 ,1,2,**, 刘景森1,2,**, 李加纳,1,2,*
,1,2,**, 刘景森1,2,**, 李加纳,1,2,*Integrating GWAS and WGCNA to screen and identify candidate genes for biological yield in Brassica napus L.
WANG Yan-Hua,1,2,**, LIU Jing-Sen1,2,**, LI Jia-Na,1,2,*通讯作者: * 李加纳, E-mail:ljn1950@swu.edu.cn
第一联系人:
收稿日期:2020-07-30接受日期:2020-12-1网络出版日期:2021-08-12
| 基金资助: |
Received:2020-07-30Accepted:2020-12-1Online:2021-08-12
| Fund supported: |
作者简介 About authors
E-mail:hawer313@163.com

摘要
生物产量是作物获得高产的重要基础, 对于甘蓝型油菜(Brassica napus L.)尤其重要。本研究利用588份甘蓝型油菜材料构成的自然群体2年生物产量表型数据的全基因组关联分析, 再结合高生物产量材料‘CQ45’和低生物产量材料‘CQ46’的转录组测序(RNA-seq)结果, 整合了6个甘蓝型油菜材料6个部位(茎秆、叶片、花后30 d主轴与侧枝种子、花后30 d主轴与侧枝角果皮)的转录组数据构建的加权共表达网络分析(WGCNA), 筛选出与生物产量相关的候选基因。通过相关分析发现, 2年间甘蓝型油菜自然群体中生物产量对大多数产量相关性状都具有正向效应; 自然群体2年生物产量分析的最佳模型均为K+PCA模型, 共检测到9个显著位点(P < 1/385691或P < 0.05/385691); 根据CQ45和CQ46共36组转录组数据, 选择MAD值为前5%的基因共计5052个用于构建WGCNA, 通过筛选合并共得到了15个模块, 其中5个基因共表达模块分别与叶片、茎秆和花后30 d种子显著性相关; 整合了WGCNA中关键模块的hub gene、GWAS分析得到的显著SNP位点和极端表型差异基因确定候选基因, 它们的拟南芥同源基因为HCEF1、HOG1、SBPASE、ACT2, 这些基因在光合作用的卡尔文循环、碳同化、物质积累等方面发挥重要作用。
关键词:
Abstract
Biomass yield is especially important for Brassica napus, as it is the basis for high yields of crops. In this study, the phenotypic data of the natural populations composed of 588 materials were used for genome-wide association analysis (GWAS). We performed the transcriptome sequencing (RNA-seq) of biomass yield using ‘CQ45’ (high biological yield material) and ‘CQ46’ (low biological yield material). A weighted gene co-expression network analysis (WGCNA) network was constructed by integrating transcriptome data of six tissues of the extreme materials, such as stalks, leaves, 30 day after flowering (DAF) seeds of main inflorescence and lateral branch, 30 DAF pod keratin of main branch and lateral branch. We finally screened the candidate genes related to biomass yield. The main results are as follows: Biomass yields in B. napus had positive effects on most yield-related traits; K + PCA model was the best model for biomass analysis of the natural population, and nine significant loci were detected in the best model (P < 1/385691 or P < 0.05/385691); according to 36 groups of transcriptome data, MAD value of each gene was calculated. A total of 5052 genes with MAD value of the top 5% were selected to construct WGCNA. Fifteen gene modules were obtained, among which, five genes co-expression modules were significantly correlated with leaves, stems, and seeds of 30 DAF. The hub genes of the key modules in WGCNA, the significant SNP loci obtained from GWAS, and the extreme phenotypic differential genes were integrated to identify the candidate genes. Their Arabidopsis homologous genes were HCEF1, HOG1, SBPASE, and ACT2, which played the important roles in the Calvin cycle, carbon assimilation, and material accumulation of photosynthesis.
Keywords:
PDF (23772KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王艳花, 刘景森, 李加纳. 整合GWAS和WGCNA筛选鉴定甘蓝型油菜生物产量候选基因. 作物学报[J], 2021, 47(8): 1491-1510 DOI:10.3724/SP.J.1006.2021.04175
WANG Yan-Hua, LIU Jing-Sen, LI Jia-Na.
油菜作为四大油料作物(大豆、油菜、向日葵、花生)之一, 由于其较强的适应性, 在世界各地广泛种植[1,2,3]。随着人类对油菜的需求不断增加, 油菜总产量增速不断加快[4], 世界油料作物中油菜的种植面积及总产量已位居第二, 仅次于大豆[5,6]。同时油菜也是我国主要农作物, 在作物种植面积中排名第五, 仅次于水稻、玉米、小麦、大豆[7,8,9,10,11,12]。近年来, 油菜多功能开发利用的持续发展, 形成了油菜的油用、菜用、花用、蜜用、饲用等“一菜多用”的模式, 大幅度的提高了油菜的种植效益[13]。在实际生产中, 我国油菜产量仍较低, 生产成本却很高, 国际竞争力严重不足[14,15]。目前, 我国油菜超过60%依赖进口, 并且种植面积和单位面积产量都停滞不前, 这对我国粮食安全战略有着严重威胁[16]。因此, 提高油菜产量是我国油菜育种不断追求的目标[17,18,19,20,21,22,23,24]。
生物产量指单位面积土地上获得的不包括根系的作物干物质的总量, 它是描述茎、叶制造并积累有机物能力强弱的量[25]。生物产量体现作物总的生产力, 或者说作物光合作用碳同化的能力。多项报道已经表明, 生物产量与经济产量密切相关, 作物提高产量的方法通常有2种: 即在生物产量不变的情况下, 提高收获指数; 或收获指数不变的情况下, 提高生物产量[26,27,28,29,30]。近年来, 高通量测序技术的不断突破、成本不断降低, 多样本的转录组测序逐渐被应用于系统研究生命科学问题[31], 传统的少量样本比较分析已经无法有效处理海量的生物信息数据。因此, 生物信息分析工具和方法应运而生[32]。其中, 加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)能够特异筛选出与目标性状高度关联的基因, 并进行模块(module)化分类, 得到具有高度生物学意义的共表达模块(co-expression module), 能够有效筛选到核心基因(hub genes)[33]。WGCNA算法作为一种精准、高效的生物信息学及生物数据挖掘方法, 被广泛应用于生物学各个领域, 利用WGCNA对马铃薯C16和C119品种构建抗逆生理性状高度关联的权重网络, 为深入探究马铃薯抗逆分子机制提供新的数据资源[34,35]。本研究以生物产量性状为主要研究对象, 通过GWAS分析, 鉴定出与生物产量性状紧密相关的SNP位点; 然后提取SNP位点前后共500 kb的序列片段作为候选区段, 提取候选区段内所有基因并结合转录组分析结果筛选获得差异候选基因; 同时, 结合转录组数据构建WGCNA共表达网络, 筛选鉴定出甘蓝型油菜生物产量关键候选基因。
1 材料与方法
1.1 供试材料
以来自世界各地的588份油菜自交系作为材料, 其中455份来自中国(大部分来自重庆、湖南、湖北等地), 11份来自亚洲其他地区, 102份来自欧洲, 13份来自北美洲, 7份来自澳大利亚。所有材料均由重庆市油菜工程技术研究中心提供。1.2 材料种植及农艺性状考察
588份材料于2016、2018年种植于重庆北碚歇马油菜基地(29o45′39.99″, 106o22′38.47″, 海拔238.57 m)。每年播种时间为9月20日左右, 按完全随机区组设计, 2个重复同时种植, 每个材料种植2行, 每行15株, 行距40 cm, 株距20 cm。田间管理选择常规生产方式, 2个重复按照相同的管理措施和施肥水平进行, 收获时间为次年5月5日左右。于成熟期, 在每个小区中选择5株长势一致的植株。将植株分成上部分枝和下部茎秆两部分, 并将上部分枝装进网袋中。上部分枝和下部茎秆分别彻底晒干后, 使用天平秤进行称量, 将两部分重量相加得到植株的总干重。每份材料的5个样本干重的平均值为该材料的生物产量(biomass yield, BY)。1.3 表型数据分析
利用Microsoft Excel软件初步整理588份自然群体的表型数据, 并计算单个环境及差值条下生物产量的平均值、标准差、变异系数等, 并用SPSS绘制生物产量的正态分布图; 利用SPSS统计分析软件对自然群体进行各性状进行相关分析; 利用R软件对多年环境下自然群体的生物产量表型数据进行最佳线性无偏预测(best linear unbiased prediction, BLUP)处理。1.4 全基因组关联分析
利用本实验室已有588份甘蓝型油菜重测序的数据, 通过基因型分析, 最终获得385,691个可利用的SNP标记数据[36]。本研究利用这些标记结合重庆2017、2019年和BLUP环境的生物产量(BY)表型数据行全基因组关联分析。本研究使用6种统计模型在Tassel 5.2.1[37]软件中执行对甘蓝型油菜生物产量的关联分析, 即: Q模型、naive模型、K模型、PCA模型、Q+K模型和PCA+K模型。Q代表群体结构, PCA代表主成分, K代表亲缘关系; naive、Q和PCA模型在一般线性模型(general linear model, GLM)下运行, K、Q+K和PCA+K模型在混合线性模型(mixed linear model, MLM)下运行。利用SAS软件对这6种模型的运算结果中的-lg(P)的观测值和-lg(P)期望值绘制QQ (Quantile-quantile)图。通过比较QQ图确定最佳模型, 并选择最佳模型下生物产量性状的全基因组关联分析结果作为进一步分析对象。本试验使用的SNP数据为385,691个, 因此规定P值小于阈值(1/385,691=2.593E-06和0.05/385,691=1.296E-07)的位点为显著关联位点。利用Haploview[38]软件制作Manhattan图, 将通过关联分析检测到的染色体组上与各性状显著相关的标记位点可视化。1.5 转录组测序分析
从供试群体中选择1个高生物产量(CQ45)和1个低生物产量(CQ46)材料, 分别选取蕾苔期茎秆(J)和叶片(Le)、采用挂花标记的方法选取开花后30 d主轴种子(30S)、主轴角果皮(30P)、侧枝种子(30CS)和侧枝角果皮(30CP) 6个时期的样品, 每个样品取2个生物重复, 送北京诺禾致源生物信息科技有限公司进行转录组测序, 在OmicShare (基迪奥)云平台完成(1.6 加权共表达网络分析
使用R软件包preprocessCore中的normalize. quantiles函数对转录组数据进行标准化。然后计算每个基因的MAD (median absolute deviation), 选择的MAD值为前5%的基因作为样本间变异大的基因群用于WGCNA构建。使用average-linkage层次聚类法对基因进行聚类, 按照混合动态剪切树的标准, 并设置每个基因网络模块最少的基因数为30。在使用动态剪切法在确定基因模块后, 我们依次计算每个模块的特征向量值(eigengenes), 然后对模块进行聚类分析, 将距离较近的模块合并成新的模块, 设置height = 0.25、deepSplit = 3、minModuleSize = 30。计算所得到模块的特征向量与样本来源组织的相关性, 选择相关系数>0.6的模块作为与组织部位显著相关模块。通过特征向量基因分析确定目标基因模块, 对所有的模块中具有代表性的基因-特征向量基因(module eigengene, ME)进行聚类分析, 进一步进行两两模块之间的ME相关性分析, ME之间的相关性越高, 其所在的模块的相关性也就越高; 将模块中的所有基因和ME基因在所有样本中分别进行表达水平分析, GO和KEGG分析。最后使用Cytoscape[39]将所关注模块的ME基因的共表达网络进行可视化。2 结果与分析
2.1 表型数据统计及各性状的相关性分析
对2017年和2019年588份自然群体的生物产量的表型变异情况进行统计分析发现, 在不同环境下自群体的生物产量呈连续变异, 近似正态分布, 说明生物产量是受多基因控制的数量性状, 符合GWAS分析的要求。各产量相关性状的相关分析结果表明, 每角果粒数在2017年和2019年与茎秆干重、经济产量、收获指数和生物产量均呈正相关, 而茎秆干重仅在2017年呈现出显著正相关; 茎秆干重与上部生物产量、经济产量、生物产量在2年间均呈现显著正相关; 上部生物产量与经济产量、收获指数、生物产量在2年间均呈现显著正相关; 而经济产量和收获指数与生物产量在2年间同样呈现正相关。多数性状间的相关性均达到显著或极显著水平, 表明产量相关的各性状间存在显著相关作用(表1)。
Table 1
表1
表1自然群体2年各产量相关性状间的相关分析
Table 1
| 性状 Trait | 环境Environment | 粒果比SWSI | 千粒重TSW | 每角粒数SNPS | 茎秆干重ST | 上部生物产量CBY | 籽粒产量SY | 收获指数HI | 生物产量BY |
|---|---|---|---|---|---|---|---|---|---|
| SWSI SWSI | 2017CQ | 1 | |||||||
| 2019CQ | 1 | ||||||||
| TSW | 2017CQ | 0.506** | 1 | ||||||
| TSW | 2019CQ | -0.008 | 1 | ||||||
| SNPS | 2017CQ | 0.513** | 0.156** | 1 | |||||
| SNPS | 2019CQ | 0.284 | 0.056** | 1 | |||||
| ST | 2017CQ | -0.023 | 0.108* | 0.104* | 1 | ||||
| ST | 2019CQ | -0.055** | 0.213** | 0.198** | 1 | ||||
| CBY | 2017CQ | 0.0247 | 0.106* | 0.072 | 0.682** | 1 | |||
| CBY | 2019CQ | -0.037 | 0.249** | 0.171** | 0.567** | 1 | |||
| SY | 2017CQ | 0.204** | 0.155** | 0.210** | 0.524** | 0.773** | 1 | ||
| SY | 2019CQ | 0.179 | 0.327** | 0.409** | 0.424** | 0.726** | 1 | ||
| HI | 2017CQ | 0.308** | 0.127** | 0.232** | -0.099* | 0.142** | 0.687** | 1 | |
| HI | 2019CQ | 0.033** | 0.292** | 0.262** | 0.556** | 0.972** | 0.866** | 1 | |
| BY | 2017CQ | 0.007 | 0.117* | 0.089 | 0.861** | 0.957** | 0.738** | 0.056 | 1 |
| BY | 2019CQ | -0.042 | 0.313** | 0.265** | 0.752** | 0.905** | 0.718** | 0.901 | 1 |
新窗口打开|下载CSV
2.2 甘蓝型油菜生物产量性状的GWAS分析
各生物产量性状在6种模型下的关联分析后得到的结果进行Quantile-Quantile散点图(QQ Plot)的绘制, 选择与预测P值最接近的一种模型作为最佳模型, 2017年、2019年和BLUP环境下最佳模型为K+PCA模型(图1)。在最佳模型下, 利用本实验室已有的385,691个SNP, 以P值小于阈值(1/385,692 = 2.593E-06)确定显著关联SNP位点, 并绘制曼哈顿图(图2)。生物产量2017、2019年和BLUP环境下共检测到9个显著关联标记位点(表2)。其中, 在2017年环境中检测到7个显著关联标记位点, 分别位于A03、A07、C01 (2个)、C04 (2个)、C06染色体, 并且位于同一条染色体的显著关联位点其置信区间高度重合; 2019年检测到2个显著关联位点, 分别位于A07和C09染色体; 而BLUP环境下共检测到4个显著关联位点, 分别位于A07、C03、C06 (2个), 位于C06染色体的2个显著关联位点其置信区间高度重合。9个SNP的贡献率在5.64%~7.98%, 其中2017年检测到的位于C06染色体的SNP (S16_ 26169577)贡献率最高。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1生物产量在各模型下的QQ图
Fig. 1Quantile-quantile plot of biomass yield in six models
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2生物产量的曼哈顿点图
Fig. 2Manhattan plots of biomass yield using the optimal model
Table 2
表2
表2最佳模型下生物产量显著位点表
Table 2
| 染色体 Chr. | 物理位置 Position | 位点 SNP | P值 P-value | 贡献率 R2 (%) | 置信区间 Confidence interval (250 kb up/downstream) |
|---|---|---|---|---|---|
| A3 | 7901059 | S3_7901059 | 9.91E-07 | 7.25 | 7651059-8151059 |
| A7 | 15487057 | S7_15487057 | 1.06E-06 | 7.70 | 15237057-15737057 |
| A7 | 16899362 | S7_16899362 | 1.46E-06 | 7.43 | 16649362-17149362 |
| A7 | 19665408 | S7_19665408 | 1.73E-06 | 5.99 | 19415408-19915408 |
| C1 | 3258674 | S11_3258674 | 1.46E-06 | 7.65 | 3008674-3508674 |
| C3 | 20691876 | S13_20691876 | 1.87E-06 | 5.64 | 20441876-20941876 |
| C4 | 34485253 | S14_34485253 | 1.39E-06 | 7.51 | 34174883-34735253 |
| C6 | 26169577 | S16_26169577 | 2.87E-07 | 7.98 | 25919577-26419577 |
| C9 | 23643765 | S19_23643765 | 1.14E-06 | 7.61 | 23393765-23893765 |
新窗口打开|下载CSV
2.3 极端表型材料RNA-Seq分析
在叶片中检测到极端材料的差异基因共6820个, CQ45对CQ46显著上调表达的基因有3516个, 显著下调表达的基因有3304个(图3-A)。生物分子功能类中相关性最高的是氧化还原酶活性(GO:0016903)、催化活性(GO:0003824)等, 而参与基因数量较多的生物过程是单体代谢过程(GO:0044710)、光合作用(GO:0015979)等(图3-B); 叶片中差异基因主要集中在碳水化合物代谢途径(图3-C)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3高生物产量材料CQ45和低生物产量材料CQ46转录组分析
A: 极端表型材料各组织差异基因数量; B: 极端表型材料各组织差异基因的GO富集分析; C: 极端表型材料各组织差异基因的KEGG分析。
Fig. 3Transcriptome profile of extreme phenotypic materials CQ45 and CQ46
A: the number of differentially expressed genes in tissues of extreme phenotypic materials; B: GO enrichment analysis of differentially expressed genes in tissues of extreme phenotypic materials; C: KEGG analysis of differentially expressed genes in tissues of extreme phenotype materials.
花后30 d主轴角果皮中检测到17,309个显著差异表达基因, CQ45对于CQ46有2682个显著上调表达基因, 3570个显著下调基因(图3-A)。差异基因中分子功能相关性较高的是葡萄酶转移(GO:0046527)等, 生物过程相关性最高的有应激反应(GO:0006950)、单体生物合成过程(GO:0044711)等(图3-B); 相关性较高的途径是氨基酸的生物合成以及脂质代谢途径(图3-C)。
花后30 d侧枝角果皮中检测到5431个显著差异表达基因, 其中CQ45对于CQ46有2173个显著上调表达基因, 3258个显著下调基因(图3-A)。侧枝角果皮中分子功能相关性最高的是营养库活性(GO:0045735)等, 参与的生物过程中相关性较高的是对含氧化合物的反应(GO:1901700)等(图3-B); 这些差异基因相关性最高的是能量代谢途径(图3-C)。
花后30 d主轴种子中检测到6948个显著差异表达基因, CQ45对于CQ46有4080个显著上调表达基因, 2868个显著下调基因(图3-A)。分子功能相关性最高的是微管运动活性(GO:0003777)、硫葡萄糖苷酶活性(GO:0019137)等, 生物过程相关性较高的是细胞循环(GO:0007049)、细胞分裂(GO:0051301) (图3-B); 相关性较高的代谢途径是碳水化合物代谢中的淀粉和蔗糖代谢(图3-C)。
花后30 d侧枝种子中检测到17,309个显著差异表达基因, CQ45对于CQ46有11489个显著上调表达基因, 5820个显著下调基因(图3-A)。分子功能相关性最高的是微管运动活性(GO:0003777)、营养库活性(GO:0045735)硫葡萄糖苷酶活性(GO:0019137)等, 参与的生物过程相关性较高的是细胞循环(GO:0007049)等, 其结果与30 d主轴角果种子比较相似(图3-B); 相关性较高的是氨基酸代谢中的丙氨酸、天冬氨酸和谷氨酸代谢(图3-C)。
茎秆中检测到极端表型材料的差异基因12,867个, CQ45对于CQ46有6452个显著上调表达基因, 6415个显著下调基因(图3-A)。分子功能相关性最高的是转移酶活性(GO:0016758)、UDP葡萄糖基转移酶活性(GO:0035251)等, 参与较多的生物过程是植物型次生细胞壁生物发生(GO:0009834)、细胞壁合成(GO:0042546)等(图3-B); 参与相关性较高的代谢途径是到次生代谢产物的生物合成等途径(图3-C)。
2.4 加权共表达网络(WGCNA)分析
2.4.1 基因共表达网络的模块生成 根据36个样本得到的RNA-Seq的结果, 计算各基因的MAD值, 取MAD为前5%共计5052个基因, 进行基因表达水平的层次聚类(图4-A); 使用R软件包WGCNA构建权重共表达网络, 选取拟合曲线第1次接近0.9时的阈值参数β (β=10)筛选共表达模块(图4-B, C),共表达网络符合无尺度网络。使用动态剪切法在确定基因模块后, 依次计算每个模块的特征向量值, 然后对模块进行聚类分析, 将距离较近的模块合并成新的模块后共得到了15个模块(图4-D), 聚类树中每个枝代表1个模块, 分别是Black、Cyan、Green、Magenta、Pink、Purple、Red、Salmon、Tan、Turquoise、Yellow、Greenyellow、Brown、Blue模块, Grey模块是无法聚集到其他模块的基因集合(包含11个基因)。在15个模块中基因数量33~1843不等, 其中包含基因数量最多的是Turquoise模块(1843个), 最少的是Cyan模块(33个), 各个基因之间的表达关系如图4-E。图4
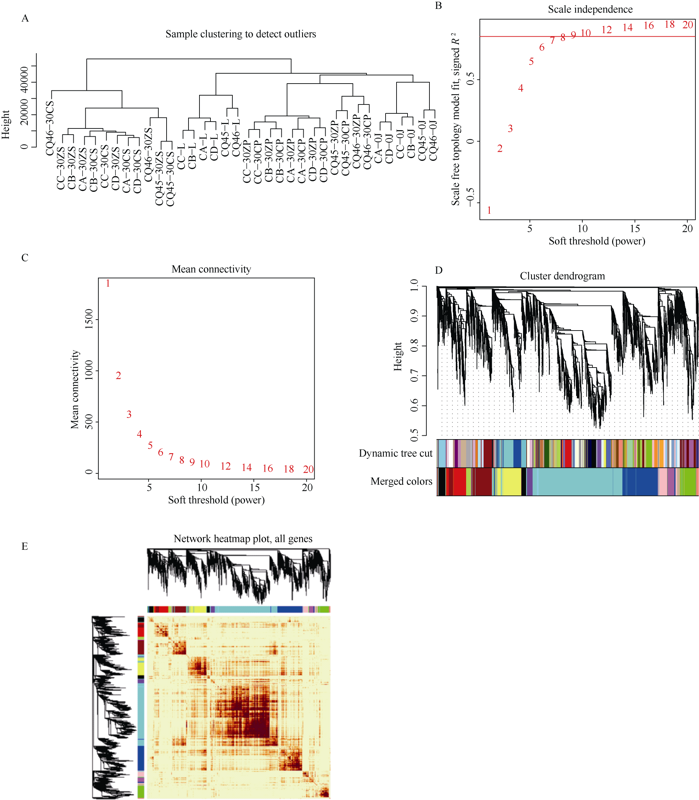 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4样本聚类与基因模块的生成
A: 样本聚类与数据矫正; B和C: 基于规模独立性和均值连通性选择软阈值; D: 已识别模块的树状聚类图; E: 已识别模块的热图。
Fig. 4Sample clustering and gene module generation
A: sample clustering to detect outliers; B and C: the selection of the soft threshold based on scale independence and mean connectivity; D: cluster dendrogram of the identified modules; E: the heatmap of identified modules.
2.4.2 模块与组织之间相关性 本研究分别计算这15个模块的特征向量与样本来源组织的相关性, 得到5个显著相关的模块, 其中与叶片(L)极显著相关的模块是Turquoise模块, 相关系数达到0.77; 与茎秆显著相关的模块为Green、Magenta、Pink模块, 其相关系数分别为0.9、0.62、0.75; 与30 d主轴种子显著相关的模块是Yellow模块, 其相关系数为0.6 (图5-A)。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5基因共表达网络模块与各组织的相关性分析
A: 基因共表达网络模块与组织部位关联热图; B: 各样本中Turquoise模块的所有基因与相应ME的表达水平; C: 不同模块两两之间ME的相关性; D: ME聚类树。
Fig. 5Correlation analysis between gene co-expression network modules and tissues
A: association analysis of gene co-expression network modules with tissues; B: expression levels of all genes and corresponding ME in turquoise module of each sample; C: ME correlation between different modules; D: ME cluster tree.
通过特征向量分析确定目标基因模块, 对所有的模块中具有代表性的基因-特征向量基因(module eigengene, ME)进行聚类分析(图5-B), ME之间的相关性越高, 其所在的模块的相关性也就越高, 进一步进行两两模块之间的ME相关性分析(图5-C); 将模块中的所有基因和ME基因在所有样本中分别进行表达水平分析发现, 各个模块内的基因其在不同组织部位中的表达情况呈现不同的特性。每个模块内的基因表达水平高度相关, ME表达水平与模块整体表达水平也高度相关(图5-D和附图1), 说明目标模块的ME可充分代表其模块中的整体基因。
附图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT附图1各样本中关键模块的所有基因与相应ME的表达水平
Fig. S1Expression levels of all genes and corresponding ME in key modules of each sample
2.4.3 目标基因模块中的关键基因 为获得与组织部位生长发育和功能高度相关的5个模块中的核心基因, 利用Cytoscape软件对基因互作调控网络进行可视化处理, 筛选出模块中共表达权重>0.1并且连通性排名前10的基因, 并结合NCBI数据库确定模块内的关键基因(图6)。Turquoise模块中确定了6个关键基因, 分别为BnaA04g26780D、BnaA08g16820D、BnaA09g12770D、BnaA09g35380D、BnaC06g09740D、BnaC08g16510D。Green模块中筛选出了4个核心基因, 分别为BnaC08g01540D、BnaC04g14330D、BnaC04g08360D、BnaA04g06420D。Magenta模块中筛选出了4个核心基因, 分别为BnaA04g20970D、BnaC03g73810D、BnaA04g26560D、BnaC07g20310D。Pink模块中筛选出了6个核心基因, 分别为BnaC04g08100D、BnaA05g05140D、BnaC02g36210D、BnaA03g21210D、BnaA05g05230D、BnaC04g04640D。Yellow模块中筛选出了8个核心基因, 分别为BnaA10g02240D、BnaC05g02160D、BnaA04g22070D、BnaA09g02110D、BnaA03g47170D、BnaC01g17050D、BnaC01g09900D、BnaC07g48660D。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6关键模块的基因共表达网络以及核心基因
Fig. 6Co-expression network of key modules and hub genes
2.5 候选基因的筛选
检测到显著性SNP位点上下游500 kb的LD置信区间内寻找甘蓝型油菜基因, 结合转录组差异表达基因, 通过WGCNA筛选到的与组织部位生长发育高度相关的关键模块中的核心基因(hub genes), 初步确定与甘蓝型油菜生物产量相关的基因, 将这些基因的蛋白质序列与拟南芥基因蛋白质序列进行BLAST对比, 结合前人已报道的拟南芥同源基因的功能以及特性, 筛选到生物产量性状中发挥重要作用的候选基因, 在拟南芥中的同源基因分别是HCEF1、SBPASE、HOG1、ACT2、CP12-2、GAPC1 (表3)。Table 3
表3
表3生物产量候选基因
Table 3
| 基因 Genes | 染色体 Chromosome | 物理位置 Physical position | 拟南芥同源基因 Homologs in A. thaliana | 功能注释 Functional annotation |
|---|---|---|---|---|
| BnaA04g04350D | A04 | 3211999-3214328 | AT3G54050 | 高循环电子流 High cyclic electron flow 1 (HCEF1) |
| BnaA04g06420D | A04 | 5053017-5055052 | AT4G13940 | 同源依赖性的基因沉默 HOMOLOGY-DEPENDENT GENE SILENCING 1 (HOG1) |
| BnaA07g19320D | A07 | 15496197-15496586 | AT3G62410 | CP12含域蛋白2 CP12 domain-containing protein 2 (CP12-2) |
| BnaA09g35380D | A09 | 25808955-25811193 | AT3G55800 | 景天庚酮糖二磷酸酶 Sedoheptulose-bisphosphatase (SBPASE) |
| BnaC03g33610D | C03 | 20464104-20466211 | AT3G04120 | 甘油醛-3-磷酸脱氢酶C亚基1 Glyceraldehyde-3-phosphate dehydrogenase C subunit 1 (GAPC1) |
| BnaC03g73810D | C03 | 1811021-1813439 | AT3G18780 | 肌动蛋白2 Actin 2 (ACT2) |
| BnaC08g48810D | C08 | 3782204-3784813 | AT3G54050 | 高循环电子流 High cyclic electron flow 1 (HCEF1) |
新窗口打开|下载CSV
BnaA04g04350D和BnaC08g48810D在拟南芥中的同源基因是HCEF1, 编码拟南芥叶绿体-1,6-二磷酸酶[40]。豌豆中叶绿体-1,6-二磷酸果糖(FBPase)干扰载体的转基因植株表现出叶鲜重明显增加、光合作用的测量结果显示具有更高的碳同化率, 表明FBPase作为二氧化碳同化中的关键酶的作用, 并且还可以协调碳和氮的代谢[41]。BnaA04g06420D在拟南芥中的同源基因是HOG1, 编码S-腺苷同型半胱氨酸水解酶(SAHH)。SAHH是维持真核生物甲基化稳态的关键酶[42], 会竞争性地抑制甲基转移酶(MT)活性, 使植株生长缓慢, 繁殖力低, 种子发芽减少[43,44]。BnaA07g19320D在拟南芥的同源基因是CP12-2, 编码在叶绿体基质中发现的一种小肽, 在黑暗中氧化的CP12与甘油醛-3-磷酸脱氢酶和磷酸布洛激酶(碳同化循环的两种酶)形成一种非活性超分子复合物[45], CP12-2的转录物在决定成熟叶片和光合作用能力方面起着重要作用[46,47,48], Marri等人发现, CP12蛋白氧化对植物不同环境条件下光合过程的整体平衡至关重要[49]。BnaA09g35380D在拟南芥中的同源基因SBPASE是一种卡尔文循环酶, 具有光合固碳作用[48]。BnaC03g33610D在拟南芥中的同源基因为GAPC1, 磷酸化甘油醛-3-P脱氢酶(GAPC-1)是一种高度保守的胞浆酶, 纯合gapc-1缺失突变株表现出生长延迟、角果形态改变和种子数量低[51]。BnaC03g73810D在拟南芥中的同源基因是ACT2, 是拟南芥营养组织中主要肌动蛋白基因, 对植物生长发育有着极其重要的作用[52]。多项研究结果表明, 甘蓝型油菜生物产量相关候选基因的同源基因功能, 在植物氧化还原、能量和碳水化合物代谢、光合作用、物质积累等生长发育过程中发挥作用。
3 讨论
生物产量体现作物总的生产力, 对作物的经济产量有着密不可分的关系。本研究所使用甘蓝型油菜自然群体中的生物产量变化范围广、变异系数大, 说明生物产量是比较复杂的性状。本研究在2017、2019年和BLUP环境下共检测到9个生物产量显著关联标记位点, 贡献率在5.64%~7.98%。与生物产量显著关联的SNP位点分布在各个染色体, Lu等[53]利用520份来自世界各地的甘蓝型油菜材料作为自然群体, 使用MLM进行全基因组关联分析, 在除了A01、A06、C02、C07以外的15条染色体上共检测到26个生物产量SNP, 贡献率在4.33%~17.03%之间; 60K SNP芯片对520分材料构成的自然群体进行全基因组关联分析, 在除了A01、A02和A10意外的16条染色体共检测到89个生物产量SNP位点, 贡献率在3.47%~8.36%之间[54]; Luo等[55]利用155分甘蓝型油菜材料构成的自然群体并使用60K SNP芯片检测到1个生物产量SNP, 其贡献率为0.76%。本研究中, 生物产量在2017、2019年和BLUP环境下共检测到9个显著关联标记位点, 分别位于A03、A07、C01、C03、C04、C06、C09染色体, 贡献率在5.64%~7.98%。前人检测到与生物产量显著关联的部分SNP位点也定位在这些染色体上, 说明生物产量的构成因素比较复杂, 且由多个SNP位点协同控制。通过RNA-Seq对极端表型材料的差异表达基因分析表明, 叶片和角果皮主要富集到光合作用、淀粉和蔗糖代谢等生物途径, 这些代谢途径可能在油菜植株的物质积累过程中发挥重要作用, 因此, 这些差异基因的不同表达模式可能是造成2个材料生物产量有显著差异的主要原因。茎秆中的差异表达基因主要富集在细胞壁合成等途径, 油菜茎秆次生细胞壁的发育对其抗倒伏有着决定性影响, 茎秆的发育状况良好对油菜碳水化合物的运输积累有着积极作用[55]。因此, 本研究极端材料茎秆中的差异基因有可能在这些方面影响其生物产量。种子是油菜主要的储存和收获器官, 其发育状况直接影响着最终产量[56], 因此也直接影响生物产量。本研究极端生物产量材料种子中的差异基因主要富集在营养库活性、蔗糖淀粉代谢等功能和途径。
WGCNA是共表达网络分析有效的分析方法, 能够特异地筛选出与目标性状具有高度生物学意义的共表达模块, 在玉米等植物中已经被证明是一种高效的数据挖掘方法。本研究得到15个生物产量加权共表达网络模块, 分析发现一些模块与部分组织有着显著的相关性, 分别是叶片与Turquoise模块, 茎秆与Green、Magenta、Pink模块, 开花后30 d主轴种子与Yellow模块相关。叶片、茎秆和种子均在作物的生物产量的构成中发挥极其重要的作用, 直接影响到农产品最终的产量。结合GWAS和转录组差异基因得到的结果、整合WGCNA挖掘得到关联模块中的核心基因(hub genes), 最终筛选到一批可能与甘蓝型油菜生物产量密切相关的基因, 分别是BnaA04g04350D、BnaA04g06420D、BnaA07g19320D、BnaA09g35380D、BnaC03g33610D、BnaC03g73810D、BnaC08g48810D。这些基因的干扰、敲除或者超量表达均引起了植株的整体重量的变化[40,41,42,43,44,45,46,47,48,49,50,51,52], 在拟南芥生物产量中发挥重要作用。
由于生物组学大数据存在复杂、多层次和信息互补的特点, 分析这些数据的一个关键目标是确定可预测表型性状的有效模型, 发现重要性状的关键调节因子并阐明其生物功能。本研究根据GWAS分析得到的与生物产量高度关联SNP标记, 同时结合转录组分析确定的差异基因以及WGCNA得到的基因富集模块进一步筛选出与甘蓝型油菜生物产量相关的基因。通过BLAST对比找到这些基因在拟南芥中的同源基因, 并根据其注释以及已有报道进一步筛选与生物产量高度相关的候选基因, 为油菜中生物产量候选基因的功能研究奠定基础。
4 结论
本研究通过2年间甘蓝型油菜自然群体中生物产量对大多数产量相关性状都具有正向效应, 说明甘蓝型油菜的生物产量是其他产量相关性状的基础和保障。结合GWAS关联基因与转录组差异基因得到178个基因, 根据拟南芥同源基因的功能, 筛选得到与生物产量性状相关的BnaA07g19320D和BnaC03g33610为关键候选基因, 它们在能量和碳水化合物代谢以及光合作用等方面中发挥作用。此外, 选取WGCNA所得到的重点模块中连通性前10的基因作为hub gene, 通过同源基因功能注释分析, 得到生物产量候选基因BnaC03g73810D、BnaA09g35380、BnaA04g06420D、BnaA04g04350D和BnaC08g48810D, 在光合作用的卡尔文循环、碳同化、物质积累等方面发挥作用。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOIURL [本文引用: 1]
PMID [本文引用: 1]

Association analyses that exploit the natural diversity of a genome to map at very high resolutions are becoming increasingly important. In most studies, however, researchers must contend with the confounding effects of both population and family structure. TASSEL (Trait Analysis by aSSociation, Evolution and Linkage) implements general linear model and mixed linear model approaches for controlling population and family structure. For result interpretation, the program allows for linkage disequilibrium statistics to be calculated and visualized graphically. Database browsing and data importation is facilitated by integrated middleware. Other features include analyzing insertions/deletions, calculating diversity statistics, integration of phenotypic and genotypic data, imputing missing data and calculating principal components.
PMID [本文引用: 1]
Research over the last few years has revealed significant haplotype structure in the human genome. The characterization of these patterns, particularly in the context of medical genetic association studies, is becoming a routine research activity. Haploview is a software package that provides computation of linkage disequilibrium statistics and population haplotype patterns from primary genotype data in a visually appealing and interactive interface.http://www.broad.mit.edu/mpg/haploview/jcbarret@broad.mit.edu
[本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
PMID [本文引用: 2]
Genes introduced into higher plant genomes can become silent (gene silencing) and/or cause silencing of homologous genes at unlinked sites (homology-dependent gene silencing or HDG silencing). Mutations of the HOMOLOGY-DEPENDENT GENE SILENCING1 (HOG1) locus relieve transcriptional gene silencing and methylation-dependent HDG silencing and result in genome-wide demethylation. The hog1 mutant plants also grow slowly and have low fertility and reduced seed germination. Three independent mutants of HOG1 were each found to have point mutations at the 3' end of a gene coding for S-adenosyl-l-homocysteine (SAH) hydrolase, and hog1-1 plants show reduced SAH hydrolase activity. A transposon (hog1-4) and a T-DNA tag (hog1-5) in the HOG1 gene each behaved as zygotic embryo lethal mutants and could not be made homozygous. The results suggest that the homozygous hog1 point mutants are leaky and result in genome demethylation and poor growth and that homozygous insertion mutations result in zygotic lethality. Complementation of the hog1-1 point mutation with a T-DNA containing the gene coding for SAH hydrolase restored gene silencing, HDG silencing, DNA methylation, fast growth, and normal seed viability. The same T-DNA also complemented the zygotic embryo lethal phenotype of the hog1-4 tagged mutant. A model relating the HOG1 gene, DNA methylation, and methylation-dependent HDG silencing is presented.
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
DOIURL [本文引用: 3]
DOIURL [本文引用: 2]
[本文引用: 1]
DOIURL [本文引用: 2]
DOIURL [本文引用: 2]
[本文引用: 1]
DOIURL [本文引用: 1]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}