在多种作物逆境胁迫下相关性状遗传机制的研究中有广泛应用[11,12]。随着甘蓝型油菜全基因组序列的公布及芸薹属60K SNP芯片的开发[13],GWAS已成功地应用于油菜复杂性状的QTL检测[14,15,16,17,18,19]。CHEN等[14]对419个甘蓝型油菜品种幼苗期进行GWAS分析,鉴定出32个与镉积累性状有关的候选基因。WAN等[15]在油菜种子苗期利用GWAS检测到与4个耐盐性状相关的75个显著SNP位点。WEI等[16]通过SNP性状关联和转录组测序分析,在347个油菜自交系的菌核病抗性中鉴定出24个基因。此外,研究者利用GWAS发现与油菜萌发活力[17]、含油量[18]、开花时间[19]等有关的显著位点并预测候选基因。陈东亮等[20]从基因组水平分析油菜的草铵膦抗性,发现18个与草铵膦的灭生机制相关的候选基因。ZHANG等[21]为研究苯达松和磺酰脲敏感性突变体BEL的遗传性质,使用生物学技术构建了水稻BEL位点的精细图,并在限制区间内发现5个基因。针对苯磺隆,近年来也相继有报道。杨倩等[22]以对苯磺隆敏感和抗性的播娘蒿为试材,利用RNA-seq分析得到了35个与苯磺隆代谢相关的功能基因。同样以播娘蒿为材料,YANG等[23]利用全局差异基因表达谱技术共鉴定出26个苯磺隆耐性差异表达的基因,并通过qRT-PCR验证了其中8个基因。GAINES等[24]使用Illumina HiSeq进行RNA-seq发现4种基因(P450s、硝酸盐单加氧酶、GST和GT)在黑燕麦代谢除草剂抗性方面发挥关键作用。【本研究切入点】磺酰脲类除草剂苯磺隆一般用作苗床和芽后除草[25],在油菜播前或移栽前使用将在土壤中造成残留,这对油菜种子萌发存在着较大影响[10],尽管前人通过资源筛选[26]、人工诱变[27]、转基因育种[28]等方法筛选出耐苯磺隆油菜植株,但是关于油菜各生育时期,尤其是萌发期对苯磺隆耐性的遗传机制和分子机理的研究较少。【拟解决的关键问题】本研究在前期筛选的适宜浓度的苯磺隆处理下,对遗传来源不同的241份甘蓝型油菜种质进行发芽期耐苯磺隆特性鉴定,并借助覆盖油菜全基因组的5.2万个SNP标记分别对相对根长、相对鲜重、相对发芽率进行全基因组关联分析,以探究油菜在苯磺隆逆境胁迫下的生理形态所反应的基因调控机制,为耐苯磺隆油菜品种的研究提供参考。1 材料与方法
1.1 试验处理与性状调查
241份甘蓝型油菜品种由西南大学油菜中心收集并提供[10],于室内进行培养皿纸上发芽试验。根据前期筛选的浓度,在铺有2层滤纸的培养皿中加入3 mL浓度为25 mg·kg-1的苯磺隆溶液,蒸馏水作为对照,每皿均匀放置20粒已清洗的饱满种子,3次重复。将培养皿置于温度25℃、相对湿度为85%的人工培养箱中培养,光照和黑暗时间为16 h/8 h。第7天统计种子发芽率,每皿随机选取生长一致的10株幼苗分别测定根长和鲜重。参考王倩等[10]方法,分别计算每个品系的相对鲜重、相对根长和相对发芽率,并进行GWAS分析。采用Excel 2016软件对表型数据进行初步整理,计算其平均数、标准差和变异系数。1.2 全基因组关联分析
利用油菜基因组60 K Illumina Infinium SNP芯片对241份种质基因型进行分析[14],参照XU等[19]方法筛选用于LD分析和关联分析的SNP标记。群体结构由Structure 2.3.4软件基于贝叶斯数学模型进行分析,并参照WAN等[15]确定最终亚群数目。亲缘关系(relative kinship)K矩阵由Tassel 5.0软件分析,其值为负时设为0[29]。利用软件Tassel5.0估算群体内连锁不平衡(linkage disequilibrium,LD)的衰减[18]。使用一般线性模型(general linear model,GLM)和混合线性模型(mixed linear model,MLM)中6种模型进行关联分析[19,30]。3个性状的最优模型根据Quantile- Quantile散点图(QQ plot)在6种模型下的结果进行分析比较获得,并在此基础上绘制曼哈顿图,显示关联分析检测到的与目标性状显著相关的标记位点[31]。同时,负对数(1/N)作为显著关联SNP阈值,其中N是关联分析中SNP的总数,本研究的阈值为4.5(-log10(1/32493))。1.3 候选基因的预测
根据与性状显著关联的SNP的位置左右延伸其所在染色体上R2=0.2时的衰减距离,为候选基因的LD区间。利用法国公布的甘蓝型油菜“Darmor-Bzh”的基因组注释信息及TAIR网站对LD区间内的候选基因进行功能注释和分析[30]。2 结果
2.1 表型统计分析
3个相对性状均表现出广泛的表型变异,其中(表1)相对根长变异幅度为0.050—0.588,平均值为0.136,变异系数为48.95%;相对鲜重变异幅度为0.411—2.357,平均值为0.980,变异系数为20.60%;相对发芽率变异幅度为0.333—1.000,平均值为0.942,变异系数为10.28%。结果(图1)表明,3个性状均呈连续性分布,符合数量性状的特点,适合进行GWAS分析。Table 1
表1
表1苯磺隆胁迫下相关性状的表型统计
Table 1
| 性状 Traits | 均值±标准差 Mean±SD | 变幅 Change amplitude | 变异系数 CV (%) |
|---|---|---|---|
| 相对根长Relative root length | 0.136±0.066** | 0.050-0.588 | 48.950 |
| 相对鲜重Relatively fresh weight | 0.980±0.202** | 0.411-2.357 | 20.593 |
| 相对发芽率Relative germination rate | 0.942±0.097** | 0.333-1.000 | 10.276 |
新窗口打开|下载CSV
图1
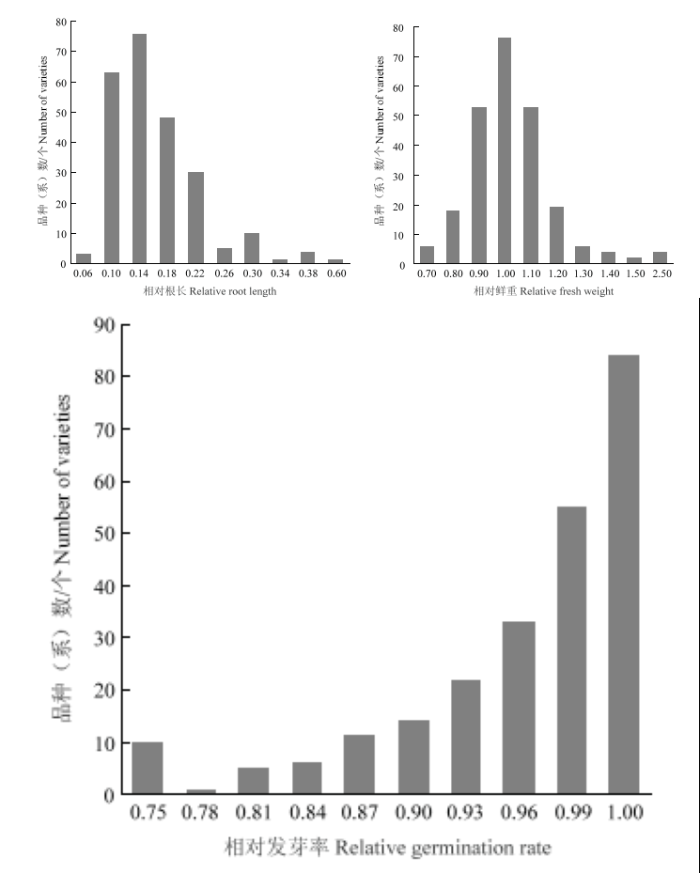 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图13 个相对性状频数分布图
Fig. 1Phenotype frequency distribution of 3 related traits
2.2 SNP标记分析
将挑选出多态性和质量较高的32 493个SNP标记均匀分配到19条染色体,其中C04染色体上SNP标记数目最多,为3 005个;而C09染色体上SNP标记数目最少,为1 006个(表2)。这些SNP标记涉及的碱基变化为A/T、T/C、A/G、A/C、T/G、T/A、C/G和G/C 8种类型,其中A/G和T/C类型最多,分别为11 691和10 864;而C/G和G/C类型最少,分别为109和114(图2)。Table 2
表2
表2SNP在染色体上的分布
Table 2
| 染色体 Chr. | SNP数目 No. | 长度 Length (bp) | SNP密度 SNP density (1 SNP/kb) | 染色体 Chr. | SNP数目 No. | 长度 Length (bp) | SNP密度 SNP density (1 SNP/kb) | |
|---|---|---|---|---|---|---|---|---|
| A01 | 1533 | 23213190 | 15.14 | C01 | 2321 | 38751959 | 16.70 | |
| A02 | 1266 | 24751063 | 19.55 | C02 | 2225 | 46171227 | 20.75 | |
| A03 | 2206 | 29727584 | 13.48 | C03 | 2707 | 60554466 | 22.37 | |
| A04 | 1444 | 19097304 | 13.23 | C04 | 3005 | 48891192 | 16.27 | |
| A05 | 1623 | 22986419 | 14.16 | C05 | 1011 | 43165527 | 42.70 | |
| A06 | 1519 | 24371484 | 16.04 | C06 | 1287 | 37161109 | 28.87 | |
| A07 | 1882 | 23921822 | 12.71 | C07 | 1683 | 44305541 | 26.33 | |
| A08 | 1085 | 18658096 | 17.20 | C08 | 1527 | 38226084 | 25.03 | |
| A09 | 1637 | 33803997 | 20.65 | C09 | 1006 | 48440856 | 48.15 | |
| A10 | 1526 | 17348295 | 11.37 | |||||
| A染色体SNP 的平均分布 Mean A genome | 1572 | 23787925 | 15.13 | C染色体SNP 的平均分布 Mean C genome | 1864 | 45074218 | 24.19 |
新窗口打开|下载CSV
图2
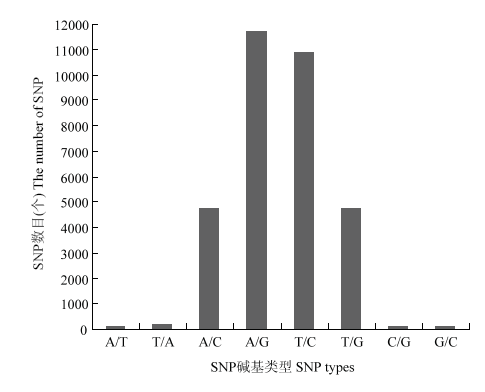 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图232493个SNP碱基类型分布图
Fig. 232493 maps of SNP base types
2.3 群体结构与亲缘关系分析
选择在染色体上均匀分布,且MAF大于0.3的10 655个SNP标记分析群体材料间亲缘关系和群体结构。利用软件Structure2.3.4对每个可能的K值模拟运算,当K=2时,显示△K有最大变化(图3-A),这241份甘蓝型油菜分为P1(94份材料)和P2(147份材料)2个亚群(图3-B)。利用Tassel5.1.0软件的Kinship模块分析任意2个材料之间的亲缘关系值(图3-C),显示约56.28%材料间的亲缘关系值为0,约12.77%材料的亲缘关系值在0—0.05,表明试验群体材料间的亲缘关系较远,符合进行GWAS分析的要求。图3
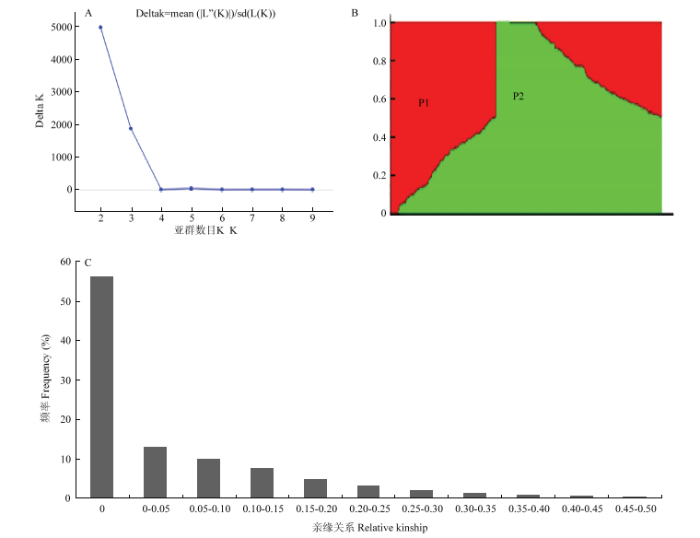 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3241份甘蓝型油菜的群体结构和亲缘关系分析
A:群体的?k值;B:群体结构示意图;C:亲缘关系分布
Fig. 3Analysis of population structure and relative kinship in 241 B.napus
A: Estimation of ?k value in population; B: Group structure diagram; C: Distribution of relative kinship values
2.4 连锁不平衡在A、C基因组中的衰减
利用Tassel 5.1.0软件分析已筛选的32 493个高密度SNP标记来估算LD在甘蓝型油菜A、C基因组中的衰减。从LD衰减散点图(图4)中可以看出A、C基因组的衰减距离各有不同,但R2均随着遗传距离的增加而衰减。以决定系数(coefficient of determination)R2=0.2作为阈值(表3),A基因组的平均衰减距离为170 kb,其中A08染色体的衰减速度最慢,衰减距离也是最大,约为550 kb;A02和A03衰减速度最快,衰减距离约为130 kb。C基因组的平均衰减距离为650 kb,其中C02的衰减距离约1 150 kb,明显高于其他LD值,说明C02可能包含较多受人工进化选择影响性状的基因[32];染色体C05的衰减距离最小约为270 kb。从中可看出甘蓝型油菜A基因组衰退距离小于C基因组,说明A基因组可能发生了更多的重组,其遗传变异更加丰富。Table 3
表3
表3连锁不平衡在A、C基因组中的衰减距离
Table 3
| 染色体 Chr. | A基因组LD衰减距离 A genome LD attenuation distance (kb) | 染色体 Chr. | C基因组LD衰减距离 C genome LD attenuation distance (kb) | |
|---|---|---|---|---|
| A01 | 150 | C01 | 750 | |
| A02 | 130 | C02 | 1150 | |
| A03 | 130 | C03 | 370 | |
| A04 | 150 | C04 | 560 | |
| A05 | 150 | C05 | 270 | |
| A06 | 150 | C06 | 370 | |
| A07 | 140 | C07 | 350 | |
| A08 | 550 | C08 | 640 | |
| A09 | 370 | C09 | 760 | |
| A10 | 240 | |||
| A基因组 A genome | 170 | C基因组 C genome | 650 |
新窗口打开|下载CSV
图4
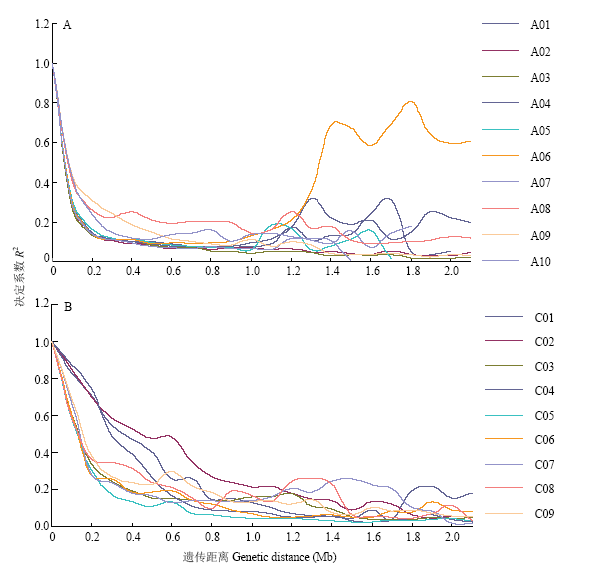 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4连锁不平衡在A、C基因组中不同染色体的衰减
A:A基因组的LD衰减距离;B:C基因组的LD衰减距离
Fig. 4The linkage disequilibrium decline in different chromosomes for A and C genome
A: LD decline A genome; B: LD decline C genome
2.5 苯磺隆胁迫下油菜萌发期相关性状的6种模型比较
采用基于一般线性模块的GLM、Q和PCA模型和混合线性模块的K、Q+K和PCA+K共6种模型来进行关联分析[19]。通过比较6种模型下Q-Q图的分布发现(图5),在3个性状中,与GLM模型相比,MLM模型下的K、K+Q和K+PCA模型均能较好地控制假阳性,但K+PCA模型检测到P值更接近期望值,因此选用K+PCA作为3个相对性状的最优模型来寻找关联位点。图5
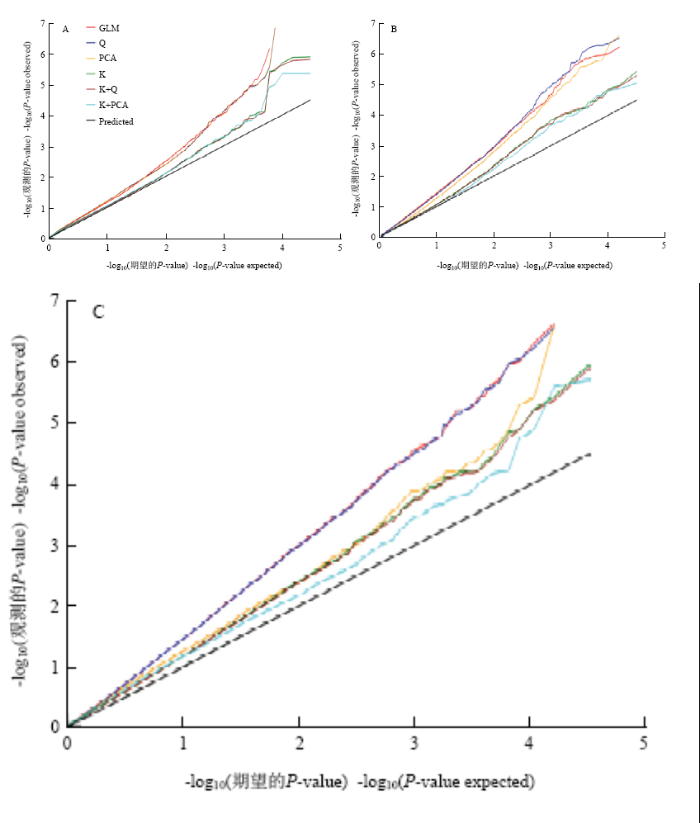 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图56种统计模型对耐苯磺隆相关性状的关联分析QQ图比较
A:相对根长;B:相对鲜重;C:相对发芽率
Fig. 5Quantile-quantile plots of estimated -log10(P) from association analysis using six models in related traits with tribenuron-methyl
A: Relative root length; B: Relative fresh weight; C: Relative germination rate
2.6 全基因组关联分析
全基因组关联分析表明(表4),与相对根长显著关联的SNP标记共检测到6个,分布于染色体A03、A05、C02、C03和C06上,可解释10.35%—12.22%的表型变异(图6-A)。与相对鲜重显著关联的SNP标记共检测到6个,分别位于A01、C02、C03和C04染色体上,这些位点可解释9.42%—11.66%的表型变异率(图6-B)。与相对发芽率显著关联的SNP标记共检测到4个,分别位于A04、A07、C02和C09染色体上,这些位点可解释10.95%—13.14%的表型变异率(图6-C)。Table 4
表4
表4苯磺隆胁迫下相关性状的显著关联标记
Table 4
| 性状 Trait | 标记 Marker | 等位基因 Allele | 染色体 Chr. | 位置 Position (bp) | 阈值 P-value | 贡献率 R2(%) |
|---|---|---|---|---|---|---|
| 相对根长 Relative root length | Bn-A03-p21744993 | A/G | A03 | 20527711 | 4.37×10-6 | 12.19 |
| Bn-A05-p23188406 | A/C | A05 | 21346681 | 1.18×10-5 | 11.22 | |
| Bn-A02-p5442618 | A/G | C02 | 5190991 | 4.35×10-6 | 12.20 | |
| Bn-A05-p9277011 | A/C | C02 | 26364667 | 2.94×10-5 | 10.35 | |
| Bn-scaff_18322_1-p2518158 | T/C | C03 | 6708755 | 4.26×10-6 | 12.22 | |
| Bn-scaff_17799_1-p1082380 | A/G | C06 | 35520328 | 9.75×10-6 | 11.42 | |
| 相对鲜重 Relative fresh weight | Bn-A01-p11163980 | A/G | A01 | 9233493 | 1.48×10-5 | 11.12 |
| Bn-scaff_22749_1-p462527 | T/C | C02 | 25111534 | 8.44×10-6 | 11.66 | |
| Bn-scaff_17457_1-p187316 | A/G | C03 | 54357603 | 1.34×10-5 | 11.22 | |
| Bn-scaff_17457_1-p82664 | T/G | C03 | 54463472 | 2.31×10-5 | 10.69 | |
| Bn-scaff_15798_1-p152131 | T/G | C04 | 36758548 | 2.28×10-5 | 9.42 | |
| Bn-scaff_15794_3-p286350 | A/C | C03 | 55318298 | 3.12×10-5 | 10.40 | |
| 相对发芽率 Relative germination rate | Bn-A04-p10628643 | T/C | A04 | 11766577 | 1.88×10-6 | 13.14 |
| Bn-A07-p5710530 | T/G | A07 | 7686960 | 2.52×10-6 | 12.85 | |
| Bn-scaff_16300_1-p1228965 | A/C | C02 | 22565646 | 1.75×10-5 | 10.95 | |
| Bn-A09-p9040117 | A/C | C09 | 11435455 | 1.28×10-5 | 11.26 |
新窗口打开|下载CSV
图6
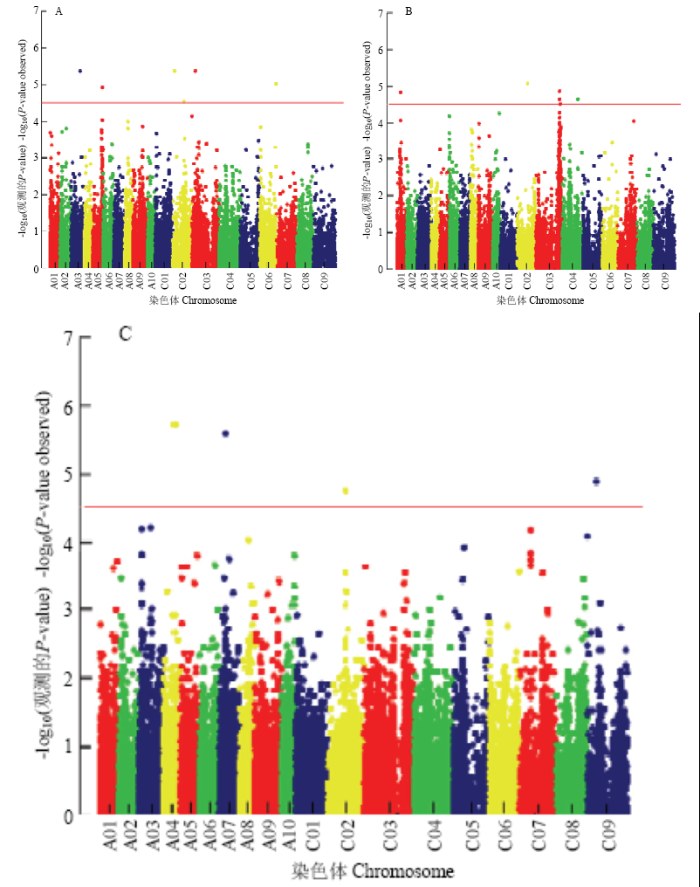 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6对苯磺隆耐性相关性状的Manhattan图
A:相对根长;B:相对鲜重;C:相对发芽率
Fig. 6The manhattan graph of tolerance related traits
A: Relative root length; B: Relative fresh weight; C: Relative germination rate
2.7 候选基因预测
通过对甘蓝型油菜3个相对性状的全基因组关联分析,利用已公布的甘蓝型油菜基因组测序结果,将与各性状显著关联的SNP标记定位到油菜的基因组上,基于甘蓝型油菜参考基因组序列,对LD区间的候选基因进行分析,筛选出了25个与苯磺隆耐性相关性状有关的候选基因(表5)。Table 5
表5
表5苯磺隆耐性相关性状的候选基因
Table 5
| 性状 Trait | 标记 Marker | 染色体 Chr. | LD区间 LD interval (bp) | 候选基因 Candidate gene | 拟南芥基因 Arabidopsis gene | 基因 Gene | 参考文献 Reference |
|---|---|---|---|---|---|---|---|
| 相对根长 Relative root length | Bn-A03-p21744993 | A03 | 20397711-20657711 | BnaA03g40900D | AT3G50740 | UGT72E1 | [33] |
| Bn-A05-p23188406 | A05 | 21196681-21496681 | BnaA05g30790D | AT3G06590 | AIF2 | [34] | |
| Bn-A02-p5442618 | C02 | 4040991-6340991 | BnaC02g07650D | AT5G17960 | 未知Unknown | ||
| BnaC02g08720D | AT5G19450 | CDPK19 | [35] | ||||
| BnaC02g10110D | AT1G01190 | CYP78A8 | [36] | ||||
| BnaC02g10430D | AT5G58860 | CYP86A1 | [37] | ||||
| Bn-A05-p9277011 | C02 | 25214667-27514667 | BnaC02g27690D | AT4G01210 | 未知Unknown | ||
| BnaC02g28290D | AT4G12320 | CYP706A6 | |||||
| Bn-scaff_17799_ 1-p1082380 | C06 | 35150328-35890328 | BnaC06g38000D | AT1G77290 | 未知Unknown | ||
| 相对鲜重 Relative fresh weight | Bn-A01-p11163980 | A01 | 9083493-9383493 | BnaA01g17570D | AT4G16190 | 未知Unknown | |
| Bn-scaff_22749 _1-p462527 | C02 | 23961534-26261534 | BnaC02g27690D | AT4G01210 | 未知Unknown | ||
| BnaC02g27750D | AT4G01070 | GT72B1 | [38] | ||||
| Bn-scaff_17457 _1-p187316 | C03 | 53987603-54727603 | BnaC03g64930D | AT4G12560 | CPR1 | [39] | |
| BnaC03g65100D | AT4G22690 | CYP706A1 | [40] | ||||
| BnaC03g65140D | AT4G23100 | GSH1 | [41] | ||||
| Bn-scaff_17457 _1-p82664 | C03 | 54093472-54833472 | BnaC03g65260D | AT1G49860 | GSTF14 | [42] | |
| Bn-scaff_15798 _1-p152131 | C04 | 36198548-37318548 | BnaC04g35220D | AT1G13080 | CYP71B2 | [43] | |
| BnaC04g35620D | AT1G78490 | CYP708A3 | |||||
| 相对发芽率 Relative germination rate | Bn-A07-p5710530 | A07 | 7546960-7826960 | BnaA07g07310D | AT1G30410 | ATMRP13 | [41] |
| BnaA07g07330D | AT1G30420 | ATMRP12 | [44] | ||||
| Bn-scaff_16300 _1-p1228965 | C02 | 21415646-23715646 | BnaC02g24410D | AT1G47620 | CYP96A8 | [45] | |
| BnaC02g24920D | AT1G78270 | UGT85A4 | [46] | ||||
| BnaC02g24980D | AT1G78380 | ATGSTU19 | [47] | ||||
| Bn-A09-p9040117 | C09 | 10675455-12195455 | BnaC09g14070D | AT3G26160 | CYP71B17 | [48] | |
| BnaC09g14150D | AT3G26170 | CYP71B19 | [49] | ||||
| BnaC09g15080D | AT1G59700 | GSTU16 | [50] |
新窗口打开|下载CSV
对相对根长的全基因组关联分析,找到了8个与耐性机制有关的候选基因。在C02染色体上的基因BnaC02g10110D、BnaC02g10430D和BnaC02g28290D分别与拟南芥基因AT1G01190、AT5G58860和AT4G12320同源,均为细胞色素P450基因家族成员。A03染色体上的基因BnaA03g40900D和C06染色体上的基因BnaC06g38000D分别与拟南芥基因AT3G50740和AT1G77290同源,前者是松柏醇葡糖基转移酶,后者是谷胱甘肽S-转移酶家族蛋白成员,均参与对有毒物质的反应。A05染色体上的基因BnaA05g30790D与拟南芥基因AT3G06590同源,是一种bHLH转录因子。位于C02染色体上的基因BnaC02g27690D的拟南芥同源基因AT4G01210,是糖基转移酶家族蛋白成员。在C02染色体上的基因BnaC02g07650D和BnaC02g08720D分别与拟南芥基因AT5G17960和AT5G19450同源,均参与植物生长发育中非生物胁迫的调控。
扫描与相对鲜重显著关联SNP标记的LD区域,找到了9个与抗性机制有关的候选基因。在A01染色体上的基因BnaA01g17570D与拟南芥基因AT4G16190同源,为木瓜蛋白酶家族半胱氨酸蛋白酶。C02染色体上的基因BnaC02g27690D和拟南芥基因AT4G01210同源, BnaC02g27750D的拟南芥同源基因AT4G01070响应于毒性物质。位于C03染色体上的基因BnaC03g64930D和拟南芥基因AT4G12560同源,参与防御反应的负调控。C04染色体上的基因BnaC04g35220D、BnaC04g35620D和C03染色体上的基因BnaC03g65100D分别与拟南芥基因AT1G13080、AT1G78490和AT4G22690同源,均为细胞色素P450家族成员。C03染色体上的基因BnaC03g65260D和BnaC03g65140D的拟南芥同源基因AT1G49860和AT4G23100分别参与编码谷胱甘肽转移酶和谷胱甘肽生物合成。
在与相对发芽率关联的SNP位点的LD区域找到了8个与苯磺隆耐性机制有关的候选基因。在A07染色体上找到的基因BnaA07g07310D和BnaA07g07330D分别与拟南芥中的基因AT1G30410和AT1G30420同源,属于ABC转运蛋白的多药抗性相关蛋白(MRP和ABCC)亚家族的基因。C09染色体上的基因BnaC09g14070D、BnaC09g14150D和C02染色体上的基因BnaC02g24410D分别与拟南芥基因AT3G26160、AT3G26170和AT1G47620同源,均为细胞色素P450基因家族。C02染色体上的基因BnaC02g24920D在拟南芥中的同源基因AT1G78270与UDP-葡糖基转移酶有关。C02染色体上的基因BnaC02g24980D和C09染色体上的基因BnaC09g15080D分别与拟南芥基因AT1G78380和AT1G59700同源,参与谷胱甘肽代谢过程。
3 讨论
3.1 苯磺隆胁迫下的萌发期鉴定
苯磺隆是中国麦田广泛使用的阔叶除草剂之一,属乙酰乳酸合成酶(acetolactate synthase,ALS)抑制剂类除草剂家族,具有杀草谱广、活性高、选择性强,对哺乳动物毒性低等优点[1],是世界上使用最多的一类除草剂[2]。油菜对苯磺隆较敏感,生产上油菜田直接利用苯磺隆化学除草较少,但是在耐广谱除草剂草甘膦杂草日益增多[51]且胺苯磺隆等使用残留药害重的情况下[7],苯磺隆与单子叶除草剂混合使用可以用于油菜播前苗床地或者大田移栽前除草[8]。因苯磺隆主要作用在杂草的茎叶,对未萌发的杂草影响较小,因此多数研究集中在作物苗期或者成株期[25,51],很少关注苯磺隆残留对后茬作物种子萌发的影响,尤其是对其敏感的阔叶型作物种子萌发的影响。王正贵[4]和杜慧平等[5]研究表明,苯磺隆在麦田土壤中的降解半衰期为10 d左右。DOWSETT等[52]研究表明在低pH土壤中喷施苯磺隆28 d后种植饲料甜菜生长活力最好,在高pH土壤中即使试验期长达56 d甜菜生长也较差。苯磺隆一方面可以作为油菜田潜在的阔叶型除草剂[8,26]并在播种或者移栽前进行除草[28],另一方面也是油菜化学杀雄剂主要的组成成分[53],筛选对苯磺隆耐性极好的种质在生产和杂种配制等方面具有非常重要的意义。针对这种情况,根据苯磺隆田间用量及其在土壤中的衰减,设计浓度梯度进行胁迫浓度筛选,最后筛选了25 mg·kg-1(相当于田间除草剂使用浓度的1/10)作为种子萌发期的胁迫浓度,并以该浓度处理241份油菜种子,筛选出3个苯磺隆耐性较强种质,并认为综合分析相对根长、相对鲜重、相对发芽率可评价油菜种质资源萌发期耐性[10]。因此本文根据这三个指标性状进行苯磺隆残留胁迫下油菜萌发期GWAS候选基因挖掘。虽然有研究[54,55]表明植物在非生物胁迫下幼苗期耐受性优于萌发期,但是由于基因表达的时空特点,这些萌发期苯磺隆耐性较好的种质是否在苗期或者成株期具有相同的表现,还有待于验证。3.2 关联分析与候选基因
不同的分析模型影响关联分析的结果[11-14,30]。为确保关联结果的准确性,最大程度降低假阳性结果,GWAS前要对每个性状进行最优模型的选择。XU等[19]在油菜开花的全基因组关联分析中发现,K+PCA模型能更好地降低假阳性的发生。本研究对每个性状进行了6种模型的分析,发现包括K模型,PCA+K模型和Q+K模型在内的MLM模型可显著降低P值,而K+PCA模型能更好地控制3个性状的假阳性。植物耐受苯磺隆除草剂主要与ALS基因突变和非靶标酶代谢解毒能力有关,其中最常见的酶分别是谷胱甘肽转移酶GSTs和UDP依赖性糖基转移酶(UGTs);最常见的代谢反应包括P450s的氧化和羧化酯酶的水解,以及通过多药耐药相关蛋白(MRP)家族的载体的代谢反应[51,56]。本研究所筛选到的25个候选基因大多与细胞色素P450酶系及谷胱甘肽转移酶活性有关。虽然本研究没有定位到ALS突变位点,但是不能简单地认为241份材料没有ALS突变基因,由于GWAS分析结果与基因表达引起的性状效应值大小有关,ALS基因如果在萌发期的相对鲜重、相对根长、相对发芽率3个指标性状中表达引起的表型值较小,那么在GWAS分析中就无法检测。有研究表明播娘蒿抗性水平大小与ALS基因表达量多少无关[51]。DéLYE等[57]通过比较除草剂敏感性生物测定与基因分型,其结果显示具有抗性的植物中约75%的植物是通过增加除草剂代谢而具有抗耐性,揭示了非靶位点抗性的重要性。本研究发现在A07染色体上2个标记相距很近的BnaA07g07310D和BnaA07g07330D为多药耐药相关蛋白(MRP)家族,其作为ATP依赖的输出泵可对谷胱甘肽结合物和葡萄糖醛酸化起作用[58]。前人研究表明大豆的CYP71A10[59]、菊芋的CYP76B1[60]、小麦的CYP71C6v1[61]以及烟草的CYP71A11和CYP81B2[62]均为可降解除草剂的植物P450酶系,证明了植物细胞色素P450酶系参与了对许多除草剂的代谢和解毒作用。本研究在C02、C03、C04、C09染色体上筛选的9个基因均为细胞色素P450基因家族成员,其中BnaC02g10430D的拟南芥同源基因AT5G58860仅在根组织中显著表达。CUMMINS等[63]研究发现使用除草剂安全剂处理可增强小麦中涉及农药解毒的葡糖基转移酶和O-甲基转移酶的表达活性,降低了作物对除草剂的敏感性。RIECHERS等[64]通过提高GST酶的活性来刺激除草剂在植物中的代谢反应,表明GST蛋白在各种解毒过程中有重要作用。CUMMINS等[65]研究发现通过转基因表达AmGSTF1的拟南芥对一些除草剂具有一定的耐受性,其抗耐性的产生是由于谷胱甘肽、花青素和类黄酮的积累增加而产生。研究证明P450s和GSTs可由多种非生物刺激诱导,对于保护植物免受氧化损伤很重要[41,66]。本文筛选到C02染色体上的基因BnaC02g24980D、C03染色体上的基因BnaC03g65260D和C09染色体上的基因BnaC09g15080D为谷胱甘肽S-转移酶家族蛋白,BnaC03g65140D编码谷氨酸半胱氨酸连接酶,均参与谷胱甘肽的生物合成代谢过程并响应多种逆境胁迫。有研究表明BnaC02g24980D的拟南芥同源基因AT1G78380在拟南芥的耐盐耐旱等胁迫中起着重要作用[47],同时也有研究[67]表明该同源基因ATGSTU19的表达被除草剂安全剂上调。此外,本研究在与3个性状相关联的SNP的LD区域找到5个拟南芥功能未知的的基因,其中在相对根长和相对鲜重中定位到C02染色体上相同基因位点BnaC02g27690D,这些基因具有重要的挖掘价值,将成为下一步研究的重要内容。
4 结论
扫描得到与相对根长显著相关的SNP位点共6个,与相对鲜重相关的SNP位点共6个,与相对发芽率相关的SNP位点共监测到4个,覆盖了A、C基因组。共找到25个与苯磺隆耐性相关的候选基因,这些候选基因可能通过P450酶或者谷胱甘肽转移酶活性的调控导致油菜对苯磺隆耐药性的差异。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1146/annurev.pp.40.060189.002301URL [本文引用: 2]

The Ge contents of plants and animals were investigated by a wet ashing procedure by hydride generation and inductively coupled plasma atomic emission spectrometry with flow injection. The analytical results obtained indicated that Ge contents widely vary in plant and animal kingdoms in the range of 8-203 ppb.
DOI:10.1614/0043-1745(2002)050[0700:RROWTA]2.0.CO;2URL [本文引用: 2]
Herbicides that target the enzyme acetolactate synthase (ALS) are among the most widely used in the world. Unfortunately, these herbicides are also notorious for their ability to select resistant (R) weed populations. Now, there are more weed species that are resistant to ALS-inhibiting herbicides than to any other herbicide group. In most cases, resistance to ALS-inhibiting herbicides is caused by an altered ALS enzyme. The frequent occurrence of weed populations resistant to ALS inhibitors can be attributed to the widespread usage of these herbicides, how they have been used, the strong selection pressure they exert, and the resistance mechanism. In several cropping systems, ALS-inhibiting herbicides were used repeatedly as the primary mechanism of weed control. These herbicides exert strong selection pressure because of their high activity on sensitive biotypes at the rates used and because of their soil residual activity. Several point mutations within the gene encoding ALS can result in a herbicide-resistant ALS. From investigations of numerous R weed biotypes, five conserved amino acids have been identified in ALS that, on substitution, can confer resistance to ALS inhibitors. Substitutions of at least 12 additional ALS amino acids can also confer herbicide resistance in plants and other organisms but, to date, have not been found in R weed populations. Mutations in ALS conferring herbicide resistance are at least partially dominant, and because the gene is nuclear inherited, it is transmitted by both seed and pollen. Furthermore, in many cases there is apparently a negligible fitness cost of the resistance gene in the absence of herbicide selection. Although resistance to ALS-inhibiting herbicides has been a bane to weed management, it has spurred many advances within and beyond the weed science discipline. As examples, resistance to ALS-inhibiting herbicides has been exploited in the development of herbicide-resistant crops, studies of weed population dynamics, and in developing protocols for targeted gene modification. Resistance to ALS-inhibiting herbicides has greatly affected weed science by influencing how we view the sustainability of our weed management practices, what we consider when developing and marketing new herbicides, and how we train new weed scientists.
DOI:10.1016/j.cropro.2003.12.003URLMagsci [本文引用: 1]
Weeds growing from seed can cause severe problems in forest nurseries and in woodland establishment by competing for resources with the young trees, leading to reduced growth and survival. Herbicides approved for use on new plantings of farm forestry and forest nurseries were usually developed originally for use in agricultural crops. As a result, information on the susceptibility of weeds that may be a problem in forestry situations rather than the rotational cropping systems used in agriculture is limited. In this series of glasshouse experiments the efficacy of residual and foliar-acting herbicides primarily used on agricultural crops was tested on 14 species of weeds. Cardamine hirsuta, Epilobium ciliatum, Poa annua, Senecio vulgaris, and Spergula arvensis were all effectively controlled by diphenamid, lenacil, metamitron, metazachlor and napropamide when applied immediately after sowing. These species were also controlled when sown into soil sprayed 1 month earlier, indicating persistent phytotoxicity of the herbicides. Cirsium vulgare, E. ciliatum, Senecio jacobaea, Trifolium repens and Urtica dioica grown from seed were well controlled by low dose pre-emergence applications of atrazine, clopyralid+cyanazine, metazachlor and simazine. Ranunculus repens was effectively controlled by pendimethalin and propyzamide. Cyanazine applied early post-emergence also controlled all these species, but their susceptibility to other herbicides varied. Pre-emergence control of the perennial weed species Agrostis stolonifera, Rubus fruticosus and Rumex obtusifolius grown from seed was given by low doses of atrazine, clopyralid+cyanazine, metazachlor and propyzamide. Isoxaben, napropamide and pendimethalin were effective on A. stolonifera and R. obtusifolius. Early post-emergence applications of atrazine and cyanazine controlled all these species; other herbicides varied in efficacy. E. ciliatum, R. repens, S. jacobaea and U. dioica as small seedlings were well controlled by recommended doses of amidosulfuron, pyridate and tribenuron-methyl. T. repens was generally unaffected even at the earliest growth stage. The doses of residual herbicides required for weed control were relatively low, indicating the greater susceptibility of weeds grown in containers compared with field conditions, probably due to restricted rooting depth. The results provide guidance on which herbicides may be worth testing in field conditions for the control of these problem weeds, however diphenamid is no longer commercially available and cyanazine is currently not being supported by the EC Pesticide Review Programme.
[D].
[本文引用: 2]
[D].
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
.
[本文引用: 1]
[本文引用: 2]
.
[本文引用: 2]
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 5]
[本文引用: 5]
DOI:10.1038/s41598-017-11318-6URLPMID:28883611 [本文引用: 2]
react-text: 431 In this study, we established two doubled haploid (DH) libraries with a total of 207 DH lines. We applied BR and GA inhibitors to all DH lines at seedling stage and measured seedling BR and GA inhibitor responses. Moreover, we evaluated field traits for each DH line (untreated). We conducted genome-wide association studies (GWAS) with 62,049 genome wide SNPs to explore the genetic control of... /react-text react-text: 432 /react-text [Show full abstract]
DOI:10.3390/genes9020087URLPMID:29443881 [本文引用: 1]
Abstract Sesame has great potential as an industrial crop but its production is challenged by drought and salt stresses. To unravel the genetic variants leading to salinity and drought tolerances at the germination stage, genome-wide association studies of stress tolerance indexes related to NaCl-salt and polyethylene glycol-drought induced stresses were performed with a diversity panel of 490 sesame accessions. An extensive variation was observed for drought and salt responses in the population and most of the accessions were moderately tolerant to both stresses. A total of 132 and 120 significant Single Nucleotide Polymorphisms (SNPs) resolved to nine and 15 Quantitative trait loci (QTLs) were detected for drought and salt stresses, respectively. Only two common QTLs for drought and salt responses were found located on linkage groups 5 and 7, respectively. This indicates that the genetic bases for drought and salt responses in sesame are different. A total of 13 and 27 potential candidate genes were uncovered for drought and salt tolerance indexes, respectively, encoding transcription factors, antioxidative enzymes, osmoprotectants and involved in hormonal biosynthesis, signal transduction or ion sequestration. The identified SNPs and potential candidate genes represent valuable resources for future functional characterization towards the enhancement of sesame cultivars for drought and salt tolerances.
DOI:10.1111/j.1439-0523.2012.01976.xURL [本文引用: 1]
High-throughput genomics technologies today offer unprecedented possibilities for gene discovery, complex trait analysis by genome-wide association studies, global gene expression analyses, genomic selection and predictive breeding strategies. Dissection of the complex Brassica napus genome using mapping-by-sequencing techniques provides a powerful bridge between genetic maps and genome sequences. The completed sequence of the Brassica rapa A genome and the expected forthcoming publication of the C genome (Brassica oleracea) will greatly accelerate the release of public reference sequences for B. napus (genome AC). Dramatically falling DNA costs for targeted or genomic resequencing and the availability of a new, high-density B. napus single-nucleotide polymorphism (SNP) array open the way for considerably more efficient mining and exploitation of genetic variation within the primary and secondary gene pools of B. napus. In this review, we outline some of the most significant recent advances in high-throughput genomics of Brassica crops and their potential impact on germplasm development and breeding of oilseed rape and canola in the coming years and decades.
DOI:10.3389/fpls.2018.00375URL [本文引用: 4]
Cadmium is a potentially toxic heavy metal to human health. Rapeseed (Brassica napusL.), a vegetable and oilseed crop, might also be a Cd hyperaccumulator, but there is little information on this trait in rapeseed. We evaluated Cd accumulation in different oilseed accessions and employed a genome-wide association study to identify quantitative trait loci (QTLs) related to Cd accumulation. A total of 419B. napusaccessions and inbred lines were genotyped with a 60K Illumina Infinium SNP array of Brassica. Wide genotypic variations in Cd concentration and translocation were found. Twenty-five QTLs integrated with 98 single-nucleotide polymorphisms (SNPs) located at 15 chromosomes were associated with Cd accumulation traits. These QTLs explained 3.49 7.57% of the phenotypic variation observed. Thirty-two candidate genes were identified in these genomic regions, and they were 0.33 497.97 kb away from the SNPs. We found orthologs ofArabidopsis thalianalocated near the significant SNPs on theB. napusgenome, including NRAMP6 (natural resistance-associated macrophage protein 6), IRT1 (iron-regulated transporter 1), CAD1 (cadmium-sensitive 1), and PCS2 (phytochelatin synthase 2). Of them, four candidate genes were verified by qRT-PCR, the expression levels of which were significantly higher after exposure to Cd than in the controls. Our results might facilitate the study of the genetic basis of Cd accumulation and the cloning of candidate Cd accumulation genes, which could be used to help reduce Cd levels in edible plant parts and/or create more efficient hyperaccumulators.
DOI:10.3389/fpls.2017.00593URLPMID:28491067 [本文引用: 3]
Soil salinity is a serious threat to agriculture sustainability worldwide. Salt tolerance at the seedling stage is crucial for plant establishment and high yield in saline soils; however, little information is available on rapeseed (Brassica napusL.) salt tolerance. We evaluated salt tolerance in different rapeseed accessions and conducted a genome-wide association study (GWAS) to identify salt tolerance-related quantitative trait loci (QTL). A natural population comprising 368B.napuscultivars and inbred lines was genotyped with aBrassica60K Illumina Infinium SNP array. The results revealed that 75 single-nucleotide polymorphisms (SNPs) distributed across 14 chromosomes were associated with four salt tolerance-related traits. These SNPs integrated into 25 QTLs that explained 4.21 9.23% of the phenotypic variation in the cultivars. Additionally, 38 possible candidate genes were identified in genomic regions associated with salt tolerance indices. These genes fell into several functional groups that are associated with plant salt tolerance, including transcription factors, aquaporins, transporters, and enzymes. Thus, salt tolerance in rapeseed involves complex molecular mechanisms. Our results provide valuable information for studying the genetic control of salt tolerance inB. napusseedlings and may facilitate marker-based breeding for rapeseed salt tolerance.
DOI:10.1111/pbi.12501URLPMID:26563848 [本文引用: 2]
Summary Brassica napus is one of the most important oil crops in the world, and stem rot caused by the fungus Sclerotinia sclerotiorum results in major losses in yield and quality. To elucidate resistance genes and02pathogenesis-related02genes, genome-wide association analysis of 347 accessions was performed using the Illumina 60K Brassica SNP (single nucleotide polymorphism) array. In addition, the detached stem inoculation assay was used to select five highly resistant (R) and susceptible (S) B.02napus lines, 4802h postinoculation with S.02sclerotiorum for transcriptome sequencing. We identified 17 significant associations for stem resistance on chromosomes A8 and C6, five of which were on A8 and 12 on C6. The SNPs identified on A8 were located in a 409-kb haplotype block, and those on C6 were consistent with previous QTL mapping efforts. Transcriptome analysis suggested that S.02sclerotiorum infection activates the immune system, sulphur metabolism, especially glutathione (GSH) and glucosinolates in both R and S genotypes. Genes found to be specific to the R genotype related to the jasmonic acid pathway, lignin biosynthesis, defence response, signal transduction and encoding transcription factors. Twenty-four genes were identified in both the SNP-trait association and transcriptome sequencing analyses, including a tau class glutathione S-transferase ( GSTU ) gene cluster. This study provides useful insight into the molecular mechanisms underlying the plant's response to S.02sclerotiorum .
DOI:10.3389/fpls.2015.00221URLPMID:4391041 [本文引用: 2]
Abstract Rapid and uniform seed germination is a crucial prerequisite for crop establishment and high yield levels in crop production. A disclosure of genetic factors contributing to adequate seed vigor would help to further increase yield potential and stability. Here we carried out a genome-wide association study in order to define genomic regions influencing seed germination and early seedling growth in oilseed rape (Brassica napus L.). A population of 248 genetically diverse winter-type B. napus accessions was genotyped with the Brassica 60k SNP Illumina genotyping array. Automated high-throughput in vitro phenotyping provided extensive data for multiple traits related to germination and early vigor, such as germination speed, absolute germination rate and radicle elongation. The data obtained indicate that seed germination and radicle growth are strongly environmentally dependent, but could nevertheless be substantially improved by genomic-based breeding. Conditions during seed production and storage were shown to have a profound effect on seed vigor, and a variable manifestation of seed dormancy appears to contribute to differences in germination performance in B. napus. Several promising positional and functional candidate genes could be identified within the genomic regions associated with germination speed, absolute germination rate, radicle growth and thousand seed weight. These include B. napus orthologs of the Arabidopsis thaliana genes SNOWY COTYLEDON 1 (SCO1), ARABIDOPSIS TWO-COMPONENT RESPONSE REGULATOR (ARR4), and ARGINYL-t-RNA PROTEIN TRANSFERASE 1 (ATE1), which have been shown previously to play a role in seed germination and seedling growth in A. thaliana.
DOI:10.1007/s00122-016-2697-zURLPMID:26912143 [本文引用: 3]
A set of additive loci for seed oil content were identified using association mapping and one of the novel loci on02the chromosome A5 was validated by linkage mapping.Increasing seed oil content is on
DOI:10.1093/dnares/dsv035URL [本文引用: 6]
Key message: A set of additive loci for seed oil content were identified using association mapping and one of the novel loci on the chromosome A5 was validated by linkage mapping. Increasing seed oil content is one of the most important goals in the breeding of oilseed crops including Brassica napus, yet the genetic basis for variations in this important trait remains unclear. By genome-wide... [Show full abstract]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s11032-006-9044-zMagsci [本文引用: 1]
<a name="Abs1"></a>Bentazon and sulfonylureas have been used for selective control of broadleaf weeds and sedges in rice fields for more than 20 years. A bentazon and sulfonylurea susceptible mutant, <i>bel</i>, was previously identified for the purpose of allowing these herbicides to be used for removing false hybrids from hybrid rice. While this mutation has been used successfully in rice breeding, the genetic nature of <i>bel</i> is not known. Using 1,776 susceptible plants from a population of 10,000 F<sub>2</sub> individuals, we constructed a fine map for the <i>Bel</i> locus and delimited it to a 36-kb DNA fragment between two restriction fragment length polymorphism markers, L5 and P17. Bioinformatic analysis indicated that there are five genes within this interval, an ethylene-responsive <i>OsER33</i> gene and four tandem repeats of cytochrome P450 genes designated as <i>CYP81A5</i>, <i>CYP81A6</i>, <i>CYP81A7</i>, and <i>CYP81A8</i>. Comparative sequencing could not find any differences in the coding regions of the <i>OsER33</i>, <i>CYP81A5, CYP81A7</i>, and <i>CYP81A8</i> genes between the mutant <i>bel</i> and its wild-type progenitor W6154S, but did identify a single base guanine deletion at position +1,332 bp downstream from the translation start codon of <i>CYP81A6</i>. This deletion introduces a premature stop codon and leads to the loss of the heme-binding motif, which is essential for cytochrome P450 function because it contains an absolutely conserved cysteine that serves as the fifth ligand to the heme iron. <i>CYP81A6</i> presumably functions as a hydroxylase for the detoxification of bentazon and sulfonylurea herbicides in rice. A gene-specific cleaved amplified polymorphic sequence marker and tightly linked flanking markers were developed that will be very useful for selection of the <i>bel</i> allele when transferred to photoperiod-/thermo-sensitive genic male sterility and CMS lines in hybrid rice breeding programs.
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12864-016-2915-8URLPMID:27495977 [本文引用: 1]
Flixweed (Descurainia sophiaL.) is a troublesome and widespread broadleaf weed in winter fields in China, and has evolved high level resistance to acetolactate synthase (ALS)-inhibiting sulfonylurea herbicide tribenuron-methyl. We identified a resistant flixweed population (N11) exhibiting 116.3-fold resistance to tribenuron-methyl relative to the susceptible population (SD8). Target-site ALS gene mutation Pro-197-Thr was identified in resistant plants. Moreover, the resistance can be reversed to 28.7-fold by the cytochrome P450 inhibitor malathion. The RNA-Sequencing was employed to identify candidate genes involved in non-target-site metabolic resistance in this population. Total 26 differentially expressed contigs were identified and eight of them (four P450s, one ABC transporter, three glycosyltransferase) verified by qRT-PCR. Consistent over-expression of the two contigs homology to CYP96A13 and ABCC1 transporter, respectively, were further qRT-PCR validated using additional plants from the resistant and susceptible populations. Tribenuron-methyl resistance in flixweed is controlled by target-site ALS mutation and non-target-site based mechanisms. Two genes, CYP96A13 and ABCC1 transporter, could play an important role in metabolic resistance to tribenuron-methyl in the resistant flixweed population and justify further functional studies. The online version of this article (doi:10.1186/s12864-016-2915-8) contains supplementary material, which is available to authorized users.
DOI:10.1111/tpj.12514URLPMID:24654891 [本文引用: 1]
population, including three CytP450, one nitronate monooxygenase (NMO), three GST, and one GT. Principal component analysis using these nine contigs differentiated F-R from F-S individuals. In a physiological validation experiment in which 2,4-D pre-treatment induced diclofop protection in S individuals due to increased metabolism, seven of the nine genetically validated contigs were induced significantly. Four contigs (two CytP450, NMO, and GT) were consistently highly expressed in nine field-evolved metabolic resistant L. rigidum populations. These four contigs were strongly associated with the resistance phenotype and are major candidates for contributing to metabolic diclofop resistance.
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 2]
[D].
[本文引用: 2]
DOI:10.1038/ng1702URLPMID:16380716 [本文引用: 1]
As population structure can result in spurious associations, it has constrained the use of association studies in human and plant genetics. Association mapping, however, holds great promise if true signals of functional association can be separated from the vast number of false signals generated by population structure. We have developed a unified mixed-model approach to account for multiple levels of relatedness simultaneously as detected by random genetic markers. We applied this new approach to two samples: a family-based sample of 14 human families, for quantitative gene expression dissection, and a sample of 277 diverse maize inbred lines with complex familial relationships and population structure, for quantitative trait dissection. Our method demonstrates improved control of both type I and type II error rates over other methods. As this new method crosses the boundary between family-based and structured association samples, it provides a powerful complement to currently available methods for association mapping.
[本文引用: 3]
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
.
DOI:10.3389/fchem.2017.00115URLPMID:5742180
Calcium dependent protein kinases (CDPKs) play significant role in regulation of plant growth and development in response to various stresses including drought. A set of 32 CDPK genes identified in maize were further used for searching of orthologs in the model plantArabidopsis(72) and major food crops such as rice (78) and sorghum (91). We comprehensively studied the phylogenetic relationship, annotations, gene duplications, gene structure, divergence time, 3-D protein structures and tissue-specific drought induced expression of CDPK genes in all four species. Variation in intron frequency in the studied species was one of the reasons for the functional diversity of CDPK genes to various stress responses. Protein kinase and protein kinase C phosphorylation site domains were the most conserved motifs identified in all species. Four groups were identified from the sequence-based phylogenetic analysis, in which maize CDPKs were clustered in group III. Expression data showed that the CDPK genes were highly expressed in leaf of maize, rice, and sorghum whereas inArabidopsisthe maximum expression was observed in root. The expression assay showed 5, 6, 11, and 9 were the commonly and differentially expressed drought-related orthologous genes in maize,Arabidopsis, rice, and sorghum, respectively. 3-D protein structure were predicted for the nine genes (Arabidopsis: 2, maize: 2, rice: 3, and sorghum: 2) showing differential expression in at least three species. The predicted 3-D structures were further evaluated and validated by Ramachandran plot, ANOLEA, ProSA, and Verify-3D. The superimposed 3-D structure of drought-related orthologous proteins retained similar folding pattern owing to their conserved nature. Functional annotation revealed the involvement of CDPK genes in various pathways such as osmotic homeostasis, cell protection, and root growth. The interactions of CDPK genes in various pathways play crucial role in imparting drought tolerance through different ABA and MAPK signaling cascades. These selected candidate genes could be targeted in development of drought tolerant genotypes in maize, rice, and sorghum through appropriate breeding approaches. Our comparative experiments of CDPK genes could also be extended in the drought stress breeding programmes of the related species.
DOI:10.1093/jxb/erq389URLPMID:3060681
\0
DOI:10.1111/pbi.12775URLPMID:28640934
Plants contain large numbers of family 1 UDP‐glucose dependent glycosyltransferases (UGTs), including members that conjugate xenobiotics. Arabidopsis contains 107 UGT genes with 99 family members successfully expressed as glutathione transferase (GST)‐fusion proteins in E.coli. A high‐throughput catalytic screen was developed based on quantification of the fusion by measuring GST activity. UGT activity using UDP‐glucose as donor was then determined using 11 synthetic acceptors bearing hydroxyl, amino and thiol groups that had been shown to undergo conjugation in plant extracts. In total, 44 UGTs, largely members of the D and E groups, were active toward xenobiotics, glucosylating phenol and thiol acceptors. In contrast, N‐glucosyltransferase activity was almost exclusively restricted to a single enzyme, UGT72B1. Using DNA microarrays, the induction of UGT transcripts following treatment with the herbicide safener fenclorim was compared in Arabidopsis and rice. D and L group members were the most safener‐inducible UGTs in both species. The respective Arabidopsis enzymes showed low conjugating activity toward xenobiotics. Using Genevestigator, a small group of safened D and L UGTs were consistently induced in response to biotic and abiotic stress suggestive of protective activities beyond xenobiotic detoxification in both species. The induction of other detoxifying gene families following treatment with fenclorim, namely cytochromes P450 and glutathione transferases, further confirmed the selective enhancement of related sub‐family members in the two species giving new insight into the safening response in cereals, which enhances herbicide tolerance compared with dicots, which are unresponsive to these treatments.
DOI:10.1111/j.1365-313X.2009.03995.xURLPMID:19682297
Arabidopsis gain-of-resistance mutants, which show HR-like lesion formation and SAR-like constitutive defense responses, were used well as tools to unravel the plant defense mechanisms. We have identified a novel mutant, designated constitutive expresser of PR genes 30 (cpr30), that exhibited dwarf morphology, constitutive resistance to the bacterial pathogen Pseudomonas syringae and the dramatic induction of defense-response gene expression. The cpr30-conferred growth defect morphology and defense responses are dependent on ENHANCED DISEASE SUSCEPTIBILITY 1 (EDS1), PHYTOALEXIN DEFICIENT 4 (PAD4), and NONRACE-SPECIFIC DISEASE RESISTANCE 1 (NDR1). Further studies demonstrated that salicylic acid (SA) could partially account for the cpr30-conferred constitutive PR1 gene expression, but not for the growth defect, and that the cpr30-conferred defense responses were NPR1 independent. We observed a widespread expression of CPR30 throughout the plant, and a localization of CPR30-GFP fusion protein in the cytoplasm and nucleus. As an F-box protein, CPR30 could interact with multiple Arabidopsis-SKP1-like (ASK) proteins in vivo. Co-localization of CPR30 and ASK1 or ASK2 was observed in Arabidopsis protoplasts. Based on these results, we conclude that CPR30, a novel negative regulator, regulates both SA-dependent and SA-independent defense signaling, most likely through the ubiquitin-proteasome pathway in Arabidopsis.
DOI:10.1021/es050385rURLPMID:16173598
Arabidopsis thaliana root transcriptome responses to the munition, hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX), were assessed using serial analysis of gene expression (SAGE). Sequencing of SAGE libraries from control and RDX-exposed root tissues revealed induction of genes known to respond to a variety of general stresses. Among the highly induced genes were several encoding molecular chaperones and transcription factors as well as vacuolar proteins and peroxidases. Strongly repressed transcripts included ones encoding ribosomal proteins, a cyclophilin, a katanin, and a peroxidase. Comparison of the transcriptional profile for the RDX response to a profile previously described for Arabidopsis roots exposed to trinitrotoluene (TNT) revealed significant differences in the inferred gene expression patterns. This suggests that Arabidopsis employs drastically different mechanisms for coping with these two compounds. With respect to the goal of engineering plants to better tolerate and degrade explosives at contaminated sites, these results suggest that enhancement of different genes and metabolic pathways may be required to deal effectively with each type of explosive. This has ramifications for phytoremediation efforts since many contaminated sites harbor both compounds.
[本文引用: 1]
DOI:10.1007/s00425-002-0890-6URLPMID:12430019
Despite the completion of the sequencing of the entire genome of Arabidopsis thaliana (L.) Heynh., the exact determination of each single gene and its function remains an open question. This is especially true for multigene families. An approach that combines analysis of genomic structure, expression data and functional genomics to ascertain the role of the members of the multidrug-resistance-related protein (MRP) gene family, a subfamily of the ATP-binding cassette (ABC) transporters from Arabidopsis is presented. We used cDNA sequencing and alignment-based re-annotation of genomic sequences to define the exact genic structure of all known AtMRP genes. Analysis of promoter regions suggested different induction conditions even for closely related genes. Expression analysis for the entire gene family confirmed these assumptions. Phylogenetic analysis and determination of segmental duplication in the regions of AtMRP genes revealed that the evolution of the extraordinarily high number of ABC transporter genes in plants cannot solely be explained by polyploidisation during the evolution of the Arabidopsis genome. Interestingly MRP genes from Oryza sativa L. (rice; OsMRP) show very similar genomic structures to those from Arabidopsis. Screening of large populations of T-DNA-mutagenised lines of A. thaliana resulted in the isolation of AtMRP insertion mutants. This work opens the way for the defined analysis of a multigene family of important membrane transporters whose broad variety of functions expands their traditional role as cellular detoxifiers.
DOI:10.1016/j.cub.2010.01.051URLPMID:20226671
The development of multicellular organisms is controlled by differential gene expression whereby cells adopt distinct fates. A spatially resolved view of gene expression allows the elucidation of transcriptional networks that are linked to cellular identity and function. The haploid female gametophyte of flowering plants is a highly reduced organism: at maturity, it often consists of as few as three cell types derived from a common precursor []. However, because of its inaccessibility and small size, we know little about the molecular basis of cell specification and differentiation in the female gametophyte. Here we report expression profiles of all cell types in the mature 3]. A comparison of human and Highlights? Transcriptomes of the cell types in the mature Arabidopsis female gametophyte ? Egg cell enrichment of PAZ/Piwi-domain genes indicates epigenetic regulation by siRNA ? Overrepresentation of three transcription factor families in the female gametophyte ? Human and Arabidopsis egg cells are enriched in similar functional groups
DOI:10.1104/pp.104.051714URLPMID:15618427
Phytoprostanes are prostaglandin/jasmonate-like products of nonenzymatic lipid peroxidation that not only occur ubiquitously in healthy plants but also increase in response to oxidative stress. In this work, we show that the two naturally occurring B1-phytoprostanes ($\text{PPB}_{1}$) regioisomers I and II (each comprising two enantiomers) are short-lived stress metabolites that display a broad spectrum of biological activities. Gene expression analysis of Arabidopsis (Arabidopsis thaliana) cell cultures treated with$\text{PPB}_{1}$-I or -II revealed that both regioisomers triggered a massive detoxification and defense response. Interestingly, expression of several glutathione S-transferases, glycosyl transferases, and putative ATP-binding cassette transporters was found to be increased by one or both$\text{PPB}_{1}$regioisomers, and hence, may enhance the plant's capacity to inactivate and sequester reactive products of lipid peroxidation. Moreover, pretreatment of tobacco (Nicotiana tabacum) suspension cells with$\text{PPB}_{1}$considerably prevented cell death caused by severe CuSO4poisoning. Several Arabidopsis genes induced by$\text{PPB}_{1}$, such as those coding for adenylylsulfate reductase, tryptophan synthase chain, and PAD3 pointed to an activation of the camalexin biosynthesis pathway that indeed led to the accumulation of camalexin in$\text{PPB}_{1}$treated leaves of Arabidopsis. Stimulation of secondary metabolism appears to be a common plant reaction in response to$\text{PPB}_{1}$. In three different plant species,$\text{PPB}_{1}$-II induced a concentration dependent accumulation of phytoalexins that was comparable to that induced by methyl jasmonate.$\text{PPB}_{1}$-I was much weaker active or almost inactive. No differences were found between the enantiomers of each regioisomer. Thus, results suggest that$\text{PPB}_{1}$represent stress signals that improve plants capacity to cope better with a variety of stresses.
[本文引用: 1]
DOI:10.1126/science.1255215URLPMID:25104393
Coordination of cell division and pattern formation is central to tissue and organ development, particularly in plants where walls prevent cell migration. Auxin and cytokinin are both critical for division and patterning, but it is unknown how these hormones converge upon tissue development. We identify a genetic network that reinforces an early embryonic bias in auxin distribution to create a local, nonresponding cytokinin source within the root vascular tissue. Experimental and theoretical evidence shows that these cells act as a tissue organizer by positioning the domain of oriented cell divisions. We further demonstrate that the auxin-cytokinin interaction acts as a spatial incoherent feed-forward loop, which is essential to generate distinct hormonal response zones, thus establishing a stable pattern within a growing vascular tissue.
DOI:10.1105/tpc.113.112151URLPMID:23723324
Nonfluorescent chlorophyll catabolites (NCCs) were described as products of chlorophyll breakdown in Arabidopsis thaliana. NCCs are formyloxobilin-type catabolites derived from chlorophyll by oxygenolytic opening of the chlorin macrocycle. These linear tetrapyrroles are generated from their fluorescent chlorophyll catabolite (FCC) precursors by a nonenzymatic isomerization inside the vacuole of senescing cells. Here, we identified a group of distinct dioxobilin-type chlorophyll catabolites (DCCs) as the major breakdown products in wild-type Arabidopsis, representing more than 90% of the chlorophyll of green leaves. The molecular constitution of the most abundant nonfluorescent DCC (NDCC), At-NDCC-1, was determined. We further identified cytochrome P450 monooxygenase CYP89A9 as being responsible for NDCC accumulation in wild-type Arabidopsis; cyp89a9 mutants that are deficient in CYP89A9 function were devoid of NDCCs but accumulated proportionally higher amounts of NCCs. CYP89A9 localized outside the chloroplasts, implying that FCCs occurring in the cytosol might be its natural substrate. Using recombinant CYP89A9, we confirm FCC specificity and show that fluorescent DCCs are the products of the CYP89A9 reaction. Fluorescent DCCs, formed by this enzyme, isomerize to the respective NDCCs in weakly acidic medium, as found in vacuoles. We conclude that CYP89A9 is involved in the formation of dioxobilin-type catabolites of chlorophyll in Arabidopsis.
DOI:10.1111/j.1365-313X.2004.02171.xURLPMID:15341629102
Arabidopsis thaliana RD26 cDNA, isolated from dehydrated plants, encodes a NAC protein. Expression of the RD26 gene was induced not only by drought but also by abscisic acid (ABA) and high salinity. The RD26 protein is localized in the nucleus and its C terminal has transcriptional activity. Transgenic plants overexpressing RD26 were highly sensitive to ABA, while RD26 -repressed plants were insensitive. The results of microarray analysis showed that ABA- and stress-inducible genes are upregulated in the RD26 -overexpressed plants and repressed in the RD26 -repressed plants. Furthermore, RD26 activated a promoter of its target gene in Arabidopsis protoplasts. These results indicate that RD26 functions as a transcriptional activator in ABA-inducible gene expression under abiotic stress in plants.
[D].
[本文引用: 4]
[D].
[本文引用: 4]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1104/pp.114.242750URLPMID:25106819Magsci [本文引用: 1]
Weedy plant species that have evolved resistance to herbicides due to enhanced metabolic capacity to detoxify herbicides (metabolic resistance) are a major issue. Metabolic herbicide resistance in weedy plant species first became evident in the 1980s in Australia (in Lolium rigidum) and the United Kingdom (in Alopecurus myosuroides) and is now increasingly recognized in several crop-weed species as a looming threat to herbicide sustainability and thus world crop production. Metabolic resistance often confers resistance to herbicides of different chemical groups and sites of action and can extend to new herbicide(s). Cytochrome P450 monooxygenase, glycosyl transferase, and glutathione S-transferase are often implicated in herbicide metabolic resistance. However, precise biochemical and molecular genetic elucidation of metabolic resistance had been stalled until recently. Complex cytochrome P450 superfamilies, high genetic diversity in metabolic resistant weedy plant species (especially cross-pollinated species), and the complexity of genetic control of metabolic resistance have all been barriers to advances in understanding metabolic herbicide resistance. However, next-generation sequencing technologies and transcriptome-wide gene expression profiling are now revealing the genes endowing metabolic herbicide resistance in plants. This Update presents an historical review to current understanding of metabolic herbicide resistance evolution in weedy plant species.
DOI:10.1111/j.1365-3180.2007.00544.xURL [本文引用: 1]
DélyeC, MenchariY, GuilleminJ-P, MatéjicekA, MichelS, CamilleriC & ChauvelB (2007) Status of black grass () resistance to acetyl-coenzyme A carboxylase inhibitors in France. , 95–105. Summary We assessed the contributions of target site- and non-target site-based resistance to herbicides inhibiting acetyl-coenzyme A carboxylase (ACC) in (black grass). A total of 243 populations collected across France were analysed using herbicide sensitivity bioassay (2465300 seedlings analysed) and ACC genotyping (1365188 seedlings analysed). Seedlings resistant to at least one ACC-inhibiting herbicide were detected in 99.2% of the populations. Mutant, resistant ACC allele(s) were detected in 56.8% of the populations. Among the five resistant ACC alleles known in , alleles containing an isoleucine-to-leucine substitution at codon 1781 were predominant (59.5% of the plants containing resistant ACC alleles). Comparison of the results from herbicide sensitivity bioassays with genotyping indicated that more than 75% of the plants resistant to ACC-inhibiting herbicides in France would be resistant via increased herbicide metabolism. Analysis of herbicide application records suggested that in 15.9% of the populations studied, metabolism-based resistance to ACC-inhibiting herbicides was mostly selected for by herbicides with other modes of action. Our study revealed the importance of non-target site-based resistance in . Using herbicides with alternative modes of action to control populations resistant to ACC-inhibiting herbicides, the recommended management approach, may thus be jeopardised by the widespread occurrence of metabolism-based resistance mechanisms conferring broad-spectrum cross-resistance.
[本文引用: 1]
DOI:10.1073/pnas.96.4.1750URLPMID:9990096 [本文引用: 1]
A strategy based on the random isolation and screening of soybean cDNAs encoding cytochrome P450 monooxygenases (P450s) was used in an attempt to identify P450 isozymes involved in herbicide metabolism. Nine full-length (or near-full-length) P450 cDNAs representing eight distinct P450 families were isolated by using PCR-based technologies. Five of the soybean P450 cDNAs were expressed successfully in yeast, and microsomal fractions generated from these strains were tested for their potential to catalyze the metabolism of 10 herbicides and 1 insecticide. In vitro enzyme assays showed that the gene product of one heterologously expressed P450 cDNA (CYP71A10) specifically catalyzed the metabolism of phenylurea herbicides, converting four herbicides of this class (fluometuron, linuron, chlortoluron, and diuron) into more polar compounds. Analyses of the metabolites suggest that the CYP71A10 encoded enzyme functions primarily as an N-demethylase with regard to fluometuron, linuron, and diuron, and as a ring-methyl hydroxylase when chlortoluron is the substrate. In vivo assays using excised leaves demonstrated that all four herbicides were more readily metabolized in CYP71A10-transformed tobacco compared with control plants. For linuron and chlorotoluron, CYP71A10-mediated herbicide metabolism resulted in significantly enhanced tolerance to these compounds in the transgenic plants.
DOI:10.1104/pp.005801URLPMID:12226498 [本文引用: 1]
The Jerusalem artichoke (Helianthus tuberosus) xenobiotic inducible cytochrome P450, CYP76B1, catalyzes rapid oxidative dealkylation of various phenylurea herbicides to yield nonphytotoxic metabolites. We have found that increased herbicide metabolism and tolerance can be achieved by ectopic constitutive expression of CYP76B1 in tobacco (Nicotiana tabacum) and Arabidopsis. Transformation with CYP76B1 conferred on tobacco and Arabidopsis a 20-fold increase in tolerance to linuron, a compound detoxified by a single dealkylation, and a 10-fold increase in tolerance to isoproturon or chlortoluron, which need successive catalytic steps for detoxification. Two constructs for expression of translational fusions of CYP76B1 with P450 reductase were prepared to test if they would yield even greater herbicide tolerance. Plants expressing these constructs had lower herbicide tolerance than CYP76B1 alone, which is apparently a consequence of reduced stability of the fusion proteins. In all cases, increased herbicide tolerance results from more extensive metabolism, as demonstrated with exogenously fed phenylurea. Beside increased herbicide tolerance, expression of CYP76B1 has no other visible phenotype in the transgenic plants. Our data indicate that CYP76B1 can function as a selectable marker for plant transformation, allowing efficient selection in vitro and in soil-grown plants. Plants expressing CYP76B1 may also be a potential tool for phytoremediation of contaminated sites.
DOI:10.1002/ps.969URLPMID:15627243 [本文引用: 1]
Glyphosate is a non-selective herbicide which acts by inhibiting 5-enolpyruvylshikimate-3-phosphate synthase. Wheat cytochrome P450 monooxygenase specifically catalyzes the metabolism of some sulfonylurea herbicides such as chlorsulfuron and triasulfuron. Here we report that glyphosate is an inhibitor of a wheat cytochrome (CYP71C6v1), the cDNA of which was amplified by RT-PCR and heterologously expressed in yeast. The microsomal fractions derived from this strain had a Soret peak at 502 nm in the reduced carbon monoxide difference spectrum, which is a typical spectral characteristic. The addition of glyphosate to the microsomal protein resulted in a Type II spectrum indicative of binding via the nitrogen group to haem of cytochrome P450 as a sixth ligand. A spectral dissociation constant, of 70 mol litrewas observed and an ICof 11 mol litrewas found for glyphosate inhibition of CYP71C6v1 P450 activity. Copyright 2005 Society of Chemical Industry
DOI:10.1006/pest.2000.2496URL [本文引用: 1]
We describe the isolation of four cDNA clones from 2,4-D-treated cultured tobacco cells encoding novel P450 species inducible by chemical treatments. RNA blot analysis showed that mRNA levels corresponding to CYP71A11, CYP81B2, CYP81C1, and CYP81C2 genes were differentially increased by 2,4-D treatment. Furthermore, mRNA levels of CYP71A11, CYP81C1, and CYP81C2 were also increased by treatments with a herbicide safener, naphthalic anhydride, and a plant signaling molecule, salicylic acid, with different kinetics. CYP81B2 and CYP71A11 expressed in yeast showed monooxygenase activities toward 7-ethoxycoumarin and the herbicide chlortoluron. These results suggested that CYP71A11, CYP81C1, and CYP81C2 are potential targets for herbicide safeners, and CYP81B2 and CYP71A11 are involved in chlortoluron metabolism. In addition, the results suggested the possibility that 2,4-D and naphthalic anhydride may influence not only xenobiotic metabolism but also salicylic acid-dependent responses in tobacco cells.
DOI:10.1016/j.phytochem.2006.01.012URLPMID:16494903 [本文引用: 1]
In wheat ( Triticum aestivum L.), treatment with herbicide safeners enhances the expression of enzymes involved in pesticide detoxification and reduces crop sensitivity to herbicides. Since these same enzymes are involved in plant secondary metabolism, it was of interest to determine whether or not the safener cloquintocet mexyl perturbed phenolic metabolism in wheat seedlings. LC/ESI/MS analysis identified 14 phenolic substrates in the shoots of young wheat plants. Fragmentation imposed by collision induced dissociation identified specific C-glycosidic conjugates of 4′,5,7-trihydroxflavone (apigenin), 3′,4′,5,7-tetrahydroxyflavone (luteolin) and 3′- O-methylluteolin. Treatment of 7-day-old wheat shoots with cloquintocet mexyl resulted in an accelerated depletion of the conjugates of all three flavones, most notably with the glycosides of luteolin. In contrast, safener treatment caused the selective accumulation of 4′,5,7-trihydroxy-3′,5′-dimethoxyflavone (tricin) and the phenylpropanoid ferulic acid. Changes in phenolic content were associated with an increase in O-methyltransferase and C-glucosyltransferase activity toward flavonoid substrates as well as the classic enhancement of detoxifying glutathione transferases. Our results suggest that in addition to altering the capacity of wheat to metabolise herbicides and other xenobiotics, safeners can also cause a selective shift in the metabolism of endogenous phenolics.
DOI:10.1021/bk-2005-0899.ch019URL [本文引用: 1]
Glutathione S -transferase (GST) enzymes catalyze the conjugation of reduced glutathione to pesticide substrates, leading to their irreversible detoxification. Glutathione conjugation of xenobiotics is a very well studied Phase II detoxification reaction in plants, and is presumed to be requisite for their transport into the vacuole (Phase III) and possible further catabolism within the vacuole (Phase IV). GST-catalyzed glutathione conjugation is thus critical for removing xenobiotics from the cytosol and preventing them from interacting with their target sites. In plants, the expression of GST genes is regulated by many stimuli, including biotic and abiotic stresses and exposure to xenobiotics, thus indicating an important role for GST proteins in various detoxification processes. Chemicals called herbicide safeners, which protect grass crops from herbicide injury, stimulate herbicide metabolism by increasing the activity of GST enzymes that detoxify herbicides. GST proteins are soluble and their subcellular localization has usually been presumed to be cytosolic. However, immunocytochemical studies in our lab have shown that GST proteins are localized in the cytosol and inside the vacuoles of epidermal and sub-epidermal cells in herbicide safener-treated coleoptiles [Planta 217:831-840]. The majority of immunoreactive GST protein was located in the outer two cell layers of safener-treated coleoptiles, indicating that these cell types may be involved in a novel form of herbicide detoxification mechanism that involves vacuolar accumulation of GST protein and xenobiotic-glutathione conjugates.
DOI:10.1073/pnas.1221179110URLPMID:23530204Magsci [本文引用: 1]
Multiple-herbicide resistance (MHR) in black-grass (Alopecurus myosuroides) and annual rye-grass (Lolium rigidum) is a global problem leading to a loss of chemical weed control in cereal crops. Although poorly understood, in common with multiple-drug resistance (MDR) in tumors, MHR is associated with an enhanced ability to detoxify xenobiotics. In humans, MDR is linked to the overexpression of a pi class glutathione transferase. (GSTP1), which has both detoxification and signaling functions in promoting drug resistance. In both annual rye-grass and black-grass, MHR was also associated with the increased expression of an evolutionarily distinct plant phi (F) GSTF1 that had a restricted ability to detoxify herbicides. When the black-grass A. myosuroides (Am) AmGSTF1 was expressed in Arabidopsis thaliana, the transgenic plants acquired resistance to multiple herbicides and showed similar changes in their secondary, xenobiotic, and antioxidant metabolism to those determined in MHR weeds. Transcriptome array experiments showed that these changes in biochemistry were not due to changes in gene expression. Rather, AmGSTF1 exerted a direct regulatory control on metabolism that led to an accumulation of protective flavonoids. Further evidence for a key role for this protein in MHR was obtained by showing that the GSTP1- and MDR-inhibiting pharmacophore 4-chloro-7-nitro-benzoxadiazole was also active toward AmGSTF1 and helped restore herbicide control in MHR black-grass. These studies demonstrate a central role for specific GSTFs in MHR in weeds that has parallels with, similar roles for unrelated GSTs in MDR in humans and shows their potential as targets for chemical intervention in resistant weed management.
DOI:10.1111/j.1365-313X.2008.03761.xURLPMID:19067976 [本文引用: 1]
Plant glutathione transferases (GSTs) are induced by diverse biotic and abiotic stimuli, and are important for protecting plants against oxidative damage. We have studied the primary transcriptional stress response of the entire Arabidopsis GST family to seven stresses, including both biotic and abiotic stimuli, with a focus on early changes in gene expression. Our results indicate that individual GST genes are highly specific in their induction patterns. Furthermore, we have been able to link individual GSTs to particular stress stimuli. Using RNAi, we successfully co-silenced a group of four phi GSTs that represent some of the most highly expressed GST genes. Despite a marked reduction in total phi GST protein levels, the transgenic plants showed no reduction in GST activity as measured using the model substrate 1-chloro-2,4-dinitrobenzene (CDNB), and appeared to have surprisingly robust physical phenotypes during stress. However, analysis of metabolite pools showed oxidation of the glutathione pool in the RNAi lines, and we observed alterations in carbon and nitrogen compounds following salicylic acid and hydrogen peroxide stress treatments, indicative of oxidative modification of primary metabolism. Thus, there appears to be a high degree of functional redundancy within the Arabidopsis GST family, with extensive disruption being required to reveal the roles of phi GSTs in protection against oxidative stress.
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}