 ,, 陈华枝,, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 杜宇, 王海朋, 耿四海, 周丁丁, 刘思亚, 陈大福,福建农林大学蜂学学院,福州 350002
,, 陈华枝,, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 杜宇, 王海朋, 耿四海, 周丁丁, 刘思亚, 陈大福,福建农林大学蜂学学院,福州 350002Analysis of Differentially Expressed Circular RNAs and Their Regulation Networks During the Developmental Process of Apis mellifera ligustica Worker’s Midgut
GUO Rui,, CHEN HuaZhi,, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, XU GuoJun, DU Yu, WANG HaiPeng, GENG SiHai, ZHOU DingDing, LIU SiYa, CHEN DaFu,College of Bee Science, Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
第一联系人:
责任编辑: 岳梅
收稿日期:2018-07-16接受日期:2018-09-10网络出版日期:2018-12-01
| 基金资助: |
Received:2018-07-16Accepted:2018-09-10Online:2018-12-01

摘要
关键词:
Abstract
Keywords:
PDF (3399KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郭睿, 陈华枝, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 杜宇, 王海朋, 耿四海, 周丁丁, 刘思亚, 陈大福. 意大利蜜蜂工蜂中肠发育过程中的差异表达环状RNA及其调控网络分析[J]. 中国农业科学, 2018, 51(23): 4575-4590 doi:10.3864/j.issn.0578-1752.2018.23.015
GUO Rui, CHEN HuaZhi, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, XU GuoJun, DU Yu, WANG HaiPeng, GENG SiHai, ZHOU DingDing, LIU SiYa, CHEN DaFu.
0 引言
【研究意义】蜜蜂是自然界最重要的授粉昆虫,也是社会行为学模式昆虫,具有非常重要的经济和生态价值[1,2]。意大利蜜蜂(Apis mellifera ligustica,简称意蜂)属于西方蜜蜂(Apis mellifera),具有优越的采集能力、造脾能力和分泌蜂王浆能力,在世界各地的养蜂生产中广泛使用[3]。目前,有关蜜蜂肠道的发育机理及调控机制的研究十分滞后。对于环状RNA(circular RNA,circRNA)在蜜蜂肠道发育过程中的作用,相关信息仍然缺失。利用circRNA-seq技术对意蜂工蜂中肠进行测序,并对中肠发育过程中的差异表达circRNA(DEcircRNA)及其调控网络进行深入分析,可揭示DEcircRNA在中肠发育过程中的作用,为关键circRNA的筛选和功能研究打下基础。【前人研究进展】CircRNA是新近发现的一类非编码RNA(non-coding RNA,ncRNA),通过外显子或/和内含子的反向剪切形成环化RNA分子[4]。CircRNA大量存在于真核细胞中,在不同物种中具有保守性、丰富性、稳定性、组织表达特异性和时序表达特异性等特点[5]。CircRNA已被证明具有多种生物学调控功能,包括作为微小RNA(microRNA,miRNA)“海绵”,形成RNA-蛋白质复合物(RNA-binding protein,RBP),以及调控靶基因的转录和可变剪接等[6]。最新的研究结果表明含有核糖体进入位点[7]的circRNA能够翻译蛋白[8]。与线性RNA相比,circRNA缺少5′帽子和3′尾巴结构,稳定性更高且能抵抗核糖核酸外切酶RNase R的消化,因而成为理想的生物标志物[9]。利用最新的高通量测序技术和生物信息学分析方法,已在人类[10,11,12]、动物[13]、植物[14]及微生物[8,15-16]中预测和鉴定出circRNA。SALZMAN等[12]对人类15种细胞类型中的circRNA进行预测分析,发现circRNA具有组织特异性、保守性及潜在的调控功能;SHEN等[13]对斑马鱼肌肉、卵巢及眼睛等不同组织进行高通量测序,利用3种算法对circRNA进行预测,共预测到3 868个circRNA,进而对具有较高可信度的176个circRNA进行实验验证,发现其中84%真实存在;LU等[14]对水稻进行深度测序及生物信息学分析,共预测出2 354个circRNA,进一步分析发现circRNA具有相当数量的亚型;GUO等[16]对蜜蜂球囊菌(Ascosphaera apis)的菌丝及孢子混合样品进行高通量测序,通过生物信息学分析发掘出551个长度介于200—600 nt的circRNA,并发现它们与miRNA存在复杂的调控关系。较之人类和哺乳动物,昆虫的circRNA研究还处于初级阶段,相关信息极为有限[17,18,19]。WESTHOLM等[17]在全基因组水平对果蝇(Drosophila melanogaster)的circRNA进行分析,发现其circRNA具有组织表达特异性,并且可作为果蝇衰老的潜在生物标志物;GAN等[18]通过对家蚕(Bombyx mori)中肠和后肠进行高通量测序,共预测出来源于1 727个基因的3 155个circRNA,同样具有组织表达特异性,进一步分析发现中肠和后肠中的来源基因的功能和代谢通路注释信息类似;CHEN等[19]对西方蜜蜂蜂王的卵巢组织中的circRNA进行预测和分析,发掘出12 211个circRNA,并通过对刚产卵蜂王、处女王和限制产卵蜂王进行比较分析推测DEcircRNA可通过竞争性结合miRNA在蜂王卵巢组织活化及产卵过程中发挥作用。较多的研究表明circRNA可作为竞争性内源RNA(competing endogenous RNA,ceRNA)与mRNA竞争性结合miRNA,间接调控基因表达[20,21,22]。CHENG等[21]对椎间盘退行性病变的病人髓核细胞和组织通过微距阵芯片分析,发现circVMA21能够通过吸附miR-200c抑制细胞凋亡相关的X蛋白基因,将circVMA21注射到患有椎间盘退行性病变的大鼠模型可使疾病得到缓解;ZHENG等[22]通过对6个正常组织和7个癌变组织进行深度测序,发现与癌变相关的CircHIPK3包含结合9个miRNA的18个潜在结合位点,可靶向结合miR-124抑制肿瘤形成。【本研究切入点】蜜蜂中肠既是消化食物、吸收营养的重要场所[23],也是多种病原微生物寄生的主要部位。笔者所在课题组前期已在mRNA组学水平探究了意蜂4、5和6日龄幼虫肠道发育过程中的基因表达谱和差异表达规律[24],并在长链非编码RNA(long non-coding RNA,lncRNA)组学水平探究了意蜂7和10日龄工蜂中肠的差异表达lncRNA及其调控网络[25]。目前,蜜蜂circRNA的信息极为有限,circRNA在蜜蜂中肠发育中的作用尚不明确。【拟解决的关键问题】通过生物信息学方法对意蜂7和10日龄工蜂中肠发育过程中的差异表达circRNA(DEcircRNA)及其调控网络进行深入分析,提供意蜂工蜂中肠的circRNA表达谱及差异表达信息,并在组学水平探究DEcircRNA在中肠发育过程中的作用。1 材料与方法
试验于2017年在福建农林大学蜂学学院蜜蜂保护实验室完成。1.1 生物材料
意蜂工蜂取自福建农林大学蜂学学院教学蜂场。1.2 测序样品及全转录组数据来源
笔者所在课题组前期已利用二代测序技术对意蜂工蜂中肠进行深度测序,获得了高质量的全转录组数据[25]。其中,测序样品的制备过程简述如下:从外观健康且群势较强的意蜂蜂群中选择蜂子状况良好的封盖子脾,迅速提至实验室,放入(34±0.5)℃培养箱,将刚出房的工蜂(记为0 d)放入四周打孔的干净塑料盒(10只/盒),每个塑料盒上方插入一支装有50%(w/v)无菌糖水的饲喂器。(34±0.5)℃饲养工蜂至10 d。每日检查工蜂存活情况,及时清理死亡工蜂。进行3次生物学重复。待意蜂工蜂7日龄和10日龄时,在超净台用干净镊子拉取工蜂中肠,放入RNA-Free的EP管,经液氮速冻后迅速转移至-80℃超低温冰箱保存备用。意蜂7日龄工蜂中肠样品的3个重复为Am7-1、Am7-2和Am7-3;意蜂10日龄工蜂中肠样品的3个重复为Am10-1、Am10-2和Am10-3。上述6个中肠样品的全转录组测序由广州基迪奥生物科技有限公司完成,测序平台为Illumina HiSeq 4000,测序策略为双端(paired-end)测序,测序数据已上传NCBI SRA数据库,BioProject号:PRJNA406998。cDNA建库过程简述如下:利用RNA抽提试剂盒AxyPrepTM Multisource Total RNA Miniprep Kit(TaKaRa公司,中国)抽提蜜蜂中肠样品的总RNA,为最大限度地保留所有非编码RNA(ncRNA),去除核糖体RNA后的mRNA和ncRNA用裂解缓冲液随机打断为小片段,作为模板用六碱基随机引物、缓冲液、dNTPs、RNase H和DNA polymerase I合成cDNA第2链;经过QiaQuick PCR试剂盒(Qiagen公司,德国)纯化并加EB缓冲液洗脱经末端修复、加碱基A,加测序接头,然后通过尿嘧啶-N-糖基化酶(UNG)降解cDNA第2链。
1.3 测序数据质控及circRNA预测
对于测序得到的原始读段(raw reads),去除N的比例大于10%的、质量值Q≤10的和碱基数占整条读段数的50%以上的读段,过滤得到的有效读段(clean reads)用于后续的数据分析。利用TopHat软件[26]将各中肠样品的有效读段与西方蜜蜂参考基因组(Amel_4.5)[27]进行比对,从比对结果中提取未比对上读段(unmapped reads),然后截取每一条未比对上读段的两端(默认20 nt),得到短序列读段(anchors reads),继而再次比对参考基因组,将得到的比对结果提交给find_circ软件[11],从而对circRNA进行筛选和鉴定,筛选条件包括:(1)breakpoints=1,即只保留有且只有1个清晰breakpoint的环状RNA;(2)anchor overlap≤2,即每条reads的两个anchors reads比对到基因组上的位置overlap不能超过2 bp;(3)edit≤2,即只允许2 bp错配;(4)n uniq>2,即uniq reads大于2条;(5)best qual A>35或best qual B>35,即每条reads的其中一个anchors reads比对到基因组上最好的mapping结果要比其排第二的结果的分值高35分以上;(6)n uniq>int (samples/2),即支持该circRNA的uniq reads要大于总样品数的1/2;(7)circRNA的长度小于100 kb。1.4 CircRNA的表达量、差异表达及来源基因分析
采用RPM(mapped back-splicing junction reads per million mapped reads)法计算circRNA的表达量。先计算Am7和Am10中所有circRNA的lg (RPM+1)值,再利用基迪奥在线工具平台Omicshare(www. omicshare.com/tools)绘制Am7和Am10的circRNA的箱线图,从而比较二者的circRNA的表达量,参数为默认参数。利用DEGseq软件[28]对circRNA进行差异表达分析,DEcircRNA的筛选条件包括:(1)差异倍数(fold change,FC)≥2;(2)P<0.05;(3)错误发现率(false discovery rate,FDR)<0.05。来源于内含子或由内含子和外显子环化的circRNA能够调节亲本基因的表达[22,29],使用短reads比对工具Bowtie[30]分别将这些circRNA两端的anchors reads比对西方蜜蜂参考基因组[27],两端都比对上同一个基因即为该circRNA的来源基因,再将来源基因通过BLAST注释到GO和KEGG数据库,按照Q值≤0.5筛选显著富集GO条目或KEGG代谢通路的DEcircRNA的来源基因注释。1.5 DEcircRNA的靶miRNA预测及调控网络构建
利用TargetFinder软件[31]预测DEcircRNA靶向结合的miRNA,得到DEcircRNA-miRNA的靶向调控关系。按照P≤0.05,自由能≤35的标准,从结果中提取DEcircRNA潜在的靶向结合miRNA的位点信息。进一步预测DEcircRNA的靶miRNA靶向结合的mRNA,根据DEcircRNA与靶miRNA、miRNA与靶mRNA的结合关系,得到DEcircRNA-miRNA-mRNA的靶向调控关系,最后通过Cytoscape v.3.2.1软件[32]对各调控网络进行可视化,参数采用默认参数。1.6 DEcircRNA的实时荧光定量PCR(RT-qPCR)验证
为了验证circRNA-seq数据的可靠性,随机选取6个DEcircRNA(novel_circ_000663、novel_circ_005547、novel_circ_014049、novel_circ_002507、novel_circ_ 012440及novel_circ_001915)进行RT-qPCR验证。参照GUO等[16]的方法,利用DNAMAN软件根据所选circRNA的碱基序列设计相应的特异性反向引物,委托上海生工生物工程有限公司进行合成,相关引物信息详见表1。利用RNA抽提试剂盒(TaKaRa公司,中国)分别提取Am7和Am10样品的总RNA,分为两份,其中一份总RNA用3 U/mg RNase R(吉赛生物公司,中国)进行消化以去除线性RNA,37℃处理15 min后以随机引物进行反转录,得到circRNA的cDNA;另一份总RNA直接作为模板,以Oligo (dT)23作为引物进行反转录,得到所有含polyA的RNA对应的cDNA,作为内参基因actin的qPCR模板[33]。反应体系(20 μL)包含正、反向引物(10.0 μmol·L-1)各1 μL,cDNA模板DNA 1 μL,SYBR Green Dye 10 μL,DEPC水7 μL。反应在QuantStudio荧光定量PCR仪(ThermoFisher公司,美国)上进行,按照SYBR Green Dye试剂盒(Vazyme公司,中国)操作说明书上的方法,每个反应进行3次重复。反应条件:95℃预变性1 min,95℃变性15 s,60℃延伸30 s,共45个循环,最后72℃延伸45 s。利用2-ΔΔCt 法对上述DEcircRNAs的相对表达量进行计算。通过Graph Prism软件进行相关数据分析及绘图。Table 1
表1
表1RT-qPCR验证的引物信息
Table 1
| 引物名称Primer name | 引物序列Primer sequence |
|---|---|
| Novel_circ_005547-F | TCTGCTACTCAAATGGAGGG |
| Novel_circ_005547-R | CCCACTGTCTCTCTTCTAAGGA |
| Novel_circ_014049-F | GGAAGGAAGGAAGTAGCGA |
| Novel_circ_014049-R | CACGAACACCACCCAATA |
| Novel_circ_002507-F | ATTTCCTTGGGCATAGCC |
| Novel_circ_002507-R | CTCGGTCAAACCATACACC |
| Novel_circ_012440-F | AGTCTGTTCGGTAATCCCG |
| Novel_circ_012440-R | CTCACCTGATACTTCACCTTTG |
| Novel_circ_001915-F | CATCATCTCCGAAACCGA |
| Novel_circ_001915-R | TTGAGGTGGCTGACTTGA |
| Actin-F | CACTCCTGCTATGTATGTCGC |
| Actin-R | GGCAAAGCGTATCCTTCA |
新窗口打开|下载CSV
2 结果
2.1 高通量测序数据的质控及评估
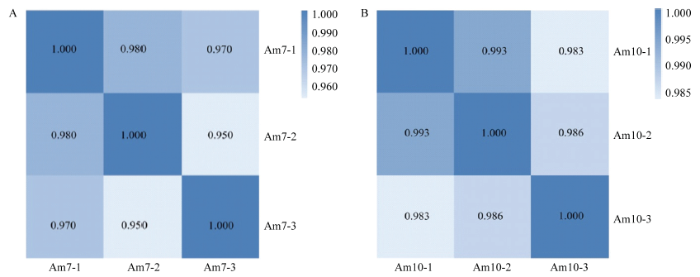
前期研究中的意蜂工蜂中肠样品Am7及Am10测序分别得到134 802 058条raw reads,经过滤得到clean reads数平均为147 051 470条,各样品的平均Q20和Q30分别为97.34%和93.86%[25]。此外,Am7与Am10的组内Pearson相关系数都≥0.950(图1)。上述结果表明本研究中的测序数据质量良好,可用于下一步分析。本研究中,各样品的anchors reads平均为181 583 825条,比对上参考基因组的anchors reads数平均为19 616 356条(表2)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1各意蜂工蜂中肠样品的不同生物学重复间的Pearson相关性
Fig. 1Pearson correlations between different biological repeats within each A. m. ligustica worker’s midgut sample
Table 2
表2
表2未比对上核糖体数据库的有效读段比对参考基因组的信息统计
Table 2
| 样品 Sample | 短序列读段 Anchors reads | 比对上的短序列读段 Mapped anchors reads | 比对率 Mapping ratio (%) |
|---|---|---|---|
| Am7-1 | 236400908 | 19006601 | 8.04 |
| Am7-2 | 168420472 | 19873468 | 11.80 |
| Am7-3 | 136779122 | 17502228 | 12.80 |
| Am10-1 | 195020440 | 22441139 | 11.51 |
| Am10-2 | 184091234 | 21109367 | 11.47 |
| Am10-3 | 168790774 | 17765330 | 10.53 |
新窗口打开|下载CSV
2.2 意蜂工蜂中肠circRNA的表达谱分析
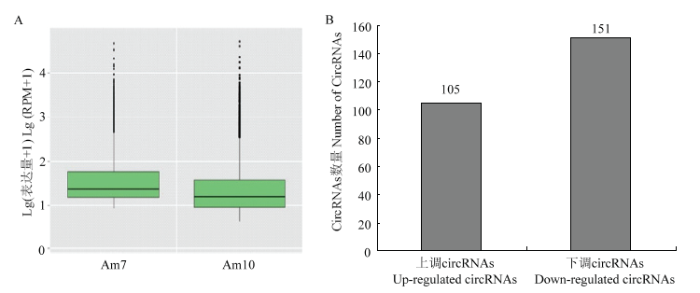
对Am7和Am10中所有circRNA进行表达量分析,结果显示前者中的circRNA的整体表达水平较高(图2-A)。Am7中表达量最高的circRNA为novel_ circ_003183、novel_circ_010717、novel_circ_012530、novel_circ_000476及novel_circ_011750(表3);Am10中表达量最高的circRNA为novel_circ_003183、novel_ circ_010717、novel_circ_012530、novel_circ_000896、novel_circ_000476(表4)。进一步分析发现,novel_ circ_003183、novel_circ_010717、novel_circ_012530及novel_circ_000476在Am7和Am10中均高量表达,推测它们在意蜂工蜂中肠的发育过程中发挥重要作用。Novel_circ_009675和novel_circ_013879分别在Am7和Am10中高量表达,推测二者分别在意蜂工蜂中肠发育的不同时期发挥特殊功能。差异分析结果显示,Am7 vs Am10中共有256个DEcircRNA,包含105个上调和151个下调circRNA(图2-B)。Table 3
表3
表3Am7样品中表达量最高的前10位circRNA
Table 3
| CircRNA名称 Name of circRNAs | RPM值 RPM value | 来源基因ID ID of source gene | 长度 Length (bp) | 环化类型 Type of circularization |
|---|---|---|---|---|
| novel_circ_003183 | 45581.85 | 410805 | 552 | annot_exons |
| novel_circ_010717 | 33244.11 | 413053 | 956 | annot_exons |
| novel_circ_012530 | 21253.62 | 725393 | 1190 | one_exon |
| novel_circ_000476 | 15898.09 | 552294 | 1742 | annot_exons |
| novel_circ_011750 | 14352.44 | 410379 | 613 | antisense |
| novel_circ_011749 | 14094.35 | 410379 | 613 | antisense |
| novel_circ_000896 | 12421 | 100576586 | 1362 | annot_exons |
| novel_circ_006484 | 9348.5 | 413332 | 419 | annot_exons |
| novel_circ_009675 | 7112.647 | 408996 | 404 | annot_exons |
| novel_circ_012115 | 6698.064 | 724592 | 727 | annot_exons |
新窗口打开|下载CSV
Table 4
表4
表4Am10中表达量最高的前10位circRNA
Table 4
| CircRNA名称 Name of circRNAs | RPM值 RPM value | 来源基因ID ID of source gene | 长度 Length (bp) | 环化类型 Type of circularization |
|---|---|---|---|---|
| novel_circ_003183 | 50507.89 | 410805 | 552 | annot_exons |
| novel_circ_010717 | 39732.13 | 413053 | 956 | annot_exons |
| novel_circ_012530 | 23025.9 | 725393 | 1190 | one_exon |
| novel_circ_000896 | 17755.41 | 100576586 | 1362 | annot_exons |
| novel_circ_000476 | 17621.29 | 552294 | 1742 | annot_exons |
| novel_circ_011750 | 13241.09 | 410379 | 613 | antisense |
| novel_circ_011749 | 12729.28 | 410379 | 613 | antisense |
| novel_circ_006484 | 9038.38 | 413332 | 419 | annot_exons |
| novel_circ_013879 | 7982.833 | 408521 | 1219 | exon_intron |
| novel_circ_012115 | 7316.428 | 724592 | 727 | annot_exons |
新窗口打开|下载CSV
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2意蜂工蜂中肠发育过程的circRNA的表达谱
A:Am7和Am10中circRNA表达量的箱线图Box plot of the circRNAs’ expression quantity in Am7 and Am10;B:Am7 vs Am10中的DEcircRNA DEcircRNAs in Am7 vs Am10
Fig. 2Expression pattern of circRNAs in the developmental process of A. m. ligustica worker’s midgut
2.3 意蜂工蜂中肠DEcircRNA来源基因的GO数据库注释
GO分类结果显示,Am7 vs Am10的DEcircRNA来源基因可注释到32个GO条目(term),涉及细胞组分、生物学进程和分子功能3大功能分类(图3)。细胞组分中,注释基因数最多的前5位分别是细胞(21个)、细胞组件(21个)、细胞膜组件(15个)、细胞膜(15个)及细胞器(15个);生物学进程中,注释基因数最多的前5位分别是单组织进程(38个)、细胞进程(38个)、代谢进程(35个)、定位(13个)及生物调控(10个),此外应激反应和发育进程注释基因数分别为7和2个;分子功能中,注释基因数最多的前5位分别是结合(50个)、催化活性(35个)、转运器活性(8个)、核苷酸结合转录因子活性(4个)及分子传感器活性(3个)。上述结果说明DEcircRNA在意蜂工蜂中肠的生长、发育、代谢、免疫和细胞生命活动中发挥功能。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3DEcircRNA来源基因的GO数据库注释
Fig. 3GO database annotations of DEcircRNAs’ source genes
2.4 意蜂工蜂中肠DEcircRNA来源基因的KEGG数据库注释
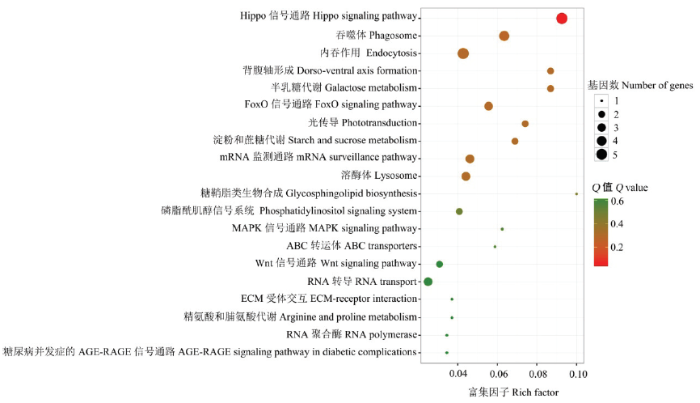
KEGG数据库注释结果显示(图4),意蜂工蜂中肠DEcircRNA的来源基因可注释到35条代谢通路(pathway),其中注释基因数最多的前10位分别是Hippo信号通路(5个,图5-A)、内吞作用(5个,图5-B)、吞噬体(4个)、FoxO信号通路(3个)、mRNA监测通路(3个)、溶酶体(3个)、RNA转导(3个)、背腹轴形成(2个)、淀粉和蔗糖代谢(2个)及半乳糖代谢(2个),说明DEcircRNA通过参与多条信号通路、免疫通路对意蜂工蜂中肠的生长发育、免疫防御进行调节。内吞作用、Hippo信号通路的概貌详见图5-A和图5-B。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4DEcircRNA来源基因的KEGG数据库注释
圆圈大小代表富集在某一通路的基因数多少,越大代表基因数越多;圆圈颜色代表某一通路的富集基因的显著性高低,越红代表显著性越高
Fig. 4KEGG database annotations of DEcircRNAs’ source genes
The size of the circle indicates the number of enriched genes in a certain pathway, and the larger the circle, the more the number of genes; the color of the circle indicates the significance of enriched genes in a certain pathway, and the redder the color, the higher the significance
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5Hippo信号通路和内吞作用的概貌
红色方框代表注释在该通路的DEcircRNA的来源基因
Fig. 5Overview map of Hippo signaling pathway and endocytosis
Red boxes indicate DEcircRNAs’ source genes annotated in certain pathway
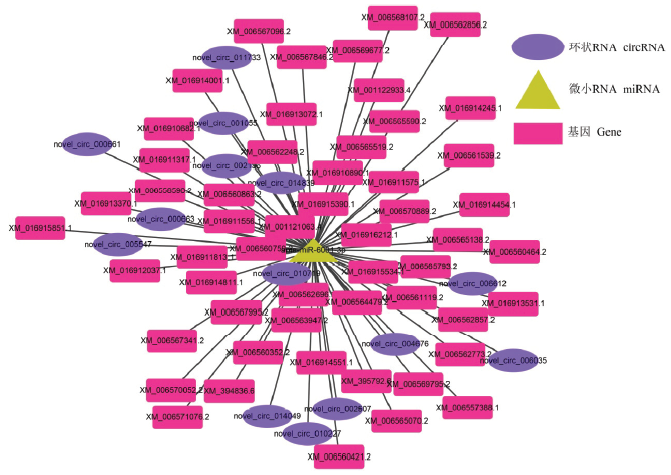
2.5 意蜂工蜂中肠的DEcircRNA-miRNA、DEcircRNA- ame-miR-6001-3p-mRNA调控网络构建及分析
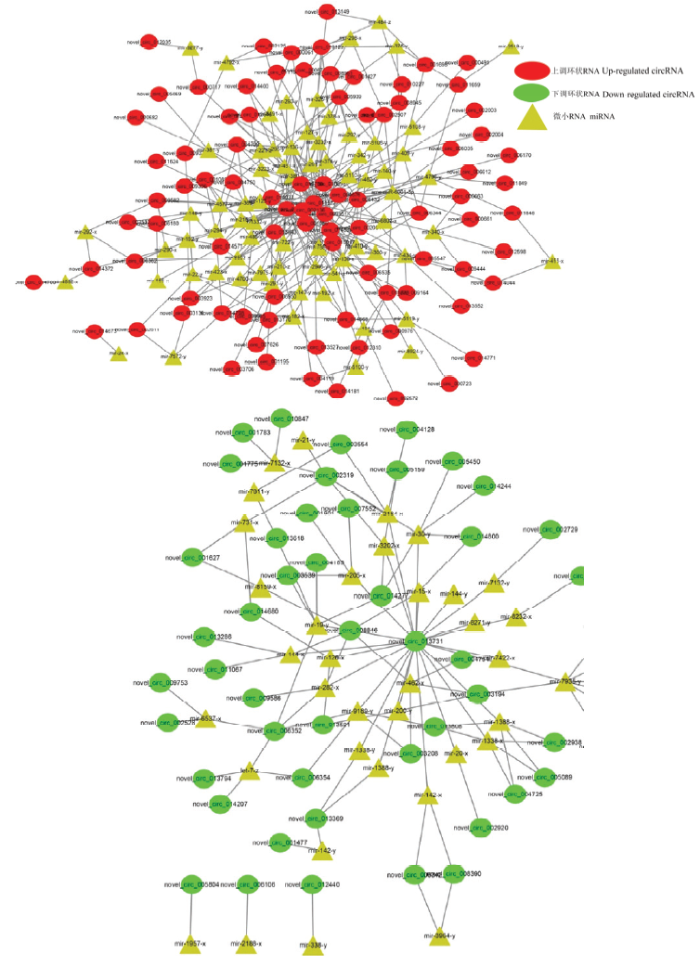
通过TargetFinder和Cytoscape软件构建及可视化意蜂工蜂中肠的DEcircRNA-miRNA调控网络,88个上调circRNA靶向结合71个miRNA,其中novel_ circ_011577、novel_circ_010719及novel_circ_ 009951结合的miRNA数最多,分别为32、28和18个;53个下调circRNA可靶向结合36个miRNA,其中novel_ circ_013731、novel_circ_002319及novel_circ_006352结合的miRNA数最多,分别为23、6和5个。多数DEcircRNA(70.21%)仅能结合1—2个miRNA。此外,mir-136-y、ame-miR-6001-3p及mir-136-y结合的circRNA数最多,分别为15、14和14个circRNA(图6)。影响miRNA与靶基因的结合,从而对下游的基因表达进行间接调控。进一步构建DEcircRNA-ame- miR-6001-3p-mRNA的调控网络(图7),分析结果显示共有14个DEcircRNA(novel_circ_011733、novel_circ_001055、novel_circ_000661、novel_circ_ 002196、novel_circ_014839、novel_circ_000663、novel_ circ_005547、novel_circ_010719、novel_circ_006612、novel_circ_004676、novel_circ_006035、novel_circ_ 002507、novel_circ_014049及novel_circ_010227)靶向结合同一个miRNA(ame-miR-6001-3p),说明这些DEcircRNA可通过竞争性结合ame-miR-6001-3p而抑制其与靶基因的结合,从而影响靶基因的表达水平。上述结果说明意蜂工蜂中肠发育过程中DEcircRNA通过作为ceRNA充当miRNA的海绵,间接调控基因表达。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6意蜂工蜂中肠的DEcircRNA-miRNA调控网络
Fig. 6DEcircRNAs-miRNAs regulation networks in A. m. ligustica worker’s midgut
图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7意蜂工蜂中肠的DEcircRNA-ame-miR-6001-3p-mRNA调控网络
Fig. 7DEcircRNAs-ame-miR-6001-3p-mRNAs regulation networks in A. m. ligustica worker’s midgut
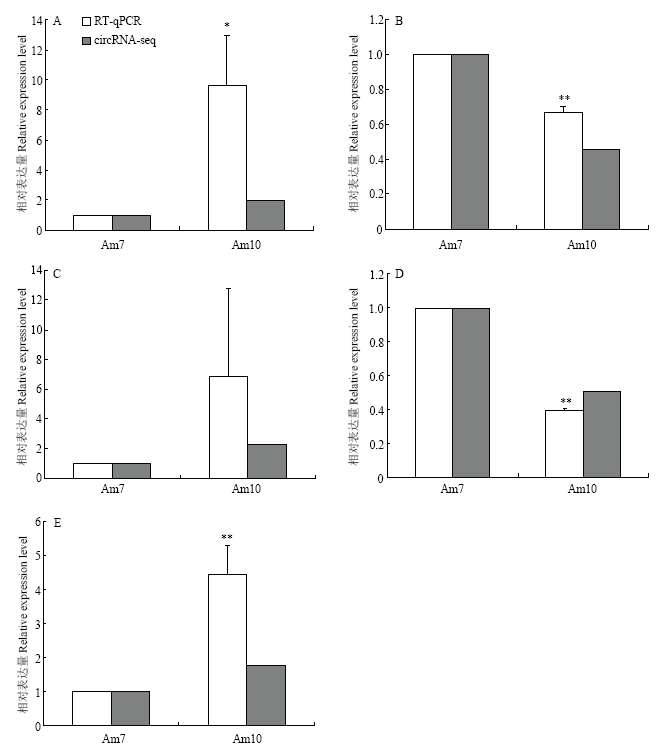
2.6 DEcircRNA的RT-qPCR验证
随机挑取6个DEcircRNA进行RT-qPCR验证,结果显示其中5个DEcircRNA的表达量变化趋势和转录组数据的结果一致(图8),说明本研究中的测序数据真实可靠。图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8DEcircRNA的RT-qPCR验证
RT-qPCR组中,*表示 P<0.05,**表示P<0.01 In RT-qPCR group, * indicates P<0.05, ** indicates P<0.01
Fig. 8RT-qPCR validation of DEcircRNAs
A: Novel_circ_005547; B: Novel_circ_012440; C: Novel_circ_014049; D: Novel_circ_001915; E: Novel_circ_002507
3 讨论
CircRNA在可变剪接、转录调控和来源基因的表达调控等方面具有重要功能[6]。随着高通量测序技术和生物信息学的发展,人们已在人类[10,11,12]、牛[34]、猪[35]、鸡[36]、老鼠[37]、斑马鱼[13]、草鱼[38]、水稻[14]、大麦[39]、拟南芥[40]、线虫[41]、蜜蜂球囊菌[16]、古细菌[15]和丁型肝炎病毒[8]等动植物及微生物中预测和鉴定出circRNA。2018年,CHEN等[19]通过高通量测序技术和生物信息学方法从西方蜜蜂蜂王的卵巢组织中预测出12 211个circRNA,这是迄今唯一有关蜜蜂circRNA的研究报道。前期研究中,笔者通过对意蜂7和10日龄工蜂中肠进行全转录组测序和全面分析,提供了中肠发育过程中lncRNA的表达谱及差异表达信息,构建并分析了差异表达lncRNA(DElncRNA)的调控网络,在组学水平揭示了DElncRNA在意蜂工蜂中肠发育过程中的作用[25]。本研究在此基础上,进一步通过生物信息学方法对意蜂工蜂中肠的circRNA进行挖掘,从意蜂7和10日龄工蜂中肠中分别预测出7 341和9 092个circRNA,为蜜蜂的ncRNA信息提供了重要补充。本研究发现,novel_circ_003183、novel_circ_010717、novel_circ_012530、novel_circ_000896、novel_circ_ 000476、novel_circ_011750、novel_circ_011749、novel_ circ_006484、novel_circ_012115及novel_circ_009675在Am7和Am10中皆高量表达,说明它们在意蜂中肠发育过程中具有重要功能,但它们是否在其他日龄的意蜂工蜂中肠中高量表达,有待于进一步研究。Novel_circ_009675和novel_circ_013879分别在Am7和Am10中高量表达,二者在意蜂中肠发育的不同时期发挥特殊功能。Am7 vs Am10中包含105个上调和151个下调circRNA,推测这些DEcircRNA与意蜂中肠发育关系密切。
来源于外显子和内含子组成的circRNA(exon- intron circRNA,EIciRNA)可通过与RNA聚合酶II、U1小核核糖核蛋白及基因启动子相互作用对其来源基因的转录进行调控[29]。蜜蜂的天然食物主要包括含葡萄糖、果糖和淀粉等成分的蜂蜜及含蛋白质的花粉[42]。蜜蜂中肠作为主要的消化和吸收器官,含有大量的消化酶如淀粉酶、蔗糖酶和蛋白酶等[43]。本研究中,DEcircRNA的来源基因中有35个可注释到催化活性;分别有1、2和2个来源基因可注释到磷酸肌醇代谢、淀粉和蔗糖代谢和半乳糖代谢等消化吸收相关通路。上述结果表明相应的DEcircRNA参与意蜂工蜂中肠的食物消化、营养吸收等过程。此外,分别有2和35个来源基因注释到发育进程和代谢进程,暗示DEcircRNA在此过程扮演重要角色。Hippo信号通路可通过抑制细胞增殖和促进细胞凋亡对器官大小进行调节[44],也能和其他信号通路相互作用共同调节肠道组织的稳态[45],还在肠道结构的维持特别是上皮细胞的分化过程起重要作用[46]。Wnt信号通路与哺乳动物胚胎形成、卵巢发育、平面细胞极化等生理学进程密切相关[47],还能够与Hippo、Notch和TGF-beta信号通路共同影响昆虫的体节形成、色素沉淀、附肢发育以及翅等器官的发育[48]。本研究发现,分别有5和2个来源基因注释到Hippo和Wnt信号通路,表明相应的DEcircRNA可能通过调节Hippo和Wnt信号通路对意蜂工蜂中肠的细胞生长、分化及凋亡以及结构和稳态维持进行调控。还发现有7个来源基因注释到应激反应,表明相应的DEcircRNA参与调控意蜂工蜂中肠对外界环境的适应过程。
蜜蜂的免疫系统包括群体免疫与个体免疫,后者又分为细胞免疫和体液免疫。其中,蜜蜂的细胞免疫包括内吞作用、吞噬作用、黑化作用等,体液免疫主要为抗菌肽的合成与释放[49]。MAPK信号通路能够通过产生免疫效应因子刺激淋巴细胞活性,对外界入侵的病原微生物进行免疫防御[50]。本研究中,对于意蜂工蜂中肠发育过程中DEcircRNA的来源基因,分别有5、4、3和1个注释到内吞作用、吞噬体、溶酶体和泛素介导的蛋白水解等细胞免疫通路,仅有1个注释在MAPK信号通路。上述结果表明相应的DEcircRNA参与意蜂工蜂中肠的细胞免疫、体液免疫的调控过程,并且可能在细胞免疫调控方面发挥更重要的作用。
2011年,SALMENA等[51]提出了“竞争性内源RNA”假说,即任何包含miRNA反应元件(miRNA response element)的RNA,例如mRNA、假基因、lncRNA和circRNA,可以竞争性地结合miRNA。此后,越来越多的研究结果都证实了ceRNA假 说[20-22,52-54]。DENG等[52]通过对患急性心肌梗塞病人和正常人的DEcircRNA进行分析,发现circRNA_081881能够靶向结合miR-548,使急性心肌梗塞相关的PPARγ表达量上调;XU等[53]分别比较分析了患恶性肿瘤的组织和正常组织的DEcircRNA,发现hsa_circ_000984通过靶向结合miR-106b,有效上调细胞生长调节相关基因CDK6的表达水平。本研究中,调控网络分析结果显示141个DEcircRNA可靶向结合107个miRNA,其中大部分DEcircRNA(70.21%)仅能结合1—2个miRNA,少数DEcircRNA可结合多个miRNA,例如novel_circ_011577和novel_circ_010719结合的miRNA数可达32和28个,表明二者作为ceRNA的重要性。还发现多个DEcircRNA可共同靶向结合同一个miRNA,例如分别有15和14个DEcircRNA共同结合mir-136-y和ame-miR-6001-3p。上述结果表明DEcircRNA可作为ceRNA吸附结合miRNA,减少其对mRNA的抑制和降解,从而影响意蜂工蜂肠道的生长发育。目前,circRNA的功能研究尚处起步阶段,绝大多数circRNA的功能还不明确。LIU等[54]通过对骨关节炎相关circRNA的深入研究,发现CircRNA-CER可通过吸附mir-136对MMP13的表达进行调控,从而参与软骨细胞外基质(EMC)的降解。本研究中,novel_circ_000976、novel_circ_005547和novel_circ_006344等23个DEcircRNA靶向结合的mir-136-y和mir-136-x与mir-136具有高度同源性,推测相应的DEcircRNA可能通过对mir-136-y、mir-136-x表达水平的调节,参与意蜂工蜂中肠细胞外基质的降解。蜕皮激素主要调控昆虫幼虫老旧组织的程序化细胞死亡与成虫干细胞的分裂分化[55],MELLO等[56]通过敲除变态发育过程中的蜜蜂个体蜕皮激素受体编码基因,发现ame-miR-6001-3p在敲除个体中上调表达,表明ame-miR-6001-3p能够抑制蜕皮激素的分泌,从而影响中肠干细胞的分裂和分化。本研究中,DEcircRNA-ame-miR-6001-3p-mRNA调控网络分析结果显示,共有novel_circ_000661、novel_circ_000663和novel_circ_001055等14个DEcircRNA能够共同靶向结合ame-miR-6001-3p,推测上述DEcircRNA可通过竞争性结合ame-miR-6001-3p调控蜕皮激素受体编码基因的表达,从而影响中肠干细胞的分裂及分化。进一步通过设计跨反向剪切位点的引物对随机挑选DEcircRNA进行RT-qPCR验证,结果显示83.33%(5/6)的DEcircRNA相对表达量与测序结果一致,证实了本研究中circRNA差异表达信息的可靠性。
本研究仅对意蜂7和10日龄工蜂中肠的circRNA进行相关分析,预测出的circRNA在其他日龄工蜂中肠、同一日龄的不同组织是否表达以及表达水平的高低仍需要进一步研究。此外,若要全面解析意蜂工蜂中肠的发育机理及调控机制,则需对更多日龄的工蜂中肠进行测序,进而在全局水平进行更加深入的分析,此为下一步的工作重点。
4 结论
通过对意蜂7和10日龄工蜂中肠的深度测序和生物信息学分析,提供了中肠发育过程的circRNA表达谱及差异表达信息。DEcircRNA可能通过调控来源基因的表达水平和作为ceRNA在意蜂工蜂中肠发育过程中发挥重要功能。靶向结合ame-miR-6001-3p的14个DEcircRNA可能参与意蜂中肠干细胞的分裂及分化。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1186/1471-2164-16-1URLPMID:4326529 [本文引用: 1]

Background The honey bee is an important model system for increasing understanding of molecular and neural mechanisms underlying social behaviors relevant to the agricultural industry and basic science. The western honey bee, Apis mellifera, has served as a model species, and its genome sequence has been published. In contrast, the genome of the Asian honey bee, Apis cerana, has not yet been sequenced. A. cerana has been raised in Asian countries for thousands of years and has brought considerable economic benefits to the apicultural industry. A cerana has divergent biological traits compared to A. mellifera and it has played a key role in maintaining biodiversity in eastern and southern Asia. Here we report the first whole genome sequence of A. cerana. Results Using de novo assembly methods, we produced a 238 Mbp draft of the A. cerana genome and generated 10,651 genes. A.cerana-specific genes were analyzed to better understand the novel characteristics of this honey bee species. Seventy-two percent of the A. cerana-specific genes had more than one GO term, and 1,696 enzymes were categorized into 125 pathways. Genes involved in chemoreception and immunity were carefully identified and compared to those from other sequenced insect models. These included 10 gustatory receptors, 119 odorant receptors, 10 ionotropic receptors, and 160 immune-related genes. Conclusions This first report of the whole genome sequence of A. cerana provides resources for comparative sociogenomics, especially in the field of social insect communication. These important tools will contribute to a better understanding of the complex behaviors and natural biology of the Asian honey bee and to anticipate its future evolutionary trajectory.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12943-017-0663-2URLPMID:28535767 [本文引用: 1]
Circular RNAs, a novel class of endogenous noncoding RNAs, are characterized by their covalently closed loop structures without a 5′ cap or a 3′ Poly A tail. Although the mechanisms of circular RNAs’ generation and function are not fully clear, recent research has shown that circular RNAs may function as potential molecular markers for disease diagnosis and treatment and play an important role in the initiation and progression of human diseases, especially in tumours. This review summarizes some information about categories, biogenesis, functions at the molecular level, properties of circular RNAs and the possibility of circular RNAs as biomarkers in cancers.
DOI:10.1016/j.canlet.2017.03.027URL [本文引用: 1]
DOI:10.1016/j.canlet.2015.06.003URLPMID:26052092 [本文引用: 2]
Circular RNAs (circRNAs) are a novel type of RNA that, unlike linear RNAs, form a covalently closed continuous loop and are highly represented in the eukaryotic transcriptome. Recent studies have discovered thousands of endogenous circRNAs in mammalian cells. CircRNAs are largely generated from exonic or intronic sequences, and reverse complementary sequences or RNA-binding proteins (RBPs) are necessary for circRNA biogenesis. The majority of circRNAs are conserved across species, are stable and resistant to RNase R, and often exhibit tissue/developmental-stage-specific expression. Recent research has revealed that circRNAs can function as microRNA (miRNA) sponges, regulators of splicing and transcription, and modifiers of parental gene expression. Emerging evidence indicates that circRNAs might play important roles in atherosclerotic vascular disease risk, neurological disorders, prion diseases and cancer; exhibit aberrant expression in colorectal cancer (CRC) and pancreatic ductal adenocarcinoma (PDAC); and serve as diagnostic or predictive biomarkers of some diseases. Similar to miRNAs and long noncoding RNAs (lncRNAs), circRNAs are becoming a new research hotspot in the field of RNA and could be widely involved in the processes of life. Herein, we review the formation and properties of circRNAs, their functions, and their potential significance in disease.
DOI:10.1016/j.molcel.2014.08.019URL [本文引用: 1]
Ashwal-Fluss et02al. provide evidence that circRNA production competes with linear mRNA splicing. They demonstrate that intronic sequences largely determine the production rate of circRNAs and identify a splice factor (muscleblind) that can promote circularization.
DOI:10.1038/323558a0URL [本文引用: 3]
DOI:10.2144/000114061URLPMID:23931592 [本文引用: 1]
Perkel JM.
DOI:10.1261/rna.035667.112URL [本文引用: 2]
Circular RNAs composed of exonic sequence have been described in a small number of genes. Thought to result from splicing errors, circular RNA species possess no known function. To delineate the universe of endogenous circular RNAs, we performed high-throughput sequencing (RNA-seq) of libraries prepared from ribosome-depleted RNA with or without digestion with the RNA exonuclease, RNase R. We identified >25,000 distinct RNA species in human fibroblasts that contained non-colinear exons (a "backsplice") and were reproducibly enriched by exonuclease degradation of linear RNA. These RNAs were validated as circular RNA (ecircRNA), rather than linear RNA, and were more stable than associated linear mRNAs in vivo. In some cases, the abundance of circular molecules exceeded that of associated linear mRNA by >10-fold. By conservative estimate, we identified ecircRNAs from 14.4% of actively transcribed genes in human fibroblasts. Application of this method to murine testis RNA identified 69 ecircRNAs in precisely orthologous locations to human circular RNAs. Of note, paralogous kinases HIPK2 and HIPK3 produce abundant ecircRNA from their second exon in both humans and mice. Though HIPK3 circular RNAs contain an AUG translation start, it and other ecircRNAs were not bound to ribosomes. Circular RNAs could be degraded by siRNAs and, therefore, may act as competing endogenous RNAs. Bioinformatic analysis revealed shared features of circularized exons, including long bordering introns that contained complementary ALU repeats. These data show that ecircRNAs are abundant, stable, conserved and nonrandom products of RNA splicing that could be involved in control of gene expression.
[本文引用: 3]
DOI:10.1371/journal.pgen.1003777URLPMID:24039610 [本文引用: 3]
Thousands of loci in the human and mouse genomes give rise to circular RNA transcripts; at many of these loci, the predominant RNA isoform is a circle. Using an improved computational approach for circular RNA identification, we found widespread circular RNA expression in Drosophila melanogaster and estimate that in humans, circular RNA may account for 1% as many molecules as poly(A) RNA. Analysis of data from the ENCODE consortium revealed that the repertoire of genes expressing circular RNA, the ratio of circular to linear transcripts for each gene, and even the pattern of splice isoforms of circular RNAs from each gene were cell-type specific. These results suggest that biogenesis of circular RNA is an integral, conserved, and regulated feature of the gene expression program. Last year, we reported that circular RNA isoforms, previously thought to be very rare, are actually a pervasive feature of eukaryotic gene expression programs; indeed, the major RNA isoform from hundreds of human genes is a circle. Previous novel RNA species that initially appeared to be special cases, of dubious biological significance, have subsequently proved to have critical, conserved biological roles. An almost universal characteristic of regulatory macromolecules is that they are themselves regulated during development and differentiation. Here, we show that the repertoire of genes expressing circular RNA, the relative levels of circular: linear transcripts from each gene, and even the pattern of splice isoforms of circular RNAs from each gene were cell-type specific, including examples of striking regulation. In humans, we estimate that circular RNA may account for about 1% as many molecules as poly(A) RNA. The ubiquity of circular RNA and its specific regulation could significantly alter our perspective on post-transcriptional regulation and the roles that RNA can play in the cell.
DOI:10.1002/1873-3468.12500URLPMID:27878987 [本文引用: 3]
Circular RNA (circRNA), a class of RNAs with circular structure, has received little attention until recently, when some new features and functions were discovered. In the present study, we sequenced circRNAs in zebrafish (Danio rerio) and identified 3868 circRNAs using three algorithms (find_circ, CIRI, segemehl). The analysis of microRNA target sites on circRNAs shows that some circRNAs may function as miRNA sponges. Furthermore, we identified the existence of reverse complementary sequences in the flanking regions of only 25 (2.64%) exonic circRNAs, indicating that the mechanism of zebrafish exonic circRNA biogenesis might be different from that in mammals. Moreover, 1122 (29%) zebrafish circRNA sequences showed homology with human, mouse and coelacanth circRNAs.
DOI:10.1261/rna.052282.115URLPMID:26464523 [本文引用: 3]
Abstract Various stable circular RNAs (circRNAs) are newly identified to be the abundance of noncoding RNAs in Archaea, Caenorhabditis elegans, mice, and humans through high-throughput deep sequencing coupled with analysis of massive transcriptional data. CircRNAs play important roles in miRNA function and transcriptional controlling by acting as competing endogenous RNAs or positive regulators on their parent coding genes. However, little is known regarding circRNAs in plants. Here, we report 2354 rice circRNAs that were identified through deep sequencing and computational analysis of ssRNA-seq data. Among them, 1356 are exonic circRNAs. Some circRNAs exhibit tissue-specific expression. Rice circRNAs have a considerable number of isoforms, including alternative backsplicing and alternative splicing circularization patterns. Parental genes with multiple exons are preferentially circularized. Only 484 circRNAs have backsplices derived from known splice sites. In addition, only 92 circRNAs were found to be enriched for miniature inverted-repeat transposable elements (MITEs) in flanking sequences or to be complementary to at least 18-bp flanking intronic sequences, indicating that there are some other production mechanisms in addition to direct backsplicing in rice. Rice circRNAs have no significant enrichment for miRNA target sites. A transgenic study showed that overexpression of a circRNA construct could reduce the expression level of its parental gene in transgenic plants compared with empty-vector control plants. This suggested that circRNA and its linear form might act as a negative regulator of its parental gene. Overall, these analyses reveal the prevalence of circRNAs in rice and provide new biological insights into rice circRNAs. 脗漏 2015 Lu et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
DOI:10.1093/nar/gkr1009URLPMID:3326292962175459219598570825 [本文引用: 2]
Circular RNA forms had been described in all domains of life. Such RNAs were shown to have diverse biological functions, including roles in the life cycle of viral and viroid genomes, and in maturation of permuted tRNA genes. Despite their potentially important biological roles, discovery of circular RNAs has so far been mostly serendipitous. We have developed circRNA-seq, a combined experimental/computational approach that enriches for circular RNAs and allows profiling their prevalence in a whole-genome, unbiased manner. Application of this approach to the archaeon Sulfolobus solfataricus P2 revealed multiple circular transcripts, a subset of which was further validated independently. The identified circular RNAs included expected forms, such as excised tRNA introns and rRNA processing intermediates, but were also enriched with non-coding RNAs, including C/D box RNAs and RNase P, as well as circular RNAs of unknown function. Many of the identified circles were conserved in Sulfolobus acidocaldarius, further supporting their functional significance. Our results suggest that circular RNAs, and particularly circular non-coding RNAs, are more prevalent in archaea than previously recognized, and might have yet unidentified biological roles. Our study establishes a specific and sensitive approach for identification of circular RNAs using RNA-seq, and can readily be applied to other organisms.
[本文引用: 4]
DOI:10.1016/j.celrep.2014.10.062URLPMID:25544350 [本文引用: 2]
Westholm et al. annotate Drosophila circular RNAs from a massive collection of total RNA-seq data, providing insights into their biogenesis and function. In particular, circularizing exons are predominantly associated with long flanking introns, are preferred locations of conserved coding miRNA sites, and accumulate to highest levels in the aging CNS.
DOI:10.1016/j.ibmb.2017.09.003URLPMID:28918159 [本文引用: 2]
Bombyx mori is an economically important holometabolous lepidopteran insect. In B. mori endogenous noncoding RNAs such as microRNAs (miRNAs) and Piwi-interacting RNAs play crucial biological functions in metamorphosis and sex determination. In addition, circular RNAs (circRNAs) have been recently identified as noncoding RNAs in most common model organisms and show potential as gene regulators. However, to date, there have been few studies on the circRNAs present in the B. mori genome conducted to date. Here, we identified 3916 circRNAs by deep circular transcriptome sequencing using the silk gland of B. mori . 3155 circRNAs were found to be derived from 1727 parental genes. The circRNAs displayed tissue-specific expression between the middle silk gland (MSG) and posterior silk gland (PSG), with 2532 and 880 being upregulated circRNAs in the MSG and PSG, respectively. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses showed that the parental genes from the MSG and PSG were generally annotated to similar categories and pathways. The interaction network of circRNAs and miRNAs showed that circRNAs might act as miRNA sponges or interact with miRNAs in some other way. Overall, the results revealed the complicated patterns of circRNAs in the B. mori silk gland providing a new angle from which to explore the mechanisms of complex gene regulation and efficient silk protein synthesis.
URLPMID:29709513 [本文引用: 3]
Abstract Circular RNAs (circRNAs) are non-coding RNAs newly identified and play important roles in RNA regulation. The mechanism and function of circRNAs have been reported in some species. However, little is known regarding circRNAs in honey bees. In this study, we analyzed circRNAs through bioinformatics, and predicted 12,211 circRNAs in the ovary of honey bee queens. 1340, 175 and 100 circRNAs were differentially expressed in comparisons of egg-laying queens vs virgin queens, egg-laying inhibited queens vs egg-laying queens and egg-laying recovery queens vs egg-laying inhibited queens. Further, functional annotation of differentially expressed circRNAs revealed several pathways that are closely related to ovary activation and oviposition, including insulin secretion and calcium signaling pathways. Moreover, the potential interactions among circRNAs, miRNAs, lncRNAs and mRNAs were investigated. Ame_circ_0005197 and ame_circ_0016640 were observed to sponge several reproductive related miRNAs. These findings demonstrate that circRNAs have potential effects in ovary activation and oviposition of honey bees.
DOI:10.1093/nar/gkw027URLPMID:26861625 [本文引用: 2]
Most RNAs generated by the human genome have no protein-coding ability and are termed non-coding RNAs. Among these include circular RNAs, which include exonic circular RNAs (circRNA), mainly found in the cytoplasm, and intronic RNAs (ciRNA), predominantly detected in the nucleus. The biological functions of circular RNAs remain largely unknown, although ciRNAs have been reported to promote gene transcription, while circRNAs may function as microRNA sponges. We demonstrate that the circular RNA circ-Foxo3 was highly expressed in non-cancer cells and were associated with cell cycle progression. Silencing endogenous circ-Foxo3 promoted cell proliferation. Ectopic expression of circ-Foxo3 repressed cell cycle progression by binding to the cell cycle proteins cyclin-dependent kinase 2 (also known as cell division protein kinase 2 or CDK2) and cyclin-dependent kinase inhibitor 1 (or p21), resulting in the formation of a ternary complex. Normally, CDK2 interacts with cyclin A and cyclin E to facilitate cell cycle entry, while p21works to inhibit these interactions and arrest cell cycle progression. The formation of this circ-Foxo3-p21-CDK2 ternary complex arrested the function of CDK2 and blocked cell cycle progression.
DOI:10.1136/annrheumdis-2017-212056URLPMID:29343508 [本文引用: 2]
ObjectivesCircular RNAs (circRNAs) have been proven to function as competing endogenous RNAs to interact with microRNAs (miRNAs) and influence the expression of miRNA target mRNAs. In this study, we investigated whether circRNAs could act as competing endogenous RNAs to regulate the pathological process of intervertebral disc degeneration (IVDD).MethodsThe role and mechanism of a circRNA, circVMA21, in IVDD were explored in nucleus pulposus (NP) cells and degenerative NP tissues from patients and rat models. The interaction between circVMA21 and miR-200c as well as the target mRNA, X linked inhibitor-of-apoptosis protein (XIAP), was examined.ResultsThe decreased expression of XIAP in the inflammatory cytokines-treated NP cells and the degenerative NP tissues was directly associated with excessive apoptosis and imbalance between anabolic and catabolic factors of extracellular matrix. miR-200c regulated NP cell viability and functions through inhibiting XIAP. circVMA21 acted as a sponge of miR-200c and functioned in NP cells through targeting miR-200c and XIAP. Intradiscal injection of circVMA21 alleviated IVDD in the rat model.ConclusionsCircVMA21 could alleviate inflammatory cytokines-induced NP cell apoptosis and imbalance between anabolism and catabolism of extracellular matrix through miR-200c-XIAP pathway. It provides a potentially effective therapeutic strategy for IVDD.
DOI:10.1038/ncomms11215URLPMID:4823868 [本文引用: 4]
Circular RNAs (circRNAs) represent a class of widespread and diverse endogenous RNAs that may regulate gene expression in eukaryotes. However, the regulation and function of human circRNAs remain largely unknown. Here we generate ribosomal-depleted RNA sequencing data from six normal tissues and seven cancers, and detect at least 27,000 circRNA candidates. Many of these circRNAs are differently expressed between the normal and cancerous tissues. We further characterize one abundant circRNA derived from Exon2 of theHIPK3gene, termed circHIPK3. The silencing of circHIPK3 but notHIPK3mRNA significantly inhibits human cell growth. Via a luciferase screening assay, circHIPK3 is observed to sponge to 9 miRNAs with 18 potential binding sites. Specifically, we show that circHIPK3 directly binds to miR-124 and inhibits miR-124 activity. Our results provide evidence that circular RNA produced from precursor mRNA may have a regulatory role in human cells. Circular RNAs are formed from exon back-splicing, the significance of these endogenous RNAs is beginning to be unraveled. Here, the authors identify thousands of circular RNAs differentially expressed between normal and cancer tissues and show that an abundant circular RNA generated fromHIPK3regulates cell growth.
[本文引用: 1]
[本文引用: 1]
URL [本文引用: 1]
为探讨意大利蜜蜂(简称意蜂)幼虫肠道发育过程中的基因表达谱和基因差异表达信息,研究基于前期已获得的高质量的意蜂幼虫肠道转录组数据,利用STEM软件对意蜂4、5和6日龄幼虫肠道的差异表达基因(DEGs)进行趋势分析,进一步通过相关生物学软件对显著上调趋势包含的DEGs进行GO与KEGG代谢通路富集分析及表达量聚类分析。趋势分析结果显示,5 920个DEGs共聚类在8个基因表达模式,其中935个DEGs聚类在1个显著上调趋势。GO富集分析结果显示,显著上调趋势包含DEGs富集于39个GO条目,富集基因数最多的为结合,其次是细胞进程和单组织进程。KEGG代谢通路富集分析结果显示,显著上调趋势包含DEGs富集于94个代谢通路,富集基因数最多的是嘌呤代谢、内质网蛋白质加工和内吞作用。表达量聚类分析结果显示,随着意蜂幼虫肠道发育时间的延长,代谢和免疫相关通路富集基因的表达水平逐渐增强。研究提供了意蜂幼虫肠道发育过程的基因表达谱和基因差异表达信息,揭示了基因表达规律,为深入研究意蜂幼虫肠道发育的分子机制打下了一定基础。
URL [本文引用: 1]
为探讨意大利蜜蜂(简称意蜂)幼虫肠道发育过程中的基因表达谱和基因差异表达信息,研究基于前期已获得的高质量的意蜂幼虫肠道转录组数据,利用STEM软件对意蜂4、5和6日龄幼虫肠道的差异表达基因(DEGs)进行趋势分析,进一步通过相关生物学软件对显著上调趋势包含的DEGs进行GO与KEGG代谢通路富集分析及表达量聚类分析。趋势分析结果显示,5 920个DEGs共聚类在8个基因表达模式,其中935个DEGs聚类在1个显著上调趋势。GO富集分析结果显示,显著上调趋势包含DEGs富集于39个GO条目,富集基因数最多的为结合,其次是细胞进程和单组织进程。KEGG代谢通路富集分析结果显示,显著上调趋势包含DEGs富集于94个代谢通路,富集基因数最多的是嘌呤代谢、内质网蛋白质加工和内吞作用。表达量聚类分析结果显示,随着意蜂幼虫肠道发育时间的延长,代谢和免疫相关通路富集基因的表达水平逐渐增强。研究提供了意蜂幼虫肠道发育过程的基因表达谱和基因差异表达信息,揭示了基因表达规律,为深入研究意蜂幼虫肠道发育的分子机制打下了一定基础。
[本文引用: 4]
[本文引用: 4]
DOI:10.1186/gb-2013-14-4-r36URLPMID:4053844 [本文引用: 1]
TopHat is a popular spliced aligner for RNA-sequence (RNA-seq) experiments. In this paper, we describe TopHat2, which incorporates many significant enhancements to TopHat. TopHat2 can align reads of various lengths produced by the latest sequencing technologies, while allowing for variable-length indels with respect to the reference genome. In addition to de novo spliced alignment, TopHat2 can align reads across fusion breaks, which can occur after genomic translocations. TopHat2 combines the ability to identify novel splice sites with direct mapping to known transcripts, producing sensitive and accurate alignments, even for highly repetitive genomes or in the presence of pseudogenes. TopHat2 is available at http://ccb.jhu.edu/software/tophat .
[本文引用: 2]
DOI:10.1093/bioinformatics/btp612URL [本文引用: 1]
DOI:10.1038/nsmb.2959URLPMID:25664725 [本文引用: 2]
Abstract Noncoding RNAs (ncRNAs) have numerous roles in development and disease, and one of the prominent roles is to regulate gene expression. A vast number of circular RNAs (circRNAs) have been identified, and some have been shown to function as microRNA sponges in animal cells. Here, we report a class of circRNAs associated with RNA polymerase II in human cells. In these circRNAs, exons are circularized with introns 'retained' between exons; we term them exon-intron circRNAs or EIciRNAs. EIciRNAs predominantly localize in the nucleus, interact with U1 snRNP and promote transcription of their parental genes. Our findings reveal a new role for circRNAs in regulating gene expression in the nucleus, in which EIciRNAs enhance the expression of their parental genes in cis, and highlight a regulatory strategy for transcriptional control via specific RNA-RNA interaction between U1 snRNA and EIciRNAs.
DOI:10.1186/gb-2009-10-3-r25URLPMID:2690996 [本文引用: 1]
Bowtie is an ultrafast, memory-efficient alignment program for aligning short DNA sequence reads to large genomes. For the human genome, Burrows-Wheeler indexing allows Bowtie to align more than 25 million reads per CPU hour with a memory footprint of approximately 1.3 gigabytes. Bowtie extends previous Burrows-Wheeler techniques with a novel quality-aware backtracking algorithm that permits mismatches. Multiple processor cores can be used simultaneously to achieve even greater alignment speeds. Bowtie is open source http://bowtie.cbcb.umd.edu .
[本文引用: 1]
DOI:10.1093/bioinformatics/btq675URLPMID:21149340 [本文引用: 1]
ABSTRACTSummary: Cytoscape is a popular bioinformatics package for biological network visualization and data integration. Version 2.8 introduces two powerful new features--Custom Node Graphics and Attribute Equations--which can be used jointly to greatly enhance Cytoscape's data integration and visualization capabilities. Custom Node Graphics allow an image to be projected onto a node, including images generated dynamically or at remote locations. Attribute Equations provide Cytoscape with spreadsheet-like functionality in which the value of an attribute is computed dynamically as a function of other attributes and network properties.Availability and implementation: Cytoscape is a desktop Java application released under the Library Gnu Public License (LGPL). Binary install bundles and source code for Cytoscape 2.8 are available for download from http://cytoscape.org.Contact: msmoot@ucsd.edu
DOI:10.1080/15476286.2017.1411461URLPMID:29268657 [本文引用: 1]
Abstract The pathogenesis of Bombyx mori cytoplasmic polyhedrosis virus (BmCPV) infection is unclear, although accumulating evidence indicates that circular RNAs (circRNAs), which act as competing endogenous RNAs or positive regulators, play important roles in regulating gene expression in eukaryotes and, thus, may play a role in BmCPV infections. To explore the expression and biological functions of circRNAs in the silkworm midgut following BmCPV infection, silkworm circRNA expression profiles of normal midgut tissue (control) and BmCPV-infected midgut tissue (test) were determined using high-through sequencing. A total of 9,753 and 7,475circRNAs were detected from the control and test samples, respectively. The two samples shared 6,085 circRNAs, while 646 and 737 circRNAs were expressed specifically in the control and test samples, respectively. A total of 3,638 circRNAs were shown to be differentially expressed, and 400 circRNAs were substantially differentially expressed with a fold-change 2.0 (p< 0.05 and a false discover rate < 0.05), of which 294 were up-regulated and 106 were down-regulated following infection. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses were conducted to determine the principal functions of the substantially differentially regulated genes. circRNA-miRNA interaction networks were constructed based on a correlation analysis between the differentially expressed circRNAs and the nature of their microRNA (miRNA) binding sites. The network inferred that 13 miRNAs interacting with 193 circRNAs were among the 300 most abundant relationships. bmo-miR-3389-5p, bmo-miR-745-3p, and bmo-miR-3262 were related to 30, 34, and 34 circRNAs, respectively. circRNA_8115, circRNA_9444, circRNA_4553, circRNA_0827, and circRNA_6649 contained six, five, four, four, and four miRNA binding sites, respectively. We further found that alternative circularization of circRNAs is a common feature in silkworms and that the junction sites of many silkworm circRNAs are flanked by canonical GT/AG splicing signals. Our study is the first to show the circRNA response to virus infection. Thus, it provides a novel perspective on circRNA-miRNA interactions during BmCPV pathogenesis, and it lays the foundation for future research of the potential roles of circRNAs in BmCPV pathogenesis.
DOI:10.3168/jds.2015-10381URLPMID:27040791 [本文引用: 1]
Recent studies have revealed that, in addition to hormones and other protein factors, noncoding RNA molecules play an important regulatory role in milk protein synthesis. Circular RNAs (circRNAs) are universally expressed noncoding RNA species that have been proposed recently to regulate the expression of their parental genes. In the present study, the deep RNA-sequencing technique known as RNA-seq was used to compare expression profiles of circRNAs from 2 pooled RNA samples from cow mammary gland on d 90 and 250 postpartum and to identify the key circRNAs involved in lactation. A total of 4,804 and 4,048 circRNAs were identified in the cow mammary gland on d 90 and 250 postpartum, respectively, of which only 2,231 circRNAs were co-expressed at both lactation stages, suggesting high stage specificity in the circRNAs. The enrichment of some Gene Ontology terms for the circRNA parental genes differed between lactation stages. Among the top 10 enriched Gene Ontology terms, vesicle, endoplasmic reticulum, and mitochondrial lumen were more common on lactation d 90. All 4 casein-coding genes (CSN1S1,CSN1S2,CSN2, andCSN3) produced circRNAs in the cattle mammary gland. In total, 80 circRNAs were identified from these 4 genes; circRNAs fromCSN1S1had very high abundance, and 3 of them accounted for 36% of all the circRNAs expressed in the mammary gland on lactation d 90. Three circRNAs fromCSN1S1, 1 circRNA fromCSN1S2, and 1 circRNA fromCSN2were all more highly expressed on lactation d 90 than on lactation d 250, as confirmed by quantitative PCR. These circRNAs had several target sites for the microRNA miR-2284 family and were predicted to targetCSN1S1andCSN2mRNA, suggesting their potential involvement in regulating expression of the casein genes.
DOI:10.5713/ajas.17.0651URLPMID:29268579 [本文引用: 1]
An experiment was conducted to identify and characterize the circular RNA expression and metabolic characteristics in the liver of Jinhua pigs and Landrace pigs. Three Jinhua pigs and three Landrace pigs respectively at 70-day were slaughtered to collect the liver tissue samples. Immediately after slaughter, blood samples were taken to detect serum biochemical indicators. Total RNA extracted from liver tissue samples were used to prepare the library and then sequence on HiSeq 2500. Bioinformatic methods were employed to analyze sequence data to identify the circRNAs and predict the potential roles of differentially expressed circRNAs between the two breeds. Significant differences in physiological and biochemical traits were observed between growing Jinhua and Landrace pigs. We identified 84,864 circRNA candidates in two breeds and 366 circRNAs were detected as significantly differentially expressed. Their host genes are involved in lipid biosynthetic and metabolic processes according to the gene ontology analysis and associated with metabolic pathways. Our research represents the first description of circRNA profiles in the porcine liver from two divergent phenotype pigs. The predicted miRNA-circRNA interaction provides important basis for miRNA-circRNA relationships in the porcine liver. These data expand the repertories of porcine circRNA and are conducive to understanding the possible molecular mechanisms involved in miRNA and circRNA. Our study provides basic data for further research of the biological functions of circRNAs in the porcine liver.
DOI:10.18632/oncotarget.16442URLPMID:5471026 [本文引用: 1]
Avian leukosis virus subgroup (ALV-J) is an oncogenic neoplasm-inducing retrovirus that causes significant economic losses in the poultry industry. Recent studies have demonstrated circular RNAs (circRNAs) are implicated in pathogenic processes; however, no research has indicated circRNAs are involved in resistance to disease. In this study, over 1800 circRNAs were detected by circRNA sequencing of liver tissues from ALV-J-resistant (n= 3) and ALV-J-susceptible chickens (n= 3). 32 differentially expressed circRNAs were selected for analyzing including 12 upregulated in ALV-J-resistant chickens and 20 upregulated in ALV-J-susceptible chickens, besides, the top five microRNAs (miRNAs) for 12 upregulated circRNAs in ALV-J-resistant chickens were analyzed. Gene ontology and KEGG pathway analyses were performed for miRNA target genes, the predicted genes were mainly involved in immune pathways. This study provides the first evidence that circRNA alterations are involved in resistance to ALV-J-induced tumor formation. We propose circRNAs may help to mediate tumor induction and development in chickens.
DOI:10.1186/s13059-015-0706-1URLPMID:26201400 [本文引用: 1]
Circular RNAs (circRNAs) are a new class of non-polyadenylated non-coding RNAs that may play important roles in many biological processes. Here we develop a single-cell universal poly(A)-independent...
DOI:10.3390/ijms18091977URLPMID:28906455 [本文引用: 1]
Grass carp hemorrhagic disease, caused by the grass carp reovirus (GCRV), is a major disease that hampers the development of grass carp aquaculture in China. The mechanism underlying GCRV infection is still largely unknown. Circular RNAs (circRNAs) are important regulators involved in various biological processes. In the present study, grass carp were infected with GCRV, and spleen samples were collected at 0 (control), 1, 3, 5, and 7 days post-infection (dpi). Samples were used to construct and sequence circRNA libraries, and a total of 5052 circRNAs were identified before and after GCRV infection, of which 41 exhibited differential expression compared with controls. Many parental genes of the differentially expressed circRNAs are involved in metal ion binding, protein ubiquitination, enzyme activity, and nucleotide binding. Moreover, 72 binding miRNAs were predicted from the differentially expressed circRNAs, of which eight targeted genes were predicted to be involved in immune responses, blood coagulation, hemostasis, and complement and coagulation cascades. Upregulation of these genes may lead to endothelial and blood cell damage and hemorrhagic symptoms. Our results indicate that an mRNA iRNA ircRNA network may be present in grass carp infected with GCRV, providing new insight into the mechanism underlying grass carp reovirus infection.
DOI:10.3389/fpls.2016.00776URLPMID:27375638 [本文引用: 1]
RNA circularization made by head-to-tail back-splicing events is involved in the regulation of gene expression from transcriptional to post-translational levels. By exploiting RNA-Seq data and down-stream analysis, we shed light on the importance of circular RNAs in plants. The results introduce circular RNAs as novel interactors in the regulation of gene expression in plants and imply the comprehensiveness of this regulatory pathway by identifying circular RNAs for a diverse set of genes. These genes are involved in several aspects of cellular metabolism as hormonal signaling, intracellular protein sorting, carbohydrate metabolism and cell-wall biogenesis, respiration, amino acid biosynthesis, transcription and translation, and protein ubiquitination. Additionally, these parental loci of circular RNAs, from both nuclear and mitochondrial genomes, encode for different transcript classes including protein coding transcripts, microRNA, rRNA, and long non-coding/microprotein coding RNAs. The results shed light on the mitochondrial exonic circular RNAs and imply the importance of circular RNAs for regulation of mitochondrial genes. Importantly, we introduce circular RNAs in barley and elucidate their cellular-level alterations across tissues and in response to micronutrients iron and zinc. In further support of circular RNAs' functional roles in plants, we report several cases where fluctuations of circRNAs do not correlate with the levels of their parental-loci encoded linear transcripts.
DOI:10.1002/1873-3468.12440URLPMID:27685607 [本文引用: 1]
A new regulatory class of small endogenous RNAs called circular RNAs (circRNAs) has been described as miRNA sponges in animals. Using 16 Arabidopsis thaliana RNA-Seq data sets, we identified 803 circRNAs in RNase R-/non-RNase R-treated samples. The results revealed the following features: Canonical and non-canonical splicing can generate circRNAs; chloroplasts are a hotspot for circRNA generation; furthermore, limited complementary sequences exist not only in introns, but also in the sequences flanking splice sites. The latter finding suggests that multiple combinations between complementary sequences may facilitate the formation of the circular structure. Our results contribute to a better understanding of this novel class of plant circRNAs. This article is protected by copyright. All rights reserved.
[本文引用: 1]
[本文引用: 1]
DOI:10.3969/j.issn.1006-267x.2017.04.013URL [本文引用: 1]
本试验旨在研究不同越冬饲料对蜜蜂中肠消化酶活性、组织发育状态以及抗氧化酶基因表达的影响,以便为广大蜂农选择越冬饲料提供参考。在2015年10月底选取群势相当的本地意大利蜜蜂(Apis mellifera L.)越冬蜂群(群内无储备越冬饲料脾)9群(5框蜂/群),随机分为3个试验组(3群/组),从11月2号开始分别以蜂蜜(蜂蜜组)、果葡糖浆(果葡糖浆组)和白砂糖水(白砂糖∶水=2∶1,蔗糖组)为越冬饲料进行饲喂,饲喂至11月下旬蜂群进入越冬期。整个试验期为2015年10月底至2016年3月初。分别于越冬前(11月初)、越冬中期(1月初)和越冬后(3月初)采集蜜蜂中肠,测定中肠消化酶(淀粉酶、蔗糖酶和蛋白酶)活性,在越冬中期采集蜜蜂中肠用于中肠组织发育状态和抗氧化酶基因[超氧化物歧化酶1(Sod1)、超氧化物歧化酶2(Sod2)和过氧化氢酶(CAT)]相对表达量等指标的检测。结果表明:在越冬中期,果葡糖浆组和蜂蜜组的蜜蜂中肠内淀粉酶活性显著高于蔗糖组(P0.05)。在越冬中期和越冬后,饲喂不同越冬饲料的蜜蜂中肠内蔗糖酶活性差异不显著(P0.05)。在越冬中期,果葡糖浆组的蜜蜂中肠内蛋白酶活性显著高于蔗糖组和蜂蜜组(P0.05)。蜂蜜组和蔗糖组的蜜蜂中肠肠壁厚度极显著大于果葡糖浆组(P0.01),且蜂蜜组和蔗糖组的蜜蜂中肠隐窝深度也极显著大于果葡糖浆组(P0.01)。在越冬中期,蔗糖组的蜜蜂中肠Sod1基因的相对表达量显著高于蜂蜜组和果葡糖浆组(P0.05)。由此得出,蜂蜜能够提高越冬蜂中肠消化酶活性,蜂蜜和白砂糖有利于越冬蜂中肠组织的发育,而且白砂糖能够提高越冬蜂中肠抗氧化酶基因的表达。因此,蜂蜜和白砂糖比果葡糖浆更适合作为蜜蜂的越冬饲料。
DOI:10.3969/j.issn.1006-267x.2017.04.013URL [本文引用: 1]
本试验旨在研究不同越冬饲料对蜜蜂中肠消化酶活性、组织发育状态以及抗氧化酶基因表达的影响,以便为广大蜂农选择越冬饲料提供参考。在2015年10月底选取群势相当的本地意大利蜜蜂(Apis mellifera L.)越冬蜂群(群内无储备越冬饲料脾)9群(5框蜂/群),随机分为3个试验组(3群/组),从11月2号开始分别以蜂蜜(蜂蜜组)、果葡糖浆(果葡糖浆组)和白砂糖水(白砂糖∶水=2∶1,蔗糖组)为越冬饲料进行饲喂,饲喂至11月下旬蜂群进入越冬期。整个试验期为2015年10月底至2016年3月初。分别于越冬前(11月初)、越冬中期(1月初)和越冬后(3月初)采集蜜蜂中肠,测定中肠消化酶(淀粉酶、蔗糖酶和蛋白酶)活性,在越冬中期采集蜜蜂中肠用于中肠组织发育状态和抗氧化酶基因[超氧化物歧化酶1(Sod1)、超氧化物歧化酶2(Sod2)和过氧化氢酶(CAT)]相对表达量等指标的检测。结果表明:在越冬中期,果葡糖浆组和蜂蜜组的蜜蜂中肠内淀粉酶活性显著高于蔗糖组(P0.05)。在越冬中期和越冬后,饲喂不同越冬饲料的蜜蜂中肠内蔗糖酶活性差异不显著(P0.05)。在越冬中期,果葡糖浆组的蜜蜂中肠内蛋白酶活性显著高于蔗糖组和蜂蜜组(P0.05)。蜂蜜组和蔗糖组的蜜蜂中肠肠壁厚度极显著大于果葡糖浆组(P0.01),且蜂蜜组和蔗糖组的蜜蜂中肠隐窝深度也极显著大于果葡糖浆组(P0.01)。在越冬中期,蔗糖组的蜜蜂中肠Sod1基因的相对表达量显著高于蜂蜜组和果葡糖浆组(P0.05)。由此得出,蜂蜜能够提高越冬蜂中肠消化酶活性,蜂蜜和白砂糖有利于越冬蜂中肠组织的发育,而且白砂糖能够提高越冬蜂中肠抗氧化酶基因的表达。因此,蜂蜜和白砂糖比果葡糖浆更适合作为蜜蜂的越冬饲料。
[本文引用: 1]
[本文引用: 1]
DOI:10.1242/dev.045500URLPMID:21138973 [本文引用: 1]
Abstract The Hippo pathway has emerged as a conserved signaling pathway that is essential for the proper regulation of organ growth in Drosophila and vertebrates. Although the mechanisms of signal transduction of the core kinases Hippo/Mst and Warts/Lats are relatively well understood, less is known about the upstream inputs of the pathway and about the downstream cellular and developmental outputs. Here, we review recently discovered mechanisms that contribute to the dynamic regulation of Hippo signaling during Drosophila and vertebrate development. We also discuss the expanding diversity of Hippo signaling functions during development, discoveries that shed light on a complex regulatory system and provide exciting new insights into the elusive mechanisms that regulate organ growth and regeneration.
DOI:10.1016/j.cub.2007.10.039URLPMID:17980593 [本文引用: 1]
The mechanisms that regulate mammalian organ size are poorly understood. It is unclear whether the pathways that control organ size also impinge on stem/progenitor cells. A highly expressed gene in stem cells is [1], the ortholog of [2]. Mutations in components of this pathway produce tissue overgrowth phenotypes in the fly whereas mammalian orthologs, like salvador [3], merlin [4], [5], and , have been implicated in tumorigenesis. We report here that YAP1 increases organ size and causes aberrant tissue expansion in mice. YAP1 activation reversibly increases liver size more than 4-fold. In the intestine, expression of endogenous YAP1 is restricted to the progenitor/stem cell compartment, and activation of YAP1 expands multipotent undifferentiated progenitor cells, which differentiate upon cessation of YAP1 expression. YAP1 stimulates Notch signaling, and administration of -secretase inhibitors suppressed the intestinal dysplasia caused by YAP1. Human colorectal cancers expressing higher levels of YAP1 share molecular aspects with YAP1-induced dysplastic growth in the mouse. Our data show that the Hippo signaling pathway regulates organ size in mammals and can act on stem cell compartments, indicating a potential link between stem/progenitor cells, organ size, and cancer.
DOI:10.1128/MCB.01034-07URLPMID:17785439 [本文引用: 1]
The Wnt signaling pathway is deregulated in over 90% of human colorectal cancers. beta-Catenin, the central signal transducer of the Wnt pathway, can directly modulate gene expression by interacting with transcription factors of the TCF/LEF family. In the present study we investigate the role of Wnt signaling in the homeostasis of intestinal epithelium by using tissue-specific, inducible beta-catenin gene ablation in adult mice. Block of Wnt/beta-catenin signaling resulted in rapid loss of transient-amplifying cells and crypt structures. Importantly, intestinal stem cells were induced to terminally differentiate upon deletion of beta-catenin, resulting in a complete block of intestinal homeostasis and fatal loss of intestinal function. Transcriptional profiling of mutant crypt mRNA isolated by laser capture microdissection confirmed those observations and allowed us to identify genes potentially responsible for the functional preservation of intestinal stem cells. Our data demonstrate an essential requirement of Wnt/beta-catenin signaling for the maintenance of the intestinal epithelium in the adult organism. This challenges attempts to target aberrant Wnt signaling as a new therapeutic strategy to treat colorectal cancer.
DOI:10.3321/j.issn:1000-3282.2003.02.004URL [本文引用: 1]
Wnt蛋白及其受体、调节蛋白等一起组成了复杂的信号通路 ,调控细胞的分化 ,参与发育的多个重要过程 .近来的研究表明 :Wnt信号通路也是调节哺乳动物生殖系统正常发育所必需 .它主要参与了缪勒氏管及其派生器官的形成 ,调控卵泡的发育、排卵及黄体化 ,另外与正常妊娠的建立以及妊娠过程中乳腺的变化也有关
DOI:10.3321/j.issn:1000-3282.2003.02.004URL [本文引用: 1]
Wnt蛋白及其受体、调节蛋白等一起组成了复杂的信号通路 ,调控细胞的分化 ,参与发育的多个重要过程 .近来的研究表明 :Wnt信号通路也是调节哺乳动物生殖系统正常发育所必需 .它主要参与了缪勒氏管及其派生器官的形成 ,调控卵泡的发育、排卵及黄体化 ,另外与正常妊娠的建立以及妊娠过程中乳腺的变化也有关
DOI:10.1016/j.ceb.2012.12.006URLPMID:23312716 [本文引用: 1]
Tissue regeneration is vital to the form and function of an organ. At the core of an organs ability to self-renew is the stem cell, which maintains homeostasis, and repopulates injured or aged tissue. Tissue damage can dramatically change the dimensions of an organ, and during regeneration, an organ must halt growth once the original tissue dimensions have been restored. Therefore, stem cells must give rise to the appropriate number of differentiated progeny to achieve homeostasis. How this tissue-size checkpoint is regulated and how tissue size information relayed to stem cell compartments is unclear, however, it is likely that these mechanisms are altered during the course of tumorigenesis. An emerging signaling cascade, the Hippo Signaling Pathway, is a broadly conserved potent organ size regulator [1]. However, this pathway does not act alone. A number of examples demonstrate crosstalk between Hippo and other signaling pathways including Wnt, Tgf尾 and Notch, with implications for stem cell biology. Here, we focus on these interactions primarily in the context of well characterized stem cell populations.
DOI:10.1016/j.jip.2009.06.018URLPMID:19909969 [本文引用: 1]
Chalkbrood is a fungal disease of honey bee brood caused by Ascosphaera apis. This disease is now found throughout the world, and there are indications that chalkbrood incidence may be on the rise. In this review we consolidate both historic knowledge and recent scientific findings. We document the worldwide spread of the fungus, which is aided by increased global travel and the migratory nature of many beekeeping operations. We discuss the current taxonomic classification in light of the recent complete reworking of fungal systematics brought on by application of molecular methods. In addition, we discuss epidemiology and pathogenesis of the disease, as well as pathogen biology, morphology and reproduction. New attempts at disease control methods and management tactics are reviewed. We report on research tools developed for identification and monitoring, and also include recent findings on genomic and molecular studies not covered by previous reviews, including sequencing of the A. apis genome and identification of the mating type locus.
DOI:10.1038/icb.2014.52URLPMID:24983458 [本文引用: 1]
Mycobacterium tuberculosis (M. tuberculosis), the causative agent of tuberculosis, is an intracellular bacterium capable of surviving and persisting within host mononuclear cells. The host response against tubercle bacilli is dominated by fine-tuned interaction of innate and adaptive immune responses. Toll-like receptors (TLRs) play a critical role in the formation of this immune response by facilitating in elaboration of protective T helper type 1 (Th1) cytokines and microbicidal molecules, but the intracellular persistence of M. tuberculosis in the phagosome and processing and presentation of TLR ligands by host antigen-presenting cell leads to continuous and chronic TLR2 signaling. The prolonged stimulation of TLR ultimately results in elaboration of immunosuppressive cytokines and downregulation of antigen presentation by major histocompatibility complex (MHC) class II and therefore becomes beneficial for M. tuberculosis, resulting in its continued survival inside macrophages. An understanding of the host-pathogen interaction in tuberculosis is important to delineate the mechanisms that can modulate the immune response toward protection. This review focuses on the role of TLRs in immune response and immune evasion and how M. tuberculosis maintains its dominance over the host during infection. A precise understanding of the TLRs and M. tuberculosis interaction will undoubtedly lead to the development of novel therapies to combat tuberculosis.
DOI:10.1016/j.cell.2011.07.014URLPMID:3235919 [本文引用: 1]
Here, we present a unifying hypothesis about how messenger RNAs, transcribed pseudogenes, and long noncoding RNAs “talk” to each other using microRNA response elements (MREs) as letters of a new language. We propose that this “competing endogenous RNA” (ceRNA) activity forms a large-scale regulatory network across the transcriptome, greatly expanding the functional genetic information in the human genome and playing important roles in pathological conditions, such as cancer.
[本文引用: 2]
DOI:10.18632/oncotarget.21748URLPMID:29207676 [本文引用: 1]
Circular RNAs (circRNAs) as a novel type of noncoding RNAs (ncRNAs) are widely studied in the development of human various diseases, including cancer. Here, we found circular RNA hsa_circ_000984 encoded by theCDK6gene was remarkably upregulated in the tissues of colorectal cancer (CRC) patients and in the CRC cell lines. Moreover, high expression level of hsa_circ_000984 was significantly associated with advanced colorectal cancer. Further analysis revealed that hsa_circ_000984 knockdown could inhibit cell proliferation, migration, invasionin vitroand tumor formationin vivoin CRC cell lines. Mechanically, we found that hsa_circ_000984 may act as a competing endogenous RNA (ceRNA) by competitively binding miR-106b and effectively upregulate the expression ofCDK6, thereby inducing a series of malignant phenotypes of tumor cells. Taken together, these observations suggest that the hsa_circ_000984 could mediate the expression of geneCDK6by acting as a ceRNA, which may contribute to a better understanding of between the regulatory miRNA network and CRC pathogenesis.
[本文引用: 2]
DOI:10.1016/j.mod.2006.05.005URL [本文引用: 1]
DOI:10.3389/fgene.2014.00445URLPMID:25566327 [本文引用: 1]
Major developmental transitions in multicellular organisms are driven by steroid hormones. In insects, these, together with juvenile hormone (JH), control development, metamorphosis, reproduction and aging, and are also suggested to play an important role in caste differentiation of social insects. Here, we aimed to determine how EcR transcription and ecdysteroid titers are related during honeybee postembryonic development and what may actually be the role of EcR in caste development of this social insect. In addition, we expected that knocking-down EcR gene expression would give us information on the participation of the respective protein in regulating downstream targets of EcR. We found that in Apis mellifera females, EcR-A is the predominantly expressed variant in postembryonic development, while EcR-B transcript levels are higher in embryos, indicating an early developmental switch in EcR function. During larval and pupal stages, EcR-B expression levels are very low, while EcR-A transcripts are more variable and abundant in workers compared to queens. Strikingly, these transcript levels are opposite to the ecdysteroid titer profile. 20-hydroxyecdysone (20E) application experiments revealed that low 20E levels induce EcR expression during development, whereas high ecdysteroid titers seem to be repressive. By means of RNAi-mediated knockdown (KD) of both EcR transcript variants we detected the differential expression of 234 poly-A(+) transcripts encoding genes such as CYPs, MRJPs and certain hormone response genes (Kr-h1 and ftz-f1). EcR-KD also promoted the differential expression of 70 miRNAs, including highly conserved ones (e.g., miR-133 and miR-375), as well honeybee-specific ones (e.g., miR-3745 and miR-3761). Our results put in evidence a broad spectrum of EcR-controlled gene expression during postembryonic development of honeybees, revealing new facets of EcR biology in this social insect.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}