 ,, 杜宇,, 童新宇, 熊翠玲, 郑燕珍, 徐国钧, 王海朋, 耿四海, 周丁丁, 郭意龙, 吴素珍, 陈大福,福建农林大学蜂学学院,福州 350002
,, 杜宇,, 童新宇, 熊翠玲, 郑燕珍, 徐国钧, 王海朋, 耿四海, 周丁丁, 郭意龙, 吴素珍, 陈大福,福建农林大学蜂学学院,福州 350002Differentially Expressed MicroRNAs and Their Regulation Networks in Apis mellifera ligustica Larval Gut During the Early Stage of Ascosphaera apis Infection
GUO Rui,, DU Yu,, TONG XinYu, XIONG CuiLing, ZHENG YanZhen, XU GuoJun, WANG HaiPeng, GENG SiHai, ZHOU DingDing, GUO YiLong, WU SuZhen, CHEN DaFu,College of Bee Science, Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
第一联系人:
收稿日期:2018-08-2接受日期:2018-10-1网络出版日期:2019-01-01
| 基金资助: |
Received:2018-08-2Accepted:2018-10-1Online:2019-01-01

摘要
关键词:
Abstract
Keywords:
PDF (5899KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郭睿, 杜宇, 童新宇, 熊翠玲, 郑燕珍, 徐国钧, 王海朋, 耿四海, 周丁丁, 郭意龙, 吴素珍, 陈大福. 意大利蜜蜂幼虫肠道在球囊菌侵染前期的 差异表达microRNA及其调控网络[J]. 中国农业科学, 2019, 52(1): 166-180 doi:10.3864/j.issn.0578-1752.2019.01.015
GUO Rui, DU Yu, TONG XinYu, XIONG CuiLing, ZHENG YanZhen, XU GuoJun, WANG HaiPeng, GENG SiHai, ZHOU DingDing, GUO YiLong, WU SuZhen, CHEN DaFu.
0 引言
【研究意义】意大利蜜蜂(Apis mellifera ligustica,简称意蜂)是我国养蜂生产中的主要蜂种,在农业生产和生态保护等方面具有重要价值[1]。白垩病是蜜蜂球囊菌(Ascosphaera apis)特异性侵染蜜蜂幼虫而引起的致死性真菌病害,对养蜂生产造成巨大损失。蜜蜂幼虫食入被球囊菌孢子污染的食物,孢子进入中肠后开始低水平萌发,到了预蛹期随着中肠和后肠的连通,孢子进入后肠后在氧气的刺激下剧烈萌发,同时菌丝大量生长并陆续穿透肠壁和体壁,从而导致幼虫死亡[2]。目前,有关蜜蜂幼虫响应球囊菌胁迫应答及分子调控机制的研究报道较少。微小RNA(microRNA,miRNA)是一类长度约为18—25个核苷酸的非编码RNA(non-coding RNA,ncRNA),通过与其靶基因3′ UTR(untranslated region)特异性结合,降解或抑制mRNA的蛋白质翻译过程[3]。作为基因表达的关键调控因子,miRNA可广泛参与细胞增殖与分化[4]、生长发育[5]、细胞凋亡[6]和抗病毒[7]等生物学过程。近年来,较多的研究结果表明miRNA参与对昆虫及其病原互作的调控[8,9]。因此,解析蜜蜂幼虫在球囊菌侵染前期的miRNA表达谱,深入研究差异表达miRNA(differentially expressed miRNA,DEmiRNA)及其调控网络,可为宿主响应球囊菌胁迫的分子调控及宿主-病原互作机制提供必要基础,也能为宿主的关键调控miRNA的筛选和功能研究提供重要的信息和线索。【前人研究进展】ZHANG等[10]研究发现,携带登革病毒(DENV)的埃及伊蚊(Aedes aegypti)被沃尔巴克氏体(Wolbachia)侵染后,aae-miR-2940可特异性表达,并显著抑制了与登革病毒复制相关的DNA甲基化转移酶基因(AaDnmt2)的表达;李盛杰[11]研究发现,果蝇(Drosophila melanogaster)被革兰氏阳性菌藤黄微球菌(Micrococcus luteus)侵染后,其miR-310家族和miR-958可通过抑制Toll信号通路产生的抗菌肽分子Drosomycin的表达而影响宿主的免疫应答。但相比于果蝇和蚊类等模式昆虫,蜜蜂与病原互作相关的miRNA研究较为滞后。LOUREN?O等[12]利用革兰氏阴性菌粘质沙雷氏菌(Serratia marcescens)和M. luteus侵染意蜂工蜂,发现miRNA可通过参与体液免疫中Toll、Imd、JNK以及Jak-STAT信号通路的调控,促进抗菌肽与黑化反应的激活来应对细菌的侵染;HUANG等[13]研究发现,东方蜜蜂微孢子虫(Nosema ceranae)可能通过影响西方蜜蜂(Apis mellifera)代谢调控相关的miRNA表达促进自身的快速繁殖。笔者所在课题组前期已通过二代测序技术对球囊菌侵染的中华蜜蜂(Apis cerana cerana,简称中蜂)和意蜂4日龄幼虫肠道进行转录组分析,在mRNA组学水平解析了病原在侵染前期与宿主间的互作[14,15]。然而,蜜蜂幼虫在球囊菌侵染过程的miRNA表达谱仍然缺失,miRNA在蜜蜂幼虫的胁迫应答中的作用仍未可知。【本研究切入点】此前的相关研究主要集中在球囊菌侵染后期,即6日龄幼虫到预蛹的过渡期,有关球囊菌侵染前期的研究还非常滞后。球囊菌在侵染前期已经开始出现低水平的孢子萌发和菌丝生长,必然伴随着复杂的基因表达和ncRNA调控。miRNA作为关键的转录后调控因子在昆虫的免疫防御过程中发挥关键的作用[16]。本研究利用small RNA-seq(sRNA-seq)技术对正常及球囊菌胁迫的意蜂4日龄幼虫肠道进行高通量测序,对宿主miRNA的表达谱、显著DEmiRNA及其调控网络进行探究,并筛选免疫防御相关的关键miRNA。【拟解决的关键问题】通过深入分析显著DEmiRNA及其调控网络,在miRNA组学水平解析意蜂幼虫在球囊菌侵染前期的胁迫应答,揭示显著DEmiRNA在宿主-病原互作中的作用,为解析意蜂幼虫-球囊菌的互作机制打下基础。1 材料与方法
试验于2017年9月至2018年7月在福建农林大学蜂学学院蜜蜂保护实验室完成。1.1 供试生物材料
供试意蜂幼虫取自福建农林大学蜂学学院教学蜂场。1.2 意蜂幼虫人工饲养及样品测序
参照前期研究建立的方法[14]活化球囊菌、纯化孢子以及人工饲养意蜂幼虫,配制含球囊菌孢子的饲料(终浓度为1×107个孢子/mL),饲喂处理组3日龄幼虫,24 h后待含孢子饲料被食尽,饲喂不含球囊菌的正常饲料;对照组幼虫饲喂不含孢子的正常饲料。肠道是球囊菌与意蜂幼虫互作的主要部位,因此选择幼虫肠道作为测序材料。分别解剖上述对照组和处理组4日龄幼虫肠道组织,剖取的幼虫肠道每9只放于1个RNA-Free的EP管中,液氮速冻后保存在-80℃的超低温冰箱。进行3次生物学重复。对照组的3个生物学重复分别为AmCK-1、AmCK-2和AmCK-3;处理组的3个生物学重复分别为AmT-1、AmT-2和AmT-3。委托广州基迪奥生物科技有限公司对上述6个肠道样品进行单端测序,测序平台为Illumina MiSeq。首先,利用Trizol法从意蜂幼虫肠道样本中提取total RNA,琼脂糖凝胶电泳切胶选择18—30 nt的片段,连接3′端接头,连接产物以15%变性PAGE胶电泳分离,切胶选择36—44 nt的目的条带;将上述回收产物连接5′端接头,进而对两侧带有接头的small RNA样本进行RT-PCR,反转录产物以3.5%琼脂糖凝胶电泳分离,切胶回收140—160 bp区域的目的条带,回收产物即为终文库,建好的文库上机测序。原始数据已上传NCBI SRA数据库,BioProject号:PRJNA408312。1.3 测序数据的质控及DEmiRNA的预测
参照前期研究建立的方法[17]对测序数据进行质控和定位,并将得到miRNA的非注释标签序列(unannotated tags)。通过Bowit软件将非注释标签序列与西方蜜蜂的参考基因组(assembly Amel 4.5)序列进行比对,得到相关tags在参考基因组上的位置信息,即为mapped tags。利用miRDeep2软件[18]将mapped tags与miRBase数据库中已知miRNA前体序列进行比对,从而鉴定已知miRNA的表达。并通过TPM(tags per million)算法对各样本中miRNA的表达量进行归一化处理。利用R软件计算各样品之间的相关性系数。以|log2 fold change|≥1且P≤0.05为筛选显著DEmiRNA的标准。1.4 显著DEmiRNA靶基因预测及分析
利用TargetFinder软件[19]对显著DEmiRNA的靶基因进行预测,并通过BLAST软件将预测的靶基因序列与Gene Ontology和KEGG数据库比对。将显著DEmiRNA的靶基因分别按照分子功能、细胞组分、生物学进程进行分类,并利用基迪奥OmicShare云平台(http://www.omicshare.com/tools/Home/Index/index. html)对靶基因进行KEGG pathway富集分析。最后,根据DEmiRNA与mRNA的靶向结合关系,利用Cytoscape软件构建miRNA-mRNA的调控网络。1.5 DEmiRNA的茎环实时荧光定量PCR(Stem-loop RT-qPCR)验证
通过Stem-loop RT-qPCR检测4个随机选取的miRNA(novel-m0031-3p、novel-m0034-5p、miR-3793-x和ame-miR-6000a-5p)在AmCK和AmT中的表达情况,以验证sRNA-seq数据的可靠性。根据所选miRNA的序列,参照CHEN等[20]的方法,利用DNAMAN软件(Lynnon Biosoft公司,美国)设计特异性的Stem-loop引物、上游引物和通用的反向引物,委托上海生工生物工程有限公司进行引物合成。选择snRNA U6作为内参。利用RNA抽提试剂盒(Axygen公司,美国)分别提取AmCK和AmT的总RNA,利用Stem-loop引物进行反转录得到相应的cDNA作为模板进行PCR和qPCR。常规PCR体系为50 μL,包括PCR mix(TaKaRa公司,日本)25 μL、无菌水17.5 μL、正、反向引物及cDNA模板各2.5 μL。反应程序如下:95℃ 5 min,95℃ 30 s,50℃ 30 s,72℃ 1 min,34个循环,72℃ 5 min。随后利用1.8%的琼脂糖凝胶电泳检测PCR的产物。qPCR反应体系(20 μL):SYBR Green Dye 10 μL,正、反向引物各1 μL,cDNA模板1 μL,Rox 0.44 μL,DEPC水补至20 μL。在ABI 7500荧光定量PCR仪(ABI公司,美国)中进行反应,反应条件:95℃ 1 min,95℃ 15 s,49—60℃ 60 s,共40个循环,熔解曲线默认系统程序。所选miRNA的相对表达量采用2-ΔΔCt法计算。每个反应进行3次生物学重复和3次技术重复。最后利用GraphPad Prism 5软件进行qPCR结果的检验及绘图。本研究使用的引物序列信息详见表1。Table 1
表1
表1本研究使用的引物
Table 1
| 引物名称 Primer name | 引物序列 Primer sequence |
|---|---|
| LOOP-miR-3793-x | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGGCCAGG |
| LOOP-ame-miR-6000a-5p | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATAGAGAC |
| LOOP-novel-m0031-3p | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCCTGCTT |
| LOOP-novel-m0034-5p | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATATCACA |
| F-miR-3793-x | ACACTCCAGCTGGGAGCGTGTTTTC |
| F-ame-miR-6000a-5p | ACACTCCAGCTGGGCAGCAGCAGCAG |
| F-novel-m0031-3p | GCATCCTCTTGAAT |
| F-novel-m0034-5p | CAGGTAACTACTGC |
| R | CTCAACTGGTGTCGTGGA |
| U6-F | GTTAGGCTTTGACGATTTCG |
| U6-R | GGCATTTCTCCACCAGGTA |
新窗口打开|下载CSV
2 结果
2.1 意蜂幼虫肠道的测序数据的质控与评估
两组幼虫肠道样品的sRNA-seq分别产生13 553 302和10 777 534条raw reads,经严格过滤后得到的clean reads数分别为13 186 921和10 480 913条(表2);此外,AmCK、AmT组内各生物学重复之间的Pearson相关性系数均在0.9822和0.9508以上,说明本研究的高通量测序数据质量良好,可用于进一步分析。Table 2
表2
表2sRNA-seq数据总览
Table 2
| 样品Sample | 原始读段Raw reads | 有效读段Clean reads |
|---|---|---|
| AmCK-1 | 15146178 | 14736731 (97.30%) |
| AmCK-2 | 13310167 | 12954535 (97.33%) |
| AmCK-3 | 12203560 | 11869496 (97.26%) |
| AmT-1 | 10741447 | 10453795 (97.32%) |
| AmT-2 | 12035887 | 11694678 (97.17%) |
| AmT-3 | 9555268 | 9294267 (97.27%) |
新窗口打开|下载CSV
2.2 意蜂幼虫肠道的DEmiRNA分析
AmCK vs AmT比较组中显著差异和非显著差异miRNA数为740个。其中,10个miRNA为显著性差异表达,包含4个上调miRNA和6个下调miRNA;显著DEmiRNA中包括3个novel miRNA和7个已知miRNA(表3)。表达谱分析结果显示,DEmiRNA的差异变化幅度差别明显(图1-A),DEmiRNA在AmT中的整体表达水平低于AmCK(图1-B)。Table 3
表3
表3AmCK vs AmT 中DEmiRNA的信息统计
Table 3
| 差异表达miRNA ID DEmiRNA ID | AmCK中的表达量 Expression in AmCK | AmT中的表达量 Expression in AmT | 以2为底miRNA的相对变化倍数的对数值 log2 fold change | 靶基因数 Number of target genes |
|---|---|---|---|---|
| miR-7085-x | 0.01 | 8.40 | 9.71 | 129 |
| miR-8440-y | 0.01 | 5.29 | 9.05 | 2265 |
| novel-m0023-5p | 0.01 | 1.79 | 7.48 | 334 |
| miR-3793-x | 33.72 | 89.76 | 1.41 | 398 |
| novel-m0034-3p | 4.06 | 1.95 | -1.05 | 768 |
| miR-4331-y | 74.07 | 27.70 | -1.42 | 77 |
| ame-miR-6000a-5p | 19.06 | 4.14 | -2.20 | 77 |
| miR-971-y | 3.25 | 0.31 | -3.37 | 100 |
| novel-m0025-3p | 0.88 | 0.01 | -6.46 | 294 |
| miR-4968-y | 11.61 | 0.01 | -10.18 | 279 |
新窗口打开|下载CSV
图1
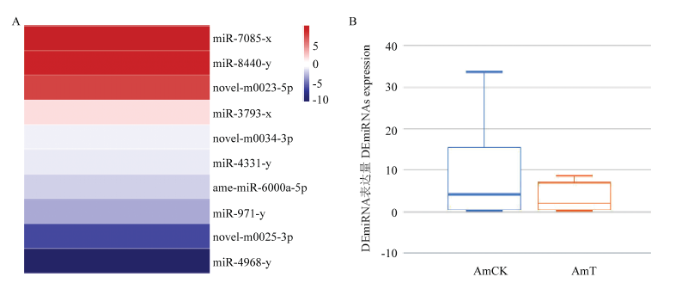 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1AmCK vs AmT中DEmiRNA的表达谱
A:DEmiRNA的表达量聚类Expression clustering of DEmiRNAs;B:不同样品中DEmiRNA的表达量比较Comparison of DEmiRNAs expression in different samples
Fig. 1Expression profile of DEmiRNAs in AmCK vs AmT comparison group
2.3 意蜂幼虫肠道DEmiRNA的靶基因预测及功能注释
对于AmCK vs AmT中的显著DEmiRNA,共预测出3 788个靶基因。GO数据库注释结果显示,上调miRNA的1 240个靶基因共涉及39个GO 条目(term),富集靶基因数最多的分别是结合(733个)、细胞进程(698个)、代谢进程(580个)、单组织进程(576个)、催化活性(439个)、细胞膜(387个)、细胞膜组件(380个)、细胞(286个)、细胞组件(286个)、定位(245个)等(图2-A);下调miRNA的749个靶基因可注释到34个GO条目,富集靶基因数最多的分别是细胞进程(533个)、结合(517个)、代谢进程(394个)、单组织进程(373个)、催化活性(240个)、细胞膜(227个)、细胞膜组件(226个)、细胞(216个)、细胞组件(216个)、细胞器(192个)等(图2-B)。进一步分析发现,对于上调和下调miRNA,分别有204和113个靶基因注释到应激反应,说明显著DEmiRNA参与宿主对球囊菌的胁迫应答的调控。图2
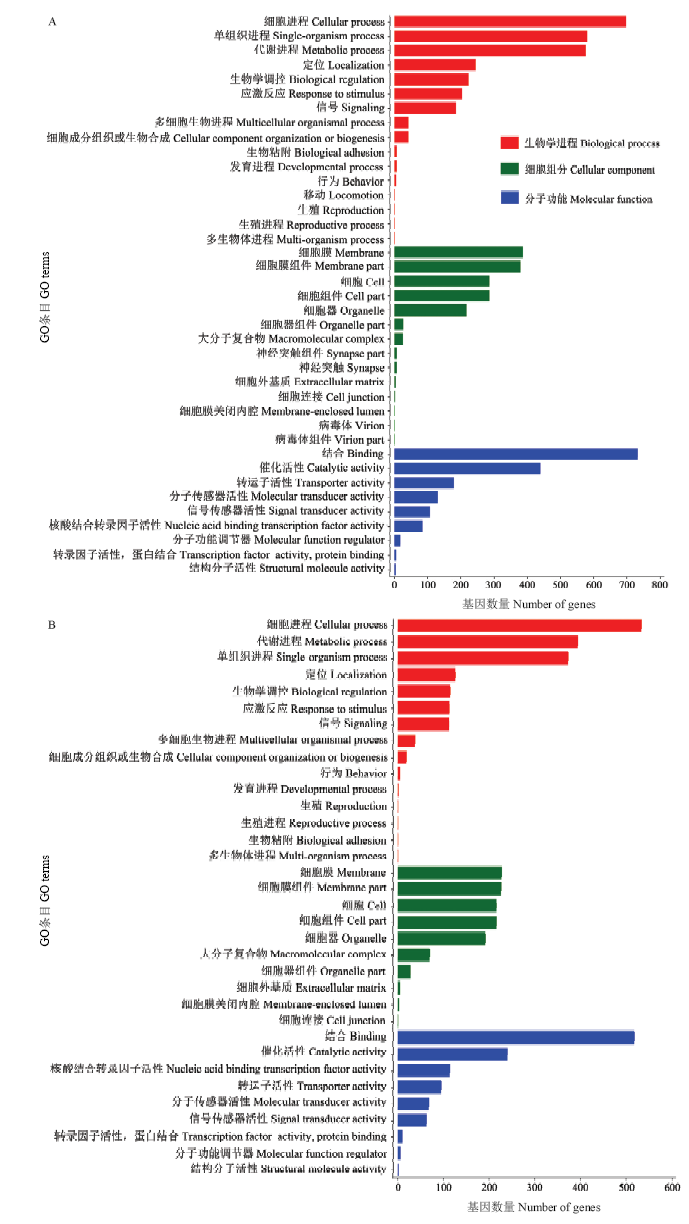 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2AmCK vs AmT中显著DEmiRNA的靶基因的GO数据库注释
A:上调miRNA的靶基因Target genes of up-regulated miRNAs;B:下调miRNA的靶基因Target genes of down-regulated miRNAs
Fig. 2GO database annotation of significant DEmiRNA’ target genes in AmCK vs AmT comparison group
对显著DEmiRNA的靶基因进行KEGG数据库注释,结果显示上调miRNA的靶基因可注释到95条代谢通路(pathway),其中富集数最多的分别是Wnt信号通路(112个)、Hippo信号通路(62个)、光传导(55个)、嘌呤代谢(48个)、Hedgehog信号通路(43个)、神经活性的配体-受体相互作用(19个)、昼夜节律(41个)、寿命调节途径(35个)、氨基糖与核苷酸糖代谢(32个)和内吞作用(31个)(图3-A);下调miRNA的靶基因注释到66条代谢通路,其中富集数最多的分别是内吞作用(52个)、磷脂酰肌醇信号系统(30个)、嘌呤代谢(28个)、磷酸肌醇代谢(25个)、Notch信号通路(20个)、神经活性的配体-受体相互作用(19个)、叶酸合成(18个)、背腹轴的形成(14个)、Wnt信号通路(13个)和mRNA监视通路(12个)(图3-B)。进一步分析结果显示,在细胞免疫相关通路中,对于上调和下调的miRNA,分别有31和52个靶基因注释到内吞作用,15和7个靶基因注释到泛素介导的蛋白水解;在体液免疫相关通路中,对于上调和下调的miRNA,分别有11和5个靶基因注释到Jak-STAT信号通路,有1和3个靶基因注释到MAPK信号通路。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3AmCK vs AmT中DEmiRNA的靶基因的KEGG数据库注释
A:上调miRNA的靶基因Target genes of up-regulated miRNAs;B:下调miRNA的靶基因Target genes of down-regulated miRNAs
Fig. 3KEGG database annotation of significant DEmiRNA’ target genes in AmCK vs AmT comparison group
2.4 意蜂幼虫肠道DEmiRNA的调控网络构建及分析
筛选具有KEGG数据库注释的靶基因,并分别构建上调miRNA和下调miRNA与靶基因的调控网络,结果显示显著DEmiRNA位于调控网络的中心,其中4个上调miRNA可结合499个靶基因(图4-A),277个靶基因可被6个下调miRNA调控(图4-B),DEmiRNA与靶基因之间形成复杂的调控网络。其中,上调miRNA中的miR-8440-y结合靶基因最多,达到419个(图4-A);下调miRNA中的novel-m0034-3p可结合149个靶基因(图4-B)。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4意蜂幼虫肠道的DEmiRNA的调控网络
A: 上调miRNA的调控网络Regulation networks of up-regulated miRNAs; B:下调miRNA的调控网络Regulation networks of down-regulated miRNAs。绿色圆形代表靶基因,红色三角代表miRNA Green circles indicate target genes, red triangles indicate miRNAs
Fig. 4Regulation networks of DEmiRNAs in A. m. ligustica larval gut
提取注释到Wnt信号通路、内吞作用的靶基因,构建它们与相关显著DEmiRNA的调控网络,分析结果显示7个显著DEmiRNA结合96个注释到Wnt信号通路的靶基因,二者形成1个较大和3个较小的调控网络,其中结合靶基因数由多到少分别为miR-8440-y(78个)、miR-3793-x(27个)、miR-4968-y(9个)、miR-7085-x(5个)、novel-m0025-3p(3个)、novel-m0023-5p(2个)和miR-4331-y(1个)(图5-A);8个显著DEmiRNA结合55个注释到内吞作用的靶基因,二者形成1个较大的和2个较小的调控网络,其中miR-8440-y、novel-m0034-3p、ame-miR-6000a- 5p、novel-m0025-3p、novel-m0023-5p、miR-4968-y、miR-4331-y和miR-971-y结合的靶基因数分别为26、26、12、10、5、2、1和1个(图5-B)。进一步分析发现,miR-4331-y、miR-4968-y、miR-8440-y、novel-m0023-5p和novel-m0025-3p共同了参与上述2条代谢通路调控,而miR-3793-x和miR-7085-x仅参与对Wnt信号通路的调控,ame-miR-6000a-5p、miR-971-y和novel-m0034-3p仅涉及对内吞作用的调控(图5-A、5-B)。
图5
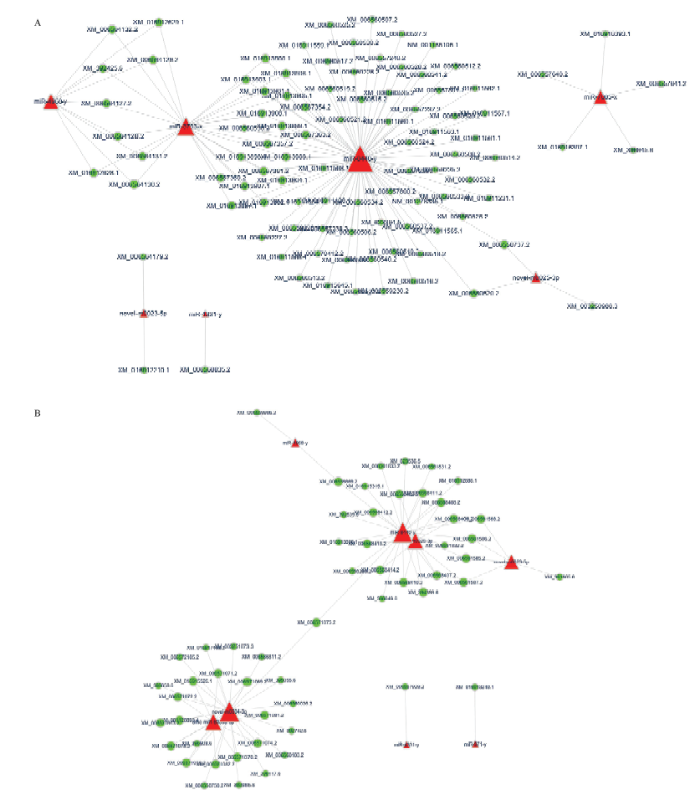 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5意蜂幼虫肠道的Wnt信号通路和内吞作用相关显著DEmiRNA的调控网络
A: Wnt信号通路相关DEmiRNA的调控网络Regulation networks of DEmiRNAs related to Wnt signaling pathway; B:内吞作用相关DEmiRNA的调控网络Regulation networks of DEmiRNAs related to endocytosis。绿色圆形代表靶基因,红色三角代表miRNA Green circles indicate target genes, red triangles indicate miRNAs
Fig. 5Regulation networks of significant DEmiRNAs associated with Wnt signaling pathway and endocytosis in A. m. ligustica larval gut
2.5 意蜂幼虫肠道DEmiRNA的Stem-loop RT-PCR和qPCR验证
为整体验证测序数据的准确性,从所有显著差异表达和非显著差异表达的740个miRNA中随机挑选2个非显著差异表达miRNA(novel-m0031-3p和novel-m0034-5p)和2个显著差异miRNA(miR-3793-x和ame-miR-6000a-5p)进行Stem-loop RT-PCR验证,电泳结果显示上述miRNA在AmCK和AmT中均可扩增出符合预期的目的条带(图6-A)。进一步对上述4个DEmiRNA进行qPCR检测,结果显示它们的表达水平的变化趋势与sRNA-seq数据中的变化趋势一致(图6-B、6-C、6-D、6-E),证实了测序结果的可靠性。图6
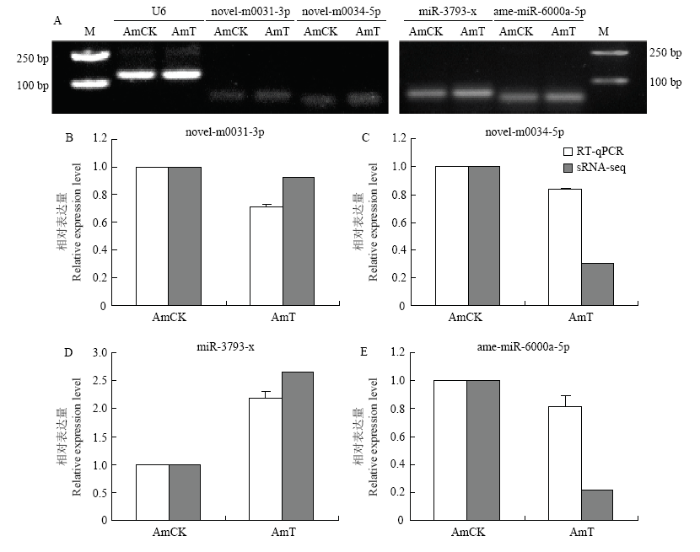 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6DEmiRNA的Stem-loop RT-PCR(A)和qPCR验证(B—E)
Fig. 6Stem-loop RT-PCR (A) and qPCR confirmation (B-E) of DEmiRNAs
3 讨论
球囊菌是一种特异性侵染蜜蜂幼虫肠道的真菌病原,能严重影响成年蜜蜂的数量和蜂群的生产力,造成较大的经济损失[21]。miRNA作为一种转录后调控的关键因子,通过调节宿主相关免疫信号通路[22],改变宿主基因的表达而影响病原复制[23],而在宿主与病原间的互作中扮演着特殊角色。笔者所在课题组前期已在mRNA组学水平全面解析意蜂及中蜂幼虫与球囊菌间的互作[24,25,26]。然而在ncRNA组学层面,至今还没有二者互作的相关研究报道。本研究利用sRNA-seq技术对正常及球囊菌侵染的意蜂4日龄幼虫肠道进行测序,预测出4个上调miRNA和6个下调miRNA,DEmiRNA在AmCK中的表达水平总体高于AmT(图1-B),表明宿主在球囊菌侵染前期,通过下调相关miRNA的表达水平降低其对相应靶基因的抑制作用,从而响应对病原的应答。3.1 球囊菌胁迫可影响与蜜蜂幼虫肠道发育、代谢和免疫相关的miRNA的表达水平
球囊菌在侵染意蜂幼虫的前期,孢子处于低水平的萌发状态,幼虫并不表现出明显的白垩病症状,但宿主与病原之间存在复杂的互作[15]。Hippo信号通路参与D. melanogaster肠道干细胞的增殖调控[27],并通过与Wnt、Notch等信号通路共同作用影响D. melanogaster的器官大小[28]。郭睿等[29,30]研究发现,意蜂工蜂中肠内的长链非编码RNA和环状RNA均可通过间接调控Hippo信号通路上的富集基因,影响蜜蜂肠道的发育过程。此外,该信号通路也被证明与蜜蜂的级型分化和卵巢发育状态密切相关[31]。本研究中,对于意蜂幼虫肠道的上调miRNA的靶基因,富集在Hippo信号通路的数量(62个),远高于下调miRNA的靶基因富集在该通路上的数量(6个),表明球囊菌在侵染前期通过调控Hippo信号通路对宿主的肠道发育产生影响。郝向伟[32]研究发现,家蚕(Bombyx mori)被大肠杆菌(Escherichia coli)和苏云金芽孢杆菌(Bacillus thuringiensis)侵染后,其肠道的BmHh表达量显著上调,作者推测Hedgehog信号通路可能参与肠干细胞的增殖和分化以及肠道损伤后的修复。本研究发现,对于富集在Hedgehog信号通路的靶基因,上调miRNA的靶基因数(43个)远多于下调miRNA的靶基因数(1个),说明多数该通路上的基因受到抑制,表明Hedgehog信号通路对不同的病原微生物存在应答差异。本研究还发现,对于上调miRNA的靶基因,富集在昼夜节律(41个)、嘌呤代谢(48个)、氨基糖与核苷酸糖代谢(32个)和嘧啶代谢(5个)等代谢通路上的数量均高于下调miRNA的靶基因富集在上述通路上的数量。HUANG等[13]研究发现,东方蜜蜂微孢子虫感染西方蜜蜂的前6 d内,宿主的17个DEmiRNA主要参与调控嘌呤代谢、嘧啶代谢和氧化磷酸化等9条代谢通路,作者推测这与N. ceranae通过改变宿主的代谢以促进其更快速的繁殖机制有关。本研究表明在球囊菌侵染前期,意蜂幼虫肠道的部分生命活动、新陈代谢受到病原的抑制,推测球囊菌通过调节宿主的上述生物学过程为自身的孢子萌发创造有利条件。Wnt信号通路广泛参与昆虫的细胞增殖、组织分化、器官形成等过程[33,34]。该通路还可通过与TGF-beta、Hippo、Notch、MAPK、FoxO等信号通路的共同作用而参与蜂王卵巢激活和产卵过程[35]。本研究中,共有7个显著DEmiRNA(miR-8440-y、miR-3793-x和miR-4968-y等)的96个靶基因注释到Wnt信号通路,表明球囊菌侵染会影响宿主的Wnt信号通路基因的表达。此外,有研究表明Wnt信号通路参与了人类的先天免疫反应[36,37],例如SMITH等[38]发现miR-34家族对Hela细胞内的Wnt/β-catenin信号通路具有抑制作用,并表现出强烈的抗黄病毒作用;乔莹[39]发现被隐核虫(Cryptocaryon irritans)感染的大黄鱼(Larimichthys crocea)体内miR-466-x、miR-1895-y和miR-8485-y等miRNA广泛参与了对Wnt和Jak-STAT信号通路以及内吞作用和溶酶体等免疫通路的调控。本研究中,miR-4331-y、miR-4968-y、miR-8440-y、novel-m0023-5p和novel-m0025-3p均参与了对Wnt信号通路和内吞作用的调控,表明它们通过调控上述免疫通路对意蜂幼虫肠道的免疫应答进行调节。
3.2 miR-4331-y作为潜在的分子靶标参与宿主与病原复杂的互作过程
miRNA在物种间具有高度的保守性、表达时序性和组织特异性[40]。郭睿等[41]对意蜂幼虫肠道发育过程中miRNA的表达谱进行分析,发现miR-4331-y仅在4和5日龄幼虫肠道内差异表达,并间接调控富集在Wnt、FoxO、TGF-beta、Notch和Jak-STAT等信号通路上的靶基因。ZHAO等[42]研究发现,猪肾细胞(PK-15)被传染性肠胃炎病毒(Transmissible gastroenteritis virus,TGEV)侵染后,其miR-4331的表达量显著上调,并通过靶向结合RB1作用于p38 MAPK信号通路,从而调控TGEV诱导的PK-15线粒体损伤。此外,在患有H1N1流感病毒的病人以及感染低致病性H5N3流感病毒的鸡的体内,均发现miR-204的表达显著提高[43,44]。由于病原种类、侵染机制以及检测时间点的差异,不同宿主的同源miRNA在响应病原侵染过程中,其表达量变化趋势可能并不完全一致。例如ZHANG等[45]发现甲型H1N1病毒(SIV-H1N1/2009)可通过抑制新生猪气管细胞(NPTr)中ssc-miR-204和ssc-miR-4331的表达促进自身在细胞内的复制;ssc-miR-204和ssc-miR-4331可抑制病毒的HA表面糖蛋白和NS1干扰素拮抗剂蛋白的表达,从而对病毒的复制进行负调控。本研究中,miR-4331-y的表达量在球囊菌胁迫后呈显著下调,并通过对靶基因的调控同时参与对内吞作用、吞噬体、Jak-STAT和Wnt信号通路等免疫通路的间接调控,表明宿主在球囊菌侵染前期通过下调miR-4331-y的表达水平降低对靶基因的抑制作用,从而激活相关免疫通路,以响应球囊菌的胁迫;球囊菌也可能抑制宿主肠道内miR-4331-y的表达量,以利于孢子的萌发。未来可通过合成相应的miRNA mimic和miRNA inhibitor对miR-4331-y进行过表达和敲减,从而深入研究其在宿主的胁迫应答中的作用。3.3 意蜂幼虫肠道响应球囊菌胁迫前期过程中mRNA与miRNA组学比较
为探究不同蜂种的蜜蜂幼虫在球囊菌侵染前期的应答及与病原间的互作,笔者课题组曾对正常及球囊菌胁迫的意蜂和中蜂4日龄幼虫肠道进行转录组测序和分析[14,15],发现意蜂幼虫的差异表达基因涉及代谢进程、免疫系统进程和应激反应等19个功能分类,并有5个角质层蛋白基因和2个抗菌肽编码基因表达水平下调,进而在mRNA组学水平解析了宿主的胁迫应答[15]。昆虫肠道围食膜是抵御经口摄入病原的重要物理屏障,角质层蛋白作为围食膜的主要成分之一,在保护肠道健康过程中发挥重要作用[46]。本研究发现,上调miRNA中的miR-8440-y靶向结合2个角质层蛋白编码基因,表明球囊菌可通过提高宿主的miR-8440-y的表达水平降低角质层蛋白编码基因的水平,以利于球囊菌的萌发和侵染过程,这与此前的研究结果一致。4 结论
在miRNA组学水平对意蜂幼虫肠道在球囊菌侵染前期的胁迫应答及宿主-病原互作进行研究,结果表明显著DEmiRNA可通过调控细胞生命活动、新陈代谢和部分细胞和体液免疫等生物学过程参与宿主的胁迫应答。其中miR-4331-y、miR-8440-y、miR-4968-y、novel-m0023-5p和novel-m0025-3p共同参与了宿主的Wnt信号通路和内吞作用的调控,且miR-8440-y结合靶基因的数量最多。筛选出的关键miRNA可用于后续的功能研究,有望为白垩病治疗提供潜在的分子靶标。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.3969/j.issn.1674-0858.2015.02.23URL [本文引用: 1]

蜜蜂授粉对开花植物的结实具有重要作用,但目前的研究主要集中在蜜蜂授粉对异花授粉作物的授粉效果上,而对自花授粉植物的授粉效果鲜有报道。本研究以设施农业中主要的自花授粉作物—辣椒为研究对象,研究了意大利蜜蜂(意蜂)和小峰熊蜂(熊蜂)为设施辣椒授粉时的出巢访花活动规律及其授粉效果,并初步估算了其可能带来的经济效益。对两种蜂为辣椒授粉过程中的出巢访花活动规律研究表明:意蜂的出巢和工作起点温度均显著高于熊蜂,表明熊蜂每天可比意蜂多工作1.0-1.5 h。对不同蜂的授粉效果的研究表明:两种蜂授粉后的结实率差异不显著,但均显著高于自花授粉的结实率;三种授粉方式的坐果时间长短依次为自花授粉意蜂授粉熊蜂授粉;花后30 d三种授粉方式的单果重和果实长度差异显著,单果重依次为43.04、57.23和67.58 g,对应果实长度依次为17.41、19.14和20.28㎝。两种蜂授粉后的果实籽粒数(121.35和127.25粒)显著高于人工授粉后的105.40粒。研究表明意蜂和熊蜂可使单位面积自花授粉的设施辣椒增产6.62%和9.79%,具有较好的增产效果,相应的,辣椒对两种蜂的依赖度D值分别为0.06和0.09,采用国际通用的生物经济的方法初步推算出这两种授粉方式在本世纪前十年每年均可使我国辣椒的经济效益得到可观的增加。
DOI:10.3969/j.issn.1674-0858.2015.02.23URL [本文引用: 1]
蜜蜂授粉对开花植物的结实具有重要作用,但目前的研究主要集中在蜜蜂授粉对异花授粉作物的授粉效果上,而对自花授粉植物的授粉效果鲜有报道。本研究以设施农业中主要的自花授粉作物—辣椒为研究对象,研究了意大利蜜蜂(意蜂)和小峰熊蜂(熊蜂)为设施辣椒授粉时的出巢访花活动规律及其授粉效果,并初步估算了其可能带来的经济效益。对两种蜂为辣椒授粉过程中的出巢访花活动规律研究表明:意蜂的出巢和工作起点温度均显著高于熊蜂,表明熊蜂每天可比意蜂多工作1.0-1.5 h。对不同蜂的授粉效果的研究表明:两种蜂授粉后的结实率差异不显著,但均显著高于自花授粉的结实率;三种授粉方式的坐果时间长短依次为自花授粉意蜂授粉熊蜂授粉;花后30 d三种授粉方式的单果重和果实长度差异显著,单果重依次为43.04、57.23和67.58 g,对应果实长度依次为17.41、19.14和20.28㎝。两种蜂授粉后的果实籽粒数(121.35和127.25粒)显著高于人工授粉后的105.40粒。研究表明意蜂和熊蜂可使单位面积自花授粉的设施辣椒增产6.62%和9.79%,具有较好的增产效果,相应的,辣椒对两种蜂的依赖度D值分别为0.06和0.09,采用国际通用的生物经济的方法初步推算出这两种授粉方式在本世纪前十年每年均可使我国辣椒的经济效益得到可观的增加。
URLMagsci [本文引用: 1]
为探究蜜蜂球囊菌<em>Ascosphaera apis</em>只侵染封盖前后蜜蜂幼虫的原因及相关侵染机制, 本研究利用实验室饲养的意大利蜜蜂<em>Apis mellifera ligustica</em>幼虫, 给其接种球囊菌孢子, 探究不同接种量(0, 1.0×10<sup>2</sup>, 1.0×10<sup>3</sup>, 1.0×10<sup>4</sup>, 1.0×10<sup>5 </sup>和1.0×10<sup>6</sup> 孢子/mL)、 接种时期(3, 4, 5和6龄幼虫)以及28℃低温处理6 h对蜜蜂球囊菌侵染的影响。同时对处于不同侵染阶段的蜜蜂幼虫做病理学切片, 探究球囊菌侵染的过程。结果显示: 球囊菌孢子接种量与蜜蜂的发病率密切相关(<em>r</em>=0.9883), 蜜蜂幼虫对低于1.0×10<sup>3</sup>孢子/mL的侵染有抗性, 与不接孢子的对照组差异不显著(<em>P</em>>0.05)。蜜蜂不同龄期接种发病率的差异是因不同龄幼虫食量不同导致的摄入孢子剂量的不同引起的, 28℃低温处理能够显著提高处于幼虫到蛹转化期蜜蜂的发病率(<em>P</em><0.05), 而对取食阶段的蜜蜂幼虫没有影响。病理学研究表明, 在整个幼虫期, 摄入的孢子因中肠没有氧气不生长, 对幼虫没有致病性, 幼虫的取食和发育过程正常, 至幼虫期结束进入蛹期后, 蜜蜂的中后肠接通, 摄入的孢子伴随蜜蜂的蛹便进入后肠并在此迅速萌发生长, 在1~2 d内菌丝即突破体表, 导致蜜蜂死亡。蜜蜂球囊菌选择营养物质储存最多而防御能力较低的幼虫到蛹转化期侵染, 降低了侵染成本, 提高成功率。该研究阐明了蜜蜂球囊菌侵染蜜蜂的机制, 丰富了昆虫与病原菌之间相互作用的内容。
URLMagsci [本文引用: 1]
为探究蜜蜂球囊菌<em>Ascosphaera apis</em>只侵染封盖前后蜜蜂幼虫的原因及相关侵染机制, 本研究利用实验室饲养的意大利蜜蜂<em>Apis mellifera ligustica</em>幼虫, 给其接种球囊菌孢子, 探究不同接种量(0, 1.0×10<sup>2</sup>, 1.0×10<sup>3</sup>, 1.0×10<sup>4</sup>, 1.0×10<sup>5 </sup>和1.0×10<sup>6</sup> 孢子/mL)、 接种时期(3, 4, 5和6龄幼虫)以及28℃低温处理6 h对蜜蜂球囊菌侵染的影响。同时对处于不同侵染阶段的蜜蜂幼虫做病理学切片, 探究球囊菌侵染的过程。结果显示: 球囊菌孢子接种量与蜜蜂的发病率密切相关(<em>r</em>=0.9883), 蜜蜂幼虫对低于1.0×10<sup>3</sup>孢子/mL的侵染有抗性, 与不接孢子的对照组差异不显著(<em>P</em>>0.05)。蜜蜂不同龄期接种发病率的差异是因不同龄幼虫食量不同导致的摄入孢子剂量的不同引起的, 28℃低温处理能够显著提高处于幼虫到蛹转化期蜜蜂的发病率(<em>P</em><0.05), 而对取食阶段的蜜蜂幼虫没有影响。病理学研究表明, 在整个幼虫期, 摄入的孢子因中肠没有氧气不生长, 对幼虫没有致病性, 幼虫的取食和发育过程正常, 至幼虫期结束进入蛹期后, 蜜蜂的中后肠接通, 摄入的孢子伴随蜜蜂的蛹便进入后肠并在此迅速萌发生长, 在1~2 d内菌丝即突破体表, 导致蜜蜂死亡。蜜蜂球囊菌选择营养物质储存最多而防御能力较低的幼虫到蛹转化期侵染, 降低了侵染成本, 提高成功率。该研究阐明了蜜蜂球囊菌侵染蜜蜂的机制, 丰富了昆虫与病原菌之间相互作用的内容。
DOI:10.1016/j.ibmb.2012.10.005URLPMID:23103375 [本文引用: 1]
MicroRNAs (miRNAs) are small non-coding RNAs that are generated in all eukaryotes and viruses. Their role as master regulators of gene expression in various biological processes has only been fully appreciated over the last decade. Accumulating evidence suggests that alterations in the expression of miRNAs may lead to disorders, including developmental defects, diseases and cancer. Here, I review what is currently known about miRNA functions in insects to provide an insight into their diverse roles in insect biology.
DOI:10.1038/ng1725URLPMID:2538576 [本文引用: 1]
Abstract Understanding the molecular mechanisms that regulate cellular proliferation and differentiation is a central theme of developmental biology. MicroRNAs (miRNAs) are a class of regulatory RNAs of approximately 22 nucleotides that post-transcriptionally regulate gene expression. Increasing evidence points to the potential role of miRNAs in various biological processes. Here we show that miRNA-1 (miR-1) and miRNA-133 (miR-133), which are clustered on the same chromosomal loci, are transcribed together in a tissue-specific manner during development. miR-1 and miR-133 have distinct roles in modulating skeletal muscle proliferation and differentiation in cultured myoblasts in vitro and in Xenopus laevis embryos in vivo. miR-1 promotes myogenesis by targeting histone deacetylase 4 (HDAC4), a transcriptional repressor of muscle gene expression. By contrast, miR-133 enhances myoblast proliferation by repressing serum response factor (SRF). Our results show that two mature miRNAs, derived from the same miRNA polycistron and transcribed together, can carry out distinct biological functions. Together, our studies suggest a molecular mechanism in which miRNAs participate in transcriptional circuits that control skeletal muscle gene expression and embryonic development.
DOI:10.1242/dev.02073URLPMID:16224045 [本文引用: 1]
Abstract Five years into the 'small RNA revolution' it is hard not to share in the excitement about the rapidly unravelling biology of microRNAs. Since the discovery of the first microRNA gene, lin-4, in the nematode Caenorhabditis elegans, many more of these short regulatory RNA genes have been identified in flowering plants, worms, flies, fish, frogs and mammals. Currently, about 2% of the known human genes encode microRNAs. MicroRNAs are essential for development and this review will summarise our current knowledge of animal microRNA function. We will also discuss the emerging links of microRNA biology to stem cell research and human disease, in particular cancer.
DOI:10.1016/j.jhep.2008.11.025URLPMID:19232449 [本文引用: 1]
To reveal the microRNA (miRNA) expression profile and related roles in rat HSCs during activation.MethodsmiRNA expression profiling was analyzed in quiescent and in culture-activated HSCs by microarray. The differentially expressed miRNAs, as verified by RT-PCR, were subjected to gene ontology (GO) analysis. Furthermore, the effects of miR-16 and miR-15b on the apoptosis of activated HSCs were investigated by Hoechst 33258, TUNEL staining and annexin-V/PI labeling flow cytometry. The underlying mechanism related to Bcl-2 and caspases was assessed.ResultsThe upregulated and downregulated miRNAs in activated HSCs were 12 miRNAs and 9 miRNAs, respectively. The differential expression of miR-16, -15b, -122, -138, -143, and -140 was validated. High-enrichment GOs containing apoptosis-related targeted genes and miRNA ene networks characterized by Bcl-2, which was targeted by the miR-15/16 family, uncovered the critical role of miR-16 and miR-15b in apoptosis. Restoring the intracellular miRNAs by miR-16 and miR-15b administration greatly reduced Bcl-2, and increased the expression of caspases 3, 8, and 9. Significantly elevated rates of apoptosis were then induced in activated HSCs.ConclusionsThe activation of HSCs relate to 21 miRNAs. Among these, mir-15b and miR-16 may be essential for apoptosis by targeting Bcl-2 and the caspase signaling pathway.
DOI:10.1186/1742-4690-3-68URLPMID:17032463 [本文引用: 1]
MicroRNAs (miRNAs) are a new class of 18???23 nucleotide long non-coding RNAs that play critical roles in a wide spectrum of biological processes. Recent reports also throw light into the role of microRNAs as critical effectors in the intricate host-pathogen interaction networks. Evidence suggests that both virus and hosts encode microRNAs. The exclusive dependence of viruses on the host cellular machinery for their propagation and survival also make them highly susceptible to the vagaries of the cellular environment like small RNA mediated interference. It also gives the virus an opportunity to fight and/or modulate the host to suite its needs. Thus the range of interactions possible through miRNA-mRNA cross-talk at the host-pathogen interface is large. These interactions can be further fine-tuned in the host by changes in gene expression, mutations and polymorphisms. In the pathogen, the high rate of mutations adds to the complexity of the interaction network. Though evidence regarding microRNA mediated cross-talk in viral infections is just emerging, it offers an immense opportunity not only to understand the intricacies of host-pathogen interactions, and possible explanations to viral tropism, latency and oncogenesis, but also to develop novel biomarkers and therapeutics.
[本文引用: 1]
[本文引用: 1]
DOI:10.1073/pnas.1303603110URLPMID:23733960 [本文引用: 1]
The endosymbiont Wolbachia is common among insects and known for the reproductive manipulations it exerts on hosts as well as inhibition of virus replication in their hosts. Recently, we showed that Wolbachia uses host microRNAs to manipulate host gene expression for its efficient maintenance in the dengue mosquito vector, Aedes aegypti. Cytosine methylation is mediated by a group of proteins called DNA (cytosine-5) methyltransferases, which are structurally and functionally conserved from prokaryotes to eukaryotes. The biological functions of cytosine methylation include host defense, genome stability, gene regulation, developmental promotion of organs, and lifespan regulation. Ae. aegypti has only one DNA methyltransferase gene (AaDnmt2) belonging to the cytosine methyltransferase family 2, which is the most deeply conserved and widely distributed gene among metazoans. Here, we show that in mosquitoes the introduced endosymbiont, Wolbachia, significantly suppresses expression of AaDnmt2, but dengue virus induces expression of AaDnmt2. Interestingly, we found that aae-miR-2940 microRNA, which is exclusively expressed in Wolbachia-infected mosquitoes, down-regulates the expression of AaDnmt2. Reversely, overexpression of AaDnmt2 in mosquito cells led to inhibition of Wolbachia replication, but significantly promoted replication of dengue virus, suggesting a causal link between this Wolbachia manipulation and the blocking of dengue replication in Wolbachia-infected mosquitoes. In addition, our findings provide an explanation for hypomethylation of the genome in Wolbachia-infected Ae. aegypti.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1016/j.ibmb.2013.03.001URL [本文引用: 1]
DOI:10.1038/srep17494URLPMID:26620304 [本文引用: 2]
In order to study the effects ofNosema ceranaeinfection on honey bee microRNA (miRNA) expression, we deep-sequenced honey bee miRNAs daily across a full 6-day parasite reproduction cycle. Seventeen miRNAs were differentially expressed in honey bees infected byN. ceranaethat potentially target over 400 genes predicted to primarily involve ion binding, signaling, the nucleus, transmembrane transport, and DNA binding. Based on Enzyme Code analysis, nine biological pathways were identified by screening target genes against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, seven of which involved metabolism. Our results suggest that differentially expressed miRNAs regulate metabolism related genes of host honey bees in response toN. ceranaeinfection.
DOI:10.3864/j.issn.0578-1752.2017.13.019URL [本文引用: 3]
[目的]白垩病是困扰养蜂生产的顽疾.目前,尚无利用二代测序技术研究中华蜜蜂(Apis Cerana cerana,简称中蜂)幼虫白垩病的报道.本研究利用RNA-seq技术对健康(AcCK)及球囊菌(Ascosphaera apis)胁迫的中蜂4日龄幼虫肠道(ACT)进行深度测序,在转录组水平研究中蜂幼虫在球囊菌胁迫早期的胁迫应答.[方法]通过I11umina HiSeq 2500平台对AcCK和AcT进行双端(PE125)测序,首先对测序数据进行质控和评估,利用edgeR软件进行差异表达基因(DEG)分析,进而对DEGs进行GO富集分析及KEGG代谢通路富集分析,最后,利用实时荧光定量PCR (qRT-PCR)验证测序数据的可靠性.[结果]AcCK和AcT的转录组测序共得到188 457 338条原始读段(raw reads),经过滤得到182 088 448条有效读段(clean reads),两端Q20与Q30均在97.96%和94.97%以上,说明测序数据质量良好;主成分分析(PCA)结果显示第一与第二主成分可分别解释基因表达总体差异的75.8%和10.7%;DEG分析结果显示上调基因与下调基因的数量分别为344和2 39个;GO富集分析结果显示DEGs共富集在36个GO分类(term)上,其中基因富集数最多的细胞(106 unigenes)、细胞组件(106 unigenes)和代谢进程(104 unigenes);KEGG代谢通路富集分析结果显示上调与下调基因分别富集在72个和45个代谢通路上,其中上调基因富集数最多的是核糖体(72 unigenes)、碳代谢(16 unigenes)和糖酵解(14 unigenes),而下调基因富集数最多的是碳代谢(9 unigenes)、二羧酸代谢(8 unigenes)和氨基酸生物合成(7 unigenes),进一步分析表明中蜂幼虫肠道的部分细胞免疫对球囊菌的胁迫产生应答,而体液免疫不产生应答,宿主的代谢相关基因受到球囊菌的显著抑制.[结论]揭示了中蜂幼虫在球囊菌入侵早期的胁迫应答,为深入解析中蜂幼虫的胁迫应答机制提供了重要信息,也为在分子水平研究关键应答基因打下了基础.
DOI:10.3864/j.issn.0578-1752.2017.13.019URL [本文引用: 3]
[目的]白垩病是困扰养蜂生产的顽疾.目前,尚无利用二代测序技术研究中华蜜蜂(Apis Cerana cerana,简称中蜂)幼虫白垩病的报道.本研究利用RNA-seq技术对健康(AcCK)及球囊菌(Ascosphaera apis)胁迫的中蜂4日龄幼虫肠道(ACT)进行深度测序,在转录组水平研究中蜂幼虫在球囊菌胁迫早期的胁迫应答.[方法]通过I11umina HiSeq 2500平台对AcCK和AcT进行双端(PE125)测序,首先对测序数据进行质控和评估,利用edgeR软件进行差异表达基因(DEG)分析,进而对DEGs进行GO富集分析及KEGG代谢通路富集分析,最后,利用实时荧光定量PCR (qRT-PCR)验证测序数据的可靠性.[结果]AcCK和AcT的转录组测序共得到188 457 338条原始读段(raw reads),经过滤得到182 088 448条有效读段(clean reads),两端Q20与Q30均在97.96%和94.97%以上,说明测序数据质量良好;主成分分析(PCA)结果显示第一与第二主成分可分别解释基因表达总体差异的75.8%和10.7%;DEG分析结果显示上调基因与下调基因的数量分别为344和2 39个;GO富集分析结果显示DEGs共富集在36个GO分类(term)上,其中基因富集数最多的细胞(106 unigenes)、细胞组件(106 unigenes)和代谢进程(104 unigenes);KEGG代谢通路富集分析结果显示上调与下调基因分别富集在72个和45个代谢通路上,其中上调基因富集数最多的是核糖体(72 unigenes)、碳代谢(16 unigenes)和糖酵解(14 unigenes),而下调基因富集数最多的是碳代谢(9 unigenes)、二羧酸代谢(8 unigenes)和氨基酸生物合成(7 unigenes),进一步分析表明中蜂幼虫肠道的部分细胞免疫对球囊菌的胁迫产生应答,而体液免疫不产生应答,宿主的代谢相关基因受到球囊菌的显著抑制.[结论]揭示了中蜂幼虫在球囊菌入侵早期的胁迫应答,为深入解析中蜂幼虫的胁迫应答机制提供了重要信息,也为在分子水平研究关键应答基因打下了基础.
DOI:10.7679/j.issn.2095-1353.2017.067URL [本文引用: 4]
[目的]球囊菌特异性地侵染蜜蜂幼虫而导致白垩病,该病造成成年蜜蜂数量的大幅下降,从而严重影响蜂蜜等产品的产量.目前,尚无利用二代测序技术研究白垩病的报道.本研究利用RNA-seq技术对正常及球囊菌胁迫后的意大利蜜蜂4日龄幼虫肠道进行深度测序,为解析宿主响应球囊菌胁迫的应答机制提供了重要的参考信息.[方法]利用Illumina Hiseq 2500平台对对照组(AmCK)和处理组(AmT)幼虫肠道进行双端(PE125)测序;利用Perl脚本进行下机数据过滤;利用R软件进行测序饱和度分析、计算各样品之间的相关性系数;利用SOAP aligner/soap2软件将未比对上核糖体的读段(Reads)比对到意大利蜜蜂参考基因组;基于GO数据库和KEGG数据库进行GO分类和KEGG代谢通路(Pathway)富集分析.[结果]RNA-seq共得到181096194条原始读段(Raw reads),经过滤得到179078764条有效读段(Clean reads).差异表达基因(DEGs)分析结果显示上调与下调基因的数量分别为4和20个.DEG的基因本体论(Gene ontology,GO)分析结果显示,这些DEGs分布于10个GO分类,除归入结合(Binding)的基因和催化活性(Catalytic activity)的部分基因外,绝大多数GO分类上的基因均表现为下调.DEGs的KEGG代谢通路分析结果显示各有1个DEG分别富集在核糖体和氧化磷酸化且均下调表达.[结论]本研究揭示了意大利蜜蜂幼虫肠道在球囊菌入侵早期的胁迫应答,为解析宿主响应球囊菌胁迫的应答机制奠定了基础.
DOI:10.7679/j.issn.2095-1353.2017.067URL [本文引用: 4]
[目的]球囊菌特异性地侵染蜜蜂幼虫而导致白垩病,该病造成成年蜜蜂数量的大幅下降,从而严重影响蜂蜜等产品的产量.目前,尚无利用二代测序技术研究白垩病的报道.本研究利用RNA-seq技术对正常及球囊菌胁迫后的意大利蜜蜂4日龄幼虫肠道进行深度测序,为解析宿主响应球囊菌胁迫的应答机制提供了重要的参考信息.[方法]利用Illumina Hiseq 2500平台对对照组(AmCK)和处理组(AmT)幼虫肠道进行双端(PE125)测序;利用Perl脚本进行下机数据过滤;利用R软件进行测序饱和度分析、计算各样品之间的相关性系数;利用SOAP aligner/soap2软件将未比对上核糖体的读段(Reads)比对到意大利蜜蜂参考基因组;基于GO数据库和KEGG数据库进行GO分类和KEGG代谢通路(Pathway)富集分析.[结果]RNA-seq共得到181096194条原始读段(Raw reads),经过滤得到179078764条有效读段(Clean reads).差异表达基因(DEGs)分析结果显示上调与下调基因的数量分别为4和20个.DEG的基因本体论(Gene ontology,GO)分析结果显示,这些DEGs分布于10个GO分类,除归入结合(Binding)的基因和催化活性(Catalytic activity)的部分基因外,绝大多数GO分类上的基因均表现为下调.DEGs的KEGG代谢通路分析结果显示各有1个DEG分别富集在核糖体和氧化磷酸化且均下调表达.[结论]本研究揭示了意大利蜜蜂幼虫肠道在球囊菌入侵早期的胁迫应答,为解析宿主响应球囊菌胁迫的应答机制奠定了基础.
DOI:10.1016/j.jinsphys.2014.08.003URLPMID:25152509 [本文引用: 1]
Insects are the most successful group of animals on earth, owing this partly to their very effective immune responses to microbial invasion. These responses mainly include cellular and humoral responses as well as RNA interference (RNAi). Small non-coding RNAs (snRNAs) produced through RNAi are important molecules in the regulation of gene expression in almost all living organisms; contributing to important processes such as development, differentiation, immunity as well as host–microorganism interactions. The main snRNAs produced by the RNAi response include short interfering RNAs, microRNAs and piwi-interacting RNAs. In addition to the host snRNAs, some microorganisms encode snRNAs that affect the dynamics of host–pathogen interactions. In this review, we will discuss the latest developments in regards to the role of microRNA in insect host–pathogen interactions and provide some insights into this rapidly developing area of research.
URL [本文引用: 1]
:本研究利用small RNA-seq技术对球囊菌的纯培养进行测序,对球囊菌的microRNAs miRNAs)进行预测、鉴定和分析,进而构建miRNAs-mRNAs的调控网络。利用Illumina Hiseq Xten平台对球囊菌菌丝与孢子进行测序,通过相关生物信息学软件对球囊菌的miRNAs进行预测和分析,通过茎环(Stem-loop)PCR对部分miRNAs进行鉴定,利用Cytoskype软件构建miRNAs-mRNAs的调控网络。本研究共获得48268696条clean reads,预测出118个球囊菌的miRNAs,它们的长度分布介于18–25 nt之间,不同长度的miRNA的首位碱基偏好性差异明显。Stem-loop PCR验证结果显示共有10个miRNAs能够扩增出符合预期的目的片段,说明多数miRNAs可能真实存在。共预测出6529个球囊菌miRNAs的靶基因,其中5725个能够注释到Nr、Swissprot、KOG、GO和KEGG数据库。进一步分析结果显示有24个靶基因注释在MAPK信号通路。Cytoskype软件分析结果显示球囊菌的miRNAs与mRNAs之间存在复杂的调控网络,绝大多数的miRNAs处于调控网络的内部且同时结合多个mRNAs。本研究率先对球囊菌的miRNAs及miRNAs-mRNAs调控网络进行全面分析,研究结果丰富了对球囊菌miRNAs的认识,为其基础生物学信息提供了有益补充,也为阐明球囊菌致病的分子机理打下了一定基础。
URL [本文引用: 1]
:本研究利用small RNA-seq技术对球囊菌的纯培养进行测序,对球囊菌的microRNAs miRNAs)进行预测、鉴定和分析,进而构建miRNAs-mRNAs的调控网络。利用Illumina Hiseq Xten平台对球囊菌菌丝与孢子进行测序,通过相关生物信息学软件对球囊菌的miRNAs进行预测和分析,通过茎环(Stem-loop)PCR对部分miRNAs进行鉴定,利用Cytoskype软件构建miRNAs-mRNAs的调控网络。本研究共获得48268696条clean reads,预测出118个球囊菌的miRNAs,它们的长度分布介于18–25 nt之间,不同长度的miRNA的首位碱基偏好性差异明显。Stem-loop PCR验证结果显示共有10个miRNAs能够扩增出符合预期的目的片段,说明多数miRNAs可能真实存在。共预测出6529个球囊菌miRNAs的靶基因,其中5725个能够注释到Nr、Swissprot、KOG、GO和KEGG数据库。进一步分析结果显示有24个靶基因注释在MAPK信号通路。Cytoskype软件分析结果显示球囊菌的miRNAs与mRNAs之间存在复杂的调控网络,绝大多数的miRNAs处于调控网络的内部且同时结合多个mRNAs。本研究率先对球囊菌的miRNAs及miRNAs-mRNAs调控网络进行全面分析,研究结果丰富了对球囊菌miRNAs的认识,为其基础生物学信息提供了有益补充,也为阐明球囊菌致病的分子机理打下了一定基础。
DOI:10.1093/nar/gkr688URLPMID:21911355 [本文引用: 1]
Abstract microRNAs (miRNAs) are a large class of small non-coding RNAs which post-transcriptionally regulate the expression of a large fraction of all animal genes and are important in a wide range of biological processes. Recent advances in high-throughput sequencing allow miRNA detection at unprecedented sensitivity, but the computational task of accurately identifying the miRNAs in the background of sequenced RNAs remains challenging. For this purpose, we have designed miRDeep2, a substantially improved algorithm which identifies canonical and non-canonical miRNAs such as those derived from transposable elements and informs on high-confidence candidates that are detected in multiple independent samples. Analyzing data from seven animal species representing the major animal clades, miRDeep2 identified miRNAs with an accuracy of 98.6-99.9% and reported hundreds of novel miRNAs. To test the accuracy of miRDeep2, we knocked down the miRNA biogenesis pathway in a human cell line and sequenced small RNAs before and after. The vast majority of the >100 novel miRNAs expressed in this cell line were indeed specifically downregulated, validating most miRDeep2 predictions. Last, a new miRNA expression profiling routine, low time and memory usage and user-friendly interactive graphic output can make miRDeep2 useful to a wide range of researchers.
DOI:10.1016/j.cell.2005.04.004URLPMID:15851028 [本文引用: 1]
Plants and animals use small RNAs (microRNAs [miRNAs] and siRNAs) as guides for posttranscriptional and epigenetic regulation. In plants, miRNAs and trans-acting (ta) siRNAs form through distinct biogenesis pathways, although they both interact with target transcripts and guide cleavage. An integrated approach to identify targets of Arabidopsis thaliana miRNAs and ta-siRNAs revealed several new classes of small RNA-regulated genes, including conventional genes such as Argonaute2 and an E2-ubiquitin conjugating enzyme. Surprisingly, five ta-siRNA-generating transcripts were identified as targets of miR173 or miR390. Rather than functioning as negative regulators, miR173- and miR390-guided cleavage was shown to set the 21-nucleotide phase for ta-siRNA precursor processing. These data support a model in which miRNA-guided formation of a 5′ or 3′ terminus within pre-ta-siRNA transcripts, followed by RDR6-dependent formation of dsRNA and Dicer-like processing, yields phased ta-siRNAs that negatively regulate other genes.
DOI:10.1093/nar/gni178URL [本文引用: 1]
DOI:10.3969/j.issn.1674-0858.2014.02.18URL [本文引用: 1]
蜜蜂白垩病(bee chalkbrood disease)是由蜜蜂球囊菌Ascosphaera apis引起的真菌性传染病,主要感染蜜蜂幼虫。白垩病是养蜂业中主要潜在病害之一,影响着世界养蜂业的发展。本文就目前国内外对蜜蜂球囊菌的研究,分别从该病的发生、分布范围、分类学、流行病学、发病机理、蜜蜂的免疫防御反应、综合防治等方面进行综述,希望为今后开展相关研究工作提供理论依据。
DOI:10.3969/j.issn.1674-0858.2014.02.18URL [本文引用: 1]
蜜蜂白垩病(bee chalkbrood disease)是由蜜蜂球囊菌Ascosphaera apis引起的真菌性传染病,主要感染蜜蜂幼虫。白垩病是养蜂业中主要潜在病害之一,影响着世界养蜂业的发展。本文就目前国内外对蜜蜂球囊菌的研究,分别从该病的发生、分布范围、分类学、流行病学、发病机理、蜜蜂的免疫防御反应、综合防治等方面进行综述,希望为今后开展相关研究工作提供理论依据。
DOI:10.1038/srep14991URLPMID:4598873 [本文引用: 1]
H5N1 influenza A virus (IAV) causes severe respiratory diseases and high mortality rates in animals and humans. MicroRNAs are being increasingly studied to evaluate their potential as therapeutic entities to combat viral infection. However, mechanistic studies delineating the roles of microRNAs in regulating host-H5N1 virus interactions remain scarce. Here, we performed microRNA microarray analysis using A549 human lung epithelial cells infected with a highly pathogenic avian influenza virus. The microRNA expression profile of infected cells identified a small number of microRNAs being dysregulated upon H5N1 influenza A virus infection. Of the differentially expressed microRNAs, miR-136 was up-regulated 5-fold and exhibited potent antiviral activityin vitroagainst H5N1 influenza A virus, as well as vesicular stomatitis virus. On the one hand, 3′-untranslated region (UTR) reporter analysis revealed a miR-136 binding site in the 3′ UTR of IL-6. However, on the other hand, we subsequently determined that miR-136 meanwhile acts as an immune agonist of retinoic acid-inducible gene 1 (RIG-I), thereby causing IL-6 and IFN-β accumulation in A549 cells. Overall, this study implicates the dual role of miRNA-136 in the regulation of host antiviral innate immunity and suggests an important role for the microRNA-activated pathway in viral infection via pattern recognition receptors.
DOI:10.1099/jgv.0.000311URLPMID:26498766 [本文引用: 1]
In order to explore the roles of microRNA(s) [miRNA(s)] in the influenza A virus life cycle, we compared the miRNA profiles of 293T and HeLa cell lines, as influenza A virus can replicate efficiently in 293T cells but only poorly in HeLa cells. We analysed differentially expressed miRNAs and identified five, including miR-33a, that could disturb influenza A virus replication significantly. Using TargetScan analysis, we found that ARCN1 could be a potential target of miR-33a. To confirm whether miR-33a could truly target ARCN1, we generated a luciferase reporter for the ARCN1 3' untranslated region (UTR) and performed a luciferase assay. The data indicated that miR-33a could suppress the luciferase activity of the reporter for the ARCN1 3' UTR but not a reporter in which the predicted miR-33a targeting sites on ARCN1 3' UTR were mutated. We performed immunoblotting to confirm that miR-33a could downregulate the protein level of ARCN1. Consistently, the level of ARCN1 protein in HeLa cells was significantly lower than that in 293T cells. We also demonstrated that ectopic expression of ARCN1 could partially rescue the inhibitory effect of miR-33a on virus replication. Furthermore, we demonstrated that miR-33a could impede virus replication at the stage of virus internalization, which was similar to the pattern for knockdown of ARCN1, indicating that miR-33a inhibits influenza virus infection by suppressing ARCN1 expression. In addition, we found that miR-33a could also weaken the viral ribonucleoprotein activity in an ARCN1-independent manner. In conclusion, we found that miR-33a is a novel inhibitory factor for influenza A virus replication.
[本文引用: 1]
DOI:10.16380/j.kcxb.2017.04.005URL [本文引用: 1]
【目的】本研究旨在通过趋势分析对胁迫意大利蜜蜂Apis mellifera ligustica(简称意蜂)幼虫肠道的球囊菌Ascosphaera apis的差异表达基因(DEGs)进行转录组分析。【方法】将纯化的球囊菌孢子配制为1×107孢子/m L的饲料饲喂意蜂3日龄幼虫,利用Illumina Hi Seq 2500平台对球囊菌胁迫的意蜂幼虫肠道c DNA进行测序,将过滤得到的有效读段(clean reads)映射(mapping)到核糖体数据库及意蜂参考基因组,最后将未映射上的reads映射到本课题组组装并注释的球囊菌参考转录组。利用STEM软件对DEGs进行趋势分析。利用WEGO软件对显著表达模式中的DEGs进行GO富集分析。利用Blastall对显著表达模式中的DEGs进行KEGG代谢通路富集分析。最后,通过对随机选取的6个DEGs进行RT-q PCR分析,对RNA-seq数据进行验证。【结果】球囊菌胁迫意蜂幼虫肠道样品的Illumina测序共得到球囊菌的25 454 076条原始读段(raw reads),经过滤得到24 909 820条clean reads,Q30均在93.46%以上。趋势分析结果显示,19 893个DEGs聚类为8个表达模式,其中有12 151个DEGs聚类为3个表现为显著上调趋势的表达模式。GO富集分析结果显示,表现上调趋势的DEGs富集在40个GO term,富集基因数最多的为细胞进程(cellular process)(2 601 unigenes),其次为代谢进程(metabolic process)(2 553 unigenes)和细胞(cell)(2 522unigenes)。KEGG代谢通路富集分析结果显示,上调趋势中的DEGs富集在119个代谢通路上,其中富集基因数最多的是核糖体(ribosome)(213 unigenes),其次为氨基酸生物合成(biosynthesis of amino acids)(154 unigenes)和内质网蛋白加工(protein processing in endoplasmic reticulum)(130unigenes)。共有48个DEGs富集在MAPK信号通路上,聚类分析结果显示,这些DEGs随着胁迫时间的延长表达水平逐渐增强。RT-q PCR结果显示,6个DEGs的表达水平变化趋势与RNA-seq数据一致,证明
DOI:10.16380/j.kcxb.2017.04.005URL [本文引用: 1]
【目的】本研究旨在通过趋势分析对胁迫意大利蜜蜂Apis mellifera ligustica(简称意蜂)幼虫肠道的球囊菌Ascosphaera apis的差异表达基因(DEGs)进行转录组分析。【方法】将纯化的球囊菌孢子配制为1×107孢子/m L的饲料饲喂意蜂3日龄幼虫,利用Illumina Hi Seq 2500平台对球囊菌胁迫的意蜂幼虫肠道c DNA进行测序,将过滤得到的有效读段(clean reads)映射(mapping)到核糖体数据库及意蜂参考基因组,最后将未映射上的reads映射到本课题组组装并注释的球囊菌参考转录组。利用STEM软件对DEGs进行趋势分析。利用WEGO软件对显著表达模式中的DEGs进行GO富集分析。利用Blastall对显著表达模式中的DEGs进行KEGG代谢通路富集分析。最后,通过对随机选取的6个DEGs进行RT-q PCR分析,对RNA-seq数据进行验证。【结果】球囊菌胁迫意蜂幼虫肠道样品的Illumina测序共得到球囊菌的25 454 076条原始读段(raw reads),经过滤得到24 909 820条clean reads,Q30均在93.46%以上。趋势分析结果显示,19 893个DEGs聚类为8个表达模式,其中有12 151个DEGs聚类为3个表现为显著上调趋势的表达模式。GO富集分析结果显示,表现上调趋势的DEGs富集在40个GO term,富集基因数最多的为细胞进程(cellular process)(2 601 unigenes),其次为代谢进程(metabolic process)(2 553 unigenes)和细胞(cell)(2 522unigenes)。KEGG代谢通路富集分析结果显示,上调趋势中的DEGs富集在119个代谢通路上,其中富集基因数最多的是核糖体(ribosome)(213 unigenes),其次为氨基酸生物合成(biosynthesis of amino acids)(154 unigenes)和内质网蛋白加工(protein processing in endoplasmic reticulum)(130unigenes)。共有48个DEGs富集在MAPK信号通路上,聚类分析结果显示,这些DEGs随着胁迫时间的延长表达水平逐渐增强。RT-q PCR结果显示,6个DEGs的表达水平变化趋势与RNA-seq数据一致,证明
DOI:10.13343/j.cnki.wsxb.20160551URL [本文引用: 1]
【目的】本研究利用RNA-seq技术对球囊菌胁迫的中华蜜蜂(中蜂)幼虫肠道进行深度测序,经趋势分析得到差异表达基因(DEGs)的显著表达模式,进而对胁迫过程中的球囊菌进行转录组学分析。【方法】利用Illumina HiSeq 2500平台对球囊菌胁迫的中蜂幼虫肠道进行深度测序,并利用相关软件进行了深入分析。最后,通过RT-qPCR对RNA-seq数据进行了验证。【结果】本研究共得到球囊菌的41133932条高质量clean reads。22865个DEGs共聚类为8个基因表达模式,其中,16769个DEGs聚类为2个显著上调趋势与2个显著下调趋势。GO富集分析结果显示,显著上调与显著下调趋势中的DEGs分别富集于40与37个GO term,基因富集数最多的为细胞进程(2486 unigenes)。KEGG代谢通路(pathway)富集分析结果显示,显著上调与显著下调趋势中的DEGs分别富集于119和112个pathway,基因富集数最多的分别是氨基酸生物合成(127 unigenes)与核糖体(98 unigenes)。进一步分析表明球囊菌在胁迫中蜂幼虫肠道的过程中通过提高物质合成促进其增殖,而宿主通过抑制球囊菌的蛋白合成抵御病原入侵。富集在MAPK信号通路的11个DEGs的表达水平随着胁迫时间的延长而逐渐下降,推测中蜂幼虫通过抑制该通路而阻遏球囊菌增殖。【结论】本研究不仅为揭示白垩病过程中的球囊菌-中蜂幼虫互作提供了重要信息,也为阐明不同抗性蜂种的球囊菌抗性差异奠定了基础。
DOI:10.13343/j.cnki.wsxb.20160551URL [本文引用: 1]
【目的】本研究利用RNA-seq技术对球囊菌胁迫的中华蜜蜂(中蜂)幼虫肠道进行深度测序,经趋势分析得到差异表达基因(DEGs)的显著表达模式,进而对胁迫过程中的球囊菌进行转录组学分析。【方法】利用Illumina HiSeq 2500平台对球囊菌胁迫的中蜂幼虫肠道进行深度测序,并利用相关软件进行了深入分析。最后,通过RT-qPCR对RNA-seq数据进行了验证。【结果】本研究共得到球囊菌的41133932条高质量clean reads。22865个DEGs共聚类为8个基因表达模式,其中,16769个DEGs聚类为2个显著上调趋势与2个显著下调趋势。GO富集分析结果显示,显著上调与显著下调趋势中的DEGs分别富集于40与37个GO term,基因富集数最多的为细胞进程(2486 unigenes)。KEGG代谢通路(pathway)富集分析结果显示,显著上调与显著下调趋势中的DEGs分别富集于119和112个pathway,基因富集数最多的分别是氨基酸生物合成(127 unigenes)与核糖体(98 unigenes)。进一步分析表明球囊菌在胁迫中蜂幼虫肠道的过程中通过提高物质合成促进其增殖,而宿主通过抑制球囊菌的蛋白合成抵御病原入侵。富集在MAPK信号通路的11个DEGs的表达水平随着胁迫时间的延长而逐渐下降,推测中蜂幼虫通过抑制该通路而阻遏球囊菌增殖。【结论】本研究不仅为揭示白垩病过程中的球囊菌-中蜂幼虫互作提供了重要信息,也为阐明不同抗性蜂种的球囊菌抗性差异奠定了基础。
[本文引用: 1]
DOI:10.1016/j.ceb.2012.12.006URLPMID:23312716 [本文引用: 1]
Tissue regeneration is vital to the form and function of an organ. At the core of an organs ability to self-renew is the stem cell, which maintains homeostasis, and repopulates injured or aged tissue. Tissue damage can dramatically change the dimensions of an organ, and during regeneration, an organ must halt growth once the original tissue dimensions have been restored. Therefore, stem cells must give rise to the appropriate number of differentiated progeny to achieve homeostasis. How this tissue-size checkpoint is regulated and how tissue size information relayed to stem cell compartments is unclear, however, it is likely that these mechanisms are altered during the course of tumorigenesis. An emerging signaling cascade, the Hippo Signaling Pathway, is a broadly conserved potent organ size regulator [1]. However, this pathway does not act alone. A number of examples demonstrate crosstalk between Hippo and other signaling pathways including Wnt, Tgf and Notch, with implications for stem cell biology. Here, we focus on these interactions primarily in the context of well characterized stem cell populations.
URL [本文引用: 1]
【目的】长链非编码RNA(lncRNA)在真核生物的基因表达、表观遗传和细胞周期调控等方面发挥重要功能。本研究旨在探究意大利蜜蜂(Apis mellifera ligustica,简称意蜂)工蜂中肠发育过程中lncRNA的表达谱及其作用。【方法】利用RNA-seq技术和链特异性建库方法对意蜂7和10日龄工蜂中肠(Am7、Am10)进行深度测序,下机的原始数据经过Perl脚本过滤得到高质量有效读段。利用bowtie工具将有效读段比对核糖体数据库,进一步利用Top Hat2软件将未比对到核糖体数据库上的数据比对到参考基因组。利用CPC和CNCI软件对转录本的编码能力进行预测。通过RT-PCR对部分lncRNA进行鉴定。利用edgeR软件进行差异表达lncRNA(DElncRNA)分析,进而预测lncRNA的上下游基因,并对上下游基因进行GO及KEGG代谢通路富集分析。联用RNAhybrid、Miranda和Target Scan软件预测DElncRNA靶向结合的mi RNA及mi RNA靶向结合的靶基因,并通过Cytoscape软件构建DElncRNAs-mi RNAs-m RNAs的调控网络。最后,通过RT-qPCR验证测序数据的可靠性。【结果】Am7和Am10的深度测序分别获得134 802 058和147 051 470条原始读段,经严格过滤分别得到134 166 157和146 293 288条有效读段;共得到3 890个DElncRNA,包括2 005个上调lncRNA与1 885个下调lncRNA。RT-PCR验证结果显示共有8个lncRNA能扩增出符合预期的目的片段,表明预测出的lncRNA真实存在。DElncRNA的上下游基因数为1 793个,它们涉及42个GO条目,包括代谢进程、发育进程、细胞进程、应激反应和免疫系统进程等;这些上下游基因还涉及251条代谢通路,包括碳代谢、嘌呤代谢和脂肪酸的生物合成等物质代谢通路,硫代谢、甲烷代谢和氧化磷酸化等能量代谢通路,Hippo信号通路、Wnt信号通路和Notch信号通路等信号通路,溶酶体、内吞作用和泛素介导的蛋白水解等细胞免疫通路,以及MAPK信号通路、Jak-STAT信号通路和NF-kappa B信号通路等体液免疫通路,上述结果表明DElncRNA在意蜂中肠发育过程中参与物质和能量代谢、细胞生命活动和免疫调控。进一步分析发现TCONS_00020918可通过调控西方蜜蜂王浆主蛋白1编码基因在意蜂工蜂中肠的营养吸收、级型分化中发挥功能。DElncRNA的调控网络分析结果显示DElncRNA与mi RNA、m RNA间存在复杂的调控关系,部分DElncRNA处于调控网络的中心位置且能靶向结合较多的mi RNA,也有部分mi RNA可被多个DElncRNA共同靶向,表明这些DElncRNA可能在中肠发育中发挥重要作用。随机挑取5个DElncRNA进行RT-qPCR验证,结果显示它们的表达量变化趋势与测序结果一致,证实了本研究测序数据的可靠性。【结论】差异表达长链非编码RNA(DElncRNA)广泛参与意蜂工蜂中肠的新陈代谢、细胞活动和免疫调控并作为竞争性内源RNA(ce RNA)发挥作用,研究结果为关键lncRNA的筛选和功能研究提供了必要的数据支持。
URL [本文引用: 1]
【目的】长链非编码RNA(lncRNA)在真核生物的基因表达、表观遗传和细胞周期调控等方面发挥重要功能。本研究旨在探究意大利蜜蜂(Apis mellifera ligustica,简称意蜂)工蜂中肠发育过程中lncRNA的表达谱及其作用。【方法】利用RNA-seq技术和链特异性建库方法对意蜂7和10日龄工蜂中肠(Am7、Am10)进行深度测序,下机的原始数据经过Perl脚本过滤得到高质量有效读段。利用bowtie工具将有效读段比对核糖体数据库,进一步利用Top Hat2软件将未比对到核糖体数据库上的数据比对到参考基因组。利用CPC和CNCI软件对转录本的编码能力进行预测。通过RT-PCR对部分lncRNA进行鉴定。利用edgeR软件进行差异表达lncRNA(DElncRNA)分析,进而预测lncRNA的上下游基因,并对上下游基因进行GO及KEGG代谢通路富集分析。联用RNAhybrid、Miranda和Target Scan软件预测DElncRNA靶向结合的mi RNA及mi RNA靶向结合的靶基因,并通过Cytoscape软件构建DElncRNAs-mi RNAs-m RNAs的调控网络。最后,通过RT-qPCR验证测序数据的可靠性。【结果】Am7和Am10的深度测序分别获得134 802 058和147 051 470条原始读段,经严格过滤分别得到134 166 157和146 293 288条有效读段;共得到3 890个DElncRNA,包括2 005个上调lncRNA与1 885个下调lncRNA。RT-PCR验证结果显示共有8个lncRNA能扩增出符合预期的目的片段,表明预测出的lncRNA真实存在。DElncRNA的上下游基因数为1 793个,它们涉及42个GO条目,包括代谢进程、发育进程、细胞进程、应激反应和免疫系统进程等;这些上下游基因还涉及251条代谢通路,包括碳代谢、嘌呤代谢和脂肪酸的生物合成等物质代谢通路,硫代谢、甲烷代谢和氧化磷酸化等能量代谢通路,Hippo信号通路、Wnt信号通路和Notch信号通路等信号通路,溶酶体、内吞作用和泛素介导的蛋白水解等细胞免疫通路,以及MAPK信号通路、Jak-STAT信号通路和NF-kappa B信号通路等体液免疫通路,上述结果表明DElncRNA在意蜂中肠发育过程中参与物质和能量代谢、细胞生命活动和免疫调控。进一步分析发现TCONS_00020918可通过调控西方蜜蜂王浆主蛋白1编码基因在意蜂工蜂中肠的营养吸收、级型分化中发挥功能。DElncRNA的调控网络分析结果显示DElncRNA与mi RNA、m RNA间存在复杂的调控关系,部分DElncRNA处于调控网络的中心位置且能靶向结合较多的mi RNA,也有部分mi RNA可被多个DElncRNA共同靶向,表明这些DElncRNA可能在中肠发育中发挥重要作用。随机挑取5个DElncRNA进行RT-qPCR验证,结果显示它们的表达量变化趋势与测序结果一致,证实了本研究测序数据的可靠性。【结论】差异表达长链非编码RNA(DElncRNA)广泛参与意蜂工蜂中肠的新陈代谢、细胞活动和免疫调控并作为竞争性内源RNA(ce RNA)发挥作用,研究结果为关键lncRNA的筛选和功能研究提供了必要的数据支持。
URL [本文引用: 1]
【目的】环状RNA(circular RNA,circRNA)在可变剪接、转录调控和来源基因的表达调控等方面具有重要功能。本研究旨在探究意大利蜜蜂(Apis mellifera ligustica,简称意蜂)工蜂中肠发育过程中circRNA的表达谱及其发育过程中的差异表达circRNA(differentially expressed circRNA,DEcircRNA),进而在转录组水平探究DEcircRNA在中肠发育中的作用。【方法】基于前期获得的意蜂7和10日龄工蜂中肠样品(Am7和Am10)的全转录组数据,利用find_circ软件从质控后的数据中预测circRNA。采用RPM算法归一化处理得到circ RNA的表达量。利用DEGseq软件对circ RNA进行差异分析,按照差异倍数(fold change)≥2、P0.05及错误发现率(false discovery rate,FDR)0.05条件筛选DEcircRNA。通过BLAST比对GO和KEGG数据库,对DEcircRNA的来源基因进行功能和代谢通路注释。利用TargetFinder软件预测DEcircRNA-miRNA及DEcircRNA-miRNA-mRNA调控网络,通过Cytoscape v.3.2.1软件对调控网络进行可视化。通过实时荧光定量PCR(RT-qPCR)验证测序数据的可靠性。【结果】意蜂中肠各样品比对上参考基因组的短序列读段数平均为19 616 356条。Am7与Am10的组内Pearson相关系数均≥0.950。共预测出256个DEcircRNA,包括105个上调circRNA和151个下调circRNA。Novel_circ_009675和novel_circ_013879分别在Am7和Am10中高量表达。DEcircRNA的来源基因可注释到包括结合、单组织进程及细胞进程在内的32个GO条目,其中分别有35、35和7个来源基因注释到催化活性、代谢进程和应激反应。上述来源基因还可注释到35条KEGG代谢通路,其中分别有5、5和4个来源基因注释到Hippo信号通路、内吞作用和吞噬体;进一步分析发现分别有1、2和2个来源基因注释到磷酸肌醇代谢、淀粉和蔗糖代谢和半乳糖代谢等物质代谢通路,5、4、3、1和1个来源基因注释到内吞作用、吞噬体、溶酶体、泛素介导的蛋白水解和MAPK信号通路等免疫通路。上述结果表明相应的DEcircRNA广泛参与意蜂工蜂中肠的生长发育、新陈代谢和免疫防御。DEcircRNA-miRNA调控网络分析结果显示,141个DEcirc RNA可靶向结合107个mi RNA,其中多数仅能结合1—2个mi RNA,但novel_circ_011577和novel_circ_010719结合的靶miRNA数可达32和28个;此外,mir-136-y、ame-miR-6001-3p及mir-136-y结合的circRNA数最多,分别为15、14和14个,表明相应的DEcircRNA可作为竞争性内源RNA在意蜂中肠发育过程发挥作用。进一步构建DEcircRNAs-ame-miR-6001-3p-mRNA调控网络,分析结果显示14个DEcircRNA可共同靶向结合ame-miR-6001-3p,表明它们可能通过调控ame-miR-6001-3p对意蜂中肠干细胞的分裂和分化进行间接调控。随机选取6个DEcircRNA进行RT-qPCR验证,结果显示5个DEcircRNA的表达量变化趋势与测序结果一致,证实了本研究测序结果的可靠性。【结论】通过对意蜂工蜂中肠发育过程中的DEcircRNA深入分析,提供了circRNA在意蜂工蜂中肠发育过程中的表达谱和差异表达信息,揭示了DEcircRNA在中肠发育过程中的作用,为中肠发育相关的关键circRNA的筛选和功能研究打下了基础。
URL [本文引用: 1]
【目的】环状RNA(circular RNA,circRNA)在可变剪接、转录调控和来源基因的表达调控等方面具有重要功能。本研究旨在探究意大利蜜蜂(Apis mellifera ligustica,简称意蜂)工蜂中肠发育过程中circRNA的表达谱及其发育过程中的差异表达circRNA(differentially expressed circRNA,DEcircRNA),进而在转录组水平探究DEcircRNA在中肠发育中的作用。【方法】基于前期获得的意蜂7和10日龄工蜂中肠样品(Am7和Am10)的全转录组数据,利用find_circ软件从质控后的数据中预测circRNA。采用RPM算法归一化处理得到circ RNA的表达量。利用DEGseq软件对circ RNA进行差异分析,按照差异倍数(fold change)≥2、P0.05及错误发现率(false discovery rate,FDR)0.05条件筛选DEcircRNA。通过BLAST比对GO和KEGG数据库,对DEcircRNA的来源基因进行功能和代谢通路注释。利用TargetFinder软件预测DEcircRNA-miRNA及DEcircRNA-miRNA-mRNA调控网络,通过Cytoscape v.3.2.1软件对调控网络进行可视化。通过实时荧光定量PCR(RT-qPCR)验证测序数据的可靠性。【结果】意蜂中肠各样品比对上参考基因组的短序列读段数平均为19 616 356条。Am7与Am10的组内Pearson相关系数均≥0.950。共预测出256个DEcircRNA,包括105个上调circRNA和151个下调circRNA。Novel_circ_009675和novel_circ_013879分别在Am7和Am10中高量表达。DEcircRNA的来源基因可注释到包括结合、单组织进程及细胞进程在内的32个GO条目,其中分别有35、35和7个来源基因注释到催化活性、代谢进程和应激反应。上述来源基因还可注释到35条KEGG代谢通路,其中分别有5、5和4个来源基因注释到Hippo信号通路、内吞作用和吞噬体;进一步分析发现分别有1、2和2个来源基因注释到磷酸肌醇代谢、淀粉和蔗糖代谢和半乳糖代谢等物质代谢通路,5、4、3、1和1个来源基因注释到内吞作用、吞噬体、溶酶体、泛素介导的蛋白水解和MAPK信号通路等免疫通路。上述结果表明相应的DEcircRNA广泛参与意蜂工蜂中肠的生长发育、新陈代谢和免疫防御。DEcircRNA-miRNA调控网络分析结果显示,141个DEcirc RNA可靶向结合107个mi RNA,其中多数仅能结合1—2个mi RNA,但novel_circ_011577和novel_circ_010719结合的靶miRNA数可达32和28个;此外,mir-136-y、ame-miR-6001-3p及mir-136-y结合的circRNA数最多,分别为15、14和14个,表明相应的DEcircRNA可作为竞争性内源RNA在意蜂中肠发育过程发挥作用。进一步构建DEcircRNAs-ame-miR-6001-3p-mRNA调控网络,分析结果显示14个DEcircRNA可共同靶向结合ame-miR-6001-3p,表明它们可能通过调控ame-miR-6001-3p对意蜂中肠干细胞的分裂和分化进行间接调控。随机选取6个DEcircRNA进行RT-qPCR验证,结果显示5个DEcircRNA的表达量变化趋势与测序结果一致,证实了本研究测序结果的可靠性。【结论】通过对意蜂工蜂中肠发育过程中的DEcircRNA深入分析,提供了circRNA在意蜂工蜂中肠发育过程中的表达谱和差异表达信息,揭示了DEcircRNA在中肠发育过程中的作用,为中肠发育相关的关键circRNA的筛选和功能研究打下了基础。
DOI:10.1038/srep18794URLPMID:4704047 [本文引用: 1]
The cellular mechanisms employed by some organisms to produce contrasting morphological and reproductive phenotypes from the same genome remains one of the key unresolved issues in biology. Honeybees (Apis mellifera) use differential feeding and a haplodiploid sex determination system to generate three distinct organismal outcomes from the same genome. Here we investigate the honeybee female and male caste-specific microRNA and transcriptomic molecular signatures during a critical time of larval development. Both previously undetected and novel miRNAs have been discovered, expanding the inventory of these genomic regulators in invertebrates. We show significant differences in the microRNA and transcriptional profiles of diploid females relative to haploid drone males as well as between reproductively distinct females (queens and workers). Queens and drones show gene enrichment in physio-metabolic pathways, whereas workers show enrichment in processes associated with neuronal development, cell signalling and caste biased structural differences. Interestingly, predicted miRNA targets are primarily associated with non-physio-metabolic genes, especially neuronal targets, suggesting a mechanistic disjunction from DNA methylation that regulates physio-metabolic processes. Accordingly, miRNA targets are under-represented in methylated genes. Our data show how a common set of genetic elements are differentially harnessed by an organism, which may provide the remarkable level of developmental flexibility required.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.4161/cc.19618URLPMID:22421157 [本文引用: 1]
Though tumor suppressor p53 and the canonical Wnt cascade have been extensively studied for the last 30 years, due to their important physiological roles, the two signaling pathways have been largely considered independent. Recently, the miR-34 family was found to directly link p53 and Wnt, revealing the tight connection between loss of tumor suppressor function and activation of oncogenic signaling. These observations demonstrate that miR-34, known to be directly downstream of p53, targets a set of highly conserved sites in the UTR of Wnt and EMT genes, specifically WNT1, WNT3, LRP6, AXIN2, -catenin, LEF1 and Snail, resulting in suppression of TCF/LEF transcriptional activity and the EMT program. The loss of p53 function increases Wnt activities and promotes the Snail-dependent EMT program at multiple levels in a miR-34/UTR-specific manner. The TCF/LEF transcriptional signature was closely associated with functionality of p53 and miR-34 in clinical samples, suggesting the pervasive impact of miR-34 loss on the oncogenic pathway in human cancer. Here, we review recent findings on ceRNA in light of novel data to elucidate the physiological relevance of the p53-miR-34-Wnt network, which encompasses sets of genes and directions of signaling. As loss of wt-p53 or hyperactivation of Wnt is critical in maintaining cancer stem cell properties and in establishing the metastatic program, these observations indicate a mechanism of miR-mediated quasi-sufficiency which connects tumor suppressor and oncogenic signaling pathways, supporting a continuum model of human cancer.
DOI:10.1126/scisignal.2001744URLPMID:3447368 [本文引用: 1]
Abstract Although loss of p53 function and activation of canonical Wnt signaling cascades are frequently coupled in cancer, the links between these two pathways remain unclear. We report that p53 transactivated microRNA-34 (miR-34), which consequently suppressed the transcriptional activity of 0205-catenin-T cell factor and lymphoid enhancer factor (TCF/LEF) complexes by targeting the untranslated regions (UTRs) of a set of conserved targets in a network of genes encoding elements of the Wnt pathway. Loss of p53 function increased canonical Wnt signaling by alleviating miR-34-specific interactions with target UTRs, and miR-34 depletion relieved p53-mediated Wnt repression. Gene expression signatures reflecting the status of 0205-catenin-TCF/LEF transcriptional activity in breast cancer and pediatric neuroblastoma patients were correlated with p53 and miR-34 functional status. Loss of p53 or miR-34 contributed to neoplastic progression by triggering the Wnt-dependent, tissue-invasive activity of colorectal cancer cells. Further, during development, miR-34 interactions with the 0205-catenin UTR affected Xenopus body axis polarity and the expression of Wnt-dependent patterning genes. These data provide insight into the mechanisms by which a p53-miR-34 network restrains canonical Wnt signaling cascades in developing organisms and human cancer.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1371/journal.ppat.1003416URLPMID:3681753 [本文引用: 1]
To identify new regulators of antiviral innate immunity, we completed the first genome-wide gene silencing screen assessing the transcriptional response at the interferon-?? (IFNB1) promoter following Sendai virus (SeV) infection. We now report a novel link between WNT signaling pathway and the modulation of retinoic acid-inducible gene I (RIG-I)-like receptor (RLR)-dependent innate immune responses. Here we show that secretion of WNT2B and WNT9B and stabilization of ??-catenin (CTNNB1) upon virus infection negatively regulate expression of representative inducible genes IFNB1, IFIT1 and TNF in a CTNNB1-dependent effector mechanism. The antiviral response is drastically reduced by glycogen synthase kinase 3 (GSK3) inhibitors but restored in CTNNB1 knockdown cells. The findings confirm a novel regulation of antiviral innate immunity by a canonical-like WNT/CTNNB1 signaling pathway. The study identifies novel avenues for broad-spectrum antiviral targets and preventing immune-mediated diseases upon viral infection.
DOI:10.1111/j.1365-2230.2012.04362.xURLPMID:22607321 [本文引用: 1]
SummaryPlasmacytoid dendritic cells (pDCs) fulfil multiple roles in immunity, and can secrete large amounts of interferon (IFN)-. However, the available evidence suggests that they may actually counteract efficient antitumour immunity. Thus in melanoma, pDCs are abundant, but they are anergic and deficient in IFN- secretion. pDC anergy is thought to be caused by immunosuppressive factors secreted by melanoma cells. One factor strongly expressed by melanoma is Wnt5a, which is implicated in cancer tissue invasion. In this paper, we show that Wnt5a is able to block the upregulation of the activation markers CD80 and CD86 on naive human pDCs stimulated by CpG oligodeoxynucleotide, and CpG-triggered secretion of IFN- by pDCs. Our results suggest that Wnt5a may not only initiate cancer invasion, but could also regulate activation of pDC. These data provide a clear rationale to investigate a role for Wnt5a in immune regulation.
DOI:10.1128/JVI.02388-16URLPMID:28148804 [本文引用: 1]
Abstract The impact of mosquito-borne flavivirus infections worldwide is significant, and many critical aspects of these viruses' biology, including virus-host interactions, host cell requirements for replication, and how virus-host interactions impact pathology, remain to be fully understood. The recent reemergence and spread of flaviviruses, including dengue virus (DENV), West Nile virus (WNV), and Zika virus (ZIKV), highlight the importance of performing basic research on this important group of pathogens. MicroRNAs (miRNAs) are small, noncoding RNAs that modulate gene expression posttranscriptionally and have been demonstrated to regulate a broad range of cellular processes. Our research is focused on identifying pro- and antiflaviviral miRNAs as a means of characterizing cellular pathways that support or limit viral replication. We have screened a library of known human miRNA mimics for their effect on the replication of three flaviviruses, DENV, WNV, and Japanese encephalitis virus (JEV), using a high-content immunofluorescence screen. Several families of miRNAs were identified as inhibiting multiple flaviviruses, including the miRNA miR-34, miR-15, and miR-517 families. Members of the miR-34 family, which have been extensively characterized for their ability to repress Wnt/ -catenin signaling, demonstrated strong antiflaviviral effects, and this inhibitory activity extended to other viruses, including ZIKV, alphaviruses, and herpesviruses. Previous research suggested a possible link between the Wnt and type I interferon (IFN) signaling pathways. Therefore, we investigated the role of type I IFN induction in the antiviral effects of the miR-34 family and confirmed that these miRNAs potentiate interferon regulatory factor 3 (IRF3) phosphorylation and translocation to the nucleus, the induction of IFN-responsive genes, and the release of type I IFN from transfected cells. We further demonstrate that the intersection between the Wnt and IFN signaling pathways occurs at the point of glycogen synthase kinase 3 (GSK3 )-TANK-binding kinase 1 (TBK1) binding, inducing TBK1 to phosphorylate IRF3 and initiate downstream IFN signaling. In this way, we have identified a novel cellular signaling network with a critical role in regulating the replication of multiple virus families. These findings highlight the opportunities for using miRNAs as tools to discover and characterize unique cellular factors involved in supporting or limiting virus replication, opening up new avenues for antiviral research. IMPORTANCE MicroRNAs are a class of small regulatory RNAs that modulate cellular processes through the posttranscriptional repression of multiple transcripts. We hypothesized that individual miRNAs may be capable of inhibiting viral replication through their effects on host proteins or pathways. To test this, we performed a high-content screen for miRNAs that inhibit the replication of three medically relevant members of the flavivirus family: West Nile virus, Japanese encephalitis virus, and dengue virus 2. The results of this screen identify multiple miRNAs that inhibit one or more of these viruses. Extensive follow-up on members of the miR-34 family of miRNAs, which are active against all three viruses as well as the closely related Zika virus, demonstrated that miR-34 functions through increasing the infected cell's ability to respond to infection through the interferon-based innate immune pathway. Our results not only add to the knowledge of how viruses interact with cellular pathways but also provide a basis for more extensive data mining by providing a comprehensive list of miRNAs capable of inhibiting flavivirus replication. Finally, the miRNAs themselves or cellular pathways identified as modulating virus infection may prove to be novel candidates for the development of therapeutic interventions. Copyright 2017 American Society for Microbiology.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1038/nrg1379URLPMID:15211354 [本文引用: 1]
Figure 2 | The current model for the biogenesis and post-transcriptional suppression of microRNAs and small interfering RNAs. The nascent pri-microRNA (pri-miRNA) transcripts are first processed into ~70-nucleotide pre-miRNAs by Drosha inside the nucleus. Pre-miRNAs are transported to the cytoplasm by Exportin 5 and are processed into miRNA:miRNA* duplexes by Dicer. Dicer also processes long dsRNA molecules into small interfering RNA (siRNA)duplexes. Only one strand of the miRNA:miRNA* duplex or the siRNA duplex is preferentiallyassembled into the RNA-induced silencing complex (RISC) , which subsequently acts on itstarget by translational repression or mRNA cleavage, depending, at least in part, on the level of complementarity between the small RNA and its target. ORF, open reading frame.
[本文引用: 1]
[本文引用: 1]
.
DOI:10.1074/mcp.RA117.000432URL [本文引用: 1]
Transmissible gastroenteritis virus (TGEV), a member of the coronaviridae family, could cause fatal diarrhea of piglets and result in numerous economic losses. Previous studies demonstrated that TGEV infection could lead to mitochondrial damage and upregulate miR-4331 level. So miR-4331 may play an important regulatory role in the control of mitochondrial function. To explore the potential role of miR-4331 in mitochondrial damage, we adopted a strategy consisting of quantitative proteomic analysis of porcine kidney (PK-15) cells in response to miR-4331 and TGEV infection. Eventually, 69 differentially expressed proteins were gained. The target of miR-4331 was identified. The effects of miR-4331 and its target RB1 on mitochondrial Ca2+level, mitochondrial membrane potential (MMP), interleukin-1 receptor accessory protein (IL1RAP), p38 MAPK signaling pathway were investigated. The results showed that miR-4331 elevated mitochondrial Ca2+level, reduced MMP, targets Retinoblastoma 1 (RB1), upregulated IL1RAP, and induced activation of p38 MAPK pathway during TGEV infection. RB1 was identified as the direct targets of miR-4331 and downregulated IL1RAP, suppressed the activation of p38 MPAK, and attenuated TGEV-induced mitochondrial damage. In addition, IL1RAP played a positive role in activating p38 MAPK signaling and negative role in TGEV-induced mitochondrial damage. The data indicate that miR-4331 aggravates TGEV-induced mitochondrial damage by repressing expression of RB1, promoting IL1RAP, and activating p38 MAPK pathway.
DOI:10.1186/1471-2164-13-278URLPMID:3496578 [本文引用: 1]
Avian influenza virus (AIV) outbreaks are worldwide threats to both poultry and humans. Our previous study suggested microRNAs (miRNAs) play significant roles in the regulation of host response to AIV infection in layer chickens. The objective of this study was to test the hypothesis if genetic background play essential role in the miRNA regulation of AIV infection in chickens and if miRNAs that were differentially expressed in layer with AIV infection would be modulated the same way in broiler chickens. Furthermore, by integrating with parallel mRNA expression profiling, potential molecular mechanisms of host response to AIV infection can be further exploited. Total RNA isolated from the lungs of non-infected and low pathogenic H5N3 infected broilers at four days post-infection were used for both miRNA deep sequencing and mRNA microarray analyses. A total of 2.6 M and 3.3 M filtered high quality reads were obtained from infected and non-infected chickens by Solexa GA-I Sequencer, respectively. A total of 271 miRNAs in miRBase 16.0 were identified and one potential novel miRNA was discovered. There were 121 miRNAs differentially expressed at the 5% false discovery rate by Fisher exact test. More miRNAs were highly expressed in infected lungs (108) than in non-infected lungs (13), which was opposite to the findings in layer chickens. This result suggested that a different regulatory mechanism of host response to AIV infection mediated by miRNAs might exist in broiler chickens. Analysis using the chicken 44 K Agilent microarray indicated that 508 mRNAs (347 down-regulated) were differentially expressed following AIV infection. A comprehensive analysis combining both miRNA and targeted mRNA gene expression suggests that gga-miR-34a, 122 1, 122 2, 146a, 155, 206, 1719, 1594, 1599 and 451, and MX1, IL-8, IRF-7, TNFRS19 are strong candidate miRNAs or genes involved in regulating the host response to AIV infection in the lungs of broiler chickens. Further miRNA or gene specific knock-down assay is warranted to elucidate underlying mechanism of AIV infection regulation in the chicken.
DOI:10.1371/journal.pone.0076811URLPMID:24116168 [本文引用: 1]
Changes in microRNA expression have been detectedin vitroin influenza infected cells, yet little is known about them in patients. microRNA profiling was performed on whole blood of H1N1 patients to identify signature microRNAs to better understand the gene regulation involved and possibly improve diagnosis. Total RNA extracted from blood samples of influenza infected patients and healthy controls were subjected to microRNA microarray. Expression profiles of circulating microRNAs were altered and distinctly different in influenza patients. Expression of highly dysregulated microRNAs were validated using quantitative PCR. Fourteen highly dysregulated miRNAs, identified from the blood of influenza infected patients, provided a clear distinction between infected and healthy individuals. Of these, expression of miR-1260, -26a, -335*, -576-3p, -628-3p and -664 were consistently dysregulated in both whole blood and H1N1 infected cells. Potential host and viral gene targets were identified and the impact of microRNA dysregulation on the host proteome was studied. Consequences of their altered expression were extrapolated to changes in the host proteome expression. These highly dysregulated microRNAs may have crucial roles in influenza pathogenesis and are potential biomarkers of influenza.
DOI:10.3390/ijms18040749URLPMID:28368362 [本文引用: 1]
The prevalence of swine pandemic H1N1/2009 influenza A virus (SIV-H1N1/2009) in pigs has the potential to generate novel reassortant viruses, posing a great threat to human health. Cellular microRNAs (miRNAs) have been proven as promising small molecules for regulating influenza A virus replication by directly targeting viral genomic RNA. In this study, we predicted potentialSus scrofa(ssc-, swine) miRNAs targeting the genomic RNA of SIV-H1N1/2009 by RegRNA 2.0, and identified ssc-miR-204 and ssc-miR-4331 to target viral HA and NS respectively through dual-luciferase reporter assays. The messenger RNA (mRNA) levels of viral HA and NS were significantly suppressed when newborn pig trachea (NPTr) cells respectively overexpressed ssc-miR-204 and ssc-miR-4331 and were infected with SIV-H1N1/2009, whereas the suppression effect could be restored when respectively decreasing endogenous ssc-miR-204 and ssc-miR-4331 with inhibitors. Because of the importance of viral HA and NS in the life cycle of influenza A virus, ssc-miR-204 and ssc-miR-4331 exhibited an inhibition effect on SIV-H1N1/2009 replication. The antiviral effect was sequence-specific of SIV-H1N1/2009, for the target sites in HA and NS of H5N1 or H9N2 influenza A virus were not conserved. Furthermore, SIV-H1N1/2009 infection reversely downregulated the expression of ssc-miR-204 and ssc-miR-4331, which might facilitate the virus replication in the host. In summary, this work will provide us some important clues for controlling the prevalence of SIV-H1N1/2009 in pig populations.
DOI:10.1002/arch.1041URLPMID:11376457 [本文引用: 1]
Peritrophic membranes (PMs) are an invertebrate-unique structure that lines the digestive tract, playing important roles in facilitating food digestion and providing protection to the gut epithelium. The importance of PMs in insects has been recognized ever since its presence was identified 200 years ago. In the last 5 years, significant progress towards understanding the PM molecular structure and the mechanism for PM formation has been made. Recent studies on Type 1 PMs from lepidopteran larvae have suggested a model for the PM molecular structure and formation. The important physiological functions of the PM suggest that PMs can be a significant structural target for insect control and the current understanding of the structure of lepidopteran larval PMs has provided us with potential opportunities for targeting the PM by various mechanisms. Arch. Insect Biochem. Physiol. 47:110-118, 2001. ? 2001 Wiley-Liss, Inc.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}