 ,, 凌辉, 刘峰, 黄宁, 王玲, 毛花英, 李聪娜, 汤翰臣, 苏炜华, 苏亚春, 阙友雄,福建农林大学农业部福建甘蔗生物学与遗传育种重点实验室/福建农林大学教育部作物遗传育种与综合利用重点实验室,福州 350002
,, 凌辉, 刘峰, 黄宁, 王玲, 毛花英, 李聪娜, 汤翰臣, 苏炜华, 苏亚春, 阙友雄,福建农林大学农业部福建甘蔗生物学与遗传育种重点实验室/福建农林大学教育部作物遗传育种与综合利用重点实验室,福州 350002Cloning and Expression Analysis of a Ⅱd Sub-Group WRKY Transcription Factor Gene from Sugarcane
ZHANG Xu,, LING Hui, LIU Feng, HUANG Ning, WANG Ling, MAO HuaYing, LI CongNa, TANG HanChen, SU WeiHua, SU YaChun, QUE YouXiong,Key Laboratory of Sugarcane Biology and Genetic Breeding (Fujian), Ministry of Agriculture, Fujian Agriculture and Forestry University/Key Laboratory of Crop Genetics and Breeding and Comprehensive Utilization, Ministry of Education, Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
第一联系人:
责任编辑: 李莉
收稿日期:2018-06-12接受日期:2018-07-29网络出版日期:2018-12-01
| 基金资助: |
Received:2018-06-12Accepted:2018-07-29Online:2018-12-01

摘要
关键词:
Abstract
Keywords:
PDF (5964KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张旭, 凌辉, 刘峰, 黄宁, 王玲, 毛花英, 李聪娜, 汤翰臣, 苏炜华, 苏亚春, 阙友雄. 一个甘蔗Ⅱd类WRKY转录因子基因的克隆和表达分析[J]. 中国农业科学, 2018, 51(23): 4409-4423 doi:10.3864/j.issn.0578-1752.2018.23.002
ZHANG Xu, LING Hui, LIU Feng, HUANG Ning, WANG Ling, MAO HuaYing, LI CongNa, TANG HanChen, SU WeiHua, SU YaChun, QUE YouXiong.
0 引言
【研究意义】甘蔗(Saccharum spp.)是生物产量巨大的C4型糖料作物,中国92%的食糖来源于甘蔗[1]。中国甘蔗主要种植于旱坡地上,其中主产区广西有90%以上的甘蔗种植在丘陵旱地[2]。甘蔗属于中度耐盐作物,盐胁迫能够降低甘蔗的产量和品质,而在干旱和半干旱地区灌溉条件下生长的甘蔗经常受到盐分胁迫[3],因此,提高甘蔗对干旱及盐胁迫的耐受性,选育高产、优质、高抗的甘蔗品种迫在眉睫。甘蔗为异源多倍体作物,遗传基础复杂,给直接利用表型性状进行传统抗性遗传研究和品种选育造成诸多困难[4];然而,甘蔗是无性繁殖作物,在组织培养过程中品种的特性稳定,因此,利用基因工程手段培育甘蔗抗性品种应用前景巨大,其中优良抗逆基因资源的发掘是首要基础。WRKY是植物中最大的转录因子家族之一,其在植物生长发育、生物及非生物逆境响应等过程起重要作用[5]。因此,对甘蔗WRKY转录因子的克隆和表达分析研究具有十分重要意义。【前人研究进展】WRKY转录因子蛋白均含有1个或2个WRKY结构域,其大约由60个氨基酸残基构成,该结构域因N端具有7个保守的“WRKYGQK”基序而得名,C端则含有1个锌指结构(CX4-7CX22-23HXH/C)[6]。根据其结构域特征,WRKY转录因子可分为3大类:第Ⅰ类含有2个WRKY结构域,锌指结构为C2H2(CX4-5-C-X22- 23-H-X1-H);第Ⅱ类含有1个WRKY结构域,锌指结构为C2H2(CX4-5-C-X22-23-H-X1-H),同时该类又可分成Ⅱa、Ⅱb、Ⅱc、Ⅱd和Ⅱe共5个亚类;第Ⅲ类含有1个WRKY结构域,锌指结构为C2-HC(C-X7-C-X23-HX)[6]。研究表明,WRKY是通过其WRKY结构域与目标基因W-BOX顺式作用元件TTGACC/T结合[7],从而调控下游靶基因的表达,最终在植物响应逆境胁迫过程中发挥作用[8]。WRKY广泛参与植物生物及非生物胁迫防御反应、生长发育、次生代谢物调控等重要生理过程[9]。如水稻OsWRKY11的过表达能调节相关防御基因的表达,从而增强水稻(Oryza sativa)对白叶枯病菌(Xanthomonas oryzae pv. oryzae)的抗性和对干旱胁迫的耐受性[10];YANG等[11]研究表明核桃(Juglans regia)JrWRKY6和JrWRKY53通过脱落酸(abscisic acid,ABA)信号传导途径提高植物对盐、渗透和热胁迫等的耐受性;拟南芥(Arabidopsis thalilana)AtWRKY75能够延缓植株叶片衰老[12],同时AtWRKY75还能通过赤霉素(gibberellin,GA)介导的信号传导途径正向调节拟南芥开花进程[13]。青蒿(Artemisia carvifolia)WRKY1参与青蒿素的合成途径[14]。自1994年ISHIGURO等[15]在甘薯中发现了第一个WRKY转录因子——SPF1后,越来越多的WRKY转录因子在植物中被发现。目前,在水稻中有109个WRKY转录因子[16],拟南芥中有72个[17],玉米(Zea mays)中有136个[18],白杨(Populus alba)中有104个[19],谷子(Setaria italica)中有105个[20]。【本研究切入点】由于甘蔗遗传背景复杂、基因组庞大,对于WRKY转录因子的研究较晚,报道较少,LAMBAIS等[21]对甘蔗26个WRKY类蛋白序列进行表达谱分析,表明其参与各种防御调控途径;LIU等[22]克隆了属于Ⅱc亚类的甘蔗Sc-WRKY,其受甘蔗黑穗病菌(Sporisorium scitamineum)、水杨酸(salicylic acid,SA)、氯化钠(sodium chloride,NaCl)和聚乙二醇(polyethylene glycol,PEG)强烈诱导。然而,对于甘蔗Ⅱd亚类WRKY转录因子的克隆及其参与非生物胁迫响应的机制尚未见报道。【拟解决的关键问题】本研究通过克隆甘蔗WRKY基因,对其进行生物信息学分析、亚细胞定位和酵母转录激活活性验证,并使用实时荧光定量PCR技术(quantitative real-time PCR,qRT-PCR)分析该基因的组织表达特异性及其在不同外源胁迫下的表达模式,以期阐述甘蔗WRKY响应非生物胁迫的作用及其机制,为深入解析该基因的功能和作用模式奠定基础,同时为甘蔗抗逆分子育种提供优良的基因资源。1 材料与方法
试验于2016年9月至2018年4月在福建农林大学农业部福建甘蔗生物学与遗传育种重点实验室完成。1.1 材料处理
组织特异性分析材料:参照黄宁等[23]方法,田间随机选取长势一致的甘蔗品种ROC22植株,连根挖出清洗干净,取幼嫩蔗根,在第七至第八节处取蔗芽、蔗髓和皮,并取甘蔗+1叶,每3株为一个样品,液氮速冻,-80℃保存备用[23]。每个组织取3个生物学重复。不同外源胁迫处理材料:参考肖新换等[24]方法,田间随机选取长势一致的甘蔗品种ROC22植株,切成单芽蔗茎,在沙中培养至4片完全展开叶。随后放入清水中培养10 d待其长出新的水生根,再用培养液培养2周左右使其生长稳定。选取上述长势一致的甘蔗苗,每3株为一个生物学重复,对其进行分组处理:第1组分别在叶面喷施5 mmol·L-1 SA(含0.01%吐温-20,v/v)、100 μmol·L-1茉莉酸甲酯(methyl jasmonate,MeJA)(含0.1%乙醇和0.05%吐温-20,v/v),均于0、3、6和12 h取样;第2组在含25% PEG水溶液中培养,取样时间点为0、3、6和12 h;第3组在含有250 mmol·L-1的NaCl水溶液中培养,于0、6、12和24 h分别取样;第4组分别在500 mmol·L-1氯化铜(cupric chloride,CuCl2)和500 mmol·L-1氯化铬(chromium chloride,CdCl2)水溶液中培养,均于0、12、24和48 h取样。每个处理各取3个生物学重复。每个样品取样完成,液氮速冻,-80℃保存。
1.2 总RNA的提取与cDNA第一链的合成
将上述甘蔗组织材料和外源胁迫材料,在液氮中研磨成粉末,再用Trizol试剂提取总RNA,用1.0%的琼脂糖凝胶电泳检测。参照RQ1 RNase-Free DNase(Promega,中国,上海)试剂使用说明书除去总RNA中含有的DNA污染,按照PrimeScript RT Reagent Kit(Perfect Real Time)(TaKaRa,中国,大连)的使用说明书反转录合成cDNA第一链,并用1.0%琼脂糖凝胶电泳检测所合成cDNA的质量,随后用于检测目的基因的相对表达量。1.3 甘蔗WRKY的RT-PCR分析
根据甘蔗转录组数据库中发现的一条WRKY的Unigene序列,使用Primer Premier 5.0软件设计扩增引物(表1),以甘蔗品种ROC22在NaCl处理6 h的cDNA为模板,进行PCR扩增。PCR体系为cDNA 1.0 μL、Ex Taq DNA酶(5 U·μL-1)0.125 μL、10×Ex Taq Buffer 2.5 μL、dNTPs(2.5 mmol·L-1)2.0 μL、上下游引物ScWRKY6 F/R(10 μmol·L-1)各1.0 μL和ddH2O 17.375 μL。PCR扩增程序为94℃ 4 min;94℃ 30 s,65℃ 30 s,72℃ 1 min 30 s,35个循环,退火温度每个循环递减0.5℃;72℃ 10 min。产物经1.5%琼脂糖凝胶电泳检测及胶回收纯化后,连接、转化,PCR筛选阳性克隆,送上海生工生物工程技术服务有限公司进行测序分析。Table 1
表1
表1引物序列及用途
Table 1
| 引物名称Primer name | 引物序列Primer sequence (5°-3°) | 用途Usage |
|---|---|---|
| ScWRKY6 | F: GCTGCTGAGGGAGTTGCATA | 扩增全长 Full length amplification |
| R: AAGCAGGGCCAAACATCTCA | ||
| GFP-ScWRKY6 | F: GGGGTACCATGGAGGAAGTGGAGGAGGC | 亚细胞定位 Subcellular localization |
| R: GCTCTAGACACCTGTGCTGACTGAGTTGGC | ||
| BD-ScWRKY6 | F: CATGCCATGGAGATGGAGGAAGTGGAGGAGGC | 转录激活 Transactivation |
| R: CGCGGATCCCACCTGTGCTGACTGAGTTGGC | ||
| qScWRKY6 | F: GCGAGGTCCTTCTTGTCGTC | 实时荧光定量PCR qRT-PCR |
| R: CGTTCACCGGATCGCTCATT | ||
| CAC | F: ACAACGTCAGGCAAAGCAAA | 内参基因 Actin gene |
| R: AGATCAACTCCACCTCTGCG | ||
| CUL | F: TGCTGAATGTGTTGAGCAGC | 内参基因 Actin gene |
| R: TTGTCGCGCTCCAAGTAGTC |
新窗口打开|下载CSV
1.4 甘蔗WRKY的生物信息学分析
将测序得到的ScWRKY6序列用NCBI中的在线工具ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/)分析开放阅读框;通过在线软件Smart(http://smart.embl-heidelberg.de/)、ExPaSy(http://web.expasy.org/protparam/)和Prabi(https://npsa-prabi.ibcp.fr/cgi-bin/secpred_gor4.pl)分析其保守结构域、一级结构和二级结构。磷酸化位点预测:NetPhos(http://www.cbs.dtu.dk/services/NetPhos/);亚细胞定位预测:Cell-PLoc 2.0(http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/)。用NCBI中BLASTP工具查找甘蔗ScWRKY6蛋白同源氨基酸序列,并用DNAMAN 6.0软件多重比对同源氨基酸序列。参照EULGEM等[6]对拟南芥WRKY的分类,选取拟南芥8个不同类群的WRKY氨基酸序列、甘蔗中已发表的Sc-WRKY氨基酸序列以及ScWRKY6蛋白同源氨基酸序列,使用MEGA6.0软件中邻位相连法(Neighbor-Joining,NJ)(Bootstrap= 1 000)构建系统进化树。1.5 甘蔗ScWRKY6蛋白的亚细胞定位分析
通过设计亚细胞定位引物GFP-ScWRKY6-F/R(表1),在目的基因ORF(去掉终止密码子)两端加入酶切位点KpnⅠ和XbaⅠ,以测序成功的阳性质粒pMD19-T-ScWRKY6为模板进行扩增、回收。连接亚细胞定位质粒pCAMBIA1300-GFP,用KpnⅠ和XbaⅠ双酶切。用T4 DNA连接酶将目的基因连接载体pCAMBIA1300-GFP,并转化大肠杆菌DH5α感受态细胞,经测序及酶切验证,获得重组质粒pCAMBIA1300-ScWRKY6-GFP。将重组质粒和对照空载质粒pCAMBIA1300-GFP分别转入农杆菌GV3101菌株中,把菌液PCR检测为阳性的单克隆菌落于含有35 μg·mL-1利福平和50 μg·mL-1卡那霉素的LB液体培养基中培养,在28℃摇床以250 r/min培养14 h;收集菌体并洗去残留LB液体培养基,用MS液体培养基稀释至OD600=0.8,加入200 μmol·L-1乙酰丁香酮后黑暗中静置1 h。选择5—7片叶且长势一致的本氏烟(Nicotiana benthamiana),用1.0 mL的针头注射含有目的基因和空载的菌液,暗培养12 h,再在光照16 h/黑暗8 h,最后在温度28℃环境中培养2 d。将含有目的基因和空载的烟草叶片剪下,叶背面朝下置于1 μg·mL-1 4',6-diamidino-2-phenylindole(DAPI)染色剂中,37℃避光温浴30 min,依次用生理盐水和无菌水清洗后制片,在激光共聚焦显微镜下观察烟草叶片细胞荧光定位情况[25]。1.6 甘蔗WRKY的酵母转录自激活活性验证
在目的基因ORF(去掉终止密码子)两端加酶切位点NcoⅠ和BamHⅠ,设计引物BD-ScWRKY6-F/R(表1),以阳性质粒pMD19-T-ScWRKY6为模板,进行扩增、回收。将胶回收产物和空载质粒pGBKT7分别用NcoⅠ和BamHⅠ进行双酶切,用T4 DNA连接酶将ScWRKY6连接到载体pGBKT7上,随后转化DH5α感受态细胞,经测序及酶切验证正确,获得酵母表达质粒pGBKT7-ScWRKY6。参考徐群刚等[26]方法,以共转载体pGBKT7-53+pGADT7-T为阳性对照,空载pGBKT7为阴性对照,重组质粒pGBKT7- ScWRKY6为试验组,分别转化酵母菌株Y2HGold,然后以100、10-1、10-2和10-3倍数稀释,分别点在含SD/-Trp、SD/-Trp(+X-α-Gal)和SD/-Trp(+X-α-Gal+ AbA) 3种平板上,将其放在30℃培养箱,培养2—3 d后拍照[26]。1.7 甘蔗WRKY的组织特异性及其在不同外源胁迫条件下的表达特性分析
使用Primer Premier 5.0软件设计定量引物qScWRKY6-F/R(表1),以clathrin adaptor complex(CAC)和cullin(CUL)为内参[27],通过qRT-PCR方法检测目的基因在甘蔗不同组织和不同外源胁迫处理样品中的相对表达量。qRT-PCR反应体系参照SYBR Green PCR Master Mix Kit (Roche,中国,上海)说明书配置,扩增程序为50℃ 2 min;95℃ 10 min;95℃ 15 s,60℃ 1 min,40个循环;增加熔解曲线,95℃ 15 s,60℃ 1 min,95℃ 15 s,60℃ 15 s;每个反应3次重复。采用2-△△Ct法计算基因的相对表达量[28],用DPS 9.50软件分析数据的显著性水平,并用软件Origin 8.0绘图。2 结果
2.1 甘蔗ScWRKY6全长cDNA序列的RT-PCR扩增及序列获得
根据甘蔗转录组数据库挖掘的WRKY基因Unigene序列设计扩增引物,通过RT-PCR扩增得到一个甘蔗WRKY类基因,命名为ScWRKY6,GenBank登录号为MH393927。序列分析结果表明(图1),该基因序列全长1 289 bp,包括1个1 059 bp的ORF(91—1 149 bp),编码一个含352个氨基酸的蛋白,并含有1个WRKY结构域和1个锌指结构域(CX5CX23HXH)(图1),推测其为第Ⅱ类WRKY转录因子。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1ScWRKY6的核酸序列及其推导的氨基酸序列
*:终止密码子。红色框部分为WRKYGQK基序;黑色框部分为C2H2基序(CX5CX23HXH)
Fig. 1The nucleotide acid sequence and deduced amino acid sequence of ScWRKY6 gene
*: Stop codon. The sequence of WRKYGQK motif was highlighted in red box, and the sequence of C2H2 motif (CX5CX23HXH) in black box
2.2 ScWRKY6及其编码蛋白的生物信息学分析
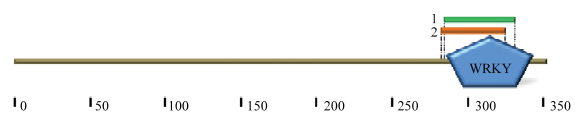
生物信息学分析显示,ScWRKY6蛋白的理论分子量为37 909.81,理论等电点为9.73,不稳定指数为50.23,是一个不稳定蛋白。此外,该蛋白含有27个酸性氨基酸残基(Asp+Glu)和46个碱性氨基酸残基(Lys+Arg),脂肪族指数为64.26,平均亲水性(GRAVY)为-0.579,由此说明ScWRKY6蛋白可能是一个亲水性蛋白。Smart分析显示(图2),该蛋白还含有EGF_CA(Calcium-binding EGF-like domain)和ChtBD2(Chitin-binding domain type 2)结构域。对ScWRKY6蛋白进行磷酸化位点预测(图3),ScWRKY6氨基酸序列含有45个磷酸化位点,包含35个丝氨酸(Serine)、9个苏氨酸(Threonine)和1个酪氨酸(Tyrosine),预测这些位点可能会发生磷酸化反应。ScWRKY6预测蛋白的二级结构分析显示,其主要具有无规则卷曲、α-螺旋和延伸链。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2ScWRKY6蛋白保守结构域分析
Fig. 2The analysis of conserved domain for ScWRKY6 protein
1:ChtBD2(286—332 bp);2:EGF_CA(282—326 bp)
图3
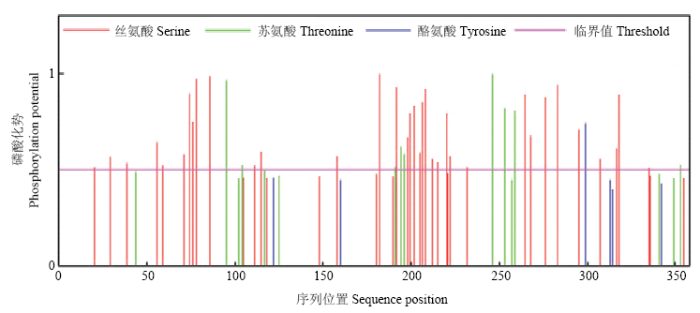 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3ScWRKY6磷酸化位点预测
Fig. 3Prediction of phosphorylation site of ScWRKY6
通过NCBI中BLASTP工具查找ScWRKY6蛋白的同源氨基酸序列,选取9个不同物种的WRKY氨基酸序列,分别为玉米(Z. mays,ACG25409.1)、高粱(Sorghum bicolor,XP_002464211.1)、黍(Panicum miliaceum,PAN46075.1)、狗尾草(Setaria viridis,XP_004982383.1)、二穗短柄草(Brachypodium distachyum,XP_010232453.1)、粗山羊草(Aegilops tauschii,XP_020149328.1)、大麦(Hordeum vulgare,BAJ85995.1)、小麦(Triticum aestivum,ACD80360.1)和水稻(O. sativa,BAT17937.1),通过多重序列比对发现(图4),其与甘蔗ScWRKY6蛋白的序列相似度分别为90%、96%、93%、90%、84%、81%、81%、80%和81%,由此推测WRKY蛋白在不同植物中有较高的保守性。系统进化树结果显示(图5),甘蔗ScWRKY6与高粱WRKY和玉米WRKY在同一分支上且遗传距离较近,而与水稻WRKY分类距离较远;另一方面,甘蔗ScWRKY6与拟南芥AtWRKY15和AtWRKY39聚在同一分支上,参照EULGEM等[6]对拟南芥WRKY的分类,其属于WRKY家族中Ⅱd亚类。
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4ScWRKY6与其他植物WRKY氨基酸序列比对
玉米:Zea mays,ACG25409.1;高粱:Sorghum bicolor,XP_002464211.1;黍:Panicum miliaceum,PAN46075.1;狗尾草:Setaria viridis,XP_004982383.1;二穗短柄草:Brachypodium distachyum,XP_010232453.1;粗山羊草:Aegilops tauschii,XP_020149328.1;大麦:Hordeum vulgare,BAJ85995.1;小麦:Triticum aestivum,ACD80360.1;水稻:Oryza sativa,BAT17937.1
Fig. 4Alignment of ScWRKY6 amino acid sequences and those from other plant species
图5
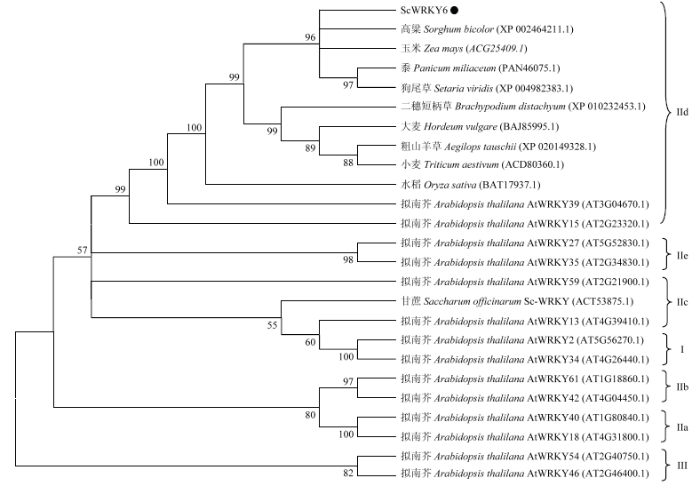 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5不同植物WRKY蛋白的系统进化树
Fig. 5Phylogenetic tree of WRKY proteins from different plant species
2.3 ScWRKY6蛋白的亚细胞定位
亚细胞定位结果如图6所示,在注射空载体pCAMBIA1300-GFP的对照组中,绿色荧光在细胞膜、细胞核、细胞质中均有分布,而注射融合载体pCAMBIA1300-ScWRKY6-GFP的细胞中,细胞核特异性染料DAPI影像与GFP绿色荧光能够完全重合,但在细胞膜和细胞质中均无荧光,说明ScWRKY6蛋白只定位在细胞核上。图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6ScWRKY6在本氏烟中下表皮细胞中的亚细胞定位
图片采用绿色荧光、明场、绿色荧光和明场叠加这3个视野拍摄。35S::GFP:携带空载pCAMBIA1300-GFP的农杆菌菌株。35S::ScWRKY6::GFP:携带重组载体pCAMBIA1300-ScWRKY6-GFP的农杆菌菌株。红色箭头1、2和3分别代表细胞膜、细胞质和细胞核。比例尺=25 μm
Fig. 6Subcellular localization of ScWRKY6 in Nicotiana benthamiana lower epidermal cells
The images are taken by green fluorescence, visible light, merged green fluorescence and visible light. 35S::GFP: The Agrobacterium tumefaciens strain carrying the empty vector pCAMBIA1300-GFP. 35S::ScWRKY6::GFP: The A. tumefaciens strain carrying the recombinant vector pCAMBIA1300-ScWRKY6- GFP. Red arrows 1, 2 and 3 indicate plasma membrane, cytoplasm and nucleus, respectively. Scale bar = 25 μm
2.4 ScWRKY6的转录激活活性分析
为了验证转录因子ScWRKY6的转录激活活性,将ScWRKY6的编码区融合到载体pGBKT7的GAL4 DNA结合域并转入酵母Y2HGold菌株中(图7),pGBKT7-ScWRKY6和阴性对照只能在SD/-Trp和SD/-Trp (+X-α-Gal)培养基上正常生长,说明外源质粒pGBKT7-ScWRKY6成功转入酵母中,但它们不能在金担子素A(AbA)缺陷型培养基SD/-Trp(+X-α-Gal+AbA)培养基上生长,且不能使X-α-Gal显蓝色,说明报告基因AUR1-C和MEL1均未被激活。而阳性对照组在3种培养基上都能正常生长,并使X-α-Gal显蓝色。结果表明,甘蔗ScWRKY6在酵母细胞中无转录自激活活性。图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7甘蔗ScWRKY6转录激活活性分析
SD/-Trp:色氨酸营养缺陷型平板培养基;SD/-Trp (+X-α-Gal):色氨酸营养缺陷型平板培养基(添加5-溴-4-氯-3-吲哚-α-D-半乳糖苷);SD/-Trp (+X-α-Gal+AbA):色氨酸营养缺陷型平板培养基(添加5-溴-4-氯-3-吲哚-α-D-半乳糖苷和金担子素A)
Fig. 7The transactivation activity assay of ScWRKY6 in sugarcane
SD/-Trp: Synthetic dropout medium plate; SD/-Trp(+X-α-Gal): Synthetic dropout medium plate without tryptophan (plus 5-bromo-4-chloro-3-indoxyl-α-D- galactopyranoside); SD/-Trp(+X-α-Gal+AbA): Synthetic dropout medium plate without tryptophan (plus 5-bromo-4-chloro-3-indoxyl-α-D-galactopyranoside and aureobasidin A)
2.5 ScWRKY6的组织特异性表达分析
组织特异性结果如图8显示,ScWRKY6在甘蔗品种ROC22的不同组织中组成型表达,其在蔗芽中的表达量最高,其后由高到低依次为叶、根、皮和蔗髓。其中,蔗芽、叶和根的表达量依次为蔗髓的2.05、1.55和1.37倍。图8
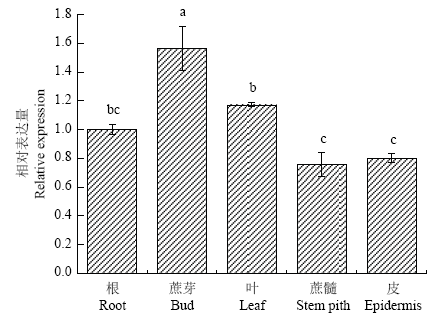 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8ScWRKY6在甘蔗不同组织中的表达情况
不同小写字母表示差异显著性(P≤0.05),误差线为每组处理的标准误差(N=3)。下同
Fig. 8The tissue-specific expression analysis of ScWRKY6 gene in sugarcane
Bars with different superscripts differ significantly (P≤0.05), and error bars represent the standard error of each treating group (N=3). The same as below
2.6 ScWRKY6在不同外源胁迫下的表达特性分析
利用qRT-PCR分析不同外源胁迫下ScWRKY6的表达情况(图9)。从图中可以看出,ScWRKY6的表达量在施加MeJA 6 h后显著上升,为对照组的2.01倍,随后其表达量无显著变化;在SA胁迫处理下,该基因的表达量无明显变化。在PEG胁迫诱导下,ScWRKY6的表达量在3 h时显著上升并达到峰值,为对照组的6.88倍,随后该基因的表达量迅速下降并恢复到对照组水平。在NaCl处理下,ScWRKY6的表达量在12 h达到峰值,为对照的4.18倍,而在处理后6 h与24 h,其表达量与对照均无显著差异。在重金属CuCl2胁迫下,ScWRKY6的表达量在处理后12 h无显著变化,而在24和48 h其表达量相对于对照组显著上调;而在CdCl2胁迫下,ScWRKY6的表达量整体处于上调的趋势,且在24 h达到最大值,约为对照组的4.86倍。图9
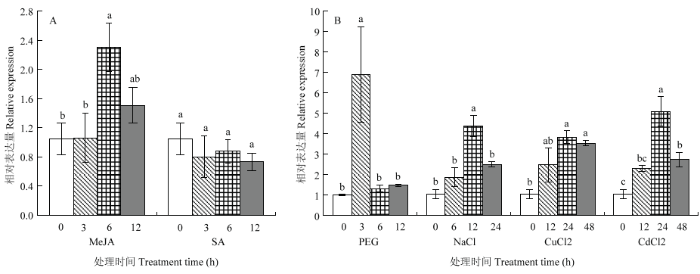 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9甘蔗ScWRKY6在不同外源胁迫下的表达特性
Fig. 9Relative expression of ScWRKY6 in sugarcane under different exogenous stresses
3 讨论
植物在生长过程中经常受到各种生物及非生物胁迫的影响,为适应这些不良环境,植物体形成了一套完善的防御调控机制[5]。转录调控是生物体控制基因表达的主要机制,对植物生长发育和应答外源胁迫具有重要意义[29]。WRKY转录因子是植物最大的转录因子家族之一[30],同时也是植物调控网络的重要组分。前人研究发现,WRKY基因家族在生物[31]及非生物[32,33]逆境应答、植物激素信号转导[9]、植物的生长发育[34]和物质代谢途径[35]等过程发挥作用,是一类广谱性调控因子。3.1 ScWRKY6蛋白结构与功能的关系
本研究从甘蔗中克隆得到ScWRKY6的cDNA全长序列,生物信息学分析显示,该基因编码的蛋白可能为碱性不稳定亲水性蛋白,这与芍药(Paeonia lactiflora Pall)PlWRKY40[36]、葡萄(Vitis vinifera)WRKY18[37]等中的分析结果相似。对ScWRKY6蛋白进行磷酸化位点预测,发现其有45个氨基酸磷酸化位点。前人研究发现蛋白质可以通过磷酸化来调控蛋白质活力与功能,进而在细胞信号转导中起重要作用[38],因此这些磷酸化位点可能与ScWRKY6蛋白的活性调控有关。甘蔗ScWRKY6蛋白含有1个WRKY结构域及1个ChtBD2几丁质结合域。前人研究表明含有几丁质结合域的几丁质酶一般具有抗菌和抗虫功能[39],由此推测ScWRKY6可能对植物应对生物胁迫有积极响应。此外,ScWRKY6还含有一个EGF_CA,即钙结合表皮生长因子类结构域。拟南芥AtWRKY17中也发现并分离出了类似的钙调蛋白结合域[40]。钙离子是细胞内的第二信使,对各种逆境信号起转导作用[41],而钙结合蛋白的含量与植物耐旱、耐盐和抗寒性有关[42],因此,EGF_CA结构域的存在,暗示ScWRKY6可能在植物逆境响应机制中发挥作用。根据EULGEM等[6]对拟南芥WRKY的分类,AtWRKY15和AtWRKY39均属于Ⅱd亚类,系统进化树聚类结果表明ScWRKY6与其在同一分支上,同样属于WRKY家族Ⅱd亚类(图5),预测ScWRKY6基因和拟南芥Ⅱd亚类WRKY基因有相似的功能。AtWRKY15已被报道参与植物生长、盐胁迫和渗透压的调节[43]。LI等[44]发现AtWRKY39受热胁迫的诱导并积极响应SA和JA的信号传导;由此为ScWRKY6的功能研究提供线索。进化树中聚类关系越相近,各成员间功能类似的可能性越大[45],因此,进化树分析结果也暗示着甘蔗与高梁的亲缘关系最近(图5)。3.2 ScWRKY6的作用方式
亚细胞定位结果显示ScWRKY6蛋白定位在细胞核上,这与生物信息学预测结果相同,同时与水稻OsWRKY11[10]、棉花(Gossypium spp.)GhWRKY40[46]和谷子WRKY36[47]等报道相同,这也与其在植物体中的转录调节功能一致。本研究发现甘蔗ScWRKY6转录因子在酵母体内不具有转录自激活活性,这与前人研究结果是类似的[48]。玉米ZmWRKY17在酵母中也未显示转录激活活性[49]。大豆(Glycine max)GmWRKY13、GmWRKY27、GmWRKY40和GmWRKY54蛋白也都不具有转录激活活性,在酵母双杂中GmWRKY13能形成蛋白二聚体,这可能与其转录功能的行使有关[48]。而对于ScWRKY6蛋白调控下游靶基因转录表达的机制有待进一步研究。WRKY基因在苹果(Malus pumila Mill.)[50]、白菜(Brassica pekinensis)[51]、大豆[52]、杨树(Populus L.)[53]等多种植物的不同器官中均有表达,且表达量存在差异。本研究中,ScWRKY6在甘蔗叶、皮、蔗髓、蔗芽和根中均有表达,其中在蔗芽中的表达量最高,在根中的表达量最低,表明该基因在甘蔗中组成型表达并具有组织特异性,这与芍药PlWRKY40在芽、叶片和萼片的表达量较高,在茎和根中表达量较低的结果类似[36]。3.3 不同外源胁迫对ScWRKY6表达特异性的影响
SA是病原菌侵染后植物防卫反应信号途径的重要组分[44],也是植物产生系统获得性抗性(systematic acquired resistance,SAR)的诱导因子[54]。SA通过诱导防御基因的表达或介导信号传导等方式参与植物抗病进程[55],SA还能作为胞外信号通过抑制过氧化氢酶活性,提高H2O2水平,进而促进木质素的合成,最终抑制病原菌的生长[56]。本研究中,ScWRKY6表达量在SA诱导下无明显变化,推测该基因可能不参与SA介导的病原菌侵染过程。茉莉酸(jasmonic acid,JA)是一种重要的植物激素,除了能调节植物开花、叶片衰老等发育过程,还能介导植物对不同环境胁迫的响应[57]。JIANG等[58]发现拟南芥AtWRKY57对灰葡萄孢菌(Botrytis cinerea)的负调控取决于JA信号传导途径,且AtWRKY57能直接结合编码JA信号通路的2个重要阻遏物(JAZ1和JAZ5)的启动子上,并激活它们的转录;香蕉(Musa nana Lour.)在冷胁迫或MeJA诱导下,其MaWRKY26被诱导表达,进而提高香蕉果实的耐寒性[59]。本研究中,ScWRKY6受MeJA诱导上调表达,推测ScWRKY6参与了由MeJA介导的逆境胁迫防御过程和代谢途径。干旱和盐胁迫是2种主要的非生物胁迫,都会导致作物产量和品质的降低[60]。PEG是一种高分子渗透剂,其本身无法穿越细胞壁进入细胞质,不会造成壁分离,能使植物细胞和组织处于类似于干旱胁迫的状态中[61]。拟南芥AtWRKY57能够赋予转基因水稻较高耐旱性,并且提高了其对PEG胁迫的耐受性,在胁迫条件下转基因植株脯氨酸含量和活性氧清除酶活性增强,胁迫响应基因(OsDREB1A和OsDREB2A)显著上调[62];本研究发现,甘蔗ScWRKY6在PEG诱导下表达量明显上调,且在3 h处表达量最高,推测其可能通过渗透物质的调节、抗氧化酶的积累及干旱胁迫响应基因的诱导等途径参与甘蔗对干旱胁迫的响应。GRUBER等[63]分析蒺藜苜蓿(Medicago truncatula)根部所有转录因子,发现只有WRKY转录因子家族显著受到盐胁迫调控,表明WRKY类转录因子更广泛地参与植物的耐盐机制。在盐胁迫下,葡萄VvWRKY30过表植株通过调节活性氧清除和渗透物质的积累来增加植株对盐胁迫的耐受性[64]。本研究中,甘蔗ScWRKY6的表达量受NaCl胁迫的诱导显著增加,其可能通过相同途径来提高甘蔗对盐胁迫的耐受性。Cd2+能植物根系的有丝分裂,阻碍根伸长生长[65];过量的Cd2+和Cu2+则会抑制植物光合和呼吸作用,破坏DNA和细胞膜完整性,最终使植物生长发育受阻甚至引起死亡[66]。WRKY转录因子也能参与植物对金属胁迫响应及转运过程,比如LIU等[67]发现拟南芥中WRKY40、WRKY18和WRKY60参与镉胁迫应答响应,且双突变体或三突变体对镉胁迫耐受性更强。本研究发现,甘蔗ScWRKY6的表达同样受CdCl2和CuCl2胁迫的诱导表达,推测其可能通过增加逆境蛋白和脯氨酸的积累和抗氧化酶活性的提高,参与甘蔗Cd2+和Cu2+金属离子胁迫响应。综上所述,ScWRKY6可能在甘蔗响应干旱、高盐及金属离子胁迫等多种非生物胁迫过程中起重要作用。4 结论
从甘蔗中克隆获得一个WRKY转录因子基因,命名为ScWRKY6,该基因序列全长1 289 bp,包含1个1 059 bp的ORF,编码352个氨基酸,预测其为碱性不稳定亲水性非分泌蛋白。该蛋白具有1个WRKY保守结构域和C2H2(CX5CX23HXH)锌指结构域,属于WRKY转录因子家族的Ⅱd亚类,定位于细胞核,没有自激活活性。ScWRKY6在甘蔗中组成型表达并具有组织特异性,在蔗芽中表达量最高,根中的表达量最低;在NaCl、PEG、MeJA、重金属Cu2+和Cd2+胁迫诱导下表达量均上调,可能在甘蔗抗旱、耐盐及响应金属离子胁迫中发挥作用。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.3864/j.issn.0578-1752.2017.01.003URL [本文引用: 1]

【目的】研究干旱胁迫对气孔导度(Gs)、PSⅡ原初光能转化效率(Fv/Fm)、叶片伸长速率(leaf elongation,LE)和叶片相对含水量(relative water content,RWC)4个甘蔗生理指标遗传变异的影响,为其在甘蔗育种程序早期阶段的应用提供参考。【方法】采用裂区设计,以自然干旱和人工灌溉为主区,不同甘蔗基因型为副区,在云南省红河州开远市和玉溪市元江县2个试验点先后对22个和18个甘蔗基因型同时开展田间试验,主要在2个生长季甘蔗拔节前期先后13次、18次、15次和10次分别对4个生理指标Gs、Fv/Fm、LE、RWC进行测量。采用软件GenStat计算各指标各次测量的遗传方差分量(σg~2)和环境方差分量(σe~2),并计算广义遗传力(hb2),采用SAS9.1对每次测量干旱和灌溉处理下获得遗传方法分量和广义遗传力进行成对t测验。【结果】四项生理指标受干旱影响极显著,13次Gs、18次Fv/Fm、15次LE和10次RWC处理间差异均为极显著。在干旱和灌溉处理下,13次Gs基因型间分别10次和11次差异显著,广义遗传力范围分别为0.19-0.68和0.19-0.82,13次平均分别为0.49和0.53,灌溉条件下的遗传方差显著高于干旱胁迫下的遗传方差;18次Fv/Fm基因型间分别17次和16次差异显著,广义遗传力范围分别为0.26-0.83和0.16-0.85,平均值分别为0.64和0.58,干旱条件下的遗传方差极显著高于灌溉条件下的遗传方差;15次LE基因型间分别14次和10次差异显著,广义遗传力范围分别为0.09—0.89和0.09—0.81,平均值分别为0.58和0.50,干旱处理下平均遗传方差和遗传力较高;10次RWC基因型间分别8次和6次差异显著,广义遗传力范围分别为0.10-0.76和0.16-0.77,平均值分别为0.57和0.47,干旱条件下平均遗传方差和广义遗传力较高。总之,除Gs,其他3个指标在干旱条件下获得广义遗传力均高于灌溉条件下的广义遗传力。【结论】干旱胁迫影响Gs、Fv/Fm、LE和R
DOI:10.3864/j.issn.0578-1752.2017.01.003URL [本文引用: 1]
【目的】研究干旱胁迫对气孔导度(Gs)、PSⅡ原初光能转化效率(Fv/Fm)、叶片伸长速率(leaf elongation,LE)和叶片相对含水量(relative water content,RWC)4个甘蔗生理指标遗传变异的影响,为其在甘蔗育种程序早期阶段的应用提供参考。【方法】采用裂区设计,以自然干旱和人工灌溉为主区,不同甘蔗基因型为副区,在云南省红河州开远市和玉溪市元江县2个试验点先后对22个和18个甘蔗基因型同时开展田间试验,主要在2个生长季甘蔗拔节前期先后13次、18次、15次和10次分别对4个生理指标Gs、Fv/Fm、LE、RWC进行测量。采用软件GenStat计算各指标各次测量的遗传方差分量(σg~2)和环境方差分量(σe~2),并计算广义遗传力(hb2),采用SAS9.1对每次测量干旱和灌溉处理下获得遗传方法分量和广义遗传力进行成对t测验。【结果】四项生理指标受干旱影响极显著,13次Gs、18次Fv/Fm、15次LE和10次RWC处理间差异均为极显著。在干旱和灌溉处理下,13次Gs基因型间分别10次和11次差异显著,广义遗传力范围分别为0.19-0.68和0.19-0.82,13次平均分别为0.49和0.53,灌溉条件下的遗传方差显著高于干旱胁迫下的遗传方差;18次Fv/Fm基因型间分别17次和16次差异显著,广义遗传力范围分别为0.26-0.83和0.16-0.85,平均值分别为0.64和0.58,干旱条件下的遗传方差极显著高于灌溉条件下的遗传方差;15次LE基因型间分别14次和10次差异显著,广义遗传力范围分别为0.09—0.89和0.09—0.81,平均值分别为0.58和0.50,干旱处理下平均遗传方差和遗传力较高;10次RWC基因型间分别8次和6次差异显著,广义遗传力范围分别为0.10-0.76和0.16-0.77,平均值分别为0.57和0.47,干旱条件下平均遗传方差和广义遗传力较高。总之,除Gs,其他3个指标在干旱条件下获得广义遗传力均高于灌溉条件下的广义遗传力。【结论】干旱胁迫影响Gs、Fv/Fm、LE和R
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/j.1439-037X.2009.00364.xURL [本文引用: 1]
Soil salinity is a major abiotic stress which adversely affects the yield and juice quality in sugarcane. However, the mineral nutrient status of plant plays a crucial role in increasing plant tolerance to salinity. We investigated the effects of K and/or Si on plant growth, yield and juice quality in two sugarcane genotypes differing in salinity tolerance. Addition of K and Si significantly (P ≤ 0.05) increased K and Si concentrations and decreased the accumulation of Na + in plants under salt stress. Cane yield and yield attributes were significantly (P ≤ 0.05) higher where K and Si were added. Juice quality characteristics like Brix (% soluble solids in juice), Pol (% sucrose in juice), commercial cane sugar (CCS) and sugar recovery in both sugarcane genotypes were also significantly (P ≤ 0.05) improved with the supplementation of K and Si. For most of the growth parameters, it was found that K either alone or in combination with Si was more effective to alleviate salt stress in both sugarcane genotypes than Si alone. Moreover, the beneficial effects of K and Si were more pronounced in salt sensitive genotype than in salt tolerant genotype. The results suggested that K and Si counteracted the deleterious effects of high salinity/sodicity in sugarcane by lowering the accumulation of Na + and increase in K + concentration with a resultant improvement in K + /Na + ratio which is a good indicator to assess plant tolerance to salinity.
DOI:10.1155/2008/458732URLPMID:18273390 [本文引用: 1]
Sugarcane is a highly productive crop used for centuries as the main source of sugar and recently to produce ethanol, a renewable bio-fuel energy source. There is increased interest in this crop due to the impending need to decrease fossil fuel usage. Sugarcane has a highly polyploid genome. Expressed sequence tag (EST) sequencing has significantly contributed to gene discovery and expression studies used to associate function with sugarcane genes. A significant amount of data exists on regulatory events controlling responses to herbivory, drought, and phosphate deficiency, which cause important constraints on yield and on endophytic bacteria, which are highly beneficial. The means to reduce drought, phosphate deficiency, and herbivory by the sugarcane borer have a negative impact on the environment. Improved tolerance for these constraints is being sought. Sugarcanes ability to accumulate sucrose up to 16% of its culm dry weight is a challenge for genetic manipulation. Genome-based technology such as cDNA microarray data indicates genes associated with sugar content that may be used to develop new varieties improved for sucrose content or for traits that restrict the expansion of the cultivated land. The genes can also be used as molecular markers of agronomic traits in traditional breeding programs.
DOI:10.3864/j.issn.0578-1752.2017.17.002URL [本文引用: 2]
【目的】WRKY转录因子是一类植物响应生物、非生物胁迫,对生长发育都起重要调控作用的转录因子。在甜菜全基因组信息分析的基础上,鉴定WRKY家族基因(Bv WRKYs),解析其组织特异性及盐、热胁迫下的表达情况,为该类基因的功能研究提供参考,为观赏甜菜和石竹目其他观赏植物的基因工程打下基础。【方法】以75条拟南芥WRKY蛋白为参考,根据WRKY保守蛋白序列(PF03106)利用hmm和BLAST同源性搜索对甜菜WRKY家族基因进行鉴定。利用Map Inspect、GSDS2.0、MEGA5.0、DNAMAN5.0、Web Logo 3、MEME生物信息学工具对甜菜WRKY家族基因染色体定位、系统发生关系、基因结构、蛋白质保守结构域、保守元件进行预测和分析。利用RNA-seq和q RT-PCR分析甜菜WRKY组织表达特异性,盐胁迫、热胁迫条件下WRKY表达情况。【结果】甜菜WRKY家族基因包含40个成员,其中39条不均匀地分布在9条染色体上,另外1条定位到随机片段上。根据WRKY保守域特征并与拟南芥WRKY蛋白进化分析,可将40个成员分为Ⅰ、Ⅱ、Ⅲ3类,Ⅰ类有9个成员,Ⅱ类有26个成员,Ⅲ类有5个成员。根据进化关系Ⅱ类可进一步分为Ⅱa(1个)、Ⅱb(4个)、Ⅱc(9个)、Ⅱd(5个)和Ⅱe(7个)5个亚类。基因结构分析发现,甜菜WRKY外显子和内含子数目具有高变异性(2—7个外显子),即使同一亚类内也都差异较大。保守元件分析显示同一类或亚类内成员具有相同的保守元件。WRKY保守域分析发现2个WRKY七肽域变型:WRKYGKK和WRKYGEK。每个WRKY至少在2个组织中表达,30个WRKY在叶中表达,40个WRKY在花序中均有表达,36个WRKY在幼叶中有表达,38个WRKY在直根中有表达,39个WRKY在幼苗中有表达,36个WRKY在种子中有表达。各WRKY表达量差异较大,可分为低表达、高表达基因两类,如Bv WRKY23、Bv WRKY3、Bv WRKY11、Bv WRKY7、Bv WRKY6、Bv WRKY26、Bv WRKY4、Bv WRKY40、Bv WRKY24、Bv WR
DOI:10.3864/j.issn.0578-1752.2017.17.002URL [本文引用: 2]
【目的】WRKY转录因子是一类植物响应生物、非生物胁迫,对生长发育都起重要调控作用的转录因子。在甜菜全基因组信息分析的基础上,鉴定WRKY家族基因(Bv WRKYs),解析其组织特异性及盐、热胁迫下的表达情况,为该类基因的功能研究提供参考,为观赏甜菜和石竹目其他观赏植物的基因工程打下基础。【方法】以75条拟南芥WRKY蛋白为参考,根据WRKY保守蛋白序列(PF03106)利用hmm和BLAST同源性搜索对甜菜WRKY家族基因进行鉴定。利用Map Inspect、GSDS2.0、MEGA5.0、DNAMAN5.0、Web Logo 3、MEME生物信息学工具对甜菜WRKY家族基因染色体定位、系统发生关系、基因结构、蛋白质保守结构域、保守元件进行预测和分析。利用RNA-seq和q RT-PCR分析甜菜WRKY组织表达特异性,盐胁迫、热胁迫条件下WRKY表达情况。【结果】甜菜WRKY家族基因包含40个成员,其中39条不均匀地分布在9条染色体上,另外1条定位到随机片段上。根据WRKY保守域特征并与拟南芥WRKY蛋白进化分析,可将40个成员分为Ⅰ、Ⅱ、Ⅲ3类,Ⅰ类有9个成员,Ⅱ类有26个成员,Ⅲ类有5个成员。根据进化关系Ⅱ类可进一步分为Ⅱa(1个)、Ⅱb(4个)、Ⅱc(9个)、Ⅱd(5个)和Ⅱe(7个)5个亚类。基因结构分析发现,甜菜WRKY外显子和内含子数目具有高变异性(2—7个外显子),即使同一亚类内也都差异较大。保守元件分析显示同一类或亚类内成员具有相同的保守元件。WRKY保守域分析发现2个WRKY七肽域变型:WRKYGKK和WRKYGEK。每个WRKY至少在2个组织中表达,30个WRKY在叶中表达,40个WRKY在花序中均有表达,36个WRKY在幼叶中有表达,38个WRKY在直根中有表达,39个WRKY在幼苗中有表达,36个WRKY在种子中有表达。各WRKY表达量差异较大,可分为低表达、高表达基因两类,如Bv WRKY23、Bv WRKY3、Bv WRKY11、Bv WRKY7、Bv WRKY6、Bv WRKY26、Bv WRKY4、Bv WRKY40、Bv WRKY24、Bv WR
DOI:10.1016/S1360-1385(00)01600-9URL [本文引用: 5]
[本文引用: 1]
.
DOI:10.3389/fpls.2016.00760URLPMID:4891567 [本文引用: 1]
Plants in their natural habitat have to face multiple stresses simultaneously. Evolutionary adaptation of developmental, physiological, and biochemical parameters give advantage over a single window of stress but not multiple. On the other hand transcription factors like WRKY can regulate diverse responses through a complicated network of genes. So molecular orchestration of WRKYs in plant may provide the most anticipated outcome of simultaneous multiple responses. Activation or repression through W-box and W-box like sequences is regulated at transcriptional, translational, and domain level. Because of the tight regulation involved in specific recognition and binding of WRKYs to downstream promoters, they have become promising candidate for crop improvement. Epigenetic, retrograde and proteasome mediated regulation enable WRKYs to attain the dynamic cellular homeostatic reprograming. Overexpression of several WRKYs face the paradox of having several beneficial affects but with some unwanted traits. These overexpression-associated undesirable phenotypes need to be identified and removed for proper growth, development and yeild. Taken together, we have highlighted the diverse regulation and multiple stress response of WRKYs in plants along with the future prospects in this field of research.
.
DOI:10.1111/jipb.12513URL [本文引用: 2]
DOI:10.1186/s12284-018-0199-0URLPMID:29330772 [本文引用: 2]
Plants are frequently subjected to abiotic and biotic stresses, and WRKY proteins play a pivotal role in the response to such stress.OsWRKY11 is induced by pathogens, drought, and heat, suggesting a function in biotic and abiotic stress responses. This study identifiedOsWRKY11, a member of WRKY group IIc. It is a transcriptional activator that localized to the nucleus. Ectopic expression ofOsWRKY11resulted in enhanced resistance to a bacterial pathogen,Xanthomonas oryzaepv.oryzae; resistance was compromised in transgenic lines under-expressingOsWRKY11. Ectopic expression ofOsWRKY11resulted in constitutive expression of defense-associated genes, whereas knock-down (kd) ofOsWRKY11reduced expression of defense-associated genes during pathogen attack, suggesting thatOsWRKY11activates defense responses.OsWRKY11 bound directly to the promoter ofCHITINASE 2, a gene associated with defense, and activated its transcription. In addition, ectopic expression ofOsWRKY11enhanced tolerance to drought stress and induced constitutive expression of drought-responsive genes. Induction of drought-responsive genes was compromised inOsWRKY11-kd plants.OsWRKY11 also bound directly to the promoter of a drought-responsive gene,RAB21, activating its transcription. In addition,OsWRKY11 protein levels were controlled by the ubiquitin-proteasome system. OsWRKY11 integrates plant responses to pathogens and abiotic stresses by positively modulating the expression of biotic and abiotic stress-related genes. The online version of this article (10.1186/s12284-018-0199-0) contains supplementary material, which is available to authorized users.
[本文引用: 1]
DOI:10.1105/tpc.17.00438URLPMID:29061866 [本文引用: 1]
Leaf senescence is a highly coordinated, complicated process involving the integration of numerous internal and environmental signals. Salicylic acid (SA) and reactive oxygen species (ROS) are two well-defined inducers of leaf senescence whose contents progressively and inter-dependently increase during leaf senescence via an unknown mechanism. Here, we characterized the transcription factor WRKY75 as a positive regulator of leaf senescence in Arabidopsis thaliana. Knock-down or knock-out of WRKY75 delayed age-dependent leaf senescence, while over-expression of WRKY75 accelerated this process. WRKY75 transcription is induced by age, SA, H2O2, and multiple plant hormones. Meanwhile, WRKY75 promotes SA production by inducing the transcription of SA INDUCTION-DEFICIENT2 (SID2) and suppresses H2O2 scavenging, partly by repressing the transcription of CATALASE2 (CAT2). Genetic analysis revealed that the mutation of SID2 or an increase in catalase activity rescued the precocious leaf senescence phenotype evoked by WRKY75 over-expression. Based on these results, we propose a tripartite amplification loop model in which WRKY75, SA, and ROS undergo a gradual but self-sustained rise driven by three interlinking positive feedback loops. This tripartite amplification loop provides a molecular framework connecting upstream signals, such as age and plant hormones, to the downstream regulatory network executed by SA- and H2O2-responsive transcription factors during leaf senescence.
DOI:10.1104/pp.17.00657URLPMID:29133369 [本文引用: 1]
Abstract Flowering time is tightly controlled by both endogenous and exogenous signals. Although several lines of evidence have suggested the involvement of WRKY transcription factors in floral initiation, the underlying mechanisms and signaling pathways involved remain elusive. Here, we newly identified Arabidopsis ( Arabidopsis thaliana ) WRKY DNA binding protein75 (WRKY75) as a positive regulator of flowering initiation. Mutation of WRKY75 resulted in a delay in flowering, whereas overexpression of WRKY75 significantly accelerated flowering in Arabidopsis. Gene expression analysis showed that the transcript abundance of the flowering time integrator gene FLOWERING LOCUS T ( FT ) was lower in wrky75 mutants than in the wild type, but greater in WRKY75 -overexpressing plants. Chromatin immunoprecipitation assays revealed that WRKY75 directly binds to the promoter of FT Both in vivo and in vitro biochemical analyses demonstrated that WRKY75 interacts with DELLA proteins. We found that both REPRESSOR OF ga1-3 (RGA) RGA-LIKE1 (RGL1) and GA INSENSITIVE (GAI) can repress the activation ability of WRKY75, thereby attenuating expression of its regulon. Genetic analyses indicated that WRKY75 positively regulates flowering in a FT-dependent manner and overexpression of RGL1 or gain-of-function of GAI could partially rescue the early flowering phenotype of WRKY75 -overexpressing plants. Taken together, our results demonstrate that WRKY75 may function as a new component of the GA-mediated signaling pathway to positively regulate flowering in Arabidopsis.
DOI:10.1111/nph.14373URLPMID:28001315 [本文引用: 1]
Abstract Artemisinin is a type of sesquiterpene lactone well known as an antimalarial drug, and is specifically produced in glandular trichomes of Artemisia annua. However, the regulatory network for the artemisinin biosynthetic pathway remains poorly understood. Exploration of trichome-specific transcription factors would facilitate the elucidation of regulatory mechanism of artemisinin biosynthesis. The WRKY transcription factor GLANDULAR TRICHOME-SPECIFIC WRKY 1 (AaGSW1) was cloned and analysed in A. annua. AaGSW1 exhibited similar expression patterns to the trichome-specific genes of the artemisinin biosynthetic pathway and AP2/ERF transcription factor AaORA. A -glucuronidase (GUS) staining assay further demonstrated that AaGSW1 is a glandular trichome-specific transcription factor. AaGSW1 positively regulates CYP71AV1 and AaORA expression by directly binding to the W-box motifs in their promoters. Overexpression of AaGSW1 in A. annua significantly improves artemisinin and dihydroartemisinic acid contents; moreover, AaGSW1 can be directly regulated by AaMYC2 and AabZIP1, which are positive regulators of jasmonate (JA)- and abscisic acid (ABA)-mediated artemisinin biosynthetic pathways, respectively. These results demonstrate that AaGSW1 is a glandular trichome-specific WRKY transcription factor and a positive regulator in the artemisinin biosynthetic pathway. Moreover, we propose that two trifurcate feed-forward pathways involving AaGSW1, CYP71AV1 and AaMYC2/AabZIP1 function in the JA/ABA response in A. annua.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.pbi.2007.04.020URLPMID:17644023 [本文引用: 1]
Members of the complex family of WRKY transcription factors have been implicated in the regulation of transcriptional reprogramming associated with plant immune responses. Recently genetic evidence directly proving their significance as positive and negative regulators of disease resistance has accumulated. WRKY genes were shown to be functionally connected forming a transcriptional network composed of positive and negative feedback loops and feed-forward modules. Within a web of partially redundant elements some WRKY factors hold central positions mediating fast and efficient activation of defense programs. A key mechanism triggering strong immune responses appears to be based on the inactivation of defense-suppressing WRKY proteins.
DOI:10.1093/dnares/dsr048URLPMID:22279089 [本文引用: 1]
The WRKY transcription factors function in plant growth and development, and response to the biotic and abiotic stresses. Although many studies have focused on the functional identification of the WRKY transcription factors, much less is known about molecular phylogenetic and global expression analysis of the complete WRKY family in maize. In this study, we identified 136 WRKY proteins coded by 119 genes in the B73 inbred line from the complete genome and named them in an orderly manner. Then, a comprehensive phylogenetic analysis of five species was performed to explore the origin and evolutionary patterns of these WRKY genes, and the result showed that gene duplication is the major driving force for the origin of new groups and subgroups and functional divergence during evolution. Chromosomal location analysis of maize WRKY genes indicated that 20 gene clusters are distributed unevenly in the genome. Microarray-based expression analysis has revealed that 131 WRKY transcripts encoded by 116 genes may participate in the regulation of maize growth and development. Among them, 102 transcripts are stably expressed with a coefficient of variation (CV) value of <15%. The remaining 29 transcripts produced by 25 WRKY genes with the CV value of >15% are further analysed to discover new organ- or tissue-specific genes. In addition, microarray analyses of transcriptional responses to drought stress and fungal infection showed that maize WRKY proteins are involved in stress responses. All these results contribute to a deep probing into the roles of WRKY transcription factors in maize growth and development and stress tolerance.<br>
[本文引用: 1]
DOI:10.3389/fpls.2015.00910URLPMID:4654423 [本文引用: 1]
Transcription factors (TFs) are major players in stress signaling and constitute an integral part of signaling networks. Among the major TFs, WRKY proteins play pivotal roles in regulation of transcriptional reprogramming associated with stress responses. In view of this, genome- and transcriptome-wide identification of WRKY TF family was performed in the C4model plants,Setaria italica(SiWRKY) andS. viridis(SvWRKY), respectively. The study identified 105 SiWRKY and 44 SvWRKY proteins that were computationally analyzed for their physicochemical properties. Sequence alignment and phylogenetic analysis classified these proteins into three major groups, namely I, II, and III with majority of WRKY proteins belonging to group II (53 SiWRKY and 23 SvWRKY), followed by group III (39 SiWRKY and 11 SvWRKY) and group I (10 SiWRKY and 6 SvWRKY). Group II proteins were further classified into 5 subgroups (IIa to IIe) based on their phylogeny. Domain analysis showed the presence of WRKY motif and zinc finger-like structures in these proteins along with additional domains in a few proteins. AllSiWRKYgenes were physically mapped on theS. italicagenome and their duplication analysis revealed that 10 and 8 gene pairs underwent tandem and segmental duplications, respectively. Comparative mapping ofSiWRKYandSvWRKYgenes in related C4panicoid genomes demonstrated the orthologous relationships between these genomes.In silicoexpression analysis ofSiWRKYandSvWRKYgenes showed their differential expression patterns in different tissues and stress conditions. Expression profiling of candidateSiWRKYgenes in response to stress (dehydration and salinity) and hormone treatments (abscisic acid, salicylic acid, and methyl jasmonate) suggested the putative involvement ofSiWRKY066andSiWRKY082in stress and hormone signaling. These genes could be potential candidates for further characterization to delineate their functional roles in abiotic stress signaling.
[本文引用: 1]
.
[本文引用: 1]
DOI:10.3969/j.issn.1000-2561.2013.11.022URL [本文引用: 2]
Using ABA99594.1 sequence from Oryza sativa. as the probe, the full-length cDNA sequence of a Diaminopimelate epimerase gene was cloned in silico from sugarcane(Saccharum comlex) and named as ScDAPE. The results of RT-PCR amplification and sequence analysis showed that the ScDAPE had a length of 1 168 bp containing the 816 bp open reading frame(ORF) for encoding 271 amino acids residues. The characteristics predicted based on the analysis of bioinformatics showed that the ScDAPE of sugarcane was a soluble acidic protein which had two conserved functional domains with the main function for amino acid synthesis, and was located in endoplasmic reticulum membrane. The mainly secondary structure elements were random roil. The electron expression analysis suggested that ScDAPE was constitutively expressed in different tissues of sugarcane, such as lateral buds, inflorescence, leaf and apical meristem, specifically higher in inflorescence. Real-time quantitative PCR(RT-qPCR) analysis revealed that the expression of ScDAPE was higher in root than in leaf sheath, pith, skin, lateral buds and leaf, and its expression could also be induced by several kinds of exogenous stresses including salicylic acid(SA), abscisic acid(ABA), methyl-jasmonate(MeJA), dehydration(PEG), high salt (NaCl)and cadmium chloride(CdCl2), while the inducible expression level of ScDAPE was most apparent after the stresses of SA and PEG, properly 12.4 times and 2.72 times higher than that of the control respectively, which suggested that ScDAPE involves in the sugarcane resistance to disease and osmotic stress. The results in this study will provide the basis for cloning and functional identification of other members of ScDAPE in sugarcane and promote the use of ScASAPE gene in sugarcane genetic engineering.
DOI:10.3969/j.issn.1000-2561.2013.11.022URL [本文引用: 2]
Using ABA99594.1 sequence from Oryza sativa. as the probe, the full-length cDNA sequence of a Diaminopimelate epimerase gene was cloned in silico from sugarcane(Saccharum comlex) and named as ScDAPE. The results of RT-PCR amplification and sequence analysis showed that the ScDAPE had a length of 1 168 bp containing the 816 bp open reading frame(ORF) for encoding 271 amino acids residues. The characteristics predicted based on the analysis of bioinformatics showed that the ScDAPE of sugarcane was a soluble acidic protein which had two conserved functional domains with the main function for amino acid synthesis, and was located in endoplasmic reticulum membrane. The mainly secondary structure elements were random roil. The electron expression analysis suggested that ScDAPE was constitutively expressed in different tissues of sugarcane, such as lateral buds, inflorescence, leaf and apical meristem, specifically higher in inflorescence. Real-time quantitative PCR(RT-qPCR) analysis revealed that the expression of ScDAPE was higher in root than in leaf sheath, pith, skin, lateral buds and leaf, and its expression could also be induced by several kinds of exogenous stresses including salicylic acid(SA), abscisic acid(ABA), methyl-jasmonate(MeJA), dehydration(PEG), high salt (NaCl)and cadmium chloride(CdCl2), while the inducible expression level of ScDAPE was most apparent after the stresses of SA and PEG, properly 12.4 times and 2.72 times higher than that of the control respectively, which suggested that ScDAPE involves in the sugarcane resistance to disease and osmotic stress. The results in this study will provide the basis for cloning and functional identification of other members of ScDAPE in sugarcane and promote the use of ScASAPE gene in sugarcane genetic engineering.
DOI:10.3724/SP.J.1145.2014.09033URL [本文引用: 1]
光合系统Ⅰ亚基O(PhotosystemⅠsubunit O,Psa O)是光合系统Ⅰ中的蛋白亚基,在两个光合系统之间平衡激发能方面起着重要的作用.为研究Psa O基因的结构和功能,对甘蔗(Saccharum offi cinarum L.)叶片全长c DNA文库进行测序,获得光合系统Ⅰ亚基O基因的全长c DNA序列,命名为Sc Psa O(Gen Bank Accession Number:KF714498).生物信息学分析表明,该基因全长708 bp,开放阅读框435 bp,编码144个氨基酸;该基因所编码的蛋白定位于叶绿体基质,无信号肽,为疏水性非分泌碱性蛋白,二级结构多为无规则卷曲,含有PJN00046家族的保守结构域,参与能量新陈代谢及脂肪酸新陈代谢.同时,该基因在不同物种间具有较强的保守性,其中与同属C4植物的同源性较C3植物高.荧光定量PCR分析结果表明,甘蔗Sc Psa O基因在叶片中的相对表达量最高,具有一定的组织特异性;在氯化钠(Na Cl)、聚乙二醇(PEG)、氯化铜(Cu Cl2)、脱落酸(ABA)、水杨酸(SA)和茉莉酸甲酯(Me JA)等外源胁迫下,其表达量均呈下调趋势,且以PEG胁迫下的下调表现最为明显.这些结果表明这几种外源胁迫可能抑制甘蔗Sc Psa O基因的转录水平表达,为其进一步功能验证以及在甘蔗基因工程中的应用积累了基础资料.
DOI:10.3724/SP.J.1145.2014.09033URL [本文引用: 1]
光合系统Ⅰ亚基O(PhotosystemⅠsubunit O,Psa O)是光合系统Ⅰ中的蛋白亚基,在两个光合系统之间平衡激发能方面起着重要的作用.为研究Psa O基因的结构和功能,对甘蔗(Saccharum offi cinarum L.)叶片全长c DNA文库进行测序,获得光合系统Ⅰ亚基O基因的全长c DNA序列,命名为Sc Psa O(Gen Bank Accession Number:KF714498).生物信息学分析表明,该基因全长708 bp,开放阅读框435 bp,编码144个氨基酸;该基因所编码的蛋白定位于叶绿体基质,无信号肽,为疏水性非分泌碱性蛋白,二级结构多为无规则卷曲,含有PJN00046家族的保守结构域,参与能量新陈代谢及脂肪酸新陈代谢.同时,该基因在不同物种间具有较强的保守性,其中与同属C4植物的同源性较C3植物高.荧光定量PCR分析结果表明,甘蔗Sc Psa O基因在叶片中的相对表达量最高,具有一定的组织特异性;在氯化钠(Na Cl)、聚乙二醇(PEG)、氯化铜(Cu Cl2)、脱落酸(ABA)、水杨酸(SA)和茉莉酸甲酯(Me JA)等外源胁迫下,其表达量均呈下调趋势,且以PEG胁迫下的下调表现最为明显.这些结果表明这几种外源胁迫可能抑制甘蔗Sc Psa O基因的转录水平表达,为其进一步功能验证以及在甘蔗基因工程中的应用积累了基础资料.
URL [本文引用: 1]
细胞色素P450基因在电子传递链、次生代谢物质合成和对外源化学药物毒性降解中发挥着重要作用,为了深入了解该基因在甘蔗中的功能,通过RT-PCR扩增获得甘蔗细胞色素P450还原酶基因的cDNA全长序列,命名为ScCPR450(Gen Bank Accession Number:KR864841).该基因全长999 bp,含有744 bp的完整开放阅读框,编码247个氨基酸.亚细胞定位结果显示,ScCPR450蛋白分布于细胞质中,与生物信息学预测结果相符.q RT-PCR表达分析表明,该基因在甘蔗中组成型表达,但有组织特异性,芽中表达量最高,其次是叶,而皮中表达量最低.在脱落酸(ABA)、水杨酸(SA)、茉莉酸甲酯(Me JA)、聚乙二醇(PEG)和氯化铜(CuCl_2)胁迫诱导过程中,该基因的表达量呈现不同变化模式,其中SA胁迫6 h下,ScCPR450基因的表达量最高,约为对照的12.21倍;在PEG胁迫下,ScCPR450基因的表达量上调且表达量稳定,推测ScCPR450基因在甘蔗响应生物和非生物胁迫中发挥一定的作用.本研究可为该基因家族其它成员的克隆以及深入解析该基因的功能特性奠定基础,进而为基于基因工程技术对甘蔗品种进行定向改良提供基因资源.
URL [本文引用: 1]
细胞色素P450基因在电子传递链、次生代谢物质合成和对外源化学药物毒性降解中发挥着重要作用,为了深入了解该基因在甘蔗中的功能,通过RT-PCR扩增获得甘蔗细胞色素P450还原酶基因的cDNA全长序列,命名为ScCPR450(Gen Bank Accession Number:KR864841).该基因全长999 bp,含有744 bp的完整开放阅读框,编码247个氨基酸.亚细胞定位结果显示,ScCPR450蛋白分布于细胞质中,与生物信息学预测结果相符.q RT-PCR表达分析表明,该基因在甘蔗中组成型表达,但有组织特异性,芽中表达量最高,其次是叶,而皮中表达量最低.在脱落酸(ABA)、水杨酸(SA)、茉莉酸甲酯(Me JA)、聚乙二醇(PEG)和氯化铜(CuCl_2)胁迫诱导过程中,该基因的表达量呈现不同变化模式,其中SA胁迫6 h下,ScCPR450基因的表达量最高,约为对照的12.21倍;在PEG胁迫下,ScCPR450基因的表达量上调且表达量稳定,推测ScCPR450基因在甘蔗响应生物和非生物胁迫中发挥一定的作用.本研究可为该基因家族其它成员的克隆以及深入解析该基因的功能特性奠定基础,进而为基于基因工程技术对甘蔗品种进行定向改良提供基因资源.
DOI:10.11926/j.issn.1005-3395.2015.05.009URLMagsci [本文引用: 2]
从香蕉(Musa acuminata)果实冷害的数字基因表达谱中筛选并分离了一个WRKY转录因子,命名为MaWRKY11。MaWRKY11具有2个WRKY保守结构域,属于I类WRKY成员。亚细胞定位证实MaWRKY11定位在细胞核,是核蛋白。转录活性分析揭示MaWRKY11具有转录激活活性,并且激活区在N端。实时荧光定量PCR分析表明MaWRKY11受冷胁迫诱导,外源茉莉酸甲酯(MeJA)处理减轻香蕉果实冷害的同时也上调了其表达。另外,利用MaWRKY11的C端作为诱饵进行酵母双杂筛库发现,MaWRKY11可与脱水诱导早期应答蛋白MaERD相互作用。以上结果表明MaWRKY11可能通过与逆境相关蛋白如MaERD互作来响应香蕉果实的冷胁迫。
DOI:10.11926/j.issn.1005-3395.2015.05.009URLMagsci [本文引用: 2]
从香蕉(Musa acuminata)果实冷害的数字基因表达谱中筛选并分离了一个WRKY转录因子,命名为MaWRKY11。MaWRKY11具有2个WRKY保守结构域,属于I类WRKY成员。亚细胞定位证实MaWRKY11定位在细胞核,是核蛋白。转录活性分析揭示MaWRKY11具有转录激活活性,并且激活区在N端。实时荧光定量PCR分析表明MaWRKY11受冷胁迫诱导,外源茉莉酸甲酯(MeJA)处理减轻香蕉果实冷害的同时也上调了其表达。另外,利用MaWRKY11的C端作为诱饵进行酵母双杂筛库发现,MaWRKY11可与脱水诱导早期应答蛋白MaERD相互作用。以上结果表明MaWRKY11可能通过与逆境相关蛋白如MaERD互作来响应香蕉果实的冷胁迫。
DOI:10.1038/srep07042URLPMID:4229666 [本文引用: 1]
Sugarcane (Saccharum spp. hybrids) is a world-wide cash crop for sugar and biofuel in tropical and subtropical regions and suffers serious losses in cane yield and sugar content under salinity and drought stresses. Although real-time quantitative PCR has a numerous advantage in the expression quantification of stress-related genes for the elaboration of the corresponding molecular mechanism in sugarcane, the variation happened across the process of gene expression quantification should be normalized and monitored by introducing one or several reference genes. To validate suitable reference genes or gene sets for sugarcane gene expression normalization, 13 candidate reference genes have been tested across 12 NaCl- and PEG-treated sugarcane samples for four sugarcane genotypes using four commonly used systematic statistical algorithms termed geNorm, BestKeeper, NormFinder and the deltaCt method. The results demonstrated that glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and eukaryotic elongation factor 1-alpha (eEF-1a) were identified as suitable reference genes for gene expression normalization under salinity/drought-treatment in sugarcane. Moreover, the expression analyses of SuSK and 6PGDH further validated that a combination of clathrin adaptor complex (CAC) and cullin (CUL) as reference should be better for gene expression normalization. These results can facilitate the future research on gene expression in sugarcane under salinity and drought stresses.
DOI:10.1006/meth.2001.1262URL [本文引用: 1]
DOI:10.1007/s12374-016-0577-3URL [本文引用: 1]
The WRKY family, a large family of transcription factors (TFs) found in higher plants, plays central roles in many aspects of biological processes and adaption to environment. However, little information is available on this family in apple ( Malus domestica ). In the present study, a total of 119 candidate WRKY genes in apple genome were identified and classified into three main groups (Group I II) based on the structure of the conserved domains. Each group or subgroup showed similar exon ntron structures and motif compositions. The evolution analysis showed that 44 MdWRKY genes clustered into 20 intensive regions (<100 kb) and 78 MdWRKY formed 85 pairs of collinear relationships, suggesting that both tandem and segmental duplications played an important role in the evolution and diversification of the WRKY gene family in apple. Furthermore, the expression of the MdWRKY genes in apple leaves in response to biotic stress ( Alternaria alternate ) and hormone treatments [salicylic acid (SA), methyl jasmonate (MeJA) and ethephon] was examined by using RNA-seq and qRT-PCR. The results showed that 63 MdWRKY genes had differential expression in their transcript abundance in response to Alternaria alternata apple pathotype infection. Moreover, most pathogen responsive MdWRKY genes were also changed significantly when apple leaves were treated by SA, MeJA or ethephon plant growth regulations, suggesting an interaction between SA, JA and ethylene (Eth) hormone signaling under biotic stress. This work may provide the basis for future studies of the genetic modification of WRKY genes for pathogen resistance in apple.
DOI:10.1155/2015/807560URLPMID:4387944 [本文引用: 1]
WRKY proteins are emerging players in plant signaling and have been thoroughly reported to play important roles in plants under biotic stress like pathogen attack. However, recent advances in this field do reveal the enormous significance of these proteins in eliciting responses induced by abiotic stresses. WRKY proteins act as major transcription factors, either as positive or negative regulators. Specific WRKY factors which help in the expression of a cluster of stress-responsive genes are being targeted and genetically modified to induce improved abiotic stress tolerance in plants. The knowledge regarding the signaling cascade leading to the activation of the WRKY proteins, their interaction with other proteins of the signaling pathway, and the downstream genes activated by them are altogether vital for justified targeting of the WRKY genes. WRKY proteins have also been considered to generate tolerance against multiple abiotic stresses with possible roles in mediating a cross talk between abiotic and biotic stress responses. In this review, we have reckoned the diverse signaling pattern and biological functions of WRKY proteins throughout the plant kingdom along with the growing prospects in this field of research.
DOI:10.1007/s13313-016-0416-5URL [本文引用: 1]
Abstract WRKY transcription factors play an important role in plant growth, development and immunity. In our study, a WRKY family member gene designated BoWRKY6 was isolated from broccoli (Brassica oleracea var. italica), and its expression was induced by downy mildew (Hyaloperonospora parasitica). Five transgenic broccoli lines over-expressing BoWRKY6 driven by the CaMV 35S promotor were obtained by Agrobacterium tumefaciens mediated transformation, and they demonstrated significant increased resistance to downy mildew, with resistant levels from low to very high. Real time-qualitative PCR analysis indicated that expressions of both BoWRKY6 and the pathogenesis-related gene 1 (BoPR1) in transgenic plants were obviously higher than those in WT plants after H. parasitica treatment. Lines of BWK14 and BWK31 exhibited very high resistance to downy mildew, and may serve as promising candidate materials for broccoli molecular breeding in the near future.
DOI:10.1016/j.bbagrm.2011.09.002URLPMID:21964328 [本文引用: 1]
78 WRKY gene super-family, one of the largest transcription factor gene families, has been suggested to play important roles in the regulation of transcriptional reprogramming associated with plant stress responses. 78 Modification of the expression patterns of WRKY genes and/or changes in their activity contributes to the elaboration of various signaling pathways and regulatory networks. 78 A single WRKY gene often responds to several stress factors, and then their proteins may participate in the regulation of several seemingly disparate processes as negative or positive regulators. 78 WRKY proteins function via protein–protein interaction and auto-regulation or cross-regulation exists extensively in the WRKY family.
DOI:10.1186/s12864-016-2677-3URLPMID:4862231 [本文引用: 1]
WRKY genes, as the most pivotal transcription factors in plants, play the indispensable roles in regulating various physiological processes, including plant growth and development as well as in response to stresses. Broomcorn millet is one of the most important crops in drought areas worldwide. However, the WRKY gene family in broomcorn millet remains unknown. A total of 32 PmWRKY genes were identified in this study using computational prediction method. Structural analysis found that PmWRKY proteins contained a highly conserved motif WRKYGQK and two common variant motifs, namely WRKYGKK and WRKYGEK. Phylogenetic analysis of PmWRKYs together with the homologous genes from the representative species could classify them into three groups, with the number of 1, 15, and 16, respectively. Finally, the transcriptional profiles of these 32 PmWRKY genes in various tissues or under different abiotic stresses were systematically investigated using qRT-PCR analysis. Results showed that the expression level of 22 PmWRKY genes varied significantly under one or more abiotic stress treatments, which could be defined as abiotic stress-responsive genes. This was the first study to identify the organization and transcriptional profiles of PmWRKY genes, which not only facilitates the functional analysis of the PmWRKY genes, and also lays the foundation to reveal the molecular mechanism of stress tolerance in this important crop. The online version of this article (doi:10.1186/s12864-016-2677-3) contains supplementary material, which is available to authorized users.
DOI:10.1105/tpc.15.00608URLPMID:26977085 [本文引用: 1]
The WD40 proteins ANTHOCYANIN11 (AN11) from petunia (Petunia hybrida) and TRANSPARENT TESTA GLABRA1 (TTG1) from Arabidopsis thaliana and associated basic helix-loop-helix (bHLH) and MYB transcription factors activate a variety of differentiation processes. In petunia petals, AN11 and the bHLH protein AN1 activate, together with the MYB protein AN2, anthocyanin biosynthesis and, together with the MYB protein PH4, distinct genes, such as PH1 and PH5, that acidify the vacuole. To understand how AN1 and AN11 activate anthocyanin biosynthetic and PH genes independently, we isolated PH3. We found that PH3 is a target gene of the AN11-AN1-PH4 complex and encodes a WRKY protein that can bind to AN11 and is required, in a feed-forward loop, together with AN11-AN1-PH4 for transcription of PH5. PH3 is highly similar to TTG2, which regulates hair development, tannin accumulation, and mucilage production in Arabidopsis. Like PH3, TTG2 can bind to petunia AN11 and the Arabidopsis homolog TTG1, complement ph3 in petunia, and reactivate the PH3 target gene PH5. Our findings show that the specificity of WD40-bHLH-MYB complexes is in part determined by interacting proteins, such as PH3 and TTG2, and reveal an unanticipated similarity in the regulatory circuitry that controls petunia vacuolar acidification and Arabidopsis hair development.
DOI:10.1007/s11240-016-0985-6URL [本文引用: 1]
Plant secondary biosynthetic pathways are exceedingly inducible by elicitors and facilitate enhanced metabolite production using plant cell tissue and organ cultures. Elicitors can regulate large number of control points and trigger the expression of key genes with increased cellular activities at biochemical and molecular level involving signal compounds. A large number of chemical elicitors viz: jasmonic acid (JA), methyl jasmonate (MeJA), salicylic acid (SA), acetyl salicylic acid (ASA), ethylene (ET) and ethrel (Ethe), heavy metals (HM), many types of chemical compounds (natural or synthetic) and their combinations are used for elicitation studies. Cell suspensions and hairy roots are commonly used culture systems followed by adventitious roots and multiple shoots for elicitation experiments. Amongst the elicitors and concentrations used 100 M MeJA was found optimum for secondary metabolite enhancement in majority of experiments compared to SA and JA. Elicitor treatments promoted yield enhancements starting from 1.0 to maximum of 2230-fold across plant species studied. Elicitors singly with media additives, combination of elicitors and elicitors other than signal compounds along with hormones were found beneficial for enhanced secondary metabolite production. Further, a combination of target gene over expression and elicitor treatment also supported higher secondary product yield. The present communication presents information exclusively about the use of chemical elicitors for secondary metabolites production in vitro covering approximately more than a decade of research at one place in one review. Further, this extensive appraisal will be useful for the understanding and manipulation of secondary metabolites for enhanced production in vitro.
URL [本文引用: 2]
WRKY转录因子在植物休眠等生长发育过程中具有重要作用。本研究基于芍药芽转录组数据,对芽休眠解除前后显著下调的编号为c22608.graphc0的序列进行了q PCR验证分析,结果表明其表达模式与转录组一致,说明其与芍药芽休眠解除密切相关。为进一步挖掘和利用该基因,从芍药‘大富贵’芽中克隆了该基因,命名为Pl WRKY40(Gen Bank登录号:KU891820)。其c DNA全长1 347 bp,开放阅读框882 bp,编码293个氨基酸,含有1个典型的WRKY结构域;基因结构分析表明,Pl WRKY40具有4个外显子和3个内含子,其内含子具有特殊结构,可能调控该基因表达;以拟南芥WRKY蛋白的分类为参照,聚类分析结果表明,Pl WRKY40属于Group IIa亚族;系统进化树分析表明,Pl WRKY40与可可Tc WRKY40具有最高的同源性;亚细胞定位观测结果显示,Pl WRKY40转录因子定位在细胞核上;半定量PCR分析显示,Pl WRKY40在芍药的不同组织器官中泛表达,其中芽表达量最高。本研究为深入探索Pl WRKY40的生理学功能奠定了基础。
URL [本文引用: 2]
WRKY转录因子在植物休眠等生长发育过程中具有重要作用。本研究基于芍药芽转录组数据,对芽休眠解除前后显著下调的编号为c22608.graphc0的序列进行了q PCR验证分析,结果表明其表达模式与转录组一致,说明其与芍药芽休眠解除密切相关。为进一步挖掘和利用该基因,从芍药‘大富贵’芽中克隆了该基因,命名为Pl WRKY40(Gen Bank登录号:KU891820)。其c DNA全长1 347 bp,开放阅读框882 bp,编码293个氨基酸,含有1个典型的WRKY结构域;基因结构分析表明,Pl WRKY40具有4个外显子和3个内含子,其内含子具有特殊结构,可能调控该基因表达;以拟南芥WRKY蛋白的分类为参照,聚类分析结果表明,Pl WRKY40属于Group IIa亚族;系统进化树分析表明,Pl WRKY40与可可Tc WRKY40具有最高的同源性;亚细胞定位观测结果显示,Pl WRKY40转录因子定位在细胞核上;半定量PCR分析显示,Pl WRKY40在芍药的不同组织器官中泛表达,其中芽表达量最高。本研究为深入探索Pl WRKY40的生理学功能奠定了基础。
URL [本文引用: 1]
以抗性品种‘左优红’组培苗为材料,克隆得到WRKY18基因,测序结果显示:VvWRKY18基因片段大小为954 bp,编码317个氨基酸序列。生物信息学分析结果显示VvWRKY18蛋白分子量约为35.2416 kDa,等电点为8.22,不稳定系数为48.61,推测为不稳定蛋白;与已知的粳稻、高粱、毛果杨及拟南芥WRKY家族蛋白高度同源;亚细胞定位预测结果显示主要存在于细胞核中,属于第二类WRKY转录因子家族成员。实时荧光定量PCR分析显示,VvWRKY18在葡萄不同组织中均有表达,在花中表达量最高;多种逆境胁迫因子如盐、干旱和低温等诱导VvWRKY18上调表达,且在低温胁迫6 h时诱导表达量最高。另外,VvWRKY18受胁迫信号分子水杨酸、脱落酸、一氧化氮和过氧化氢诱导上调表达,且VvWRKY18在一氧化氮处理下的表达模式与低温诱导的类似,推测VvWRKY18可能通过调控一氧化氮信号分子代谢途径来调节葡萄对低温胁迫应答。
URL [本文引用: 1]
以抗性品种‘左优红’组培苗为材料,克隆得到WRKY18基因,测序结果显示:VvWRKY18基因片段大小为954 bp,编码317个氨基酸序列。生物信息学分析结果显示VvWRKY18蛋白分子量约为35.2416 kDa,等电点为8.22,不稳定系数为48.61,推测为不稳定蛋白;与已知的粳稻、高粱、毛果杨及拟南芥WRKY家族蛋白高度同源;亚细胞定位预测结果显示主要存在于细胞核中,属于第二类WRKY转录因子家族成员。实时荧光定量PCR分析显示,VvWRKY18在葡萄不同组织中均有表达,在花中表达量最高;多种逆境胁迫因子如盐、干旱和低温等诱导VvWRKY18上调表达,且在低温胁迫6 h时诱导表达量最高。另外,VvWRKY18受胁迫信号分子水杨酸、脱落酸、一氧化氮和过氧化氢诱导上调表达,且VvWRKY18在一氧化氮处理下的表达模式与低温诱导的类似,推测VvWRKY18可能通过调控一氧化氮信号分子代谢途径来调节葡萄对低温胁迫应答。
DOI:10.7554/eLife.04805URLPMID:26083713 [本文引用: 1]
10.7554/eLife.04805.001Plants generally respond to herbivore attack by increasing resistance and decreasing growth. This prioritization is achieved through the regulation of phytohormonal signaling networks. However, it remains unknown how this prioritization affects resistance against non-target herbivores. In this study, we identify WRKY70 as a specific herbivore-induced, mitogen-activated protein kinase-regulated rice transcription factor that physically interacts with W-box motifs and prioritizes defence over growth by positively regulating jasmonic acid (JA) and negatively regulating gibberellin (GA) biosynthesis upon attack by the chewing herbivore Chilo suppressalis. WRKY70-dependent JA biosynthesis is required for proteinase inhibitor activation and resistance against C. suppressalis. In contrast, WRKY70 induction increases plant susceptibility against the rice brown planthopper Nilaparvata lugens. Experiments with GA-deficient rice lines identify WRKY70-dependent GA signaling as the causal factor in N. lugens susceptibility. Our study shows that prioritizing defence over growth leads to a significant resistance trade-off with important implications for the evolution and agricultural exploitation of plant immunity.DOI: http://dx.doi.org/10.7554/eLife.04805.001
DOI:10.3969/j.issn.1004-0374.2002.02.007URL [本文引用: 1]
对植物中的几丁质结合蛋白类型及各类的初级结构进行了总结,并对其所含的几丁质结合域的特征,功能及其作用进行了简述。
DOI:10.3969/j.issn.1004-0374.2002.02.007URL [本文引用: 1]
对植物中的几丁质结合蛋白类型及各类的初级结构进行了总结,并对其所含的几丁质结合域的特征,功能及其作用进行了简述。
DOI:10.1016/j.febslet.2005.01.057URL [本文引用: 1]
DOI:10.1038/35067109URLPMID:11283728 [本文引用: 1]
Abstract Plants have a very different lifestyle to animals, and one might expect that unique molecules and processes would underpin plant-cell signal transduction. But, with a few notable exceptions, the list is remarkably familiar and could have been constructed from animal studies. Wherein, then, does lifestyle specificity emerge?
DOI:10.1104/pp.104.046490URL [本文引用: 1]
DOI:10.1073/pnas.1217516109URLPMID:23169634 [本文引用: 1]
Environmental stresses adversely affect plant growth and development. A common theme within these adverse conditions is the perturbation of reactive oxygen species (ROS) homeostasis. Here, we demonstrate that the ROS-inducible Arabidopsis thaliana WRKY15 transcription factor (AtWRKY15) modulates plant growth and salt/osmotic stress responses. By transcriptome profiling, a divergent stress response was identified in transgenic WRKY15-overexpressing plants that linked a stimulated endoplasmic reticuKim-to-nucleus communication to a disrupted mitochondrial stress response under salt-stress conditions. We show that mitochondrial calcium-flux sensing might be important for regulating an active mitochondrial retrograde signaling and launching an appropriate defense response to confer salt-stress tolerance.
[本文引用: 2]
DOI:10.1007/s11427-016-5001-1URLPMID:26803306 [本文引用: 1]
-acting elements in the promoters of target genes, subsequently activating or repressing their transcription. In the present study, we systematically examined the functional diversification of the NAC transcription factor (NAC-TFs) family by analyzing their chromosomal location, structure, phylogeny, and expression pattern in genes identified in the Gr and Ga genomes, respectively, were annotated and divided into 18 subfamilies, which showed distinct divergence in gene structure and expression patterns during fiber development. In addition, when the functional parameters were examined, clear divergence was observed within tandem clusters, which suggested that subfunctionalization had occurred among duplicate genes. The expression patterns of homologous gene pairs also changed, suggestive of the diversification of gene function during the evolution of diploid cotton. These findings provide insights into the mechanisms underlying the functional differentiation of duplicated NAC-TFs genes in two diploid cotton species.
DOI:10.1371/journal.pone.0093577URLPMID:3991585 [本文引用: 1]
Abstract WRKY transcription factors form one of the largest transcription factor families and function as important components in the complex signaling processes that occur during plant stress responses. However, relative to the research progress in model plants, far less information is available on the function of WRKY proteins in cotton. In the present study, we identified the GhWRKY40 gene in cotton (Gossypium hirsutum) and determined that the GhWRKY40 protein is targeted to the nucleus and is a stress-inducible transcription factor. The GhWRKY40 transcript level was increased upon wounding and infection with the bacterial pathogen Ralstonia solanacearum. The overexpression of GhWRKY40 down-regulated most of the defense-related genes, enhanced the wounding tolerance and increased the susceptibility to R. solanacearum. Consistent with a role in multiple stress responses, we found that the GhWRKY40 transcript level was increased by the stress hormones salicylic acid (SA), methyl jasmonate (MeJA) and ethylene (ET). Moreover, GhWRKY40 interacted with the MAPK kinase GhMPK20, as shown using yeast two-hybrid and bimolecular fluorescence complementation systems. Collectively, these results suggest that GhWRKY40 is regulated by SA, MeJA and ET signaling and coordinates responses to wounding and R. solanacearum attack. These findings highlight the importance of WRKYs in regulating wounding- and pathogen-induced responses.
DOI:10.3864/j.issn.0578-1752.2015.05.03URL [本文引用: 1]
【目的】干旱等非生物胁迫严重影响了植物的生长和作物的产量。WRKY转录因子广泛参与了植物生长发育、形态建成和代谢调控等过程,在调控非生物胁迫响应中也扮演着十分重要的角色。分析谷子SiWRKY36的分子特性和功能,解析谷子转录因子的抗逆调控机制。【方法】通过对干旱胁迫谷子转录组测序结果分析,获得了一个WRKY转录因子SiWRKY36;利用生物信息学的方法分析谷子SiWRKY36的分子特性;根据SiWRKY36蛋白序列进行同源性搜索,得到与谷子SiWRKY36蛋白序列相似度较高的其他物种的蛋白序列;使用MEGA5对谷子SiWRKY36蛋白序列及其同源序列进行多序列比对分析并构建同源物种间系统进化树;利用MEME和SMART在线工具进行蛋白序列分析;利用GSDS和PHYRE2在线工具分别对谷子SiWRKY36基因结构和三级结构进行分析;从谷子基因组数据库Phytozome获取谷子SiWRKY36上游2 000 bp作为启动子;用PLACE数据库对SiWRKY36启动子顺式作用元件进行分析;利用实时荧光定量PCR检测SiWRKY36在不同胁迫条件下(PEG、低温、NaCl、MeJA、ABA、GA和SA、H2O2)的表达模式;分别以8种胁迫处理的谷子cDNA作为模板,以谷子Si001873m.g为内参,以SYBR Green染料法进行real-time PCR。用实时荧光定量PCR仪进行PCR扩增;将SiWRKY36的cDNA序列连入带有CaMV 35S启动子的pBI121表达载体中,构建表达载体pBI121-SiWRKY36,转入农杆菌,侵染野生型拟南芥得到转基因株系。用T3转SiWRKY36拟南芥植株进行抗性鉴定。【结果】谷子SiWRKY36全长1 485 bp,基因编码区包含UTR区和3个内含子以及4个外显子,与柳枝稷亲缘性最高,属于WRKY转录因子家族的第一类。SiWRKY36编码蛋白包含2个WRKY保守域,预测的SiWRKY36蛋白三级结构包含2个α螺旋结构和3个β折叠结构。启动子元件分析表明SiWRKY36包含ABA-responsive element(ABRE)、MYB、MYC、low-temperature-responsive element(LTRE)、GT-1等多17
DOI:10.3864/j.issn.0578-1752.2015.05.03URL [本文引用: 1]
【目的】干旱等非生物胁迫严重影响了植物的生长和作物的产量。WRKY转录因子广泛参与了植物生长发育、形态建成和代谢调控等过程,在调控非生物胁迫响应中也扮演着十分重要的角色。分析谷子SiWRKY36的分子特性和功能,解析谷子转录因子的抗逆调控机制。【方法】通过对干旱胁迫谷子转录组测序结果分析,获得了一个WRKY转录因子SiWRKY36;利用生物信息学的方法分析谷子SiWRKY36的分子特性;根据SiWRKY36蛋白序列进行同源性搜索,得到与谷子SiWRKY36蛋白序列相似度较高的其他物种的蛋白序列;使用MEGA5对谷子SiWRKY36蛋白序列及其同源序列进行多序列比对分析并构建同源物种间系统进化树;利用MEME和SMART在线工具进行蛋白序列分析;利用GSDS和PHYRE2在线工具分别对谷子SiWRKY36基因结构和三级结构进行分析;从谷子基因组数据库Phytozome获取谷子SiWRKY36上游2 000 bp作为启动子;用PLACE数据库对SiWRKY36启动子顺式作用元件进行分析;利用实时荧光定量PCR检测SiWRKY36在不同胁迫条件下(PEG、低温、NaCl、MeJA、ABA、GA和SA、H2O2)的表达模式;分别以8种胁迫处理的谷子cDNA作为模板,以谷子Si001873m.g为内参,以SYBR Green染料法进行real-time PCR。用实时荧光定量PCR仪进行PCR扩增;将SiWRKY36的cDNA序列连入带有CaMV 35S启动子的pBI121表达载体中,构建表达载体pBI121-SiWRKY36,转入农杆菌,侵染野生型拟南芥得到转基因株系。用T3转SiWRKY36拟南芥植株进行抗性鉴定。【结果】谷子SiWRKY36全长1 485 bp,基因编码区包含UTR区和3个内含子以及4个外显子,与柳枝稷亲缘性最高,属于WRKY转录因子家族的第一类。SiWRKY36编码蛋白包含2个WRKY保守域,预测的SiWRKY36蛋白三级结构包含2个α螺旋结构和3个β折叠结构。启动子元件分析表明SiWRKY36包含ABA-responsive element(ABRE)、MYB、MYC、low-temperature-responsive element(LTRE)、GT-1等多17
[本文引用: 2]
DOI:10.1007/s00425-017-2766-9URLPMID:28861611 [本文引用: 1]
Main conclusion We cloned and characterized the ZmWRKY17 gene from maize. Overexpression of ZmWRKY17 in Arabidopsisled to increased sensitivity to salt stress and decreased ABA sensitivity through...
DOI:10.7685/j.issn.1000-2030.2015.01.007URL [本文引用: 1]
[目的]CAX(Ca2+/H+exchanger antiporter)是一类重要的跨膜转运蛋白,在植物体内阳离子转运上起着非常重要的作用。本文对苹果MdCAXs基因进行了生物信息学和组织特异性表达分析,以期为该类基因的功能研究奠定基础。[方法]利用生物信息学的方法对苹果MdCAXs基因家族染色体定位、蛋白质等电点、亲疏水性、跨膜区域、亚细胞定位及保守结构域进行了预测和分析,同时还分析了MdCAXs基因与其他物种CAXs基因的进化关系。采用实时荧光定量PCR法对苹果MdCAXs基因在不同盐处理下根、茎、叶中的表达变化进行分析。[结果]苹果MdCAXs家族包含8个成员,分别属于CAXsⅠ型中的A亚型和B亚型,编码436~817个氨基酸,等电点4.97~7.12,且都为疏水性蛋白;所有MdCAXs基因编码的氨基酸都有11个及以上的跨膜区域,含有CAXs基因5个典型的功能域:N-端自抑制区域(NRR)、C-端功能区域、Ca2+功能域(CaD)、C功能域和D功能域。亚细胞定位预测MdCAXs主要是定位在质膜和内质网上。苹果MdCAXs基因具有组织特异性表达特点,不同种类盐处理根、茎、叶中表达发生不同程度的变化,反映了MdCAXs存在功能上的差异,对不同阳离子有特异性的应答反应。[结论]苹果MdCAXs是一类跨膜转运蛋白,具有植物CAXs基因典型的结构特征,根、茎、叶对不同的盐离子具有不同的表达响应。
DOI:10.7685/j.issn.1000-2030.2015.01.007URL [本文引用: 1]
[目的]CAX(Ca2+/H+exchanger antiporter)是一类重要的跨膜转运蛋白,在植物体内阳离子转运上起着非常重要的作用。本文对苹果MdCAXs基因进行了生物信息学和组织特异性表达分析,以期为该类基因的功能研究奠定基础。[方法]利用生物信息学的方法对苹果MdCAXs基因家族染色体定位、蛋白质等电点、亲疏水性、跨膜区域、亚细胞定位及保守结构域进行了预测和分析,同时还分析了MdCAXs基因与其他物种CAXs基因的进化关系。采用实时荧光定量PCR法对苹果MdCAXs基因在不同盐处理下根、茎、叶中的表达变化进行分析。[结果]苹果MdCAXs家族包含8个成员,分别属于CAXsⅠ型中的A亚型和B亚型,编码436~817个氨基酸,等电点4.97~7.12,且都为疏水性蛋白;所有MdCAXs基因编码的氨基酸都有11个及以上的跨膜区域,含有CAXs基因5个典型的功能域:N-端自抑制区域(NRR)、C-端功能区域、Ca2+功能域(CaD)、C功能域和D功能域。亚细胞定位预测MdCAXs主要是定位在质膜和内质网上。苹果MdCAXs基因具有组织特异性表达特点,不同种类盐处理根、茎、叶中表达发生不同程度的变化,反映了MdCAXs存在功能上的差异,对不同阳离子有特异性的应答反应。[结论]苹果MdCAXs是一类跨膜转运蛋白,具有植物CAXs基因典型的结构特征,根、茎、叶对不同的盐离子具有不同的表达响应。
DOI:10.1007/s11105-013-0672-2URL [本文引用: 1]
The WRKY transcription factor (TF) plays an important role in plant developmental processes and stress responses. However, little is known about the WRKY TF family in Chinese cabbage ( Brassica rapa ssp. pekinensis ), although its genome has been completely sequenced. In this study, 145 genes of Chinese cabbage were identified that were anchored onto chromosomes 1 10 and further fractionated into three subgenomes. Organization and syntenic analysis indicated genomic distributions and collinear relationships of the BrWRKY s. Simultaneously, the selection pressures and evolutionary divergence of duplicated gene pairs were analyzed using nonsynonymous substitutions (Ka)/synonymous substitutions (Ks). Phylogenetic analyses showed that 145 BrWRKY s were clustered into three large groups and shared typical characters of WRKY TFs. In addition, Illumina RNA-Seq transcriptome of different tissues (i.e., roots, stems, and leaves) revealed tissue-specific and differential expression profiles of the BrWRKY s, and quantitative real-time polymerase chain reaction analysis showed the distinct and corresponsive expression patterns of the BrWRKY s in response to abiotic and biotic stresses in leaves. This study showed that these gene family members might play several roles in plant development, and abiotic and biotic stress responses might benefit from their functional characterization and utilization in the resistance engineering of Chinese cabbage.
DOI:10.1186/1471-2229-13-148URLPMID:3850935 [本文引用: 1]
Background WRKY genes encode one of the most abundant groups of transcription factors in higher plants, and its members regulate important biological process such as growth, development, and responses...
DOI:10.1093/jxb/eru381URLPMID:4246191 [本文引用: 1]
This study presents the genome-wide characterization of the Populus WRKY family under biotic and abiotic stresses. Overexpression of an SA-inducible gene, PtrWRKY89, enhanced resistance to pathogens in transgenic poplar. WRKY proteins are a large family of regulators involved in various developmental and physiological processes, especially in coping with diverse biotic and abiotic stresses. In this study, 100 putative PtrWRKY genes encoded the proteins contained in the complete WRKY domain in Populus. Phylogenetic analysis revealed that the members of this superfamily among poplar, Arabidopsis, and other species were divided into three groups with several subgroups based on the structures of the WRKY protein sequences. Various cis-acting elements related to stress and defence responses were found in the promoter regions of PtrWRKY genes by promoter analysis. High-throughput transcriptomic analyses identified that 61 of the PtrWRKY genes were induced by biotic and abiotic treatments, such as Marssonina brunnea, salicylic acid (SA), methyl jasmonate (MeJA), wounding, cold, and salinity. Among these PtrWRKY genes, transcripts of 46 selected genes were observed in different tissues, including roots, stems, and leaves. Quantitative RT-PCR analysis further confirmed the induced expression of 18 PtrWRKY genes by one or more stress treatments. The overexpression of an SA-inducible gene, PtrWRKY89, accelerated expression of PR protein genes and improved resistance to pathogens in transgenic poplar, suggesting that PtrWRKY89 is a regulator of an SA-dependent defence-signalling pathway in poplar. Taken together, our results provided signi铿乧ant information for improving the resistance and stress tolerance of woody plants.
DOI:10.1007/s11033-009-9823-9URLPMID:19757158 [本文引用: 1]
-1,3-Glucanases are a group of pathogenesis-related proteins that have been reported to be involved in plant defense against pathogens in many other plant-pathogen systems. However, it was not clear if these genes play similar role in wheat ( Triticum aestivum L.) against Puccinia striiformis f. sp. tritici ( Pst ), the stripe rust pathogen. To investigate the role of -1,3-glucanase (EC 3.2.1.39) in the resistance response of wheat (cv. Suwon 11) to stripe rust, a wheat -1,3-glucanase gene induced by Pst , designated as TaGlu , was cloned and characterized. TaGlu was predicted to encode a basic protein of 334 amino acids. Quantitative real-time PCR analyses revealed that the transcription of TaGlu was induced during both compatible and incompatible interactions with Pst , but the transcription level was much higher in the incompatible interaction than that in the compatible interaction. TaGlu also showed noticeable induction of gene expression in young green leaf tissues treated with salicylic acid, methyl jasmonate or ethylene. Immunogold labeling assays showed that the enzyme were localized mainly in the host cell wall and over the extrahaustorial matrix, and the labeling densities were found significantly higher in the incompatible interaction than those in the compatible interaction.
DOI:10.1146/annurev.phyto.050908.135202URLPMID:19400653 [本文引用: 1]
Abstract For more than 200 years, the plant hormone salicylic acid (SA) has been studied for its medicinal use in humans. However, its extensive signaling role in plants, particularly in defense against pathogens, has only become evident during the past 20 years. This review surveys how SA in plants regulates both local disease resistance mechanisms, including host cell death and defense gene expression, and systemic acquired resistance (SAR). Genetic studies reveal an increasingly complex network of proteins required for SA-mediated defense signaling, and this process is amplified by several regulatory feedback loops. The interaction between the SA signaling pathway and those regulated by other plant hormones and/or defense signals is also discussed.
DOI:10.3969/j.issn.1009-2196.2009.12.013URL [本文引用: 1]
Salicylic acid (SA) is a new resistance inducer, and has been a focus in development of SA-in-duced resistance of plants to diseases and pests. The basic properties and the function of salicylic acid in-volved in plant resistance to diseases and pests are introduced. The mechanism of salicylic acid in induc-ing resistance of plants to pest insects and diseases is discussed in terms of interaction of salicylic acid with salicylic acid-binding protein, and salicylic acid-dependent signaling pathway. The prospects for application of SA in inducing resistance of plants to pests and diseases are also made.
DOI:10.3969/j.issn.1009-2196.2009.12.013URL [本文引用: 1]
Salicylic acid (SA) is a new resistance inducer, and has been a focus in development of SA-in-duced resistance of plants to diseases and pests. The basic properties and the function of salicylic acid in-volved in plant resistance to diseases and pests are introduced. The mechanism of salicylic acid in induc-ing resistance of plants to pest insects and diseases is discussed in terms of interaction of salicylic acid with salicylic acid-binding protein, and salicylic acid-dependent signaling pathway. The prospects for application of SA in inducing resistance of plants to pests and diseases are also made.
DOI:10.1038/srep37674URLPMID:5121592 [本文引用: 1]
Jasmonates (JAs) play important roles in plant growth, development and defense. Comprehensive metabolomics profiling of plants under JA treatment provides insights into the interaction and regulation network of plant hormones.
DOI:10.1104/pp.16.00747URLPMID:27268959 [本文引用: 1]
Although necrotrophic pathogens cause many devastating plant diseases, our understanding of the plant defense response to them is limited. Here, we found that loss of function of WRKY57 enhanced the resistance of Arabidopsis (Arabidopsis thaliana) against Botrytis cinerea infection. Further investigation suggested that the negative regulation of WRKY57 against B. cinerea depends on the jasmonic acid (JA) signaling pathway. Chromatin immunoprecipitation experiments revealed that WRKY57 directly binds to the promoters of JASMONATE ZIM-DOMAIN1 (JAZ1) and JAZ5, encoding two important repressors of the JA signaling pathway, and activates their transcription. In vivo and in vitro experiments demonstrated that WRKY57 interacts with nuclear-encoded SIGMA FACTOR BINDING PROTEIN1 (SIB1) and SIB2. Further experiments display that the same domain, the VQ motif, of SIB1 and SIB2 interact with WRKY33 and WRKY57. Moreover, transient transcriptional activity assays confirmed that WRKY57 and WRKY33 competitively regulate JAZ1 and JAZ5, SIB1 and SIB2 further enhance these competitions of WRKY57 to WRKY33. Therefore, coordinated regulation of Arabidopsis against B. cinerea by transcription activators and repressors would benefit plants by allowing fine regulation of defense
DOI:10.1038/srep23632URLPMID:27004441 [本文引用: 1]
Most harvested fruits and vegetables are stored at low temperature but many of them are highly sensitive to chilling injury. Jasmonic acid (JA), a plant hormone associated with various stress responses, is known to reduce chilling injury in fruits. However, little is known about the transcriptional regulation of JA biosynthesis in relation to cold response of fruits. Here, we show the involvement of a Group I WRKY transcription factor (TF) from banana fruit, MaWRKY26, in regulating JA biosynthesis. MaWRKY26 was found to be nuclear-localized with transcriptional activation property.MaWRKY26was induced by cold stress or by methyl jasmonate (MeJA), which enhances cold tolerance in banana fruit. More importantly, MaWRKY26 transactivated JA biosynthetic genesMaLOX2, MaAOS3andMaOPR3via binding to their promoters. Further, MaWRKY26 physically interacted with a VQ motif-containing protein MaVQ5, and the interaction attenuated MaWRKY26-induced transactivation of JA biosynthetic genes. These results strongly suggest that MaVQ5 might act as a repressor of MaWRKY26 in activating JA biosynthesis. Taken together, our findings provide new insights into the transcriptional regulation of JA biosynthesis in response to cold stress and a better understanding of the molecular aspects of chilling injury in banana fruit.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00438-008-0392-8URLPMID:18987888 [本文引用: 1]
The root apex contains meristematic cells that determine root growth and architecture in the soil. Specific transcription factor (TF) genes in this region may integrate endogenous signals and external cues to achieve this. Early changes in transcriptional responses involving TF genes after a salt stress in Medicago truncatula (Mt) roots were analysed using two complementary transcriptomic approaches. Forty-six salt-regulated TF genes were identified using massive quantitative real-time RT-PCR TF profiling in whole roots. In parallel, Mt16K+ microarray analysis revealed 824 genes (including 84 TF sequences) showing significant changes ( p < 0.001) in their expression in root apexes after a salt stress. Analysis of salt-stress regulation in root apexes versus whole roots showed that several TF genes have more than 30-fold expression differences including specific members of AP2/EREBP, HD-ZIP, and MYB TF families. Several salt-induced TF genes also respond to other abiotic stresses as osmotic stress, cold and heat, suggesting that they participate in a general stress response. Our work suggests that spatial differences of TF gene regulation by environmental stresses in various root regions may be crucial for the adaptation of their growth to specific soil environments.
DOI:10.1016/j.plantsci.2018.03.018URL [本文引用: 1]
High salinity severely inhibits the growth and productivity of grape plants. However, knowledge of salt-stress regulation remains limited in WRKY members of grapes. Here, we isolated a novel VvWRKY30 gene from Vitis vinifera L. and studied its role in salt-stress resistance. The VvWRKY30 protein fused with green fluorescent protein localized to the nucleus and the transcriptional activation activity of VvWRKY30 was confirmed in yeast. Moreover, VvWRKY30 showed key transcriptional activity domain at the N-terminal and specifically binds to the W-BOX. VvWRKY30 showed the highest expression in the shoot tip and functional leaves of grape plants. VvWRKY30 expression was induced by salt as well as stress signaling molecules H 2 S and H 2 O 2 . Overexpression of VvWRKY30 in Arabidopsis increased its resistance to salt stress at different stages of growth. Under salinity stress, VvWRKY30 overexpressing lines had higher antioxidant activities and lower reactive oxygen species contents. Soluble sugar and proline concentrations also increased in VvWRKY30 overexpressing lines in the presence of NaCl. In addition, the transcription of genes related to antioxidant biosynthesis, glyco-metabolism and proline biosynthesis increased in the VvWRKY30 overexpressing lines. Taken together, this study confirmed the important role of VvWRKY30 in increasing salt stress resistance by regulating reactive oxygen species-scavenging and the accumulation of osmoticum.
DOI:10.3864/j.issn.0578-1752.2016.22.006URL [本文引用: 1]
一定程度的镉胁迫严重影响了作物的生长发育和农产品的产量及品质。文中全面综述了重金属镉胁迫对作物和人类的危害,以及镉在作物体内的吸收、转运和积累特征及其相关的主要调控基因和功能。简要概述了作物抗镉耐镉机制,重点讨论了其中的根系镉滞留作用的生理和生化机制。重金属镉主要通过根部吸收进入植株,在根中,Cd^2+首先进入由细胞间隙、细胞壁微孔以及细胞壁到质膜之间的空隙等构成的"自由空间",然后通过主动或被动吸收跨膜进入胞质,再经共质体或质外体途径运输到木质部导管中。水稻等作物主要通过下列途径来适应镉胁迫:细胞壁的滞留作用、原生质体的螯合作用、液泡的区室化作用、逆境蛋白和脯氨酸的积累、抗氧化酶系统活性的提高、根系的滞留作用。根系镉滞留作用作为一种重要的抗耐镉毒害的方式,在调控作物对镉的吸收、转运和分配积累,阻碍镉进入植株地上部和原生质体,减少镉对作物自身生长发育及农产品产量和品质的影响等方面起着非常重要的作用。主要包括根茎间低转运量导致的镉滞留、根系细胞壁滞留和液泡滞留。(1)根茎间低转运量导致的镉滞留。该种滞留作用主要受到根系木质部的镉装载能力和镉长距离运输载体——植物螯合肽(PCs)含量的影响;它们主要受到质膜上跨膜离子转运蛋白HMA2和HMA4以及细胞中的PCs合成酶及其相应基因(如HMA2、HMA4、PCs1等)的调控。这些蛋白和基因对木质部的镉滞留起到负调控作用。(2)细胞壁滞留作用。根系细胞壁滞留发生在质外体部分(包括细胞壁和胞间层),主要与质外体的组成成分和结构相关,其中起关键作用的是果胶多糖,半纤维素也起到一定作用。根据果胶和半纤维素滞留镉的作用方式的不同,细胞壁滞留作用可分为物理滞留和化学滞留。物17
DOI:10.3864/j.issn.0578-1752.2016.22.006URL [本文引用: 1]
一定程度的镉胁迫严重影响了作物的生长发育和农产品的产量及品质。文中全面综述了重金属镉胁迫对作物和人类的危害,以及镉在作物体内的吸收、转运和积累特征及其相关的主要调控基因和功能。简要概述了作物抗镉耐镉机制,重点讨论了其中的根系镉滞留作用的生理和生化机制。重金属镉主要通过根部吸收进入植株,在根中,Cd^2+首先进入由细胞间隙、细胞壁微孔以及细胞壁到质膜之间的空隙等构成的"自由空间",然后通过主动或被动吸收跨膜进入胞质,再经共质体或质外体途径运输到木质部导管中。水稻等作物主要通过下列途径来适应镉胁迫:细胞壁的滞留作用、原生质体的螯合作用、液泡的区室化作用、逆境蛋白和脯氨酸的积累、抗氧化酶系统活性的提高、根系的滞留作用。根系镉滞留作用作为一种重要的抗耐镉毒害的方式,在调控作物对镉的吸收、转运和分配积累,阻碍镉进入植株地上部和原生质体,减少镉对作物自身生长发育及农产品产量和品质的影响等方面起着非常重要的作用。主要包括根茎间低转运量导致的镉滞留、根系细胞壁滞留和液泡滞留。(1)根茎间低转运量导致的镉滞留。该种滞留作用主要受到根系木质部的镉装载能力和镉长距离运输载体——植物螯合肽(PCs)含量的影响;它们主要受到质膜上跨膜离子转运蛋白HMA2和HMA4以及细胞中的PCs合成酶及其相应基因(如HMA2、HMA4、PCs1等)的调控。这些蛋白和基因对木质部的镉滞留起到负调控作用。(2)细胞壁滞留作用。根系细胞壁滞留发生在质外体部分(包括细胞壁和胞间层),主要与质外体的组成成分和结构相关,其中起关键作用的是果胶多糖,半纤维素也起到一定作用。根据果胶和半纤维素滞留镉的作用方式的不同,细胞壁滞留作用可分为物理滞留和化学滞留。物17
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}