 ,, 秦心儿, 张文亮, 董雪, 代明球, 岳兵,*华中农业大学作物遗传改良国家重点实验室, 湖北武汉 430070
,, 秦心儿, 张文亮, 董雪, 代明球, 岳兵,*华中农业大学作物遗传改良国家重点实验室, 湖北武汉 430070Genetic analysis and characterization of male sterile mutant mi-ms-3 in maize
TIAN Shi-Ke,, QIN Xin-Er, ZHANG Wen-Liang, DONG Xue, DAI Ming-Qiu, YUE Bing,*National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan 430070, Hubei, China通讯作者:
收稿日期:2020-05-7接受日期:2020-08-19网络出版日期:2020-08-31
| 基金资助: |
Received:2020-05-7Accepted:2020-08-19Online:2020-08-31
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (2737KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
田士可, 秦心儿, 张文亮, 董雪, 代明球, 岳兵. 玉米雄性不育突变体mi-ms-3的遗传分析及分子鉴定[J]. 作物学报, 2020, 46(12): 1991-1996. doi:10.3724/SP.J.1006.2020.03025
TIAN Shi-Ke, QIN Xin-Er, ZHANG Wen-Liang, DONG Xue, DAI Ming-Qiu, YUE Bing.
植物雄性不育是指雄性器官不能正常发育, 产生功能异常的雄配子, 但雌性器官能够正常发育, 可以与其他有功能的雄配子受精结实, 且植株不育性能够稳定遗传的现象。很多原因都会导致雄性不育现象的产生, 如基因漂移、基因突变、光照周期及温度的改变等[1]。雄性不育按照遗传方式的不同, 可以分为3类: 细胞质雄性不育、细胞核雄性不育以及核质互作雄性不育, 其中细胞核雄性不育还分为显性核不育和隐形核不育, 且以后者为主。
花器官作为高等植物的主要特征, 其发育模型由经典ABC模型逐步完善为之后的ABCD模型、ABCDE模型及四因子模型。玉米具有2类单性花序, 雄穗和雌穗作为成熟花的基本单位分别位于顶生花序和侧生花序上[2]。目前在玉米中已经鉴定出多个花器官发育突变体, 这些突变体对玉米花器官发育机理的研究、遗传育种的应用和提高杂种优势利用效率具有重要价值。玉米中B类基因有SILKY1 (SI1)、ZAG4、ZMM16/sterile tassel silky ear1 (sts1)、ZMM18和ZMM29[3]。SILKY1是拟南芥AP3同源基因, 它在花药和浆片原基中表达, 在siky突变体中心皮替代雄蕊, 稃状结构替代浆片[4]。ZAG4、ZMM16、ZMM18和ZMM29是拟南芥PI同源基因。zmm16 (sts1)突变体的雄穗及浆片转化为内外稃结构, 同时本应在雌穗发育过程中退化的雄蕊转化为心皮结构, 导致雌穗花丝增多。
通过创制玉米雄性不育系对大程度的降低人工成本、提高作物育种效率及种子纯度具有重要意义。本研究在B73与W22::Mu突变体杂交后代中筛选得到1个雄性不育突变体, 针对该突变体进行遗传分析及基因定位, 并进一步对该突变体在杂种优势中的利用进行了探讨。
1 材料与方法
1.1 试验材料
试验材料包括玉米自交系B73、Mo17和W22::Mu突变体(美国玉米种质中心引进)。以B73与W22::Mu突变体杂交, 杂交后代经多代自交筛选得到雄性不育突变体, 将该不育材料命名为mi-ms-3雄性不育系。上述材料均种植于华中农业大学校内试验基地。1.2 突变体的表型鉴定和遗传分析
野生型和突变体的表型调查情况如下: 散粉情况在雄穗开花后第2天上午9:00—12:00进行统计, 并根据散粉能力分为4个等级(0级, 不散粉; 1级, 少量散粉; 2级, 较多散粉; 3级, 正常散粉); 花药外露在雄穗开花结束后进行调查(突变体植株, 在其雌穗吐丝2~3 d后进行统计), 根据花药外露情况分为4个等级(0级, 不外露; 1级, 外露少于1/2; 2级, 外露多余1/2; 3级, 全部外露); 部分植株花药退化形成丝状突出物, 对雄穗上有丝状突出物的植株进行统计; 采用1% I2-KI对花粉染色进行育性调查, 每株重复3次; 取雄穗中部小穗, 剥离上位花花药, 用卡诺氏固定液固定, 送拜意尔生物公司进行花药切片观察。以mi-ms-3为母本, Mo17为父本, 杂交得到F1代, F1自交获得F2分离群体。对F2群体进行遗传分析, 在散粉期调查各单株表型, 统计各群体总株数、可育株数及不育株数, 利用卡方测验分析遗传模式。
1.3 目的基因初定位
构建的F2群体中于散粉期分别选取30株可育株和不育株, 提取DNA用于构建“野生型池”和“突变体池”, 利用BSA (Bulk Segregant Analysis)方法对进行初定位。根据定位结果在MaizeGDB数据库(http://www.maizegdb.org/)中选取多对的SSR标记(表1), 另外12份可育株和不育株组成的“野生型池”和“突变体池”进行聚丙烯酰胺凝胶电泳, 进行SSR标记多态性筛选及突变位点定位。Table 1
表1
表1基因定位及基因表达分析的引物
Table 1
| 引物名称 Primer name | 正向引物 Forward sequence (5′-3′) | 反向引物 Reverse sequence (5′-3′) | 染色体位置 Chromosome position |
|---|---|---|---|
| umc1973 | CAGGCAGAAAAGGAACGGAAC | GTGCGAGAGAAGATGGATGATTG | 3.05 |
| umc1027 | AACTCTGTCTCCGTCACCGTGT | GACCTCATCTCGGTGGAAATTG | 3.06 |
| S-1 | CCGTCATCGACATTTTACTCAG | CACAAGAGTTGGTTCGCTG | 3.06 |
| S-2 | CCTTGTACCCGCTCATCCAT | CTCCTACCGACCTGCCTG | 3.06 |
| S-3 | GTGTTACAAGAACTCAGCGT | AGCCTTCGTTCAGTTTCGGT | 3.06 |
| S-4 | TGACTGGCTTTCGTCCTACT | CACTCTCCAGCTGCTTCTCT | 3.06 |
| S-5 | ACAATTAGCTTAAGAACGC | AGCGGTGTTCATGGTACTCA | 3.06 |
| S-6 | TGGCTACTAGCTAGCTGTGCT | CTAAACCTTCACGCACAGCC | 3.06 |
| S-7 | CCAACTGAGCCGAATCGTTC | ATCATCCCATATCCCACGCC | 3.06 |
| S-8 | GTGGCCCATATGGTCTCAAC | CGGCCAGTCGATTAAAGCTC | 3.06 |
| S-9 | CGACGAAGAACTGACTTGACC | TTGCTTGATCATTCCTCCGC | 3.06 |
| S-10 | GTTTGGGCCGCATTCGAGAGA | TATTCTTGCCTCTCCCACCG | 3.06 |
| TIR-1 | CGGCCTCCATTTCGTCGAATCCCTT | ||
| 42618 | TGCCGGTACTGTGACTGTTC | CCTGAGCTCGATCTGCATGG | 3.06 |
| 42618-2 | TCTTTACTCCTCCCCTCCCA | CTTCTTGAGGATCCCGTTGC | 3.06 |
| q42618 | GCCTTAGTGCAGAGATTGACC | GCCTTTCCCAGTGCTCCA | 3.06 |
新窗口打开|下载CSV
1.4 DNA的提取、分子标记的开发及候选基因预测
在抽雄前完成对亲本和分离群体取样, CTAB法提取植株DNA[6]。待散粉结束完成育性调查后, 用于基因定位。根据初步定位结果, 在MaizeGDB上下载B73参考基因组序列, 用Blast与Mo17序列进行比对, 找出单拷贝区段中有InDel差异的位点, 根据这些差异位点设计显性标记(表1), 来源于Mo17的序列可以扩增出产物, 而来源于B73的序列则不能, 再通过琼脂糖凝胶电泳来筛选交换单株, 并通过标记加密进一步缩小定位区间。由于该突变表型是Mu转座子发生转座造成插入突变导致, 且大多数Mu转座子末端包含反向重复序列, 因此利用Mu转座子末端反向重复序列来设计引物[7], 通过该引物与候选基因特异性引物组合扩增的方法来确定候选基因。
1.5 PCR扩增及序列分析
利用Primer 3在线设计引物, 由武汉天一辉远生物科技有限公司合成引物。PCR反应总体积为15 μL, 含2 × PCR Mix 7.5 μL、 模板1.5 μL (50 ng μL-1)、引物各1 μL (5 μmol L-1), ddH2O补至15 μL。PCR反应条件为预变性(94℃, 5 min); 变性(94℃, 30 s), 退火(根据Tm值而定, 30 s), 延伸(72℃, 1 min), 34个循环; 72℃延伸5 min。在EnsemblPlants网站(
1.6 RNA提取及表达量分析
取抽雄前雄穗幼穗, 一部分放入液氮中速冻后于-80℃保存, 另外一部分用于表型观察, 待后期表型和基因型鉴定后, 挑选出相应的野生型和突变体提取RNA。参照TRI pure Reagent总RNA提取试剂盒(北京艾德莱生物科技有限公司)操作手册提取RNA, 参照反转录试剂盒(北京全式金生物技术有限公司)说明书合成cDNA。采用RT-PCR检测目的基因在野生型和突变体中的表达量。2 结果与分析
2.1 mi-ms-3的表型鉴定
突变体植株在营养生长阶段与野生型植株无差异; 而在生殖生长阶段, 突变体植株虽能正常抽雄, 但花药基本不外露(图1-A)。肉眼及花药石蜡切片观察发现, 突变体花药瘦小, 只有2个药室, 其余2个药室退化(图1-B1, B2)。突变体雄穗花药数目与野生型相比, 下位花无花药,上位花有0~3数目不等的花药, 部分花药退化为膜状, 并在其末端形成丝状物(图1-C1~C4)。1% I2-KI染色发现, 突变体花药中包含花粉, 且花粉粒能正常着色(图1-D1, D2), 但花药不开裂、不散粉。与野生型植株雌穗相比, 突变体雌穗在开花期表现为花丝增多, 每个子房对应2~4个花丝(图1-E1, E2); 授粉后, 突变体成熟果穗的籽粒两侧存在败育的籽粒。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1mi-ms-3和野生型表型
A: 野生型(左)和mi-ms-3 (右)雄穗表型, mi-ms-3花药不外露; B1~B2: 花药发育后期, 野生型和mi-ms-3花药横切片, mi-ms-3花药只有2个药室; C1~C4: mi-ms-3突变体小穗, 分别包含0~3个花药; D1~D2: 野生型和mi-ms-3花粉粒; E1: mi-ms-3 (左)和野生型(右)雌穗表型, mi-ms-3雌穗花丝增多; E2: mi-ms-3 (左)和野生型(右)籽粒表型, mi-ms-3每个籽粒对应多个花丝。
Fig. 1Phenotypic of the mi-ms-3 and wild type (WT)
A: the tassel of WT(wild type) (left) and mi-ms-3 (right), mi-ms-3 anther not exserted; B1-B2: transverse sections of the entire anther of WT and mi-ms-3 in the late microspore developmental stage, mi-ms-3 anthers with only two pollen sacs; C1-C4: the spikelet of mi-ms-3 contain 0-3 anthers respectively; D1-D2: pollen grains of WT and mi-ms-3; E1: the ear of mi-ms-3 (left) and WT (right), the number of silks in the ear of the mi-ms-3 increased; E2: the kernel of mi-ms-3 (left) and WT (right), mi-ms-3 each seed corresponds to multiple silks.
2.2 mi-ms-3的遗传分析
通过对突变体与Mo17杂交得到的F2分离群体进行遗传分析发现, F1代雄穗与雌穗均表现正常, F2群体野生型和突变体单株分别为73株和30株, 符合3﹕1 (χ2 = 0.94, P = 0.33), 说明该突变表型是由隐性单基因控制。2.3 突变基因的定位和克隆
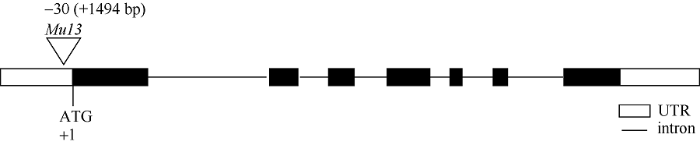
利用本实验室已合成的全基因组SSR引物对构建的2个极端混池进行扩增, 筛选多态性标记, 并初步将目的基因定位在3号染色体长臂上。根据育性调查结果, 在F2群体中随机选取野生型和突变体各12株, 构成另一组混池。在MaizeGDB数据库中选取29对SSR标记在“野生型池”和“突变体池”之间进行多态性标记筛选。对聚丙烯酰胺凝胶电泳结果分析发现, 其中8对SSR标记在“野生型池”和“突变体池”中具有多态性, 用这8对SSR标记对分离群体进行检测, 初步将目的基因定位在 umc1973和umc1027两个SSR标记之间, 物理距离为5.9 Mb。为了进一步精细定位, 根据B73和Mo17参考基因组在umc1973和umc1027两个标记间的序列差异开发InDel标记, 并对2个亲本进行多态性分析, 共筛选出10对Indel标记(表1), 最终将候选基因定位到S-6和umc1027两个标记之间, 物理距离为1.5 cM (表2)。对定位区间内的21个基因进行分析, 根据基因表达模式, 选择在花药中表达量较高的基因作为候选基因, 包括Zm00001d042609、Zm00001d042617、Zm00001d042618、Zm00001d042620和Zm00001d042624。根据Mu转座子标签法, 通过利用Mu转座子末端反向重复序列设计的引物TIR-1与候选基因特异性引物42618-R组合进行PCR扩增, 最终确定发生插入突变的基因为Zm00001d042618 (图2), 该基因编码MADS-box蛋白, 是参与花器官发育模型的B类功能基因。通过对野生型和突变体中该基因全长扩增比对发现, 突变体中该基因扩增片段长度要比野生型长。测序分析表明, 在Zm00001d042618基因起始密码子ATG上游30 bp处有1494 bp序列插入, 在插入位点处形成9 bp靶位点重复(TTCCTTGGG) (图3)。对1494 bp序列进行分析发现, 其为1个完整的Mu13转座子, 该转座子具有转座活性。
Table 2
表2
表2重要重组单株基因型及表型
Table 2
| umc1973 | S-1 | S-2 | S-3 | S-4 | S-5 | S-6 | S-7 | S-8 | S-9 | S-10 | umc1027 | 表型Phenotype | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2-2 | H | H | H | H | A | A | A | A | A | A | A | A | 突变体Mutant |
| 11-5 | H | H | H | H | H | A | A | A | A | A | A | A | 突变体Mutant |
| 9-3 | H | H | H | H | H | H | H | A | A | A | A | A | 突变体Mutant |
| 14-8 | A | A | A | A | A | A | A | A | A | A | A | H | 突变体Mutant |
| 5-4 | H | H | H | H | H | H | H | H | H | H | H | A | 野生型Wild type |
| 20-9 | A | H | H | H | H | H | H | H | H | H | H | H | 野生型Wild type |
新窗口打开|下载CSV
图2
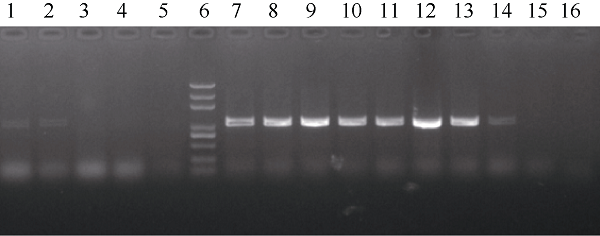 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2Mu转座子插入突变体的鉴定
1~2: 杂合野生型植株; 3~5: 纯合野生型植株; 6: marker; 7~14: 纯合突变体植株; 15: B73; 16: Mo17。
Fig. 2Identification of insertion mutant of mu transposon
1-2: heterozygous plants; 3-5: homozygous wild type plants; 6: marker; 7-14: homozygous mutant plants; 15: B73; 16: Mo17.
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3zmm16基因序列分析
Mu13转座子插入到zmm16基因ATG上游30 bp处。
Fig. 3Sequence analysis of zmm16
Mu13 transposon occurred at 30 bp upstream of ATG of zmm16.
2.4 zmm16的启动子分析
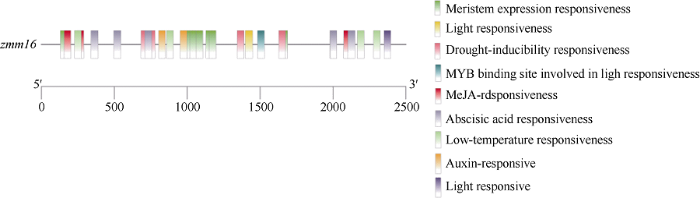
启动子预测分析结果显示, zmm16基因的启动子区包括茉莉酸甲酯响应元件、参与防御和应激反应响应元件、生长素响应元件、脱落酸响应元件、干旱胁迫响应元件及厌氧响应元件(图4)。研究表明, 茉莉酸甲酯响应元件与玉米雄穗和雌穗的发育有关[9], 特别是与玉米单性花形成密切相关[10]。除此之外, zmm16基因启动子区还存在与分生组织表达相关的顺式作用元件, 这也进一步说明Mu13转座子插入是导致mi-ms-3花药发育异常的原因。另外, 还发现低温响应顺式作用元件及多个光响应元件, 这2类响应元件对玉米zmm16基因功能的影响值得在未来深入探讨。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4zmm16启动子元件预测分析
Fig. 4Prediction and analysis of promoter elements of zmm16
2.5 zmm16表达量分析
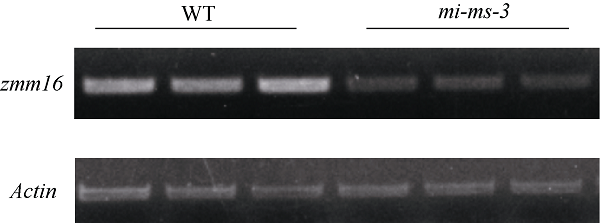
分别取突变体和野生型幼穗各3株提取RNA, 进行RT-PCR。结果表明, mi-ms-3中Zm00001d042618基因表达量降低。这说明Mu13转座子的插入突变并非导致Zm00001d042618完全不表达, 这一结果也对应了突变体中花药未完全退化的表型(图5)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5zmm16表达量分析
相对于野生型, mi-ms-3中zmm16基因表达量降低。
Fig. 5Expression level of zmm16
The expression of zmm16 in mi-ms-3 decreased compared with wild type.
3 讨论
与模式植物拟南芥相比, 玉米基因组更为复杂, 是拟南芥的几十倍。BSA法能够快速高效获得与目的基因连锁的分子标记, 发掘更多控制作物农艺性状的遗传位点。Zhao等[11]发现了一个玉米雌穗无花丝突变体sk-A7110, 利用BSA法筛选到与目的基因连锁的2个SSR标记umc1555和umc1448, 并通过精细定位及序列分析, 确定目的基因为Zm000010002970。Sun等[12]通过BSA与测序技术相结合的方法定位到水稻耐冷性相关qPSST6基因的候选区间, 并进一步通过GO分析确定了qPSST6的候选基因。当Mu转座子插入到功能基因内部或基因附近后能够破坏基因的功能从而产生突变表型。Mu转座子均具有约220 bp保守末端反向重复序列[13], 因此, Mu可作为插入标签用以进行基因定位。根据TIRs序列设计引物, 当Mu转座子插入到某一基因后产生插入突变, 通过突变表型的筛选以及TIRs相关引物, 分离得到Mu插入位点侧翼序列, 进而获得基因序列。Wang等[14]利用定位区间内25个候选基因的特异性引物与TIR引物组合扩增的方法, 成功克隆到一个雄性不育基因Ms8, 它编码一个半乳糖基转移酶, 主要在花药表皮和绒毡层中表达。Cui等[15]通过rf2a基因特异性引物和TIR引物组合扩增及测序的方法, 鉴定了rf2a四个等位基因插入的Mu转座子的种类及其插入位点。本研究采用BSA结合转座子标签法的定位策略, 成功定位到突变基因zmm16。BSA能够快速找到与目的基因连锁的分子标记, 使从整个基因组上定位突变基因更为快速。相较于通过测序来确定突变基因的方法, 转座子标签法的利用可以直接通过PCR确定目的基因, 降低了试验成本。Bartlett等[5]利用BSA法将sts1基因定位到3号染色体长臂上, 并发现突变表型与umc1266完全连锁, 根据突变体表型推定该基因属于B类基因, 在umc1266标记3.39 Mb处发现1个B类基因zmm16, 测序发现sts1-1突变体第3内含子上G突变为A, sts1-2突变体第4内含子上G突变为A, 导致转录本不能发生正常剪接, 影响基因转录过程, 造成雄花不育, 花药完全退化为膜状结构, 雌花可育, 花丝增多。本研究获得了一个新的sts1等位突变体mi-ms-3, 该突变体表型与sts1相似, 但相对于sts1花药完全退化, mi-ms-3则表现为部分花药未完全退化, 雄穗上部分小穗含有花药, 与野生型植株花药不同之处在于mi-ms-3的花药只有2个药室, 同时雄穗上其他部分花药退化为膜状并在其末端形成丝状物。在sts1突变体中, zmm16内含子发生点突变, 转录本不能进行正常剪接, 产生不完整的转录本, 导致zmm16基因功能缺失。而mi-ms-3突变表型是由Mu13转座子的插入突变造成的。取雄穗进行表达量分析发现, mi-ms-3突变体zmm16基因的表达量下降而非完全不表达, 这一突变表现为渗漏突变, 即基因编码的蛋白功能不完全失活, 仍然保留了一些功能。与sts1相同之处在于, mi-ms-3不育基因也与雌穗多花丝的标记性状[16]连锁, mi-ms-3在开花期也表现为雌穗花丝增多, 每个子房对应2~4个花丝, 成熟果穗籽粒基部有败育种子。
高等植物的基因转录起始位点一般位于起始密码子(ATG)上游-40~ -70 bp处。一般而言, 转录起始位点的缺失会影响转录, 但是也有****认为转录位点并不是转录发生所必须的[17]。植物启动子中含有许多顺式作用元件, 通过转录因子与其结合来调控基因表达, 进而影响植物的生长发育过程。研究表明, MADS-box蛋白能与基因启动子区域中的顺式作用元件特异性结合, 使靶基因在特定的时间和空间以特定的强度表达[18]。mi-ms-3中Mu13插入到zmm16基因上游, 可能导致zmm16基因编码蛋白不能与启动子区顺式作用元件结合来调控基因的表达, 而以一种低表达量的状态体现在突变体雄穗中部分小穗含有花药的表型上。zmm16基因启动子序列中存在低温响应元件及大量光响应元件, 这说明其表达可能受温度和光照环境因素的影响。在花药发育的特定阶段, 通过调节zmm16基因表达来适应一定范围内的光、温等环境变化。对玉米zmm16基因启动子区进行初步分析, 为将来进行基因功能的验证奠定了基础。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URL [本文引用: 1]

玉米雄性不育材料是玉米遗传育种中的一个极其主要的种质资源,笔者简述了玉米胞质雄性不育材料近期以来的研究和利用情况。玉米育种工作者将从多方位、多层次加以研究和利用雄性不育材料,随着生物技术的迅速发展,玉米雄性不育材料在玉米育种和制种中将得到更为广泛的利用。
[本文引用: 1]
DOI:10.1093/pcp/pct016URL [本文引用: 1]
Plant development depends on the activity of various types of meristems that generate organs such as leaves and floral organs throughout the life cycle. Grass species produce complex inflorescences and unique flowers. The grass inflorescence is composed of different types of branches, including a specialized branch called a spikelet. The spikelet is a special unit of the inflorescence and forms one to several florets, depending on the species. In the floret, floral organs such as perianth organs, carpels and stamens are formed. In Arabidopsis, because the inflorescence meristem (IM) forms the floral meristems (FMs) directly on its flanks, the change of meristem fate is relatively simple. In contrast, in grasses, different types of meristem, such as the IM, the branch meristem (BM), the spikelet pair meristem (SPM) in some grasses, the spikelet meristem (SM) and the FM, are responsible for the elaboration of their complex inflorescences and flowers. Therefore, sequential changes of meristem fate are required, and a number of genes involved in the specification of the fate of each meristem have been identified. In this review, we focus on the following issues concerning the fate of the reproductive meristems in two grass species, maize (Zea mays) and rice (Oryza sativa): (i) meristem regulation during inflorescence development; (ii) specification and fate change of the BM and the SM; (iii) determinacy of the FM; and (iv) communication between the meristem and lateral organs.
DOI:10.1007/s00425-016-2607-2URLPMID:27770199 [本文引用: 1]
MAIN CONCLUSION: The determining process of pistil fate are central to maize sex determination, mainly regulated by a genetic network in which the sex-determining genes SILKLESS 1 , TASSEL SEED 1 , TASSEL SEED 2 and the paramutagenic locus Required to maintain repression 6 play pivotal roles. Maize silks, which emerge from the ear shoot and derived from the pistil, are the functional stigmas of female flowers and play a pivotal role in pollination. Previous studies on sex-related mutants have revealed that sex-determining genes and phytohormones play an important role in the regulation of flower organogenesis. The processes determining pistil fate are central to flower development, where a silk identified gene SILKLESS 1 (SK1) is required to protect pistil primordia from a cell death signal produced by two commonly known genes, TASSEL SEED 1 (TS1) and TASSEL SEED 2 (TS2). In this review, maize flower developmental process is presented together with a focus on important sex-determining mutants and hormonal signaling affecting pistil development. The role of sex-determining genes, microRNAs, phytohormones, and the paramutagenic locus Required to maintain repression 6 (Rmr6), in forming a regulatory network that determines pistil fate, is discussed. Cloning SK1 and clarifying its function were crucial in understanding the regulation network of sex determination. The signaling mechanisms of phytohormones in sex determination are also an important research focus.
DOI:10.1016/s1097-2765(00)80450-5URLPMID:10882141 [本文引用: 1]
The degree to which the eudicot-based ABC model of flower organ identity applies to the other major subclass of angrosperms, the monocots, has yet to be fully explored. We cloned silky1 (si1), a male sterile mutant of Zea mays that has homeotic conversions of stamens into carpels and lodicules into palea/lemma-like structures. Our studies indicate that si1 is a monocot B function MADS box gene. Moreover, the si1 zag1 double mutant produces a striking spikelet phenotype where normal glumes enclose reiterated palea/lemma-like organs. These studies indicate that B function gene activity is conserved among monocots as well as eudicots. In addition, they provide compelling developmental evidence for recognizing lodicules as modified petals and, possibly, palea and lemma as modified sepals.
DOI:10.1105/tpc.15.00679URLPMID:26518212 [本文引用: 1]
In monocots and eudicots, B class function specifies second and third whorl floral organ identity as described in the classic ABCE model. Grass B class APETALA3/DEFICIENS orthologs have been functionally characterized; here, we describe the positional cloning and characterization of a maize (Zea mays) PISTILLATA/GLOBOSA ortholog Zea mays mads16 (Zmm16)/sterile tassel silky ear1 (sts1). We show that, similar to many eudicots, all the maize B class proteins bind DNA as obligate heterodimers and positively regulate their own expression. However, sts1 mutants have novel phenotypes that provide insight into two derived aspects of maize flower development: carpel abortion and floral asymmetry. Specifically, we show that carpel abortion acts downstream of organ identity and requires the growth-promoting factor grassy tillers1 and that the maize B class genes are expressed asymmetrically, likely in response to zygomorphy of grass floral primordia. Further investigation reveals that floral phyllotactic patterning is also zygomorphic, suggesting significant mechanistic differences with the well-characterized models of floral polarity. These unexpected results show that despite extensive study of B class gene functions in diverse flowering plants, novel insights can be gained from careful investigation of homeotic mutants outside the core eudicot model species.
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/nar/gnh052URLPMID:15060129 [本文引用: 1]
High-copy transposon mutagenesis is an effective tool for creating gene disruptions in maize. In order to molecularly define transposon-induced disruptions on a genome-wide scale, we optimized TAIL-PCR to amplify genomic DNA flanking maize Robertson's Mutator insertions. Sample sequencing from 43 Mutator stocks and the W22 inbred line identified 676 non-redundant insertions, and only a small fraction of the flanking sequences showed significant similarity to maize repetitive sequences. We further designed and tested 79 arbitrary primers to identify 12 primers that amplify all Mutator insertions within a DNA sample at 3.1-fold redundancy. Importantly, the products are of sufficient size to use as substrates or probes for hybridization-based identification of gene disruptions. Our adaptation simplifies previously published TAIL-PCR protocols and should be transferable to other high-copy insertional mutagens.
DOI:10.1016/j.molp.2020.06.009URLPMID:32585190 [本文引用: 1]
The rapid development of high-throughput sequencing techniques has led biology into the big-data era. Data analyses using various bioinformatics tools rely on programming and command-line environments, which are challenging and time-consuming for most wet-lab biologists. Here, we present TBtools (a Toolkit for Biologists integrating various biological data-handling tools), a stand-alone software with a user-friendly interface. The toolkit incorporates over 130 functions, which are designed to meet the increasing demand for big-data analyses, ranging from bulk sequence processing to interactive data visualization. A wide variety of graphs can be prepared in TBtools using a new plotting engine (
DOI:10.1126/science.1164645URLPMID:19131630 [本文引用: 1]
Sex determination in maize is controlled by a developmental cascade leading to the formation of unisexual florets derived from an initially bisexual floral meristem. Abortion of pistil primordia in staminate florets is controlled by a tasselseed-mediated cell death process. We positionally cloned and characterized the function of the sex determination gene tasselseed1 (ts1). The TS1 protein encodes a plastid-targeted lipoxygenase with predicted 13-lipoxygenase specificity, which suggests that TS1 may be involved in the biosynthesis of the plant hormone jasmonic acid. In the absence of a functional ts1 gene, lipoxygenase activity was missing and endogenous jasmonic acid concentrations were reduced in developing inflorescences. Application of jasmonic acid to developing inflorescences rescued stamen development in mutant ts1 and ts2 inflorescences, revealing a role for jasmonic acid in male flower development in maize.
[本文引用: 1]
DOI:10.3389/fpls.2018.01127URLPMID:30116254 [本文引用: 1]
Worldwide consumption of oil is increasing with the growing population in need for edible oil and the expansion of industry using biofuels. Then, demand for high yielding varieties of oil crops is always increasing. Brassica napus (rapeseed) is one of the most important oil crop in the world, therefore, increasing rapeseed yield through breeding is inevitable in order to cater for the high demand of vegetable oil and high-quality protein for live stocks. Quantitative trait loci (QTL) analysis is a powerful tool to identify important loci and which is also valuable for molecular marker assisted breeding. Seed-yield (SY) is a complex trait that is controlled by multiple loci and is affected directly by seed weight, seeds per silique and silique number. Some yield-related traits, such as plant height, biomass yield, flowering time, and so on, also affect the SY indirectly. This study reports the assembly of QTLs identified for seed-yield and yield-related traits in rapeseed, in one unique map. A total of 972 QTLs for seed-yield and yield-related were aligned into the physical map of B. napus Darmor-bzh and 92 regions where 198 QTLs overlapped, could be discovered on 16 chromosomes. Also, 147 potential candidate genes were discovered in 65 regions where 131 QTLs overlapped, and might affect nine different traits. At the end, interaction network of candidate genes was studied, and showed nine genes that could highly interact with the other genes, and might have more influence on them. The present results would be helpful to develop molecular markers for yield associated traits and could be used for breeding improvement in B. napus.
DOI:10.1186/s12284-018-0218-1URLPMID:29671148 [本文引用: 1]
BACKGROUND: Cold stress can cause serious abiotic damage that limits the growth, development and yield of rice. Cold tolerance during the booting stage of rice is a key factor that can guarantee a high and stable yield under cold stress. The cold tolerance of rice is controlled by quantitative trait loci (QTLs). Based on the complex genetic basis of cold tolerance in rice, additional efforts are needed to detect reliable QTLs and identify candidate genes. In this study, recombinant inbred lines (RILs) derived from a cross between a cold sensitive variety, Dongnong422, and strongly cold-tolerant variety, Kongyu131, were used to screen for cold-tolerant loci at the booting stage of rice. RESULTS: A novel major QTL, qPSST6, controlling the percent seed set under cold water treatment (PSST) under the field conditions of 17 degrees C cold water irrigation was located on the 28.4 cM interval on chromosome 6. Using the combination of bulked-segregant analysis (BSA) and next-generation sequencing (NGS) technology (Seq-BSA), a 1.81 Mb region that contains 269 predicted genes on chromosome 6 was identified as the candidate region of qPSST6. Two genes, LOC_Os06g39740 and LOC_Os06g39750, were annotated as
DOI:10.1080/07352689309382356URL [本文引用: 1]
DOI:10.1007/s00497-013-0230-yURL [本文引用: 1]
Precise somatic and reproductive cell proliferation and differentiation in anthers are crucial for male fertility. Loss of function of the Male sterile 8 (Ms8) gene causes male sterility with multiple phenotypic defects first visible in the epidermal and tapetal cells. Here, we document the cloning of Ms8, which is a putative beta-1,3-galactosyltransferase. Ms8 transcript is abundant in immature anthers with a peak at the meiotic stage; RNA expression is highly correlated with protein accumulation. Co-immuno-precipitation coupled with mass spectrometry sequencing identified several MS8-associated proteins, including arabinogalactan proteins, prohibitins, and porin. We discuss the hypotheses that arabinogalactan protein might be an MS8 substrate and that MS8 might be involved in maintenance of mitochondrial integrity.
URLPMID:12618406 [本文引用: 1]
Even in the absence of excisional loss of the associated Mu transposons, some Mu-induced mutant alleles of maize can lose their capacity to condition a mutant phenotype. Three of five Mu-derived rf2a alleles are susceptible to such Mu suppression. The suppressible rf2a-m9437 allele has a novel Mu transposon insertion (Mu10) in its 5' untranslated region (UTR). The suppressible rf2a-m9390 allele has a Mu1 insertion in its 5' UTR. During suppression, alternative transcription initiation sites flanking the Mu1 transposon yield functional transcripts. The suppressible rf2a-m8110 allele has an rcy/Mu7 insertion in its 3' UTR. Suppression of this allele occurs via a previously unreported mechanism; sequences in the terminal inverted repeats of rcy/Mu7 function as alternative polyadenylation sites such that the suppressed rf2a-m8110 allele yields functional rf2a transcripts. No significant differences were observed in the nucleotide compositions of these alternative polyadenylation sites as compared with 94 other polyadenylation sites from maize genes.
URL [本文引用: 1]
1992年在玉米族远缘杂交组合3402F3(丹340×403-2)中首次发现带标记性状的基因雄性不育(GMS)材料。 遗传分析结果表明, 不育性受一对隐性基因控制。 当不育株(A)与可育株(B)进行兄妹交, 育性分离比例接近1∶1; 而可育株(B)自交的后代, 可育株与不育株的分离比例为3∶1。 连锁遗传分析结果证明, 不育基因(ms°)与标记性状
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/s1055-7903(03)00207-0URLPMID:14615187 [本文引用: 1]
MADS-box genes encode a family of transcription factors which control diverse developmental processes in flowering plants ranging from root to flower and fruit development. Sequencing of (almost) the complete Arabidopsis genome enabled the identification of (almost) all of the Arabidopsis MADS-box genes. MADS-box genes have been divided in two large groups, termed type I and type II genes. The type II genes comprise the MEF2-like genes of animals and fungi and the MIKC-type genes of plants. The majority of MIKC-type genes are of the MIKC(c)-type, which includes all plant MADS-box genes for which expression patterns or mutant phenotypes are known. By phylogeny reconstruction, almost all of the MIKC(c)-type genes can be subdivided into 12 major gene clades, each clade comprising 1-6 paralogs from Arabidopsis and putative orthologs from other seed plants. Here we first briefly describe the deep branching of the MADS-box gene tree to place the MIKC(c)-type genes into an evolutionary context. For every clade of MIKC(c)-type genes we then review what is known about its members from Arabidopsis and well-studied members from other phylogenetically informative plant species. By gene sampling and phylogeny reconstructions we provide minimal estimates for the ages of the different clades. It turns out that 7 of the 12 major gene clades, i.e., AG-, AGL6-, AGL12-, DEF+GLO- (B), GGM13- (B(s)), STMADS11- and TM3-like genes very likely existed already in the most recent common ancestor of angiosperms and gymnosperms about 300MYA. Three of the other clades, i.e., AGL2-, AGL17-, and SQUA-like genes, existed at least already in the most recent common ancestor of monocots and eudicots about 200 MYA. Only for two gene clades, AGL15-like genes (2 genes in Arabidopsis) and FLC-like genes (6 genes) members from plants other than Brassicaceae have not been reported yet. Similarly, only one ancient clade known from other flowering plant species, TM8-like genes, is not represented in Arabidopsis. These findings reveal that the diversity of MADS-box genes in Arabidopsis is rather ancient and representative for other flowering plants. Our studies may thus help to predict the set of MADS-box genes in all other flowering plants, except for relatively young paralogs. For the different gene clades we try to identify ancestral and derived gene functions and review the importance of these clades for seed plant development and evolution. We put special emphasis on gene clades for which insights into their importance has rapidly increased just recently.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}