 ,1, 杜宇,1, 熊翠玲1, 郑燕珍1, 付中民1, 徐国钧1, 王海朋1, 陈华枝1, 耿四海1, 周丁丁1, 石彩云1, 赵红霞2, 陈大福,1
,1, 杜宇,1, 熊翠玲1, 郑燕珍1, 付中民1, 徐国钧1, 王海朋1, 陈华枝1, 耿四海1, 周丁丁1, 石彩云1, 赵红霞2, 陈大福,1Differentially Expressed MicroRNA and Their Regulation Networks During the Developmental Process of Apis mellifera ligustica Larval Gut
GUO Rui,1, DU Yu,1, XIONG CuiLing1, ZHENG YanZhen1, FU ZhongMin1, XU GuoJun1, WANG HaiPeng1, CHEN HuaZhi1, GENG SiHai1, ZHOU DingDing1, SHI CaiYun1, ZHAO HongXia2, CHEN DaFu,1通讯作者:
第一联系人:
责任编辑: 岳梅
收稿日期:2018-05-22接受日期:2018-06-28网络出版日期:2018-11-01
| 基金资助: |
Received:2018-05-22Accepted:2018-06-28Online:2018-11-01

摘要
关键词:
Abstract
Keywords:
PDF (10339KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
郭睿, 杜宇, 熊翠玲, 郑燕珍, 付中民, 徐国钧, 王海朋, 陈华枝, 耿四海, 周丁丁, 石彩云, 赵红霞, 陈大福. 意大利蜜蜂幼虫肠道发育过程中的差异表达 microRNA及其调控网络[J]. 中国农业科学, 2018, 51(21): 4197-4209 doi:10.3864/j.issn.0578-1752.2018.21.018
GUO Rui, DU Yu, XIONG CuiLing, ZHENG YanZhen, FU ZhongMin, XU GuoJun, WANG HaiPeng, CHEN HuaZhi, GENG SiHai, ZHOU DingDing, SHI CaiYun, ZHAO HongXia, CHEN DaFu.
0 引言
【研究意义】微小RNA(microRNA,miRNA)是一类长度约为18—25个核苷酸的非编码RNA(non-coding RNA,ncRNA),通过对mRNA的负调控作用而广泛参与动物[1,2]、植物[3,4]和微生物[5,6]的各类生物学过程。意大利蜜蜂(Apis mellifera ligustica,简称意蜂)是我国养蜂生产中的主要蜂种,在经济创收和生态维持等方面具有重要价值。利用small RNA-seq(sRNA-seq)技术对意蜂4、5和6日龄幼虫肠道进行测序,通过生物信息学分析方法对肠道发育过程中的差异表达miRNA(differentially expressed miRNA,DEmiRNA)进行全面分析,并分别构建上调和下调miRNA的调控网络,对揭示意蜂幼虫肠道发育过程中miRNA的差异表达规律、肠道发育相关关键miRNA的筛选和功能研究以及阐明肠道发育的机理具有重要意义。【前人研究进展】西方蜜蜂(Apis mellifera)基因组[7]的公布为其组学和分子生物学研究奠定了基础。此后,随着高通量测序技术、生物信息学分析方法和软件的革新、融合及应用,蜜蜂的转录组学研究取得了较大进展[8,9]。对蜜蜂的miRNA开展了一些研究,如ASHBY等[10]研究发现,miR-184、miR-bantam和miR315参与了蜜蜂的细胞分化、组织结构重塑以及级型分化;陈晓[11]对蜂王卵巢激活、产卵抑制和产卵恢复过程中的DEmiRNA进行靶基因预测和分析,发现多个DEmiRNA与卵巢激活和产卵密切相关;石元元[12]研究发现,ame-bantam、ame-let-7和ame-miR-8参与了东方蜜蜂(Apis cerana)4日龄蜂王幼虫和工蜂幼虫的Wnt信号通路,并与雌性蜜蜂级型分化的分子机理密切相关。此外,蜜蜂肠道不仅是食物消化、营养吸收和利用的主要场所,也是抵御病原入侵的重要免疫器官。前人对蜜蜂肠道的研究主要集中在肠道菌群的群落结构和功能预测方面[13,14],但有关蜜蜂及其幼虫肠道发育的研究极为滞后,肠道发育的分子机理仍不明确。笔者所在课题组前期已在mRNA组学水平对意蜂幼虫肠道进行了全面的转录组学研究,通过差异表达基因(DEG)和趋势分析揭示了意蜂幼虫肠道发育过程中的基因表达谱和差异表达规律[15],意蜂幼虫肠道响应球囊菌(Ascosphaera apis)胁迫的免疫应答[16],以及意蜂幼虫与球囊菌之间的互作[17]。【本研究切入点】目前为止,意蜂幼虫肠道发育过程中的miRNA表达谱仍然缺失,相关miRNA在幼虫肠道发育过程中的作用还不明确。【拟解决的关键问题】结合sRNA-seq技术对意蜂4、5和6日龄幼虫肠道样品进行测序和分析,通过DEmiRNA及其靶基因的预测和分析、调控网络的构建和分析,提供意蜂幼虫肠道发育过程中的miRNA表达谱和差异表达信息,揭示DEmiRNA在肠道发育中的作用。1 材料与方法
试验于2017年9月至2018年5月在福建农林大学蜂学学院蜜蜂保护实验室进行。1.1 生物材料
供试意蜂幼虫取自福建农林大学蜂学学院教学蜂场。1.2 意蜂幼虫人工饲养及样品测序
按照王倩等[18]的方法配制幼虫饲料,从健康意蜂蜂群中提取巢脾,将2日龄幼虫移至已预置50 μL饲料的24孔细胞培养板中,在35℃,相对湿度(RH)为90%的培养箱中饲养,每隔24 h更换饲料。分别剖取4、5和6日龄意蜂幼虫肠道(分别记为Am4、Am5和Am6),装入EP管后放入液氮速冻,随后保存在-80℃的超低温冰箱。进行3次生物学重复。Am4的3个生物学重复分别为Am4-1、Am4-2和Am4-3,Am5的3个生物学重复分别为Am5-1、Am5-2和Am5-3,Am6的3个生物学重复分别为Am6-1、Am6-2和Am6-3。上述9个肠道样品委托广州基迪奥生物科技有限公司对进行单端测序,测序平台为Illumina MiSeq。原始数据已上传NCBI SRA数据库,BioProject号:PRJNA408312。1.3 测序数据的质控及定位
对原始下机数据,利用Perl脚本剔除衔接子(adaptor)、未知碱基N和低质量的reads,从而得到高质量的序列(clean reads),保证后续数据分析的准确性。通过Bowite软件将各样品的tags序列比对到GenBank及Rfam(11.0)数据库,过滤核糖体RNA(rRNA)、胞质小RNA(scRNA)以及核内小RNA(snoRNA)等ncRNA和重复序列,得到miRNA的非注释标签序列(unannotated tags)。继而利用Bowit 软件将非注释标签序列与西方蜜蜂的参考基因组(assembly Amel 4.5)的序列进行比对,得到相关tags在参考基因组上的位置信息,即为mapped tags。1.4 DEmiRNA的预测
利用miRDeep2软件[19]将mapped tags与miRBase数据库中已知miRNA前体序列进行比对,从而鉴定已知miRNA的表达,同时得到可能的前体序列。对各样本中miRNA进行表达量的统计,并通过TPM(tags per million)算法公式(TPM=T×106/N,T表示miRNA的tags,N表示总miRNA的tags)对全部miRNA的表达量进行归一化处理。利用R软件计算各样品之间的相关性系数。DEmiRNA(Am4 vs Am5、Am5 vs Am6和Am4 vs Am6)的筛选标准为P-value≤0.05且|log2 Fold change|≥1。1.5 DEmiRNA靶基因预测及分析
根据DEmiRNA与对应物种的基因序列信息,利用TargetFinder软件[20]进行靶基因预测,并利用Blast软件将预测靶基因序列与GO(Gene Ontology)、KEGG数据库比对,获得靶基因的注释信息。利用RNAhybrid、TargetScan和Miranda软件预测DEmiRNA结合的靶基因,根据上述靶向结合关系构建miRNA-mRNA的调控网络并通过Cytoscape软件[21]将其可视化。1.6 DEmiRNA的茎环实时荧光定量PCR(Stem-loop RT-qPCR)验证
为了验证sRNA-seq数据的可靠性,随机选取3个DEmiRNA(miR-7964-y、miR-8516-x和miR-3747-x)进行Stem-loop RT-qPCR验证。根据所选DEmiRNA的序列,参照CHEN等[22]的方法,利用DNAMAN软件(Lynnon Biosoft公司,美国)设计特异性的Stem-loop引物、上游引物和下游引物,委托上海生工生物工程有限公司进行引物合成。选择snRNA U6作为内参。利用RNA抽提试剂盒(Axygen公司,美国)分别提取Am4、Am5和Am6的总RNA,利用Stem-loop引物进行反转录得到相应的cDNA,作为模板进行qPCR。反应体系(20 μL)中含有SYBR Green Dye 10 μL,上下游引物各1 μL,cDNA模板DNA 1 μL,Rox 0.44 μL,DEPC水补至20 μL。在ABI 7500荧光定量PCR仪(ABI公司,美国)中进行反应,反应条件:95℃预变性1 min,95℃变性15 s,48℃延伸30 s,共40个循环,最后72℃延伸45 s。所选miRNA的相对表达量采用2-△△Ct法计算。每个反应进行3个生物学重复和3次平行重复。2 结果
2.1 数据质控与评估
本研究中,3个幼虫肠道样品的sRNA-seq分别产生13 186 921、14 255 967和10 921 897条clean reads,经严格过滤后得到的clean tags数分别为10 841 644(82.22%)、12 037 678(84.44%)和9 230 496(84.51%)条(表1)。Am4、Am5和Am6组内各生物学重复之间的Pearson相关系数均在0.9734以上,说明各样品的重复性较好(图1)。上述结果说明本研究的测序数据质量良好,可用于进一步分析。Table 1
表1
表1sRNA-seq数据统计
Table 1
| 样品Sample | 有效读段Clean reads | 有效标签序列Clean tags |
|---|---|---|
| Am4-1 | 14736731 | 11802658 (80.09%) |
| Am4-2 | 12954535 | 10940303 (84.45%) |
| Am4-3 | 11869496 | 9781972 (82.41%) |
| Am5-1 | 13907726 | 11653319 (83.79%) |
| Am5-2 | 13844605 | 11812820 (85.32%) |
| Am5-3 | 15015570 | 12646896 (84.23%) |
| Am6-1 | 11260874 | 9324373 (82.80%) |
| Am6-2 | 10434013 | 9038930 (86.63%) |
| Am6-3 | 11070804 | 9328185 (84.26%) |
新窗口打开|下载CSV
图1
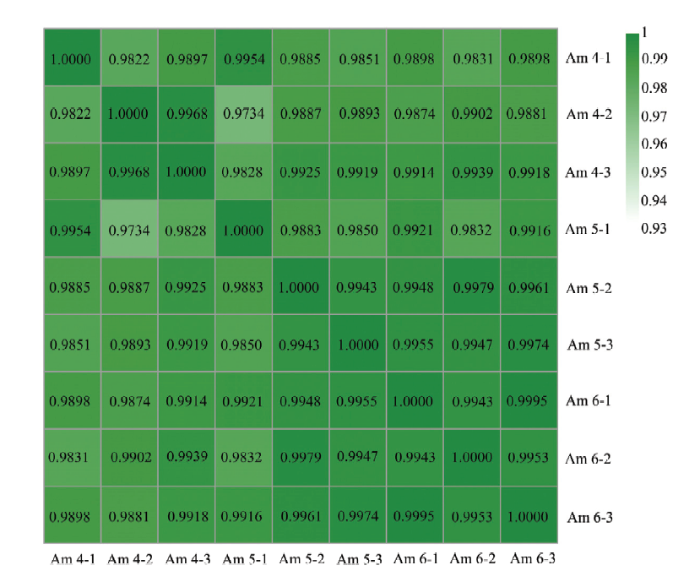 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1各意蜂幼虫肠道样品的不同生物学重复间的Pearson相关性
Fig. 1Pearson correlation between different biological repeats within each A. m. ligustica larval gut sample
2.2 意蜂幼虫肠道的DEmiRNA分析
Am4 vs Am5比较组中有26个miRNA差异表达,包括16个上调miRNA和10个下调miRNA。Am5 vs Am6 比较组共有12个DEmiRNA,包括5个上调miRNA和7个下调miRNA。Am 4 vs Am6比较组中有41个miRNA差异表达,包括15个上调miRNA和26个下调miRNA。分别对Am4 vs Am5和Am5 vs Am6的DEmiRNA进行表达量聚类分析,结果显示2个比较组中DEmiRNA的变化倍数差异明显,多数DEmiRNA的差异表达幅度较小(图2-A、2-B)。进一步分析发现,Am4 vs Am5和Am5 vs Am6中DEmiRNA分别包含3个和2个novel miRNA,其中novel-m0031-3p为2个比较组所共有。利用miRDeep2软件对novel miRNA的前体二级结构进行预测,结果显示它们均有标志性的茎环结构(图2-C—2-F)。图2
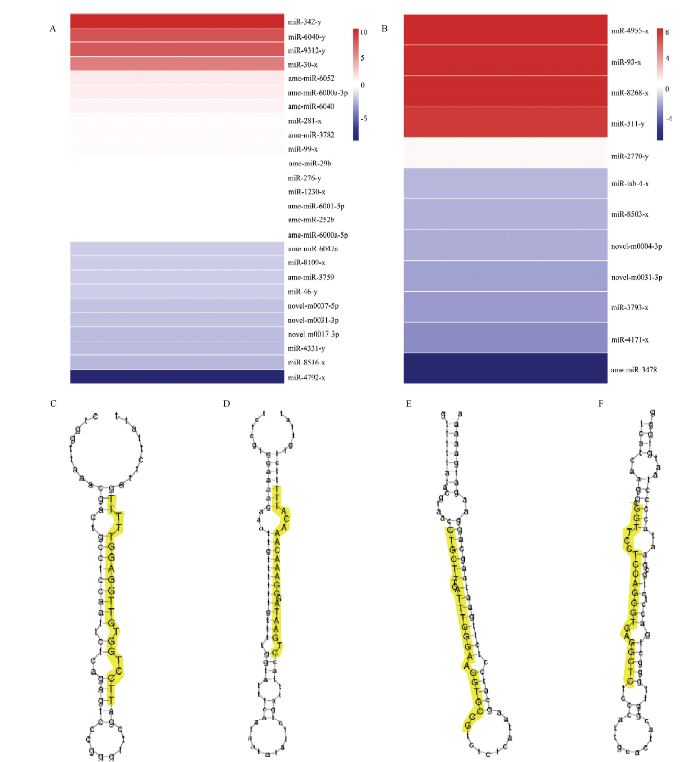 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2DEmiRNA的表达量聚类及novel miRNA前体的二级结构
A:Am4 vs Am5中DEmiRNA的表达量聚类Expression clustering of DEmiRNA in Am4 vs Am5;B:Am5 vs Am6中DEmiRNA的表达量聚类Expression clustering of DEmiRNA in Am5 vs Am6;C:novel-m0004-3p前体的二级结构Secondary structure of precursor of novel-m0004-3p;D:novel-m0017-3p前体的二级结构Secondary structure of precursor of novel-m0017-3p;E:novel-m0031-3p前体的二级结构Secondary structure of precursor of novel-m0031-3p;F:novel-m0037-5p前体的二级结构Secondary structure of precursor of novel-m0037-5p。黄色区域为novel miRNA的成熟序列Yellow regions indicate mature sequences of novel miRNA
Fig. 2Expression clustering of DEmiRNA and secondary structure of novel miRNA’ precursors
2.3 意蜂幼虫肠道的DEmiRNA的靶基因预测及功能注释
利用TargetFinder软件对意蜂幼虫肠道的DEmiRNA进行靶基因预测,Am4 vs Am5、Am5 vs Am6的DEmiRNA可分别预测出5 742和3 733个靶基因。对上述靶基因进行GO数据库注释,结果显示Am4 vs Am5的DEmiRNA的2 725个靶基因共涉及46个GO term,富集基因数最多的是结合(1 612 gene)、细胞进程(1 537 gene)、代谢进程(1 232 gene)、单组织进程(1 197 gene)、催化活性(1 020 gene)、细胞膜(761 gene)、细胞(757 gene)、细胞组件(757 gene)、细胞膜组件(745 gene)、细胞器(578 gene)等(图3);Am5 vs Am6的DEmiRNA的1 785个靶基因可注释到41个GO term,富集基因数最多的是结合(1 068 gene)、细胞进程(1 026 gene)、单组织进程(847 gene)、代谢进程(799 gene)、催化活性(591 gene)、细胞膜(533 gene)、细胞膜组件(529 gene)、细胞(448 gene)、细胞组件(448 gene)、生物学调控(348 gene)等(图3)。说明随着发育时间的延长,意蜂幼虫肠道的DEmiRNA数、靶基因数及其涉及的GO term数逐渐减少;DEmiRNA广泛参与意蜂幼虫肠道的新陈代谢、细胞生命活动及免疫防御。图3
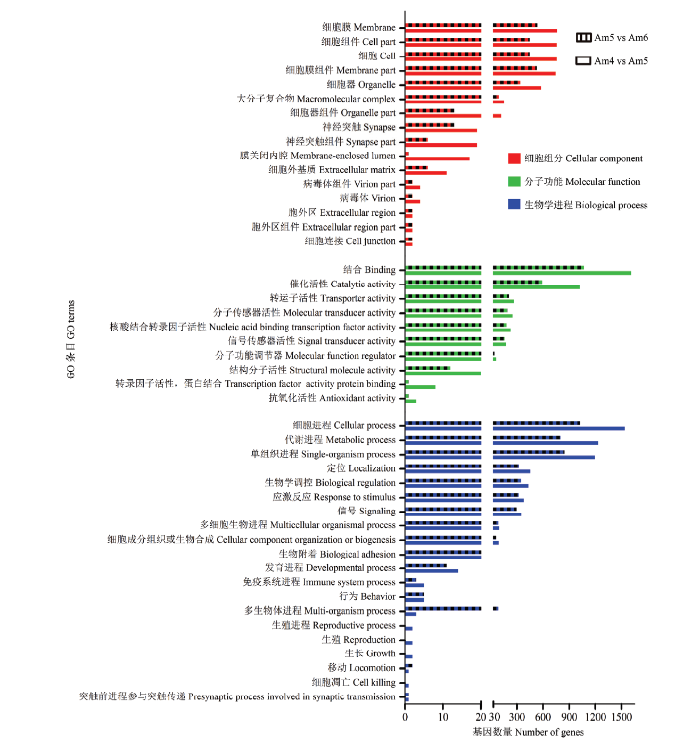 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3意蜂幼虫肠道DEmiRNA的靶基因的GO数据库注释
Fig. 3GO database annotation of DEmiRNA target genes in the larval guts of A. m. ligustica
进一步对DEmiRNA的靶基因进行KEGG代谢通路(pathway)富集分析,结果显示Am4 vs Am5中的1 046个DEmiRNA的靶基因可注释到116条pathway,其中富集基因数最多的是Wnt信号通路(140 gene)、嘌呤代谢(87 gene)、Hippo信号通路(80 gene)、内吞作用(64 gene)、光传导(60 gene)、神经活性配体-受体相互作用(57 gene)、FoxO信号通路(43 gene)、内质网中蛋白质的加工(41 gene)、磷脂酰肌醇信号系统(38 gene)、泛素介导的蛋白水解(36 gene)等(图4-A),说明相应的miRNA参与到意蜂4和5日龄幼虫肠道发育过程中的新陈代谢、蛋白质合成、免疫防御以及相关信号通路的调控。
图4
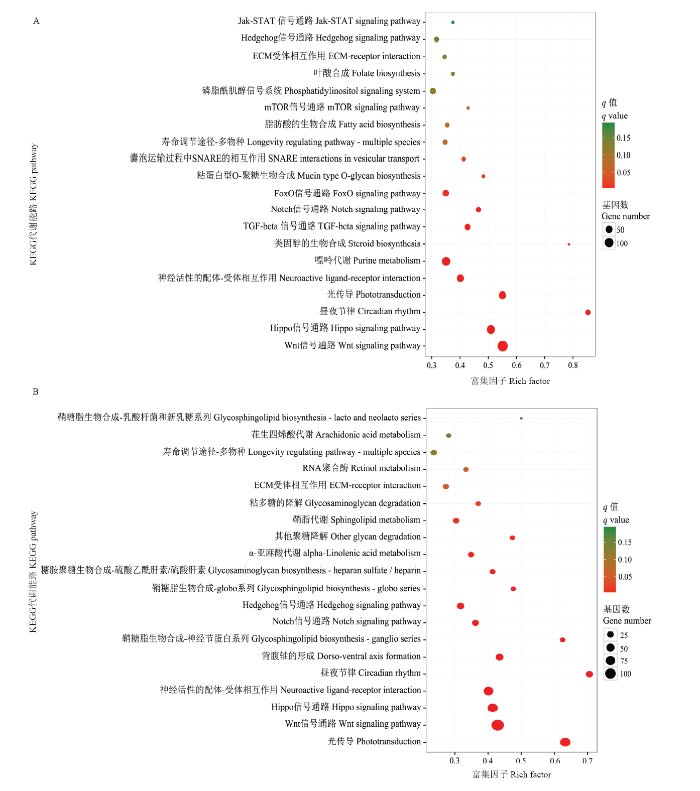 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4意蜂幼虫肠道DEmiRNA的靶基因的KEGG数据库注释
A:Am4 vs Am5中的DEmiRNA的靶基因Target genes of DEmiRNA in Am4 vs Am5;B:Am5 vs Am6中的DEmiRNA的靶基因Target genes of DEmiRNA in Am5 vs Am6
Fig. 4KEGG database annotation of DEmiRNA target genes in the larval guts of A. m. ligustica
Am5 vs Am6中的676个DEmiRNA的靶基注释到92条pathway,并主要富集在Wnt信号通路(109 gene)、光传导(69 gene)、Hippo信号通路(66 gene)、神经活性配体-受体相互作用(57 gene)、嘌呤代谢(42 gene)、内吞作用(38 gene)、背腹轴形成(30 gene)、mRNA监视(28 gene)、Hedgehog信号通路、昼夜节律(24 gene)等(图4-B),说明相应的miRNA同样广泛参与了意蜂幼虫5和6日龄肠道的生长发育过程中各类新陈代谢以及信号通路的调控过程。
2.4 意蜂幼虫肠道DEmiRNA的调控网络分析
利用软件预测DEmiRNA结合的靶基因并通过Cytoscape软件进行可视化,结果显示Am4 vs Am5中上调miRNA可结合611个靶基因,下调miRNA可结合85个靶基因,上调(或下调)miRNA与靶基因形成较为复杂的调控网络,DEmiRNA居于调控网络的中心位置,而靶基因处于调控网络的外周;其中,所有DEmiRNA均可连接2个靶基因以上,ame-miR- 6052结合的靶基因数多达204个(图5-A、5-B)。Am5 vs Am6中上调和下调miRNA可分别结合43和431个靶基因,上调(或下调)miRNA同样与靶基因形成复杂的调控网络,所有DEmiRNA均可连接3个以上的靶基因,其中miR-iab-4-x结合的靶基因数最多,达到125个(图5-C、5-D)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5意蜂幼虫肠道发育过程中DEmiRNA的调控网络
A:Am4 vs Am5中上调miRNA的miRNA-mRNA网络miRNA-mRNA networks of up-regulated miRNA in Am4 vs Am5;B:Am4 vs Am5中下调miRNA的miRNA-mRNA网络miRNA-mRNA networks of down-regulated miRNA in Am4 vs Am5;C:Am5 vs Am6中上调miRNA的miRNA-mRNA网络miRNA-mRNA networks of up-regulated miRNA in Am5 vs Am6;D:Am5 vs Am6中下调miRNA的miRNA-mRNA网络miRNA-mRNA networks of down-regulated miRNA in Am5 vs Am6
Fig. 5Regulation networks of DEmiRNA during the developmental process of A. m. ligustica larval gut
2.5 意蜂幼虫肠道DEmiRNA的RT-qPCR验证
随机挑取3个DEmiRNA(miR-7964-y、miR-8516-x和miR-3747-x)进行RT-qPCR验证,结果显示它们的表达水平的变化趋势和测序数据中相应DEmiRNA的表达水平的变化趋势一致(图6),说明本研究中的测序数据真实可靠。图6
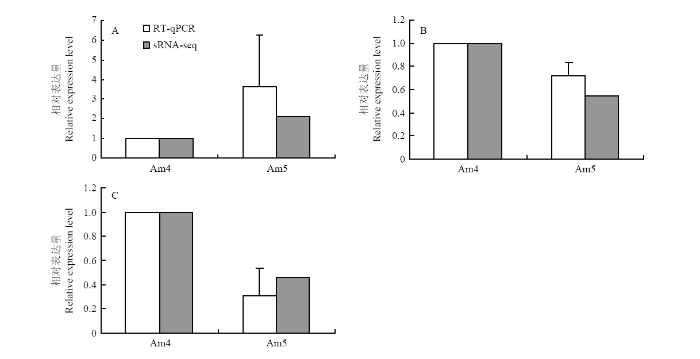 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6DEmiRNA的RT-qPCR验证
Fig. 6RT-qPCR validation of DEmiRNA
A:miR-7964-y;B:miR-8516-x;C:miR-3747-x
3 讨论
MiRNA作为一种重要的基因表达调控因子,在昆虫的各种生物学进程发挥关键作用[23]。家蚕(Bombyx mori)和小菜蛾(Plutella xylostella)不同发育阶段和不同组织中的miRNA表达谱研究揭示了miRNA在其生长发育[24]、变态[25]及行为反应[26]等过程中的重要调控功能。相比于果蝇、家蚕等模式昆虫,蜜蜂的miRNA研究相对滞后。陈璇等[27,28]曾对三型蜂不同发育阶段的混合小RNA文库进行了测序,发现了267个novel miRNA,并进一步对蜂王和工蜂4和5日龄幼虫的DEmiRNA及其靶基因进行了分析,为蜜蜂发育和级型分化关键时期的miRNA调控网络研究打下了基础。目前,有关蜜蜂幼虫肠道发育过程miRNA表达谱的研究十分滞后,miRNA在肠道发育中的调控机理仍不明确。本研究利用sRNA-seq技术对意蜂4、5和6日龄幼虫肠道进行测序,通过生物信息学方法对邻近日龄幼虫肠道的miRNA进行差异表达分析,分别预测出26和12个DEmiRNA,说明DEmiRNA的数量在意蜂幼虫肠道发育过程中有逐渐减少的趋势;进一步分析发现Am4 vs Am5和Am5 vs Am6中的特有DEmiRNA数分别为25(miR-342-y、ame-miR-3759和miR-4331-y等)和11个(miR-4955-x、miR-46-y和ame-miR-3478等),二者包含1个共有DEmiRNA(novel-m0031-3p),且novel-m0031-3p的表达水平随日龄的增加呈逐渐下调趋势,推测其通过下调表达量减少对靶基因的抑制作用,从而在意蜂幼虫肠道的不同发育阶段均发挥基础性的调控作用,而特有DEmiRNA在幼虫肠道的不同发育阶段发挥特殊的调控功能。蜜蜂幼虫肠道内存在简单的共生菌,为尽量减小意蜂幼虫肠道内共生菌对测序数据的影响,本研究一方面在人工饲喂过程中尽量减少环境微生物的带入,例如对所用器具进行高压蒸汽灭菌,用75%酒精对人工气候箱进行擦洗消毒,更换饲料时在酒精灯旁操作等;另一方面通过对原始数据进行严格的过滤和质控、比对西方蜜蜂基因组以消除肠道共生菌数据的影响,昆虫和微生物的物种亲缘关系较远,二者的基因保守性很低,因而比对西方蜜蜂基因组的数据理论上应为意蜂幼虫肠道本身的数据。
蜜蜂肠道不仅是食物消化、营养吸收和利用的主要场所,也是抵御病原入侵的重要免疫器官。本研究发现,Am5 vs Am6中的12个DEmiRNA的靶基因富集在了各类能量和物质代谢通路,如氮代谢(1 gene)、氨基酸代谢(32 gene)、碳水化合物代谢(69 gene)和脂质代谢(74 gene)等,表明相应的DEmiRNA参与了意蜂幼虫肠道能量和物质代谢的调控;还发现部分靶基因注释到与生长、发育相关的代谢通路和信号通路,包括背腹轴形成(30 gene)、Hippo信号通路(66 gene)、Wnt信号通路(109 gene)等,表明相应的DEmiRNA在生长和发育过程中发挥重要的调控作用。此外,分别有38和13个靶基因注释到内吞作用和泛素介导的蛋白质水解等细胞免疫通路,分别有6和4个靶基因注释到MAPK和Jak-STAT等体液免疫通路,表明相应的DEmiRNA参与了意蜂幼虫肠道的免疫防御过程。本研究中,Am4 vs Am5中的26个DEmiRNA的靶基因同样也广泛参与生长发育、新陈代谢以及免疫防御相关的代谢通路。Am4 vs Am5中的DEmiRNA及其靶基因数均高于Am5 vs Am6,意蜂5日龄幼虫体积近乎4日龄幼虫的2倍,但6日龄幼虫体积稍大于5日龄幼虫,推测意蜂4—5日龄幼虫的发育时期需要更多的DEmiRNA参与到生长发育、新陈代谢等方面的调控。
蜕皮激素(ecdysteroid,Ec)是昆虫体内的重要激素之一,参与昆虫生长、变态和生殖的整个生命活动,其滴度的阶段性增加是昆虫生长和发育的必要条件[29]。novel-m0031-3p在Am4 vs Am5和Am5 vs Am6中均有表达,且其结合的5个靶基因(XM_006564318.2、XM_006564319.2、XM_006564320.2、XM_006564321.2和XM_016914663.1)均涉及Ec诱导蛋白的调控,有研究表明蜕皮触发激素基因可在家蚕幼虫蜕皮和变态发育过程中起到关键调控作用[30],因此推测novel-m0031-3p通过调控Ec滴度使其达到动态平衡,从而保证意蜂幼虫肠道的正常发育。miR-281是一种高度保守的miRNA,在家蚕幼虫阶段中呈高量表达,与神经发育、组织生长密切相关[25]。周艳河[31]研究发现,受到高剂量登革病毒侵染的白纹伊蚊(Aedes albopictus)中肠内miR-281会靶向调控自身基因从而影响病毒的复制水平;熊慧萍[32]对位于可变内含子区的黑腹果蝇(Drosophila melanogaster)的miR-281-1/2基因转录和启动子进行分析,发现miR-281只在3龄幼虫、蛹和成虫期表达,且在不同发育阶段miR-281转录起始位点的转录效率存在差异。本研究中,miR-281-x是Am4 vs Am5中的特有DEmiRNA,其结合的49个靶基因涉及TGF-β信号通路(8 gene)、Hippo信号通路(7 gene)、背腹轴的形成(3 gene)和组氨酸代谢(3 gene)等代谢通路,推测其参与对意蜂幼虫肠道早期生长和发育的调控,但miR-281-x的时空表达谱需进一步研究。miR-iab-4可通过调控Hox编码蛋白的Ubx基因表达间接影响黑腹果蝇的翅形成过程,miR-iab-4的突变会影响果蝇幼虫的自身调节能力[33]。本研究中,miR-iab-4-x为Am5 vs Am6中的特有DEmiRNA,其结合的125个靶基因涉及Hippo信号通路(14 gene)、Wnt信号通路(9 gene)、FoxO信号通路(8 gene)、Notch信号通路(7 gene)、mTOR信号通路(6 gene)和背腹轴的形成(5 gene)等与生长发育相关的代谢通路,推测其参与意蜂幼虫肠道发育后期的调控,miR-iab-4-x的下调表达可能对意蜂幼虫肠道发育过程发挥重要作用。
一个miRNA可以同时靶向调控多个mRNA,反之亦然。为进一步揭示DEmiRNA的作用,本研究通过序列匹配关系预测DEmiRNA结合的靶基因,并构建了二者的调控网络(图5),发现部分上调和下调的miRNA位于调控网络的中心位置且结合较多的mRNA,如ame-miR-6052与miR-iab-4-x可分别结合204和125个靶基因,具有很高的连通性,表明二者可能在意蜂幼虫肠道的生长和发育过程中发挥关键的调控功能。细胞色素P450作为一种重要的解毒酶,广泛存在于昆虫的脂肪体、马氏管和中肠内,且在中肠的含量最高;其也可参与内源性物质代谢,在生物体内发挥重要的作用[34,35]。本研究发现Am4 vs Am5中特有的ame-miR-6052表达量上调,其结合的5个靶基因(XM_006559340.2、XM_006559341.1、XM_016912202.1、XM_016916903.1和XM_623618.5)与细胞色素P450的调节有关,推测ame-miR-6052可参与意蜂幼虫肠道对异源物质的降解、新陈代谢过程的调控,下一步可通过合成miRNA mimic、miRNA inhibitor对其进行过表达和敲减,从而深入探究其功能。
4 结论
结合高通量测序技术和生物信息学分析方法对意蜂幼虫肠道的DEmiRNA进行了全基因组水平的预测和分析,并对DEmiRNA-mRNA调控网络进行构建及分析,发现意蜂幼虫通过调节包括ame-miR-6052、miR-iab-4-x、miR-281-x和novel-m0031-3p在内的多个miRNA的表达水平对肠道生长和发育进行调控,研究结果不仅提供了意蜂肠道发育过程的miRNA表达谱及差异表达信息,也为阐明意蜂幼虫肠道的发育机理打下了基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/j.ydbio.2012.03.005URLPMID:3763516 [本文引用: 1]

78 Ago1 protein is highly enriched in Drosophila oocytes and follicle cells. 78 Many Ago1 mutant egg chambers contain only 8 nurse cells without an oocyte. 78 dicer-1, pasha and drosha exhibit similar effects on oogenesis as ago1. 78 miRNAs play roles in oocyte formation and germline cell division in Drosophila.
DOI:10.1016/j.ibmb.2012.10.005URL [本文引用: 1]
[本文引用: 1]
DOI:10.1186/s12864-017-3869-1URLPMID:28651543 [本文引用: 1]
Abiotic stresses cause severe loss of crop production. Among them, drought is one of the most frequent environmental stresses, which limits crop growth, development and productivity. Plant drought tolerance is fine-tuned by a complex gene regulatory network. Understanding the molecular regulation of this polygenic trait is crucial for the eventual success to improve plant yield and quality. Recent studies have demonstrated that microRNAs play critical roles in plant drought tolerance. However, little is known about the microRNA in drought response of the model plant tomato. Here, we described the profiling of drought-responsive microRNA and mRNA in tomato using high-throughput next-generation sequencing. Drought stress was applied on the seedlings of M82, a drought-sensitive cultivated tomato genotype, and IL9–1, a drought-tolerant introgression line derived from the stress-resistant wild speciesSolanum pennelliiLA0716 and M82. Under drought, IL9–1 performed superior than M82 regarding survival rate, H2O2elimination and leaf turgor maintenance. A total of four small RNA and eight mRNA libraries were constructed and sequenced using Illumina sequencing technology. 105 conserved and 179 novel microRNAs were identified, among them, 54 and 98 were differentially expressed upon drought stress, respectively. The majority of the differentially-expressed conserved microRNAs was up-regulated in IL9–1 whereas down-regulated in M82. Under drought stress, 2714 and 1161 genes were found to be differentially expressed in M82 and IL9–1, respectively, and many of their homologues are involved in plant stress, such as genes encoding transcription factor and protein kinase. Various pathways involved in abiotic stress were revealed by Gene Ontology and pathway analysis. The mRNA sequencing results indicated that most of the target genes were regulated by their corresponding microRNAs, which suggested02that microRNAs may play essential roles in the drought tolerance of tomato. In this study, numerous microRNAs and mRNAs involved in the drought response02of tomato were identified using high-throughput sequencing, which will provide new insights into the complex regulatory network of plant adaption to drought stress. This work will also help to exploit new players functioning in plant drought-stress tolerance. The online version of this article (doi:10.1186/s12864-017-3869-1) contains supplementary material, which is available to authorized users.
DOI:10.1007/s00253-016-8060-0URLPMID:28032193 [本文引用: 1]
Reticuloendotheliosis virus (REV) is an avian retrovirus that causes immunosuppression, growth retardation, and oncogenesis in a variety of birds. REV infection is epidemic in many countries. In this
DOI:10.1186/s12929-016-0292-xURLPMID:27784307 [本文引用: 1]
MicroRNAs (miRNAs), which are small non-coding RNAs expressed by almost all metazoans, have key roles in the regulation of cell differentiation, organism development and gene expression. Thousands of miRNAs regulating approximately 60 % of the total human genome have been identified. They regulate genetic expression either by direct cleavage or by translational repression of the target mRNAs recognized through partial complementary base pairing. The active and functional unit of miRNA is its complex with Argonaute proteins known as the microRNA-induced silencing complex (miRISC). De-regulated miRNA expression in the human cell may contribute to a diverse group of disorders including cancer, cardiovascular dysfunctions, liver damage, immunological dysfunction, metabolic syndromes and pathogenic infections. Current day studies have revealed that miRNAs are indeed a pivotal component of host-pathogen interactions and host immune responses toward microorganisms. miRNA is emerging as a tool for genetic study, therapeutic development and diagnosis for human pathogenic infections caused by viruses, bacteria, parasites and fungi. Many pathogens can exploit the host miRNA system for their own benefit such as surviving inside the host cell, replication, pathogenesis and bypassing some host immune barriers, while some express pathogen-encoded miRNA inside the host contributing to their replication, survival and/or latency. In this review, we discuss the role and significance of miRNA in relation to some pathogenic viruses.
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
URL [本文引用: 1]
蜜蜂的级型分化被证实是由蜂王浆中的Royalactin决定,工蜂和蜂王幼虫在级型分化时编码基因的表达差异也被广泛研究.我们发现,在蜜蜂幼虫的级型分化过程中,lncRNA也有着显著的表达差异,因此认为,lncRNA也参与了蜜蜂的级型分化过程.进一步的分析显示,lncRNA可能通过影响上下游基因的转录和功能执行的方式,在蜜蜂早期发育的多细胞组织发育、神经系统发育和转录调控的过程中起到重要的调控作用.
URL [本文引用: 1]
蜜蜂的级型分化被证实是由蜂王浆中的Royalactin决定,工蜂和蜂王幼虫在级型分化时编码基因的表达差异也被广泛研究.我们发现,在蜜蜂幼虫的级型分化过程中,lncRNA也有着显著的表达差异,因此认为,lncRNA也参与了蜜蜂的级型分化过程.进一步的分析显示,lncRNA可能通过影响上下游基因的转录和功能执行的方式,在蜜蜂早期发育的多细胞组织发育、神经系统发育和转录调控的过程中起到重要的调控作用.
DOI:10.1038/srep18794URLPMID:4704047 [本文引用: 1]
The cellular mechanisms employed by some organisms to produce contrasting morphological and reproductive phenotypes from the same genome remains one of the key unresolved issues in biology. Honeybees (Apis mellifera) use differential feeding and a haplodiploid sex determination system to generate three distinct organismal outcomes from the same genome. Here we investigate the honeybee female and male caste-specific microRNA and transcriptomic molecular signatures during a critical time of larval development. Both previously undetected and novel miRNAs have been discovered, expanding the inventory of these genomic regulators in invertebrates. We show significant differences in the microRNA and transcriptional profiles of diploid females relative to haploid drone males as well as between reproductively distinct females (queens and workers). Queens and drones show gene enrichment in physio-metabolic pathways, whereas workers show enrichment in processes associated with neuronal development, cell signalling and caste biased structural differences. Interestingly, predicted miRNA targets are primarily associated with non-physio-metabolic genes, especially neuronal targets, suggesting a mechanistic disjunction from DNA methylation that regulates physio-metabolic processes. Accordingly, miRNA targets are under-represented in methylated genes. Our data show how a common set of genetic elements are differentially harnessed by an organism, which may provide the remarkable level of developmental flexibility required.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.16380/j.kcxb.2017.04.005URL [本文引用: 1]
【目的】本研究旨在通过趋势分析对胁迫意大利蜜蜂Apis mellifera ligustica(简称意蜂)幼虫肠道的球囊菌Ascosphaera apis的差异表达基因(DEGs)进行转录组分析。【方法】将纯化的球囊菌孢子配制为1×107孢子/m L的饲料饲喂意蜂3日龄幼虫,利用Illumina Hi Seq 2500平台对球囊菌胁迫的意蜂幼虫肠道c DNA进行测序,将过滤得到的有效读段(clean reads)映射(mapping)到核糖体数据库及意蜂参考基因组,最后将未映射上的reads映射到本课题组组装并注释的球囊菌参考转录组。利用STEM软件对DEGs进行趋势分析。利用WEGO软件对显著表达模式中的DEGs进行GO富集分析。利用Blastall对显著表达模式中的DEGs进行KEGG代谢通路富集分析。最后,通过对随机选取的6个DEGs进行RT-q PCR分析,对RNA-seq数据进行验证。【结果】球囊菌胁迫意蜂幼虫肠道样品的Illumina测序共得到球囊菌的25 454 076条原始读段(raw reads),经过滤得到24 909 820条clean reads,Q30均在93.46%以上。趋势分析结果显示,19 893个DEGs聚类为8个表达模式,其中有12 151个DEGs聚类为3个表现为显著上调趋势的表达模式。GO富集分析结果显示,表现上调趋势的DEGs富集在40个GO term,富集基因数最多的为细胞进程(cellular process)(2 601 unigenes),其次为代谢进程(metabolic process)(2 553 unigenes)和细胞(cell)(2 522unigenes)。KEGG代谢通路富集分析结果显示,上调趋势中的DEGs富集在119个代谢通路上,其中富集基因数最多的是核糖体(ribosome)(213 unigenes),其次为氨基酸生物合成(biosynthesis of amino acids)(154 unigenes)和内质网蛋白加工(protein processing in endoplasmic reticulum)(130unigenes)。共有48个DEGs富集在MAPK信号通路上,聚类分析结果显示,这些DEGs随着胁迫时间的延长表达水平逐渐增强。RT-q PCR结果显示,6个DEGs的表达水平变化趋势与RNA-seq数据一致,证明
DOI:10.16380/j.kcxb.2017.04.005URL [本文引用: 1]
【目的】本研究旨在通过趋势分析对胁迫意大利蜜蜂Apis mellifera ligustica(简称意蜂)幼虫肠道的球囊菌Ascosphaera apis的差异表达基因(DEGs)进行转录组分析。【方法】将纯化的球囊菌孢子配制为1×107孢子/m L的饲料饲喂意蜂3日龄幼虫,利用Illumina Hi Seq 2500平台对球囊菌胁迫的意蜂幼虫肠道c DNA进行测序,将过滤得到的有效读段(clean reads)映射(mapping)到核糖体数据库及意蜂参考基因组,最后将未映射上的reads映射到本课题组组装并注释的球囊菌参考转录组。利用STEM软件对DEGs进行趋势分析。利用WEGO软件对显著表达模式中的DEGs进行GO富集分析。利用Blastall对显著表达模式中的DEGs进行KEGG代谢通路富集分析。最后,通过对随机选取的6个DEGs进行RT-q PCR分析,对RNA-seq数据进行验证。【结果】球囊菌胁迫意蜂幼虫肠道样品的Illumina测序共得到球囊菌的25 454 076条原始读段(raw reads),经过滤得到24 909 820条clean reads,Q30均在93.46%以上。趋势分析结果显示,19 893个DEGs聚类为8个表达模式,其中有12 151个DEGs聚类为3个表现为显著上调趋势的表达模式。GO富集分析结果显示,表现上调趋势的DEGs富集在40个GO term,富集基因数最多的为细胞进程(cellular process)(2 601 unigenes),其次为代谢进程(metabolic process)(2 553 unigenes)和细胞(cell)(2 522unigenes)。KEGG代谢通路富集分析结果显示,上调趋势中的DEGs富集在119个代谢通路上,其中富集基因数最多的是核糖体(ribosome)(213 unigenes),其次为氨基酸生物合成(biosynthesis of amino acids)(154 unigenes)和内质网蛋白加工(protein processing in endoplasmic reticulum)(130unigenes)。共有48个DEGs富集在MAPK信号通路上,聚类分析结果显示,这些DEGs随着胁迫时间的延长表达水平逐渐增强。RT-q PCR结果显示,6个DEGs的表达水平变化趋势与RNA-seq数据一致,证明
DOI:10.3969/j.issn.1001-4942.2009.11.037URL [本文引用: 1]
以中华蜜蜂(Apis cerana cerana,简称中蜂)幼虫为试验材料,研究了不同饲粮配方、饲养模式和培养板对中蜂幼虫发育的影响。得出以下结果:幼虫饲粮LD1和LD3对幼虫的饲喂效果显著优于LD2;比较不同移虫模式下幼虫发育成功率:日转移法为61.1%,不转移法为70.8%;其中,在不转移饲喂模式下,使用新培养板培养的发育成功率(69.8%)显著高于重复利用的培养板培养的发育成功率(39.6%)(P〈0.01)。表明室内以LD1或LD3饲喂,采用"不转移法"模式,并使用新培养板,更利于中蜂幼虫的发育。
DOI:10.3969/j.issn.1001-4942.2009.11.037URL [本文引用: 1]
以中华蜜蜂(Apis cerana cerana,简称中蜂)幼虫为试验材料,研究了不同饲粮配方、饲养模式和培养板对中蜂幼虫发育的影响。得出以下结果:幼虫饲粮LD1和LD3对幼虫的饲喂效果显著优于LD2;比较不同移虫模式下幼虫发育成功率:日转移法为61.1%,不转移法为70.8%;其中,在不转移饲喂模式下,使用新培养板培养的发育成功率(69.8%)显著高于重复利用的培养板培养的发育成功率(39.6%)(P〈0.01)。表明室内以LD1或LD3饲喂,采用"不转移法"模式,并使用新培养板,更利于中蜂幼虫的发育。
DOI:10.1093/nar/gkr688URLPMID:21911355 [本文引用: 1]
Abstract microRNAs (miRNAs) are a large class of small non-coding RNAs which post-transcriptionally regulate the expression of a large fraction of all animal genes and are important in a wide range of biological processes. Recent advances in high-throughput sequencing allow miRNA detection at unprecedented sensitivity, but the computational task of accurately identifying the miRNAs in the background of sequenced RNAs remains challenging. For this purpose, we have designed miRDeep2, a substantially improved algorithm which identifies canonical and non-canonical miRNAs such as those derived from transposable elements and informs on high-confidence candidates that are detected in multiple independent samples. Analyzing data from seven animal species representing the major animal clades, miRDeep2 identified miRNAs with an accuracy of 98.6-99.9% and reported hundreds of novel miRNAs. To test the accuracy of miRDeep2, we knocked down the miRNA biogenesis pathway in a human cell line and sequenced small RNAs before and after. The vast majority of the >100 novel miRNAs expressed in this cell line were indeed specifically downregulated, validating most miRDeep2 predictions. Last, a new miRNA expression profiling routine, low time and memory usage and user-friendly interactive graphic output can make miRDeep2 useful to a wide range of researchers.
DOI:10.1016/j.cell.2005.04.004URLPMID:15851028 [本文引用: 1]
Plants and animals use small RNAs (microRNAs [miRNAs] and siRNAs) as guides for posttranscriptional and epigenetic regulation. In plants, miRNAs and trans-acting (ta) siRNAs form through distinct biogenesis pathways, although they both interact with target transcripts and guide cleavage. An integrated approach to identify targets of Arabidopsis thaliana miRNAs and ta-siRNAs revealed several new classes of small RNA-regulated genes, including conventional genes such as Argonaute2 and an E2-ubiquitin conjugating enzyme. Surprisingly, five ta-siRNA-generating transcripts were identified as targets of miR173 or miR390. Rather than functioning as negative regulators, miR173- and miR390-guided cleavage was shown to set the 21-nucleotide phase for ta-siRNA precursor processing. These data support a model in which miRNA-guided formation of a 5′ or 3′ terminus within pre-ta-siRNA transcripts, followed by RDR6-dependent formation of dsRNA and Dicer-like processing, yields phased ta-siRNAs that negatively regulate other genes.
DOI:10.1093/bioinformatics/btq675URLPMID:21149340 [本文引用: 1]
ABSTRACTSummary: Cytoscape is a popular bioinformatics package for biological network visualization and data integration. Version 2.8 introduces two powerful new features--Custom Node Graphics and Attribute Equations--which can be used jointly to greatly enhance Cytoscape's data integration and visualization capabilities. Custom Node Graphics allow an image to be projected onto a node, including images generated dynamically or at remote locations. Attribute Equations provide Cytoscape with spreadsheet-like functionality in which the value of an attribute is computed dynamically as a function of other attributes and network properties.Availability and implementation: Cytoscape is a desktop Java application released under the Library Gnu Public License (LGPL). Binary install bundles and source code for Cytoscape 2.8 are available for download from http://cytoscape.org.Contact: msmoot@ucsd.edu
DOI:10.1093/nar/gni178URLPMID:16314309 [本文引用: 1]
A novel microRNA (miRNA) quantification method has been developed using stem-loop RT followed by TaqMan PCR analysis. Stem-loop RT primers are better than conventional ones in terms of RT efficiency and specificity. TaqMan miRNA assays are specific for mature miRNAs and discriminate among related miRNAs that differ by as little as one nucleotide. Furthermore, they are not affected by genomic DNA contamination. Precise quantification is achieved routinely with as little as 25 pg of total RNA for most miRNAs. In fact, the high sensitivity, specificity and precision of this method allows for direct analysis of a single cell without nucleic acid purification. Like standard TaqMan gene expression assays, TaqMan miRNA assays exhibit a dynamic range of seven orders of magnitude. Quantification of five miRNAs in seven mouse tissues showed variation from less than 10 to more than 30,000 copies per cell. This method enables fast, accurate and sensitive miRNA expression profiling and can identify and monitor potential biomarkers specific to tissues or diseases. Stem-loop RT-PCR can be used for the quantification of other small RNA molecules such as short interfering RNAs (siRNAs). Furthermore, the concept of stem-loop RT primer design could be applied in small RNA cloning and multiplex assays for better specificity and efficiency.
DOI:10.1016/j.cois.2015.06.004URLPMID:26251827 [本文引用: 1]
MicroRNAs (miRNAs) are small non-coding RNAs that function in gene regulatory processes in plants and animals by targeting sites within messenger RNA. In insects, miRNAs have been shown to regulate a variety of physiological processes throughout insect development, including molting, metamorphosis, oogenesis, embryogenesis, behavior and host athogen interactions. The roles of miRNAs in the model organism, Drosophila melanogaster, have been studied extensively due to the conserved nature of miRNA function among highly divergent species. However, seeking to understand miRNA function in non-drosophilid insect species has become a growing trend in insect science. Here, we highlight the recent discoveries regarding miRNA function in insect physiology and development.
DOI:10.1371/journal.pone.0004677URLPMID:2650705 [本文引用: 1]
MicroRNAs (miRNAs) are endogenous non-coding genes that participate in post-transcription regulation by either degrading mRNA or blocking its translation. It is considered to be very important in regulating insect development and metamorphosis. We conducted a large-scale screening for miRNA genes in the silkwormBombyx moriusing sequence-by-synthesis (SBS) deep sequencing of mixed RNAs from egg, larval, pupal, and adult stages. Of 2,227,930 SBS tags, 1,144,485 ranged from 17 to 25 nt, corresponding to 256,604 unique tags. Among these non-redundant tags, 95,184 were matched to the silkworm genome. We identified 3,750 miRNA candidate genes using a computational pipeline combining RNAfold and TripletSVM algorithms. We confirmed 354 miRNA genes using miRNA microarrays and then performed expression profile analysis on these miRNAs for all developmental stages. While 106 miRNAs were expressed in all stages, 248 miRNAs were egg- and pupa-specific, suggesting that insect miRNAs play a significant role in embryogenesis and metamorphosis. We selected eight miRNAs for quantitative RT-PCR analysis; six of these were consistent with our microarray results. In addition, we searched for orthologous miRNA genes in mammals, a nematode, and other insects and found that most silkworm miRNAs are conserved in insects, whereas only a small number of silkworm miRNAs has orthologs in mammals and the nematode. These results suggest that there are many miRNAs unique to insects.
DOI:10.1186/1471-2164-11-85URLPMID:2835664 [本文引用: 2]
pAbstract/p pBackground/p pMicroRNAs (miRNAs) repress target genes at the post-transcriptional level, and function in the development and cell-lineage pathways of host species. Tissue-specific expression of miRNAs is highly relevant to their physiological roles in the corresponding tissues. However, to date, few miRNAs have been spatially identified in the silkworm./p pResults/p pWe establish for the first time the spatial expression patterns of nearly 100 miRNAs in multiple normal tissues (organs) of itBombyx mori /itfemales and males using microarray and Northern-blotting analyses. In all, only 10 miRNAs were universally distributed (including bmo-let-7 and bmo-bantam), while the majority were expressed exclusively or preferentially in specific tissue types (e.g., bmo-miR-275 and bmo-miR-1). Additionally, we examined the developmental patterns of miRNA expression during metamorphosis of the body wall, silk glands, midgut and fat body. In total, 63 miRNAs displayed significant alterations in abundance in at least 1 tissue during the developmental transition from larvae to pupae (e.g., bmo-miR-263b and bmo-miR-124). Expression patterns of five miRNAs were significantly increased during metamorphosis in all four tissues (e.g., bmo-miR-275 and bmo-miR-305), and two miRNA pairs, bmo-miR-10b-3p/5p and bmo-miR-281-3p/5p, showed coordinate expression./p pConclusions/p pIn this study, we conducted preliminary spatial measurements of several miRNAs in the silkworm. Periods of rapid morphological change were associated with alterations in miRNA expression patterns in the body wall, silk glands, midgut and fat body during metamorphosis. Accordingly, we propose that corresponding ubiquitous or tissue-specific expression of miRNAs supports their critical roles in tissue specification. These results should facilitate future functional analyses./p
DOI:10.1371/journal.pone.0078787URLPMID:3827265 [本文引用: 1]
MicroRNAs (miRNAs) are a group of small RNAs involved in various biological processes through negative regulation of mRNAs at the post-transcriptional level. Although miRNA profiles have been documented in over two dozen insect species, few are agricultural pests. In this study, both conserved and novel miRNAs in the diamondback moth,Plutella xylostellaL., a devastating insect pest of cruciferous crops worldwide, were documented. High-throughput sequencing of a small RNA library constructed from a mixed life stages ofP. xylostella,including eggs, 1st to 4th (last) instar larvae, pupae and adults, identified 384 miRNAs, of which 174 wereP. xylostellaspecific. In addition, temporal expressions of 234 miRNAs at various developmental stages were investigated using a customized microarray analysis. Among the 91 differentially expressed miRNAs, qRT-PCR analysis was used to validate highly expressed miRNAs at each stage. The combined results not only systematically document miRNA profiles in an agriculturally important insect pest, but also provide molecular targets for future functional analysis and, ultimately, genetic-based pest control practice.
Magsci [本文引用: 1]
<P><FONT face=Verdana>【目的】提取及扩增蜜蜂(Apis mellifera L)sRNA,并构建文库检测富集结果是否满足高通量测序研究要求。【方法】取蜜蜂3个级型不同发育阶段个体作为材料,分别提取总RNA后混合,从中分离出15~40nt的sRNA,反转成cDNA后构建文库,进行蓝白斑筛选。挑选288个单克隆进行测序,对测序结果进行分析。【结果】有效序列为214条,插入的cDNA片段大小范围为15~39 bp。其中,sme-miR-71c miRNA 65条,ncRNA(包括tm-RNA、intron_ghI、5.8s rRNA)5条,tRNA 28条,siRNA及其他sRNA 33条, CDS 1条,未知序列82条。【结论】本实验采用的方法能有效富集蜜蜂sRNA,能够满足高通量测序从中识别出蜜蜂miRNA的研究。<BR></FONT></P>
Magsci [本文引用: 1]
<P><FONT face=Verdana>【目的】提取及扩增蜜蜂(Apis mellifera L)sRNA,并构建文库检测富集结果是否满足高通量测序研究要求。【方法】取蜜蜂3个级型不同发育阶段个体作为材料,分别提取总RNA后混合,从中分离出15~40nt的sRNA,反转成cDNA后构建文库,进行蓝白斑筛选。挑选288个单克隆进行测序,对测序结果进行分析。【结果】有效序列为214条,插入的cDNA片段大小范围为15~39 bp。其中,sme-miR-71c miRNA 65条,ncRNA(包括tm-RNA、intron_ghI、5.8s rRNA)5条,tRNA 28条,siRNA及其他sRNA 33条, CDS 1条,未知序列82条。【结论】本实验采用的方法能有效富集蜜蜂sRNA,能够满足高通量测序从中识别出蜜蜂miRNA的研究。<BR></FONT></P>
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1101/gad.1372505URLPMID:16357215 [本文引用: 1]
Abstract The Drosophila Bithorax Complex encodes three well-characterized homeodomain proteins that direct segment identity, as well as several noncoding RNAs of unknown function. Here, we analyze the iab-4 locus, which produces the microRNAs iab-4-5p and iab-4-3p. iab-4 is analogous to miR-196 in vertebrate Hox clusters. Previous studies demonstrate that miR-196 interacts with the Hoxb8 3' untranslated region. Evidence is presented that miR-iab-4-5p directly inhibits Ubx activity in vivo. Ectopic expression of mir-iab-4-5p attenuates endogenous Ubx protein accumulation and induces a classical homeotic mutant phenotype: the transformation of halteres into wings. These findings provide the first evidence for a noncoding homeotic gene and raise the possibility that other such genes occur within the Bithorax complex. We also discuss the regulation of mir-iab-4 expression during development.
DOI:10.1007/s00018-010-0600-7URLPMID:21184128 [本文引用: 1]
Abstract Cytochrome P450 enzymes (P450s) are important targets in cancer, due to their role in xenobiotic metabolism. Since P450s are the "bridges" between the environment and our body, their function can be linked in many ways to carcinogenesis: they activate dietary and environmental components to ultimate carcinogens (i), the cancer tissue maintains its drug resistance with altered expression of P450s (ii), P450s metabolize (sometimes activate) drugs used for cancer treatment (iii) and they are potential targets for anticancer therapy (iiii). These highly polymorphic enzymes are regulated at multiple molecular levels. Regulation is as important as genetic difference in the existing individual variability in P450 activity. In this review, examples of the transcriptional (DNA methylation, histone modification, modulation by xenosensors) and post-transcriptional (miRNA) regulation will be presented and thereby introduce potential molecular targets at which the metabolism of anticancer drugs, the elimination of cancerogenes or the progress of carcinogenesis could be affected.
[D].
[本文引用: 1]
[D].
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}