 ,**, 赖振光,**, 孙祖东,*, 杨守臻, 陈怀珠, 唐向民广西农业科学院经济作物研究所/农业农村部西南玉米大豆间套作区农业科学观测实验站, 广西南宁 530007
,**, 赖振光,**, 孙祖东,*, 杨守臻, 陈怀珠, 唐向民广西农业科学院经济作物研究所/农业农村部西南玉米大豆间套作区农业科学观测实验站, 广西南宁 530007Identification of the candidate genes of soybean resistance to bean pyralid (Lamprosema indicata Fabricius) by BSA-Seq and RNA-Seq
ZENG Wei-Ying,**, LAI Zhen-Guang,**, SUN Zu-Dong,*, YANG Shou-Zhen, CHEN Huai-Zhu, TANG Xiang-MinInstitute of Economic Crops, Guangxi Academy of Agricultural Sciences/Southwest Experimental Station of Maize-Soybean Intercrop, Ministry of Agriculture and Rural Affairs, Nanning 530007, Guangxi, China通讯作者: * 孙祖东, E-mail:sunzudong639@163.com
第一联系人:
收稿日期:2020-08-25接受日期:2021-01-13网络出版日期:2021-08-12
| 基金资助: |
Received:2020-08-25Accepted:2021-01-13Online:2021-08-12
| Fund supported: |
作者简介 About authors
曾维英,E-mail:zengweiying_1981@163.com
赖振光,E-mail:519229671@qq.com

摘要
豆卷叶螟(Lamprosema indicata Fabricius)是重要的大豆食叶性害虫, 挖掘大豆抗豆卷叶螟相关基因对大豆抗虫品种选育和遗传改良至关重要。本研究用大豆高抗豆卷叶螟材料赶泰-2-2和高感豆卷叶螟材料皖82-178进行杂交构建F2代分离群体, 从303个F2代单株中挑选出高抗豆卷叶螟和高感豆卷叶螟的单株各30株, 分别构建2个极端性状的DNA混合池用于全基因组重测序以分析控制豆卷叶螟相关的候选基因。结果表明, 4个样本中共有11,963,077个单核苷酸多态性(SNPs)标记, 根据SNP-index方法关联分析, 共有329个基因位于99%置信区间外, 这些基因主要集中在7号染色体5,601,065~5,865,237 bp区间(总长为0.26 Mb)、16号染色体2,975,110~6,336,096 bp区间(总长为3.36 Mb)、18号染色体44,366,115~54,297,600 bp区间(总长为9.93 Mb)等区域内。将BSA-Seq结果与转录组测序结果进行联合分析发现, 有12个基因相关联; 最后, 结合生物信息学分析、候选基因的表达模式和基因的同源注释, 锁定CNGC4、WRKY16转录因子、AAP7、丝氨酸/苏氨酸蛋白激酶、ZPR1B等12个基因为控制豆卷叶螟性状相关的候选基因。本研究结果不仅为解析大豆抗豆卷叶螟的分子机理奠定重要的基础, 也将为大豆抗虫基因的克隆奠定坚实的理论基础。
关键词:
Abstract
Bean pyralid is an important leaf-feeding insect in soybean. Identification of insect-tolerant genes from soybean has great significant to the crop insect-tolerant breeding and genetic improvement. In this study, an F2 population with 303 individuals was constructed using insect-resistant line Gantai-2-2 and insect-sensitive line Wan 82-178. 30 F2 insect-resistant individuals and 30 insect-sensitive individuals were selected respectively to construct two DNA pools which were used for the whole-genome re-sequencing. The results showed that there were a total of 11,963,077 single nucleotide polymorphism (SNPs) markers identified in two parental lines and two mixed pools. According to the association analysis of SNP-index method, a total of 329 genes were located outside the 99% confidence interval. These genes were mainly concentrated in the regions of 5,601,065-5,865,237 bp with a total of 0.26 Mb on chromosome 7, 2,975,110-6,336,096 bp with a total of 3.36 Mb on chromosome 16, and 44,366,115-54,297,600 bp with a total of 9.93 Mb on chromosome 18. Correlation analysis of BSA-Seq and transcriptome sequencing showed that 12 genes were correlated. Then, 12 candidate genes, including CNGC4, WRKY transcription factor 16, AAP7, serine/threonine protein kinase and ZPR1B were identified by bioinformatics analysis, differential expression analysis, and homologous annotation. This study laid an important foundation for the analysis of the molecular mechanism of soybean resistance to bean pyralid and the cloning of anti-insect genes.
Keywords:
PDF (3901KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
曾维英, 赖振光, 孙祖东, 杨守臻, 陈怀珠, 唐向民. 基于BSA-Seq和RNA-Seq方法鉴定大豆抗豆卷叶螟候选基因. 作物学报[J], 2021, 47(8): 1460-1471 DOI:10.3724/SP.J.1006.2021.04195
ZENG Wei-Ying, LAI Zhen-Guang, SUN Zu-Dong, YANG Shou-Zhen, CHEN Huai-Zhu, TANG Xiang-Min.
大豆是我国植物蛋白和食用油的主要来源, 虫害是影响大豆产量和品质的重要因素。豆卷叶螟(Lamprosema indicata Fabricius)分布于东北、黄淮海及南方地区[1], 是南方和黄淮大豆产区主要食叶性害虫[2,3], 一年可发生4~5代, 有世代重叠现象, 在温度、湿度适宜时会大面积发生, 一般造成大豆减产15%~20%, 严重时可达30%以上。为害严重的年份, 叶片被取食后只剩叶脉和叶柄, 造成产量的严重损失[3,4,5]。
鉴于豆卷叶螟为害的严重性, 国内外在大豆抗豆卷叶螟方向展开了深入研究, 已鉴定出高抗豆卷叶螟材料赶泰-2-2、丰平黑豆等, 也鉴定出了高感材料皖82-178、山东大豆等[6,7]; 控制豆卷叶螟的抗性位点主要位于A2、C2、D1a、D1b、H、K和O等连锁群上[8,9,10]; 可溶性糖含量、茉莉酸含量、过氧化氢酶和多酚氧化酶活性只与豆卷叶螟诱导有关, 而超氧化物岐化酶活性、乙烯和脱落酸含量与虫害诱导和材料基因型都有关[11]; 大豆胰蛋白酶抑制剂、查尔酮异构酶4、脂氧合酶9等可能是大豆抗豆卷叶螟潜在的靶标蛋白(基因)[12,13,14], gma-miR156q、gma-miR166u、gma-miR166b、gma-miR319d、gma-miR394a-3p和gma-miR396e等miRNA可能参与大豆抗豆卷叶螟的抗性调控[15]。
目前, 大豆抗虫基因主要源于微生物的苏云金芽孢杆菌(Bacillus thuringiensis, Bt)杀虫晶体蛋白基因(Cry), 并转入大豆中获得了抗虫转基因大豆。郭东全等[16]将构建的CryIA和CpTI高效双价杀虫基因转入到吉林20与吉林27大豆品种中, 获得了高效抗虫且基本稳定遗传的转基因抗虫大豆; 陈秀华等[17]将Cry1Iem基因转化到绥农14中, 得到抗虫效果较好的转化植株; 武小霞等[18]将Cry1Ial基因转入黑农35中, 检测得到抗大豆食心虫转基因植株; 朱延明等[19]将具有豆荚特异性启动子Pmsg的Cry2Aa9m基因转入绥农28中, 获得了对鳞翅目和双翅目都具有抗性植株; 蓝岚等[20]将CryIA抗虫基因转入东农50中, 证明转基因植株对大豆食心虫具有明显抗性; 高嵩[21]将Cry1Ab13抗虫基因转入到吉农28中发现, 转基因植株对大豆食心虫具有明显抗性; 美国孟山都公司、陶氏公司将Cry1Ac、Cry1F等目的基因导入大豆中, 获得对鳞翅目类昆虫具有强烈杀虫活性的转基因抗虫大豆品种, 并在加拿大、日本、美国展开商业化种植[22,23,24]。但Bt抗虫基因属于外源基因, 转Bt抗虫基因大豆对非靶标生物造成了潜在的威胁。因此, 需鉴定出大豆自身含有的抗虫目的基因并进行遗传转化, 获得抗虫转基因材料。
随着高通量测序技术的兴起, 基于全基因组测序的BSA方法在不构建遗传图谱的情况下能高效快速地挖掘目标性状基因, BSA-Seq方法已被广泛应用在水稻、黄瓜、番茄、大豆等作物中, 并且成功地定位出水稻耐盐[25]、水稻稻瘟病[26]、黄瓜早花[27]、番茄果实重量[28]、大豆疫霉病[29]等基因。本研究利用高抗豆卷叶螟材料赶泰-2-2与高感材料皖82-178杂交构建F2代分离群体, 通过对亲本和2个极端表型子代混合池进行BSA-Seq全基因组重测序, 定位目标性状关联区域, 推测与抗豆卷叶螟有关的基因; 再结合前期的转录组测序结果, 关注抗虫性状候选区域的相关基因的表达水平, 进一步缩小候选基因范围, 鉴定出控制豆卷叶螟性状的候选基因, 旨在为更深入的阐明大豆抗豆卷叶螟的分子机制开拓新思路, 为大豆的抗虫分子育种奠定理论基础。
1 材料与方法
1.1 试验材料
2018年利用皖82-178 (高感豆卷叶螟)为母本和赶泰-2-2 (高抗豆卷叶螟)为父本进行杂交, 试验材料(F2代群体和亲本材料)于2020年3月种植在广西壮族自治区农业科学院院本部试验田防虫网室中(东经108.1146°、北纬22.5059°, 红壤土), 垄长1.5 m、行距0.5 m, 3行区播种, 每行10株, 在大豆整个生育期期间不喷施农药, 不施用肥料。待植株长至10片复叶时, 按每株3虫的密度分别接豆卷叶螟4龄幼虫3头(用镊子取3头虫接到第9片复叶的中间小叶上), 48 h后以卷包数和卷叶率为评价指标进行抗性鉴定(卷叶率0~10%为高抗、10%~30%为抗、30%~50%为中抗、50%~70%为感、>70%为高感)[7,8]。从303个F2分离群体中挑选30株极端抗豆卷叶螟单株和30株极端感豆卷叶螟单株构建子代高抗混合池和高感混合池, 用于抗豆卷叶螟的基因定位。田间采集相应植株幼嫩叶片。1.2 试验方法
1.2.1 DNA提取 采用改良CTAB法[30]分别提取亲本材料(赶泰-2-2和皖82-178)和F2代群体极端个体的DNA, 将极端抗豆卷叶螟植株幼嫩叶片和极端感豆卷叶螟植株幼嫩叶片各30株的DNA等量混合, 制备总DNA 3 μg, 样品浓度≥20 ng μL-1, 构建高抗子代混合池、高感子代混合池。1.2.2 文库构建及测序 由深圳华大基因科技服务有限公司完成样本建库和测序。将检测合格的基因组DNA样本用Covaris仪超声波随机打断成特定大小的片段, 打断后的样本用Agencourt AMPure XP-Medium kit进行片段选择, 使得样本条带集中在200~300 bp左右, 使用试剂盒Qubit dsDNA HS AssayKit, 500 assays检测纯化后DNA样本量; 对打断的DNA片段进行末端修复, 在3°端连接A碱基, 将特定测序引物连接到DNA片段上; 纯化连接产物, 选择合适大小的DNA片段; 将DNA片段在cBot仪器上扩增制备文库; 对构建的文库进行质量检测; 将质量检测合格的文库上机测序。对亲本池和子代混合池分别开展20×和50×覆盖度的全基因组重测序。
1.2.3 生物信息学分析 对测序得到的Raw reads进行过滤, 将过滤后的Clean reads运用BWA软件与大豆参考基因组进行比对[31,32], 将比对结果进行排序、质量过滤和标记duplicate后进行变异类型检测。使用GATK软件查找样本相对于参考基因组之间的SNP, 用Breakdancer[33]软件来检测样本与参考基因组之间的SV位点, 用Control-FREEC[34]软件来查找样本相对于参考基因组之间的CNV区域。为得到更准确的变异信息, 对变异位点分别进行进一步的过滤, 使用变异位点注释软件Annovar[35]将变异位点在基因组上的位置区域注释出来。
1.2.4 SNP-index计算 子代群体与亲本之间的序列差异程度用SNP-index来表示, 计算经过质量控制后的每一个变异位点在高抗子代池和高感子代池中的SNP-index, 过滤掉2个子代极端混合池里面SNP-index都小于0.3的位点, 只保留双亲纯合且有差异、子代混池都为非缺少的SNP位点。同时, 将2个子代池的SNP-index相减得到Delta SNP-index。Delta SNP-index为1或者-1, 说明相对应的SNP来源于其中一个亲本, 这一位点所在的基因很有可能是与目的性状相关的基因。
1.2.5 目的基因的筛选与功能注释 使用滑窗法对滑窗内的Delta SNP-index进行计算, 取滑窗内所有SNP位点的Delta SNP-index的平均值, 滑窗大小为1.00 Mb, 步长为0.05 Mb, 对Delta SNP-index在各染色体上的分布进行作图。基于基因组Gff注释文件, 对所有候选位点进行来源注释, 挑选出位于基因区的SNP位点, 为缩小筛选范围, 选取99%置信区间作为筛选阈值, 单独把Delta SNP-index大于99%置信区间的SNP位点筛选出来进行注释。利用Blast2go_v2.5软件对所鉴定的基因进行GO注释分析, 计算得到的P-value通过Bonferroni校正之后, 以Corrected P-value≤0.05为阈值。利用Blast_v2.2.26软件在KEGG pathway数据库对所鉴定的基因进行Pathway富集分析, 计算得到的P- value通过Bonferroni校正之后, Corrected P-value≤0.05为阈值。
1.2.6 转录组分析 利用TRIzol Kit提取叶片总RNA, 采用Truseq RNA试剂盒进行mRNA纯化并构建cDNA文库, 对获得的cDNA文库进行PCR扩增富集, 2%琼脂糖凝胶电泳检测, 回收目的片段。然后使用Illumina Hiseq 2000测序仪进行对样本测序。以上试验均由深圳华大基因科技服务有限公司完成。测序后的数据经Base calling转化为Raw data或Raw reads, 随后对Raw reads进行质控。利用SOAPfuse软件[36]对原始测序数据进行去除处理得到Clean reads。再利用Tophat将Clean reads比对到大豆参考基因组, 允许有2个碱基的错配[37,38]。表达定量的结果以FPKM (Fragments per kilobase of exon model per million mapped fragments)为单位, 在得到差异检验的FDR值同时, 根据基因的表达量(FPKM值)计算该基因在不同样本间的差异表达倍数。以“FDR≤0.001和|log2 Ratio (FC, Fold Change)|≥1”作为阈值来判断基因差异表达的显著性[39]。
2 结果与分析
2.1 测序质量评估
利用高通量测序技术对亲本及子代混合池共4个样本进行全基因组重测序, 共产生194.54 G原始测序数据, 经过SOAPnuke软件对数据进行接头过滤、低质量过滤和去N过滤后, 最终得到191.35 G高质量的Clean Reads数据。GC含量在35.46%~ 35.60%之间, 测序数据质量较高(Q20≥96.29%, Q30≥91.70%) (表1)。由此可知4个样本的数据量充足, 测序数据可靠, 测序质量合格, GC分布正常, 可进行下一步分析。Table 1
表1
表1原始测序数据统计表
Table 1
| 样本 Sample | 原始序列数 Raw reads | 原始碱基数 Raw bases | 过滤后序列数 Clean reads | 过滤后碱基数 Clean bases | 有效率 Clean_data/ raw_data (%) | GC含量 Clean_GC_rate (%) | Q20含量 Q20 (%) | Q30含量Q30 (%) |
|---|---|---|---|---|---|---|---|---|
| 皖82-178 Wan 82-178 | 438,210,500 | 65,731,575,000 | 432,474,442 | 64,871,166,300 | 98.69 | 35.57 | 96.50 | 92.13 |
| 赶泰-2-2 Gantai-2-2 | 441,716,184 | 66,257,427,600 | 433,990,902 | 65,098,635,300 | 98.25 | 35.60 | 96.34 | 91.79 |
| 高感子代池 Highly susceptible pool | 206,898,280 | 31,034,742,000 | 202,406,834 | 30,361,025,100 | 97.83 | 35.54 | 96.44 | 92.01 |
| 高抗子代池 Highly resistant pool | 210,122,884 | 31,518,432,600 | 206,805,618 | 31,020,842,700 | 98.42 | 35.46 | 96.29 | 91.70 |
新窗口打开|下载CSV
使用BWA软件(
Table 2
表2
表2质控后测序数据与参考基因组比对结果统计分析
Table 2
| 样本 Sample | 过滤后序 列数 Clean reads | 比对上数据 Mapping reads | 比对率 Mapping rate (%) | 优质比对率 Properly paired ratio (%) | 平均测序深度 Mean depth | 1×覆盖度Coverage ≥ 1× (%) | 5×覆盖度 Coverage ≥ 5× (%) | 10×覆盖度 Coverage ≥ 10× (%) | 20×覆盖度 Coverage ≥ 20× (%) |
|---|---|---|---|---|---|---|---|---|---|
| 皖82-178 Wan 82-178 | 43,4152,986 | 433,232,376 | 99.79 | 99.73 | 65.78 | 96.67 | 96.12 | 95.67 | 94.62 |
| 赶泰-2-2 Gantai-2-2 | 435,703,158 | 434,715,984 | 99.77 | 99.72 | 65.98 | 96.68 | 96.13 | 95.68 | 94.60 |
| 高感子代池 Highly susceptible pool | 203,202,650 | 202,820,220 | 99.81 | 99.76 | 30.78 | 95.75 | 94.55 | 93.29 | 83.88 |
| 高抗子代池 Highly resistant pool | 207,33,600 | 207,143,542 | 99.76 | 99.71 | 31.44 | 95.76 | 94.56 | 93.32 | 84.44 |
新窗口打开|下载CSV
将测序Reads比对到参考基因组以后, 统计参考基因组上不同染色体区域的覆盖度和测序深度分布情况。1×覆盖率百分比超过95.75%, 5×覆盖率百分比超过94.55%, 10×覆盖率百分比超过93.29%, 20×覆盖率百分比超过83.88% (表2)。通过Qualimap软件对比对结果进行统计, 参考基因组被均匀覆盖, 随机性良好, 有利于SNP过滤和筛选。
2.2 SNP检测和注释
SNP分析结果显示, 4个样本中共获得11,963,077个SNP位点, 其中高感子代混合池中最少, 皖82-178 (高感亲本)中最多。SNP位置和编码信息统计显示, 同类型材料间(亲本间, 子代池间)相同类型的SNP数和比例大体相当(表3)。纯合突变SNP比率明显高于杂合突变SNP比率, 纯合突变SNP 比率均高于99.73%。Table 3
表3
表3多态性位点注释统计表
Table 3
| 变异位点信息 Variation sites information | 皖82-178 Wan 82-178 | 赶泰-2-2 Gantai-2-2 | 高感子代池 Highly susceptible pool | 高抗子代池 Highly resistant pool |
|---|---|---|---|---|
| 内含子Intron | 324,202 | 324,333 | 230,606 | 231,332 |
| 基因区间Intergenic region | 2,589,457 | 2,588,540 | 1,851,943 | 1,867,897 |
| 可变剪切位点Splicing | 612 | 620 | 449 | 444 |
| 基因上游Upstream | 192,755 | 192,753 | 133,797 | 136,203 |
| 基因下游Downstream | 162,024 | 161,574 | 113,186 | 114,671 |
| 基因上游/基因下游Upstream/downstream | 12,038 | 12,088 | 8296 | 8733 |
| 5°非翻译区UTR5° | 23,658 | 23,746 | 68,071 | 16,818 |
| 3°非翻译区UTR3° | 29,843 | 29,595 | 20,949 | 21,579 |
| 终止子提前Stop gain | 1652 | 1649 | 1177 | 1225 |
| 终止子丢失Stop loss | 270 | 276 | 197 | 204 |
| 同义突变Synonymous | 48,462 | 48,433 | 35,671 | 35,492 |
| 非同义突变Non-synonymous | 67,206 | 67,040 | 49,333 | 49,653 |
| SNP总数SNP numbers | 3,487,363 | 3,485,674 | 2,483,855 | 2,506,185 |
| 纯合突变SNP Hom SNP number | 3,480,115 | 3,478,433 | 2,477,251 | 2,499,464 |
| 杂合突变SNP Hete SNP number | 7248 | 7241 | 6604 | 6721 |
| 纯合突变SNP比率Hom SNP rate (%) | 99.79 | 99.79 | 99.73 | 99.73 |
| 杂合突变SNP比率Het SNP rate (%) | 0.21 | 0.21 | 0.27 | 0.27 |
| 转换Ts | 2,264,913 | 2,264,110 | 1,606,918 | 1,617,797 |
| 颠换Tv | 1,218,400 | 1,217,609 | 872,951 | 884,443 |
| 转换/颠换Ts/Tv | 1.86 | 1.86 | 1.84 | 1.83 |
新窗口打开|下载CSV
2.3 候选基因筛选
利用SNP-index方法对检测到的SNP进行分析, 4个样本共鉴定出高质量的可信SNP位点11,963,077个。根据SNP-index方法关联阈值判断, 所有候选位点对应的基因数量为56,909个, 当拟合后Delta SNP-index置信度为99%时, 20条染色体上共有329个基因位于99%置信区间外, 主要集中在7号染色体5,601,065~5,865,237 bp区间(总长为0.26 Mb, 区间内基因数量为25个)、16号染色体2,975,110~6,336,096 bp区间(总长为3.36 Mb, 区间内基因数量为36个)、18号染色体44,366,115~ 54,297,600 bp区间(总长为9.93 Mb, 区间内基因数量为52个)等区域内, 区间内共包含113个基因(图1)。1号染色体和19号染色体上没有检测到基因, 2号、6号、8号、9号、10号、11号和12号染色体上检测到的基因数少于10个。图1
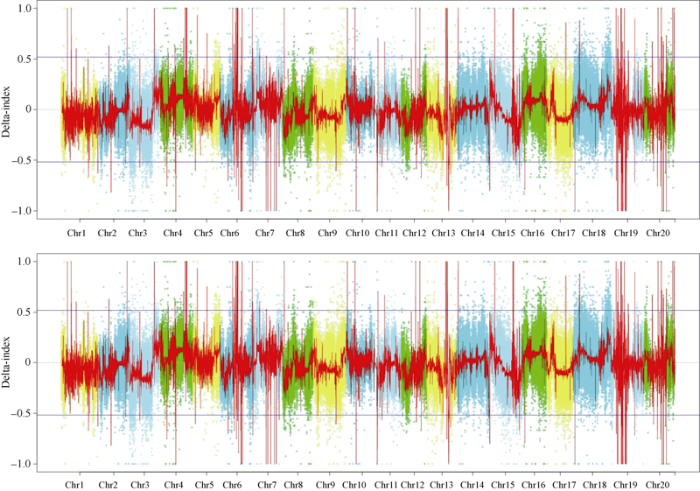 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1所有染色体Delta SNP-index图
横坐标为染色体编号, 纵坐标为Delta SNP-index值。不同颜色的点表示不同染色体上筛选后的SNP, 红色曲线表示滑窗后的Delta SNP-index值, 蓝色直线表示99%置信区间值。
Fig. 1Delta SNP-index map of all chromosomes
The X-axis is the chromosome number, Y-axis is the Delta SNP-index value. Different colored dots represent SNPs screened on different chromosomes, the red curve represents the Delta SNP-index value after the sliding window, the blue line represents the 99% confidence interval.
为进一步分析基因所在的亚细胞定位、分子功能及参与的生物学过程, 对所鉴定出的329个基因进行GO功能注释分析表明, 329个基因中有181个被注释到20个功能组中(图2), 包括9个生物学进程、7个细胞组分和4个分子功能, 在生物学进程中, 这些基因主要参与刺激反应、单一生物体过程、代谢过程、细胞过程等生物学过程; 在细胞组分中, 这些基因主要集中在细胞、细胞组分、膜、膜组分等组分; 在分子功能中, 这些基因主要参与结合、催化活性等功能。说明虫害胁迫下大豆对逆境的响应可能与细胞组织、细胞膜组织等的修复功能有关, 且一些催化活性的酶可能参与到虫害逆境胁迫中。
图2
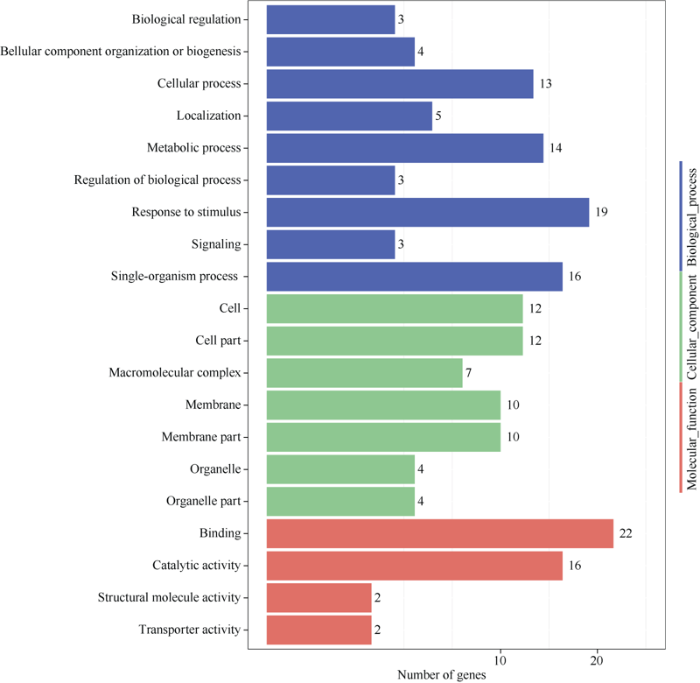 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2候选基因GO富集分析情况
Fig. 2GO enrichment of the candidate genes
为鉴别329个基因参与富集的代谢途径, 利用KEGG通路数据库对候选基因进行Pathway富集分析表明, 329个基因中有30个基因主要富集在植物病原菌互作、代谢途径、次生代谢物的生物合成、RNA降解等通路中(图3)。表明当大豆受到豆卷叶螟危害以后, 植株体内的防御系统会立即响应刺激,适当地增加体内的代谢活动, 产生防御物质, 如防御酶、蛋白质抑制酶等物质, 而且增强各种酶的活性来促进防御。
图3
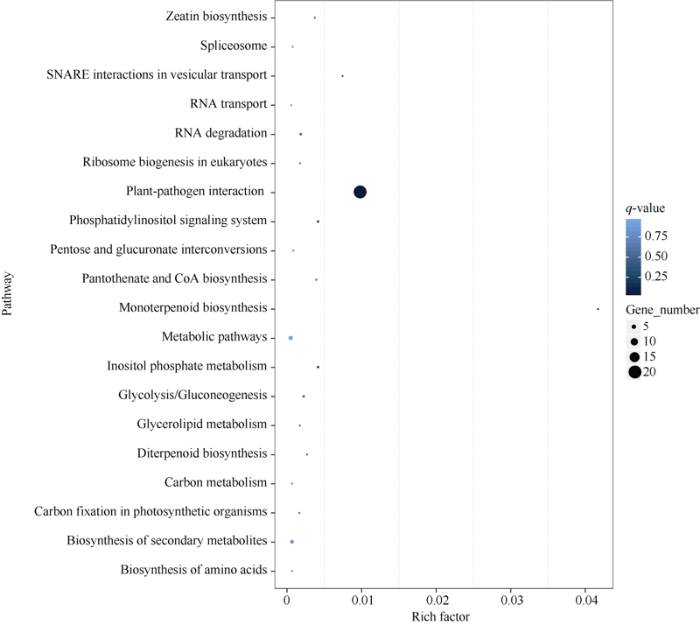 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3前20条富集通路注释情况
Fig. 3Top 20 pathway annotations of the candidate genes
将BSA-Seq结果预测的基因和转录组测序结果[13]进行联合分析, 将位于99%置信区间以外SNP对应的基因与转录组数据进行比对, 获得转录组中的同源序列及基因的表达水平。比对发现共有12个基因在4个比较组中呈显著性差异表达, 这些基因分别位于2号、12号、16号、18号等9条染色体上(表4), 其中响应豆卷叶螟取食胁迫诱导的基因有8个, 这8个基因中共有7个为显著上调基因, 分别为环核苷酸门控离子通道4 (CNGC4)、WRKY16转录因子、赤霉素2-β加双氧酶8、氨基酸透性酶7 (AAP7)、Mdis1相互受体激酶2、丝氨酸/苏氨酸蛋白激酶、ZPR1B, 只有纤维素合成酶A催化亚基4 (CesA4)为显著下调基因; 类枯草杆菌蛋白酶、抗病蛋白RGA3和硫氰酸酶结构域蛋白酶10等3个基因在高感材料中的表达量显著高于高抗材料, 1个未知基因在在高感材料中的表达量显著低于高抗材料。最后, 结合基因的同源注释、基因差异表达分析和生物信息学分析等, 推测环核苷酸门控离子通道4 (CNGC4)、WRKY16转录因子、AAP7、丝氨酸/苏氨酸蛋白激酶、ZPR1B等12个基因为抗豆卷叶螟有关的候选基因。
Table 4
表4
表4候选基因信息表
Table 4
| 序号 No. | 基因ID Gene ID | 基因注释 Gene annotation | 染色体 Chr. | 比较组 Comparison group | 上调或下调a Up or down a |
|---|---|---|---|---|---|
| 1 | XM_003526153.4 | 环核苷酸门控离子通道4 Cyclic nucleotide-gated ion channel 4 (CNGC4) | Chr. 6 | HR0/HR48 HS0/HS48 | Up |
| 2 | XM_003533631.4 | 纤维素合成酶A催化亚基4 (CesA4) Cellulose synthase A catalytic subunit 4 [UDP-forming] | Chr. 9 | HR0/HR48 HS0/HS48 | Down |
| 3 | NM_001250658.2 | WRKY16转录因子 WRKY transcription factor 16 | Chr. 12 | HR0/HR48 | Up |
| 4 | NM_001255802.1 | 赤霉素2-β加双氧酶8 Gibberellin 2-beta-dioxygenase 8-like | Chr. 11 | HR0/HR48 | Up |
| 5 | XM_003519602.4 | 氨基酸透性酶7 (AAP7) Probable amino acid permease 7 | Chr. 2 | HR0/HR48 | Up |
| 6 | XM_026126143.1 | Mdis1相互受体激酶2 MDIS1-interacting receptor like kinase 2 | Chr. 16 | HR0/HR48 | Up |
| 7 | XM_006601656.3 | 丝氨酸/苏氨酸蛋白激酶 Serine/threonine protein kinase-like protein | Chr. 18 | HR0/HR48 | Up |
| 8 | NM_001248669.2 | ZPR1B Protein LITTLE ZIPPER 2-like | Chr. 18 | HS0/HS48 | Up |
| 9 | XM_026126905.1 | 类枯草杆菌蛋白酶 Subtilisin-like protease Glyma18g48580 | Chr. 18 | HS0/HR0 | Down |
| 10 | XM_026125778.1 | 抗病蛋白RGA3 Putative disease resistance protein RGA3 | Chr. 15 | HS0/HR0 | Down |
| 11 | XM_026126161.1 | 无特征LOC100809946 Uncharacterized LOC100809946 | Chr. 16 | HS0/HR0 | Up |
| 12 | XM_026127992.1 | 硫氰酸酶结构域蛋白酶10 Rhodanese-like domain-containing protein 10 | Chr. 3 | HS48/HR48 HS0/HR0 | Down |
新窗口打开|下载CSV
3 讨论
基于BSA-Seq的方法挖掘新基因在作物农艺性状相关的基因定位中的应用日益广泛, 该方法在构建遗传图谱的情况下高效快速地挖掘目的基因[40], 是一种简单快速、准确的基因定位方法, 已成为育种家进行快速QTL定位和挖掘各种不同作物目标性状基因的一种有效方法。如Takagi等[25]利用BSA-Seq方法找到控制水稻耐盐性的候选基因OsRR22, 仅用2年时间培育出水稻耐盐性新品种; Lu等[27]利用BSA-Seq方法定位到84个控制黄瓜早花性状基因并确定CsFT基因为候选基因; Illa- Berenguer等[28]利用BSA-Seq方法定位到66个控制番茄果实重量性状基因; Guo等[41]利用BSA-Seq方法快速定位到29个辣椒抗黄瓜花叶病毒性状基因并挖掘出2个候选基因; Ma等[42]利用BSA-Seq方法定位到控制水稻低柱头外露突变候选基因; Zhao等[43]利用BSA-Seq方法鉴定到20个棉花细胞质雄性不育的育性恢复候选基因; 张尧锋等[44]利用BSA-Seq方法在甘蓝型油菜中定位到8个控制有限花序性状候选基因。BSA-Seq方法也被用于农作物的抗虫基因定位, Liang等[45]利用BSA-Seq方法定位到1个与黄瓜蚜虫抗性相关的区域, 该区域内包含40个基因, 其中16个基因可能与蚜虫抗性有关。利用全基因组重测序与BSA相结合同样能够高效和快速鉴定大豆中的目标性状基因[46], 本研究利用BSA-Seq分析和转录组测序分析对控制抗豆卷叶螟性状相关基因进行了初步定位, 筛选出12个可能与抗豆卷叶螟胁迫相关的候选基因, 再结合生物信息学分析、基因差异表达分析和基因同源性分析发现, 环核苷酸门控离子通道4 (CNGC4)、WRKY16转录因子、氨基酸透性酶7 (AAP7)、丝氨酸/苏氨酸蛋白激酶、ZPR1B、类枯草杆菌蛋白酶等6个基因已有报道与抗逆有关。而赤霉素2-β加双氧酶8、Mdis1相互受体激酶2、抗病蛋白RGA3、硫氰酸酶结构域蛋白酶10和1个未知基因可能是与抗大豆卷叶螟有关的新基因。环核苷酸门控离子通道(CNGC)是一种非选择性的阳离子通道, 对一价和二价阳离子均有通透性, 是动植物细胞中非常重要的离子通道, 在植物生长发育、生物与非生物胁迫中都发挥着重要作用[47,48]。植物细胞中, CNGC是信号转导级联系统的组成部分, 是环核苷酸作用的一个主要靶标, 是连接环核苷酸和Ca2+的纽带。在病原菌入侵的早期, 它能够参与Ca2+内流的调控[49,50], 将细胞外信号通过阳离子流转变为胞内信号, 对细胞的生理活动进行调控[51]。当细胞受到外界刺激时, 定位于细胞膜上的受体蛋白识别相应刺激, 并激活胞质内腺苷酸环化酶, 生成的环核苷酸(cAMP)使CNGCs通道打开, 胞外Ca2+内流[52]。WRKY转录因子是植物中最大的转录因子家族之一, 在调控植物的生长发育、生物及非生物胁迫等过程中具有十分重要的作用[53,54,55]。植物遭受昆虫胁迫后, 植物体内WRKY转录因子的表达水平会发生变化[56], 如拟南芥受到线虫危害时, AtWRKY23基因被诱导[57]。氨基酸透性酶(AAP)是植物中的氨基酸转运蛋白, 在氨基酸转运过程中发挥着重要作用[58,59]。AAP基因在植物抵御病原体侵害方面扮演着重要的角色, 如含AtAAP3基因和AtAAP6基因突变体植株能维持根结线虫生长率低于野生型[60]。丝氨酸/苏氨酸蛋白激酶是一类重要的信号分子, 当植物遭遇到如害虫取食、盐害、干旱等胁迫, 或是受到创伤以及其他细胞因子、激素刺激时, 丝氨酸/苏氨酸蛋白激酶迅速在丝氨酸、苏氨酸残基部位磷酸化而激活, 并进一步通过级联磷酸化作用活化下游的信号分子, 激活特定的信号传导途径, 最终将外界信号传递到细胞核, 激活或抑制特定基因的表达[61,62,63]。ZPR1基因的表达对植物株型发育有重要的调控作用, 该基因的表达水平受植物激素和光照的调节, 最终导致植物株型的变化[64]。类枯草杆菌蛋白酶是一类在植物发育和信号级联中实现高度特异性功能的丝氨酸蛋白酶, 在植物和病原物互作过程中起到重要的免疫激发作用[65]。推测以上候选基因的功能和参与的代谢途径可能与大豆抗豆卷叶螟有关, 当大豆受豆卷叶螟诱导胁迫后, 一些候选基因能发生信号转导、相关基因表达及防御物质生成等一系列的反应来抵御虫害。大豆抗豆卷叶螟是一个复杂的生理过程, 目前尚缺乏遗传调控方面的研究, 本研究中通过BSA-Seq技术与RNA-Seq技术相结合, 挖掘出一些与抗豆卷叶螟相关的候选基因, 对于这些基因是否是调控豆卷叶螟的关键基因, 以及其调控机理将在后续的工作中进行分析和验证。
4 结论
通过BSA-Seq全基因组重测序发现共有329个基因位于99%置信区间以外, 主要集中在7号、16号和18号等染色体上。通过结合转录组测序结果分析、基因的同源注释、生物信息学分析等, 初步推测CNGC4、WRKY16转录因子、AAP7、丝氨酸/苏氨酸蛋白激酶、ZPR1B等12个基因为大豆抗豆卷叶螟相关的候选基因, 这些基因可能在大豆抗豆卷叶螟过程中起着重要作用, 为大豆抗豆卷叶螟基因图位克隆奠定了基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOIURL [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
PMID [本文引用: 1]

The calcium ion is firmly established as a ubiquitous intracellular second messenger in plants. At their simplest, Ca(2+)-based signaling systems are composed of a receptor, a system for generating the increase in [Ca(2+)]cyt, downstream components that are capable of reacting to the increase in [Ca(2+)]cyt, and other cellular systems responsible for returning [Ca(2+)]cyt to its prestimulus level. Here we review the various mechanisms responsible for generating the stimulus-induced increases in [Ca(2+)]cyt known as Ca(2+) signals. We focus particularly on the mechanisms responsible for generating [Ca(2+)]cyt oscillations and transients and use Nod Factor signaling in legume root hairs and stimulus-response coupling in guard cells to assess the physiological significance of these classes of Ca(2+) signals.
PMID [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
PMID [本文引用: 1]
Parsley WRKY proteins comprise a family of plant-specific zinc-finger-type factors implicated in the regulation of genes associated with pathogen defence. In vitro, these proteins bind specifically to functionally defined TGAC-containing W box promoter elements within the Pathogenesis-Related Class10 (PR-10) genes. Here we present in vivo data demonstrating that WRKY1 is a transcriptional activator mediating fungal elicitor-induced gene expression by binding to W box elements. In situ RNA hybridization revealed that the WRKY1 gene is rapidly and locally activated in parsley leaf tissue around fungal infection sites. Transient expression studies in parsley protoplasts showed that a specific arrangement of W box elements in the WRKY1 promoter itself is necessary and sufficient for early activation and that WRKY1 binds to such elements. Our results demonstrate that WRKY transcription factors play an important role in the regulation of early defence-response genes including regulation of WRKY1.
PMID [本文引用: 1]
The WRKY proteins are a superfamily of transcription factors with up to 100 representatives in Arabidopsis. Family members appear to be involved in the regulation of various physio-logical programs that are unique to plants, including pathogen defense, senescence and trichome development. In spite of the strong conservation of their DNA-binding domain, the overall structures of WRKY proteins are highly divergent and can be categorized into distinct groups, which might reflect their different functions.
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIPMID [本文引用: 1]
During the interaction between sedentary plant-parasitic nematodes and their host, complex morphological and physiological changes occur in the infected plant tissue, finally resulting in the establishment of a nematode feeding site. This cellular transformation is the result of altered plant gene expression most likely induced by proteins injected in the plant cell by the nematode. Here, we report on the identification of a WRKY transcription factor expressed during nematode infection. Using both promoter-reporter gene fusions and in situ reverse transcription-polymerase chain reaction, we could show that AtWRKY23 is expressed during the early stages of feeding site establishment. Knocking down the expression of WRKY23 resulted in lower infection of the cyst nematode Heterodera schachtii. WRKY23 is an auxin-inducible gene and in uninfected plants WRKY23 acts downstream of the Aux/IAA protein SLR/IAA14. Although auxin is known to be involved in feeding site formation, our results suggest that, during early stages, auxin-independent signals might be at play to activate the initial expression of WRKY23.
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIPMID [本文引用: 1]
Perception of pathogen-associated molecular patterns (PAMPs) constitutes the first layer of plant innate immunity and is referred to as PAMP-triggered immunity (PTI). For a long time, part of the plant community was sceptical about the importance of PAMP perception in plants. Genetic and biochemical studies have recently identified pattern-recognition receptors (PRRs) involved in the perception of bacteria, fungi and oomycetes. Interestingly, some of the structural domains present in PRRs are similar in plants and animals, suggesting convergent evolution. Lack of PAMP perception leads to enhanced disease susceptibility, demonstrating the importance of PAMP perception for immunity against pathogens in vivo. Recently, proteins with known roles in development have been shown to control immediate PRR-signalling, revealing unexpected complexity in plant signalling. Although many PAMPs recognised by plants have been described and more are likely to be discovered, the number of PRRs known currently is limited. The study of PTI is still in its infancy but constitutes a highly active and competitive field of research. New PRRs and regulators are likely to be soon identified.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}