 ,1,*
,1,*Identification of indeterminate domain protein family genes associated with flowering time in maize
LI Yun-Fu1,2, WANG Jing-Xian1, DU Yan-Fang1, ZOU Hua-Wen2, ZHANG Zu-Xin,1,*通讯作者:
收稿日期:2018-11-1接受日期:2019-01-12网络出版日期:2019-01-31
| 基金资助: |
Received:2018-11-1Accepted:2019-01-12Online:2019-01-31
| Fund supported: |
作者简介 About authors

摘要
关键词:
Abstract
Keywords:
PDF (602KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
李云富, 王静贤, 杜艳芳, 邹华文, 张祖新. 玉米开花期相关的Indeterminate domain (IDD)蛋白家族基因的鉴定[J]. 作物学报, 2019, 45(4): 499-507. doi:10.3724/SP.J.1006.2019.83068
LI Yun-Fu, WANG Jing-Xian, DU Yan-Fang, ZOU Hua-Wen, ZHANG Zu-Xin.
开花期是玉米适应当地环境的重要性状之一。大约一万年前, 玉米起源于墨西哥西南部, 由其野生祖先大刍草驯化而来[1]。早期的玉米地方品种往往表现出明显的光周期敏感性, 在短日照条件下开花。持续不断的人工改良, 使得现代玉米栽培品种光周期敏感性下降以适应在不同光周期条件下栽培[2]。由营养生长向生殖生长转换是玉米生长发育的关键时期, 这一转换不仅直接决定植株开花期, 也与植株生殖发育密切相关, 进而影响产量[3]。因而, 开花期是玉米育种和生产的重要目标性状。玉米开花期性状一般包括抽雄期(days to tassel, DTT)、散粉期(days to anthesis, DTA)、吐丝期(days to silking, DTS)和散粉—吐丝间隔期(anthesis-silking interval, ASI)。这4个性状均属于数量性状, 由多基因控制[4]。
近年来, 随着基因组学和分子数量遗传学的理论与技术的发展, 科学家对玉米开花期的遗传基础已有初步了解, 并且已有多个玉米生育期相关基因被鉴定和分离。这些基因主要涉及花器官发育、光周期响应、激素合成与信号、成花素调控等途径[5]。参与玉米光周期响应调控的基因主要有ZCN2、ZCN8和ZmCCT家族基因。ZCN2和ZCN8均编码一种磷脂酰乙醇胺结合蛋白, 分别与拟南芥TFL和FT同源; ZCN2和ZCN8蛋白在叶片中表达分别经木质部和韧皮部运输到顶端分生组织, 参与调控玉米生殖转换[6,7]。玉米CCT家族基因ZmCCT9和ZmCCT10也是光周期响应调控的重要基因。ZmCCT9负调控ZCN8表达, 延迟玉米在长日照条件的开花期[8]。ZmCCT10是OsGhd7的同源基因, 也是玉米重要的光周期调节基因[9,10,11]。在玉米改良过程中, ZmCCT9和ZmCCT10这2个位点上的转座子插入等位基因被选择利用, 导致现代玉米对光周期的敏感性下降, 适应性种植纬度扩大[8,9,10,11]。植物激素信号也参与玉米开花期的调控。DWARF8 (D8)、 DWARF9 (D9)和GA2ox1是赤霉素合成和信号途径上的重要基因[12,13,14]。D8和D9均编码DELLA蛋白, 关联分析发现D8与长日照条件下玉米开花相关, 一个编码区6 bp缺失d8突变体提前开花, 而过表达D9也会导致开花提前[12,13]。GA2ox1编码一个赤霉素合成抑制酶, 通过调控赤霉素的水平而影响开花[14]。生殖转换和花器官发育是植物开花的生物学基础, 生殖转换和花器官发育相关基因也是开花期相关基因。如玉米DLF1是拟南芥FLOWERING LOCUS D (FD)的同源基因, 参与玉米生殖转换[15]; ZmRAP2.7是拟南芥中TARGET OF EAT1 (TOE1)的同源基因, 抑制生殖转换[16]; ZmRAP2.7上游70 kb非编码区可作为顺式因子调节功能基因ZmRap2.7的表达, 进而调控开花期[17]; MADS-BOX基因也参与玉米生育期调控, 如ZMM1、ZMM4和ZMM69正调控玉米开花期[18,19,20]。
Buckler等[21]利用NAM (Nested Association Mapping population)群体鉴定到100多个与开花期相关的QTL, 但已分离的开花期相关基因仍然十分有限。在植物中, INDETERMINATE-domain (IDD)家族蛋白是一类锌指蛋白转录因子, 也参与植物开花期的调控。如拟南芥的AtIDD8和AtIDD14通过改变植物体内含糖量和淀粉代谢调控开花[22]。水稻的RID1是IDD蛋白家族基因成员之一, 其突变体在适宜的条件下也无法开花, 表明RID1是水稻开花期控制的关键因子[23]。ID1 (INDETERMINATE1)是玉米中第1个被克隆的IDD蛋白家族基因, 其突变体id1表现为叶片增多、花序发育异常、开花期延迟且不受光周期的影响[24], 但玉米IDD蛋白家族其他成员的功能仍有待研究。
本研究利用生物信息学技术、基于IDD同源结构域序列, 在玉米基因组中鉴定并分离了37个IDD 家族基因(ZmIDD), 分析了ZmIDD基因的表达模式和ZmIDD蛋白质的结构; 关联分析发现了7个与开花期显著关联的ZmIDD基因, 鉴定了1个新的花期相关ZmIDD基因, 并进一步分析了该基因自然变异及其单倍型的遗传效应。研究结果为深入解析玉米IDD家族基因对开花期的调控奠定了基础。
1 材料与方法
1.1 试验材料与表型鉴定
2017年春将全球收集的172份玉米自交系分别种植于华中农业大学武汉试验基地(30°N, 114°E)、鄂州试验基地(30°N, 114°E)和襄阳(32°N, 112°E), 采用随机区组设计, 双行区, 3次重复, 行长3.0 m, 行距 0.6 m, 株距0.3 m。以田间系内50%的个体抽雄(植株雄穗尖端露出顶叶3~5 cm)、散粉(雄穗主轴开始散粉)和吐丝(植株雌穗的花丝从苞叶中伸出2 cm左右)分别记作该自交系的抽雄日期、散粉日期和吐丝日期分别减去播种日期, 即为抽雄期、散粉期和吐丝期。利用一般线性模型对3个环境下各性状的均值进行最优无偏估计分析(BLUP, best linear unbiased prediction), 获得各自交系不同表型性状的表型值用于关联分析。1.2 IDD家族基因序列获取及进化树构建
从TAIR (The Arabidopsis Information Resource)网站上提取拟南芥IDD家族基因的氨基酸序列, 以 FASTA格式保存。以已知的IDD (Zm00001d032922) 氨基酸序列作为查询(Query)序列, 利用BlastP搜索MaizeGDB (Maize Genetic and Genomics Database) 数据库, 初步筛选玉米IDD蛋白序列。将IDD家族特有的C2H2 (E-value<0.001)锌指蛋白序列用hmmbuild (hmmer3.1 b1)生成HMM文件, 搜索玉米B73 Pfam (http://pfam.sanger.ac.uk/)数据库, 进一步获得玉米的IDD家族蛋白的基因序列和氨基酸序列, 以FASTA 格式保存。将基因序列分别导入基因结构显示系统(GSDS, http://gsds.cbi.pku.edu.cn/)[25], 绘制基因结构图, 对个别显示错误的结构图进行手工修正。用软件CLC Sequence Viewer (http://www.Qiagenbioin formatics.com/products/clc-sequence-viewer/)进行氨基酸多序列比对, 采用邻近算法(Neighbor-Joining, NJ)构建系统进化树, Bootstrap检验1000次。将玉米IDD蛋白氨基酸序列导入在线蛋白结构域分析系统(SMART)绘制基因蛋白结构图。然后, 根据所预测的基因序列设计引物, 以PCR扩增cDNA序列, 并测序。比较扩增序列与预测序列, 验证玉米IDD家族基因预测序列的准确性。1.3 表达模式分析
从qteller下载玉米B73自交系转录组数据, 从中提取根、茎、叶、雌穗、雄穗、花丝、种子、花药8个组织的表达数据, 用heatmap.2制作heatmap图。1.4 关联分析
本实验室前期获得了50万个高质量SNP所鉴定的368份自交系的基因型数据[26]。我们从中提取了172份自交系在37个IDD家族基因位点上的SNP基因型。在此基础上, 结合基因型和表型数据, 利用Tassel 3.0软件和混合线性模型(MLM, mixed linear model)并以Q和K作为协变量开展关联分析。参照Yang等[27]的研究结果进行关联群体的结构分析(Q, population structure)和亲缘关系分析(K, kinship coefficient)。采用 P<0.01作为宽松的显著关联的阈值, 设置 Bonferroni 校验阈值为更为严格的显著阈值, 即 P<1/n (n为所用到的标记数)[28]。1.5 重测序和候选基因关联分析
为了研究Zm00001d020683在自交系群体的遗传多样性, 探究潜在的功能变异位点, 在172份自交系中重测序了该基因。首先, 根据B73参考基因组设计基因特异引物(R1-F: 5'-GTGTGGCTGCTTT TGCATTA-3'; R1-R: 5'-TCCTTGCACAGCAGTAA- 3'; R2-F: 5'-CTAAGCGTCCATCCAGTTCC-3'; R2-R: 5'-TTGGAGAAGCTCGTTGCTTT-3'), 以基因特异引物PCR扩增Zm00001d020683序列; 其次, 通过PCR产物测序以获得各个自交系中Zm00001d0206 83序列; 将所有自交系的Zm00001d020683序列导入Bioedit (https://www.bioedit.com/)软件进行序列比对, 提取各自交系在该基因座上的多态性位点; 最后, 利用各自交系多态性位点的基因型结合开花期表型, 使用Tassel3.0软件(https://www.tassel.com/), 以Q和K为协变量进行候选基因关联分析。2 结果与分析
2.1 玉米IDD家族基因蛋白结构域和系统进化分析
已知IDD家族蛋白含有保守IDD结构域, 该结构域包含一个核定位信号和两类锌指蛋白结构域(C2H2和C2HC), 其中2个C2H2结构域为 TFIIIA 类型的锌指结构。基于IDD家族蛋白的保守结构域, 在玉米基因组数据库中共筛选到37个玉米IDD蛋白(ZmIDD) (图1), 其中, 34个ZmIDD都包含3个保守基序, 仅Zm00001d034783含有2个保守基序, Zm00001d005871和Zm00001d048722各含1个保守基序, 推测这3个基因的生物学功能和作用机理可能有别于与其他IDD家族成员。 进一步利用B73 RefGen V4 cDNA 序列设计引物, 以本实验保存的B73为材料, PCR扩增ZmIDD基因序列并测序。序列分析发现大多数基因序列与参考基因组序列一致, 仅有少数基因存在单个碱基替换, 这些变异可能由PCR扩增所致或者由扩增材料与参考基因组B73有别所致。使用玉米和拟南芥 IDD 家族蛋白的氨基酸序列, 通过CLC Sequence Viewer V8软件构建玉米和拟南芥IDD家族蛋白的系统进化树(图2)。可以看出, 玉米IDD家族蛋白可以分为3个分支, 每一分支上玉米IDD蛋白都有与之同源的拟南芥IDD蛋白。在拟南芥中, IDD家族基因的生物学功能和作用机理研究比较深入; 在玉米中, 除ID1外的ZmIDD基因生物学功能和作用鲜为人知。因此, ZmIDD与拟南芥IDD家族基因的系统进化分析可为ZmIDD的生物学功能及其调控网络解析提供借鉴和指导。
图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1玉米IDD家族基因及其保守基序特征
通过GSDS2.0软件展示基因结构。直线表示内含子, 黑色矩形表示外显子。通过MEME程序分析保守基序。黑色方框: motif 1; 灰色方框: motif 2; 白色方框: motif 3。
Fig. 1Gene structures and putative conserved motifs of IDD family genes in maize
Gene structures are showed by the GSDS2.0. Putative conserved motifs of IDD family proteins are predicted by the MEME program. Black box: motif 1; gray box: motif 2; white box: motif 3.
图2
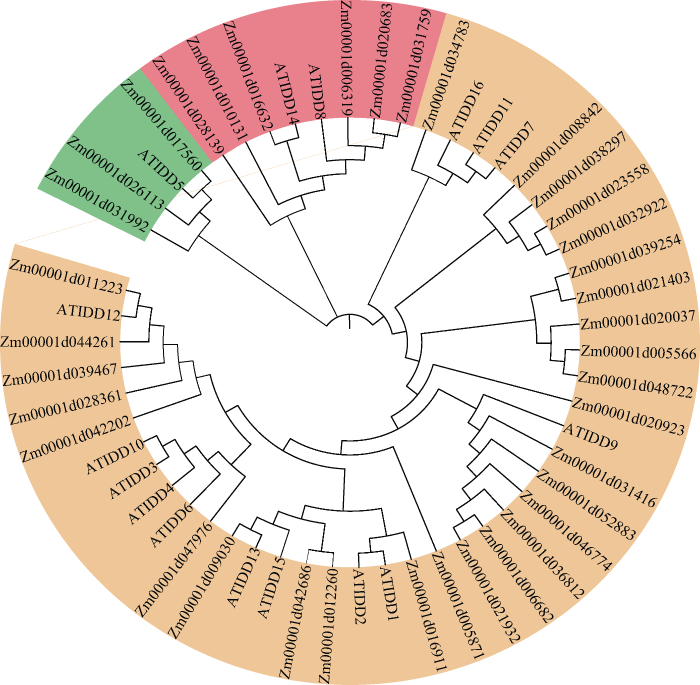 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2玉米和拟南芥IDD家族蛋白的系统进化树
系统进化树采用邻接法构建, 自举检验1000次。
Fig. 2Phylogenetic tree of IDD proteins in Zea mays and Arabidopsis thaliana
The phylogenetic tree is constructed by Neighbor-Joining method with 1000 bootstraps based on the amino acid sequence of IDD proteins in Zea mays and Arabidopsis thaliana.
2.2 IDD家族基因的表达模式
为了解IDD家族基因的表达模式, 提取了公共数据库中B73自交系8个不同组织的转录组数据, 构建了37个ZmIDD的表达模式图(图3)。总体上看, 多数基因在所研究的8个组织中低水平表达, 仅11个(29.7%)基因在至少1个组织中的mRNA水平均高于10 FPKM (fragments per kilobase of exon per million fragments mapped)。表达水平较高的基因也表现出明显的组织特异性, 所有37个ZmIDD均在茎顶端分生组织、花药和花粉中低表达, 2个基因(Zm 00001d042686和Zm00001d012260)则在除花药和种子外的6个组织中表达水平较高, 3个基因(Zm 00001d011223、Zm00001d023558和Zm00001d04 426) 在种子中特异性表达, 而Zm00001d021403在叶片中特异表达。这些基因的特异性表达模式暗示着各自发挥其功能的组织特异性以及玉米IDD家族基因发挥其作用的组织广泛性。图3
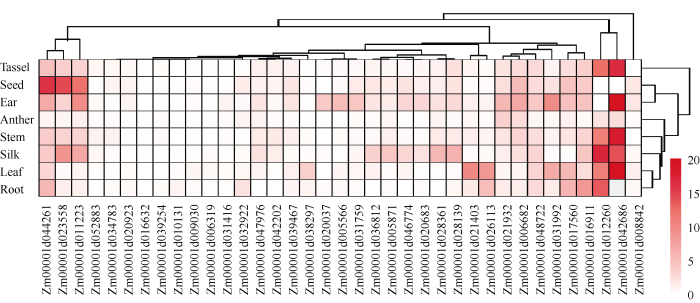 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图337个玉米IDD家族基因表达模式
方框内颜色显示基因表达水平, RNA-seq数据用FPKM表示。
Fig. 3Expression patterns of 37 maize IDD family genes
Color boxes show the expression level of maize IDD family genes. RNA-sequencing is showed by FPKM (fragments per kilobase of exon per million fragments mapped).
2.3 玉米IDD家族基因的自然变异与开花期的关联分析
分析了37个玉米IDD家族基因在172份自交系中的遗传变异(表1)。由于标记密度和分布的原因, 2个ZmIDD (Zm00001d010131和Zm00001d046774) 基因没有标记覆盖, 其他35个ZmIDD基因分别由5~97个SNP所标记, 单个基因平均SNP数为37.8个。结合基因型和3个开花期性状(抽雄期、吐丝期、散粉期)表型的关联分析发现, 在P<0.01和P<0.00075(P = 1/1323)下均鉴定到7个ZmIDD基因与开花期性状显著关联(表1), 其中, Zm00001d032922、Zm0000 1d005566、Zm00001d020683和Zm00001d023558在3个环境和两种阈值下均被检测到。另外, Zm00001 d044261、Zm00001d011223和Zm00001d03 9467在2个环境下检测到与开花期性状显著关联, 这3个基因可能受环境诱导表达进而影响开花期。因此, 我们推测这7个基因可能是玉米开花期相关基因, 具有进一步深入研究的价值。Table 1
表1
表1玉米IDD家族基因的遗传变异及其与开花期的关联分析
Table 1
| 基因 Gene | 染色体 Chr. | 数目 No. | 显著性水平Significance level P<0.01 | 显著性水平Significance level P<0.00075 P<0.00075 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 环境1 Env1 | 环境2 Env2 | 环境2 Env2 | BLUP | 环境1 Env1 | 环境2 Env2 | 环境2 Env2 | BLUP | ||||
| Zm00001d032922 | 1 | 18 | 5/7/5 | 7/7/7 | 7/8/8 | 3/3/3 | 1/1/1 | 1/1/1 | 5/3/5 | 2/2/1 | |
| Zm00001d005566 | 2 | 14 | 7/7/7 | 7/7/6 | 6/7/6 | 1/1//1 | 6/2/6 | 4/2/5 | 5/0/5 | 0 | |
| Zm00001d039467 | 3 | 36 | 7/4/4 | 6/3/3 | 6/2/2 | 2/2/0 | 6/2/0 | 5/1/1 | 0 | 0 | |
| Zm00001d044261 | 3 | 25 | 4/2/2 | 3/3/3 | 3/3/3 | 0 | 2/2/0 | 2/2/2 | 0 | 0 | |
| Zm00001d020683 | 7 | 31 | 4/3/3 | 3/5/5 | 7/4/5 | 3/3/3 | 2/2/2 | 3/3/3 | 1/1/1 | 1/1/1 | |
| Zm00001d011223 | 8 | 39 | 0 | 2/3/3 | 4/4/4 | 0 | 0 | 1/1/0 | 0/1/1 | 0 | |
| Zm00001d023558 | 10 | 80 | 12/11/13 | 10/9/9 | 9/9/9 | 1/1/2 | 4/4/2 | 3/4/2 | 7/2/5 | 0/1/1 | |
新窗口打开|下载CSV
2.4 Zm00001d020683遗传多样性及其与开花期的关联分析
由于Zm00001d020683与2个已知的拟南芥开花期相关基因AtIDD8和 AtIDD14的序列相似性高, 位于同一个聚类分支中, 推测Zm00001d020683在玉米中可能行使类似AtIDD8和AtIDD14的生物学功能, 调控开花期。为此, 进一步分析了Zm00001d 020683与开花期关联的功能位点。对该基因2 kb的启动子和600 bp的基因编码区共2.6 kb序列进行了重测序分析, 在172份自交系中, 共鉴定到45个SNP位点和19个Insertion/Deletion (InDel)位点。结合这64个多态性位点与开花期表型进行关联分析, 发现启动子区2个位点分别与抽雄期、吐丝期和散粉期显著关联, 其中, -1456 bp处为一个0/3 bp的InDel, -999 bp 处为一个0/2 bp的InDel (图4)。图4
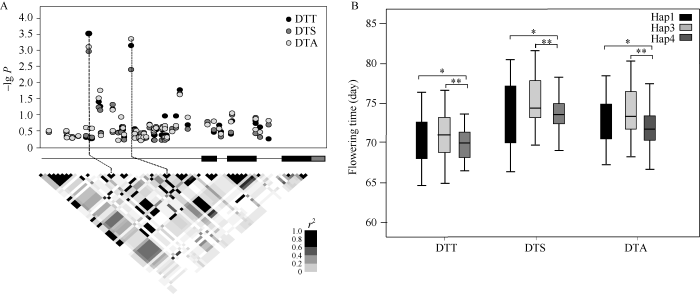 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4候选基因关联分析
A) Zm00001d020683变异位点与开花期的关联及多态性位点的连锁不平衡关系。B) 3种单倍型自交系的开花期比较。Hap1为0 + 0单倍型; Hap3为3 bp + 0单倍型; Hap4为3 bp + 2 bp单倍型。DTT为抽雄期; DTS为吐丝期; DTA为散粉期。*P < 0.05; **P < 0.01.
Fig. 4Association analysis of candidate gene
A) Association of variants at Zm00001d020683 with maize flowering time in the association panel, and linkage disequilibrium block among variants at Zm00001d020683. B) Comparison of flowering time between haplotypes. Hap1: 0 + 0 haplotype. Hap3: 3 bp + 0 haplotype. Hap4: 3 bp + 2 bp haplotype. DTT: days to tassel; DTA: days to anthesis; DTA: days to silking. * P < 0.05; **P < 0.01.
在172份自交系材料中, 这2个位点位于不同的LD block中, 共组成4种单倍型(haplotype), 即单倍型1 (Hap1) 0 + 0、单倍型2 (Hap2) 0 + 2 bp、单倍型3 (Hap3) 3 bp + 0和单倍型4 (Hap4) 3 bp + 2 bp, 各单倍型所占的比例分别为30.43%、1.45%、48.55%和19.57%。由于 Hap2频率低小于5%, 不纳入单倍型遗传效应评估。比较不同单倍型自交系的开花期差异发现, 相较于具有Hap3的自交系, 具有Hap4自交系的抽雄期、吐丝期和散粉期分别提前了0.9 d、1.2 d和1.7 d (P<0.01); 相较于具有Hap1的自交系, Hap4自交系的抽雄期、吐丝期和散粉期也有显著的提前(P<0.05)。由于Hap4在自交系群体中相对较低的频率和提早开花的遗传效应, 表明该等位基因在玉米开花期遗传改良中具有一定的应用价值。
3 讨论
IDD家族基因编码一类具有锌指蛋白的转录因子, 在植物生长发育过程中发挥着广泛作用[28,29,30,31,32]。目前的研究大多集中于模式植物拟南芥IDD基因对开花期的调控, 而挖掘玉米开花期相关的IDD家族基因具有重要的理论意义和应用价值。本研究鉴定并克隆了37个玉米IDD家族基因, 这些基因都有非常保守的结构域, 推测它们在功能上具相似性。我们发现玉米和拟南芥IDD家族基因的序列相似性很高, 推测玉米IDD家族基因可能具有拟南芥同源基因的相似功能。另外, 在已经克隆的开花期相关基因中, 拟南芥的AtIDD8和水稻中的OsID1/RID1/Ehd2以及玉米中的ID1都属于IDD家族基因[22-24,33-34]。我们通过关联分析发现, 有7个ZmIDD基因在多个环境下均能够检测到与开花期性状显著关联, 其中一个基因是前人已经报道的、控制玉米开花期的关键基因ID1[24], 因此我们可以推测其他6个基因也可能参与玉米开花期的调控。特别是Zm00001d020683基因, 其启动子区存在2个变异位点与开花期显著关联。而生物信息学预测这2个插入缺失位点可导致CGGTGCCCC 顺式元件的改变。许多研究发现, CGGTGCCCC顺式元件为ABI4(AP2转录因子的一种)转录因子直接结合位点, 顺式元件的变异则可能导致基因表达改变。因此, 一个可能的假设是, 自交系间Zm00001 d020683基因启动子的插入缺失变异会导致基因表达水平的变异, 进而导致玉米的开花期变异。调节Zm00001d020683基因的表达水平, 可能改变玉米开花期。这一假设有待进一步研究证实。通过多类群体和多种方法已鉴定到大量玉米开花期相关的遗传位点。Buckler等[21]利用NAM (Nested Association Mapping population)群体鉴定到36个DTS QTL、39个DTS QTL和29个ASI QTL以及数十个微效位点, 比较物理位置发现, 包含ID1在内的7个开花期相关基因均位于Buckler等所鉴定到的开花期相关QTL内或紧密连锁。李玉玲等[2]利用玉米自交系丹233和自交系N04构建了F2:3和BC2S2家系群体, 共鉴定到22个与开花期相关联的QTL, 分布在玉米10条染色体上。其中, QSS2-1、QBSS3-2、QSS7-1和QBAS10-1分别覆盖了本研究鉴定到的Zm00001d005566、Zm00001d044261、Zm 00001d020683和Zm00001d023558。Li等[35]利用NAM的BC2S3群体, 共鉴定到19个与开花期相关的QTL, 其中qDTA1-2、qDTA3-2和qDTA7-1分别覆盖本研究中的Zm00001d032922、Zm00001d044261和Zm00001d020683。Huang等(2012)利用NAM和IBM群体共鉴定到14个与开花期相关的QTL[11], 其中第1、第3、第7和第8染色体上的QTL分别覆盖Zm00001d032922、Zm00001d044261、Zm00001d 02 0683和Zm00001d011223。这些QTL定位结果间接支持了本研究所鉴定到的开花期相关ZmIDD基因。
IDD基因不仅调控植物开花期, 也参与植物生长发育的调控。在拟南芥中, Welch等[36]发现IDD转录因子成员JKD和MGP参与构成SCR-SHR复合体。JKD和MGP在根的干细胞中特异表达, 受SCR和SHR两个基因的调控。另外, SGR5在花序茎的内皮层表达, 参与调控拟南芥茎早期的重力反应[37]。因此, 玉米IDD家族基因也有可能调控除开花期外的其他植物生物过程。基因表达模式分析发现, 该家族基因表达具有明显的组织特异性, 暗示了各自发挥其功能的组织特异性。因此, 对IDD家族基因的深入研究也将为我们提供更多的植物个体发育和形态建成的调控信息。
4 结论
分析了玉米IDD家族基因的基本特性、遗传变异及其与玉米开花期的相关性。在B73基因组中鉴定了37个IDD家族基因, 分析了它们的结构特征和表达模式, 鉴定到包括ID1在内的7个ZmIDD基因及其自然变异与开花期的关联位点, 为这些基因的深入功能解析和育种应用奠定了基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1073/pnas.052125199URLPMID:11983901 [本文引用: 1]

There exists extraordinary morphological and genetic diversity among the maize landraces that have been developed by pre-Columbian cultivators. To explain this high level of diversity in maize, several authors have proposed that maize landraces were the products of multiple independent domestications from their wild relative (teosinte). We present phylogenetic analyses based on 264 individual plants, each genotyped at 99 microsatellites, that challenge the multiple-origins hypothesis. Instead, our results indicate that all maize arose from a single domestication in southern Mexico about 9,000 years ago. Our analyses also indicate that the oldest surviving maize types are those of the Mexican highlands with maize spreading from this region over the Americas along two major paths. Our phylogenetic work is consistent with a model based on the archaeological record suggesting that maize diversified in the highlands of Mexico before spreading to the lowlands. We also found only modest evidence for postdomestication gene flow from teosinte into maize.
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
DOI:10.1534/genetics.104.032375URLPMID:15611184 [本文引用: 1]
Genetic architecture of flowering time in maize was addressed by synthesizing a total of 313 quantitative trait loci (QTL) available for this trait. These were analyzed first with an overview statistic that highlighted regions of key importance and then with a meta-analysis method that yielded a synthetic genetic model with 62 consensus QTL. Six of these displayed a major effect. Meta-analysis led in this case to a twofold increase in the precision in QTL position estimation, when compared to the most precise initial QTL position within the corresponding region. The 62 consensus QTL were compared first to the positions of the few flowering-time candidate genes that have been mapped in maize. We then projected rice candidate genes onto the maize genome using a synteny conservation approach based on comparative mapping between the maize genetic map and japonica rice physical map. This yielded 19 associations between maize QTL and genes involved in flowering time in rice and in Arabidopsis. Results suggest that the combination of meta-analysis within a species of interest and synteny-based projections from a related model plant can be an efficient strategy for identifying new candidate genes for trait variation.
[本文引用: 1]
[本文引用: 1]
DOI:10.1104/pp.110.154211URL [本文引用: 1]
TERMINAL FLOWER1 (TFL1)-like genes are highly conserved in plants and are thought to function in the maintenance of meristem indeterminacy. Recently, we described six maize (Zea mays) TFL1-related genes, named ZEA CENTRORADIALIS1 (ZCN1) to ZCN6. To gain insight into their functions, we generated transgenic maize plants overexpressing their respective cDNAs driven by a constitutive promoter. Overall, ectopic expression of the maize TFL1-like genes produced similar phenotypes, including delayed flowering and altered inflorescence architecture. We observed an apparent relationship between the magnitude of the transgenic phenotypes and the degree of homology between the ZCN proteins. ZCN2, -4, and -5 form a monophylogenetic clade, and their overexpression produced the strongest phenotypes. Along with very late flowering, these transgenic plants produced a "bushy" tassel with increased lateral branching and spikelet density compared with nontransgenic siblings. On the other hand, ZCN1, -3, and -6 produced milder effects. Among them, ZCN1 showed moderate effects on flowering time and tassel morphology, whereas ZCN3 and ZCN6 did not change flowering time but still showed effects on tassel morphology. In situ hybridizations of tissue from nontransgenic plants revealed that the expression of all ZCN genes was associated with vascular bundles, but each gene had a specific spatial and temporal pattern. Expression of four ZCN genes localized to the protoxylem, whereas ZCN5 was expressed in the protophloem. Collectively, our findings suggest that ectopic expression of the TFL1-like genes in maize modifies flowering time and inflorescence architecture through maintenance of the indeterminacy of the vegetative and inflorescence meristems.
[本文引用: 2]
DOI:10.1534/genetics.109.106922URLPMID:19822732 [本文引用: 2]
Flowering time is a major adaptive trait in plants and an important selection criterion for crop species. In maize, however, little is known about its molecular basis. In this study, we report the fine mapping and characterization of a major quantitative trait locus located on maize chromosome 10, which regulates flowering time through photoperiod sensitivity. This study was performed in near-isogenic material derived from a cross between the day-neutral European flint inbred line FV286 and the tropical short-day inbred line FV331. Recombinant individuals were identified among a large segregating population and their progenies were scored for flowering time. Combined genotypic characterization led to delimit the QTL to an interval of 170 kb and highlighted an unbalanced recombination pattern. Two bacterial artificial chromosomes (BACs) covering the region were analyzed to identify putative candidate genes, and synteny with rice, sorghum, and brachypodium was investigated. A gene encoding a CCT domain protein homologous to the rice Ghd7 heading date regulator was identified, but its causative role was not demonstrated and deserves further analyses. Finally, an association study showed a strong level of linkage disequilibrium over the region and highlighted haplotypes that could provide useful information for the exploitation of genetic resources and marker-assisted selection in maize.
[本文引用: 2]
.
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
DOI:10.1105/tpc.109.068221URL [本文引用: 2]
KNOTTED1 (KN1)-like homeobox (KNOX) transcription factors are involved in the establishment and maintenance of plant meristems; however, few direct targets of KNOX proteins have been recognized. Using a combination of double mutant analysis and biochemistry, we found that in maize (Zea mays), KN1 negatively modulates the accumulation of gibberellin (GA) through the control of ga2ox1, which codes for an enzyme that inactivates GA. The ga2ox1 mRNA level is elevated in immature leaves of dominant KNOX mutants and downregulated in reproductive meristems of the null allele kn1-e1. KN1 binds in vivo to an intron of ga2ox1 through a cis-regulatory element containing two TGAC motifs. VP16-KN1 activates transcription in planta from a chimeric promoter containing this binding site. The domains of expression of kn1 and ga2ox1 mRNAs overlap at the base of the shoot apical meristem and the base of newly initiated leaves, suggesting that KN1-mediated activation of ga2ox1 maintains a boundary between meristem cell identity and rapidly elongating cells of the shoot. The KN1 binding site is conserved in ga2ox1 genes of different grasses, suggesting that the local regulation of bioactive GA levels through KNOX proteins is a common theme in grasses.
DOI:10.1104/pp.106.088815URLPMID:17071646 [本文引用: 1]
Separation of the life cycle of flowering plants into two distinct growth phases, vegetative and reproductive, is marked by the floral transition. The initial floral inductive signals are perceived in the leaves and transmitted to the shoot apex, where the vegetative shoot apical meristem is restructured into a reproductive meristem. In this study, we report cloning and characterization of the maize (Zea mays) flowering time gene delayed flowering1 (dlf1). Loss of dlf1 function results in late flowering, indicating dlf1 is required for timely promotion of the floral transition. dlf1 encodes a protein with a basic leucine zipper domain belonging to an evolutionarily conserved family. Three-dimensional protein modeling of a missense mutation within the basic domain suggests DLF1 protein functions through DNA binding. The spatial and temporal expression pattern of dlf1 indicates a threshold level of dlf1 is required in the shoot apex for proper timing of the floral transition. Double mutant analysis of dlf1 and indeterminate1 (id1), another late flowering mutation, places dlf1 downstream of id1 function and suggests dlf1 mediates floral inductive signals transmitted from leaves to the shoot apex. This study establishes an emergent framework for the genetic control of floral induction in maize and highlights the conserved topology of the floral transition network in flowering plants.
DOI:10.1073/pnas.0704145104URLPMID:17595297 [本文引用: 1]
Flowering time is a fundamental trait of maize adaptation to different agricultural environments. Although a large body of information is available on the map position of quantitative trait loci for flowering time, little is known about the molecular basis of quantitative trait loci. Through positional cloning and association mapping, we resolved the major flowering-time quantitative trait locus, Vegetative to generative transition 1 (Vgt1), to an 2-kb noncoding region positioned 70 kb upstream of an Ap2-like transcription factor that we have shown to be involved in flowering-time control. Vgt1 functions as a cis-acting regulatory element as indicated by the correlation of the Vgt1 alleles with the transcript expression levels of the downstream gene. Additionally, within Vgt1, we identified evolutionarily conserved noncoding sequences across the maize-sorghum-rice lineages. Our results support the notion that changes in distant cis-acting regulatory regions are a key component of plant genetic adaptation throughout breeding and evolution.
DOI:10.1534/g3.114.010686URLPMID:4025479 [本文引用: 1]
One of the major quantitative trait loci for flowering time in maize, the Vegetative to generative transition 1 (Vgt1) locus, corresponds to an upstream (70 kb) noncoding regulatory element of ZmRap2.7, a repressor of flowering. At Vgt1, a miniature transposon (MITE) insertion into a conserved noncoding sequence was previously found to be highly associated with early flowering in independent studies. Because cytosine methylation is known to be associated with transposons and to influence gene expression, we aimed to investigate how DNA methylation patterns in wild-type and mutant Vgt1 correlate with ZmRap2.7 expression. The methylation state at Vgt1 was assayed in leaf samples of maize inbred and F1 hybrid samples, and at the syntenic region in sorghum. The Vgt1-linked conserved noncoding sequence was very scarcely methylated both in maize and sorghum. However, in the early maize Vgt1 allele, the region immediately flanking the highly methylated MITE insertion was significantly more methylated and showed features of methylation spreading. Allele-specific expression assays revealed that the presence of the MITE and its heavy methylation appear to be linked to altered ZmRap2.7 transcription. Although not providing proof of causative connection, our results associate transposon-linked differential methylation with allelic state and gene expression at a major flowering time quantitative trait locus in maize.
[本文引用: 1]
DOI:10.1104/pp.16.00285URLPMID:27457125 [本文引用: 1]
Abstract Flowering time (FTi) control is well examined in the long-day plant Arabidopsis (Arabidopsis thaliana), and increasing knowledge is available for the short-day plant rice (Oryza sativa). In contrast, little is known in the day-neutral and agronomically important crop plant maize (Zea mays). To learn more about FTi and to identify novel regulators in this species, we first compared the time points of floral transition of almost 30 maize inbred lines and show that tropical lines exhibit a delay in flowering transition of more than 3 weeks under long-day conditions compared with European flint lines adapted to temperate climate zones. We further analyzed the leaf transcriptomes of four lines that exhibit strong differences in flowering transition to identify new key players of the flowering control network in maize. We found strong differences among regulated genes between these lines and thus assume that the regulation of FTi is very complex in maize. Especially genes encoding MADS box transcriptional regulators are up-regulated in leaves during the meristem transition. ZmMADS1 was selected for functional studies. We demonstrate that it represents a functional ortholog of the central FTi integrator SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) of Arabidopsis. RNA interference-mediated down-regulation of ZmMADS1 resulted in a delay of FTi in maize, while strong overexpression caused an early-flowering phenotype, indicating its role as a flowering activator. Taken together, we report that ZmMADS1 represents a positive FTi regulator that shares an evolutionarily conserved function with SOC1 and may now serve as an ideal stating point to study the integration and variation of FTi pathways also in maize. 2016 American Society of Plant Biologists. All rights reserved.
.
[本文引用: 1]
DOI:10.1126/science.1174276URLPMID:19661422 [本文引用: 2]
Flowering time is a complex trait that controls adaptation of plants to their local environment in the outcrossing species Zea mays (maize). We dissected variation for flowering time with a set of 5000 recombinant inbred lines (maize Nested Association Mapping population, NAM). Nearly a million plants were assayed in eight environments but showed no evidence for any single large-effect quantita...
DOI:10.1111/j.1365-313X.2010.04432.xURLPMID:202020202020202020202020202020202020 [本文引用: 2]
There has been a long-standing interest in the role played by sugars in flowering. Of particular interest is how sugar-related signals are integrated into flowering genetic pathways. Here, we demonstrate that the INDETERMINATE DOMAIN transcription factor AtIDD8 regulates photoperiodic flowering by modulating sugar transport and metabolism. We found that whereas AtIDD8-deficient idd8 mutants exhibit delayed flowering under long days, AtIDD8-overexpressing plants (35S:IDD8) show early flowering. In addition, the sucrose synthase genes SUS1 and SUS4 were upregulated in 35S:IDD8 plants but downregulated in idd8 mutants, in which endogenous sugar levels were altered. AtIDD8 activates the SUS4 gene by binding directly to its promoter, resulting in promoted flowering in SUS4-overexpressing plants. SUS4 expression also responds to photoperiodic signals. Notably, the AtIDD8 gene is suppressed by sugar deprivation. Therefore, we conclude that AtIDD8 regulation of sugar transport and metabolism is linked to photoperiodic flowering.
DOI:10.1073/pnas.0806019105URL [本文引用: 1]
Transition from the vegetative phase to reproductive phase is a crucial process in the life cycle of higher plants. Although the molecular mechanisms of flowering regulation have been extensively characterized in a number of plant species, little is known regarding how the transition process initiates. Here, we show that the Rice Indeterminate 1 (RID1) gene acts as the master switch for the transition from the vegetative to reproductive phase. RID1 encodes a Cys-2/His-2-type zinc finger transcription factor that does not have an ortholog in Arabidopsis spp. A RID1 knockout (rid1), mutated by T-DNA insertion, never headed after growing for >500 days under a range of growth conditions and is thus referred to as a never-flowering phenotype. This mutation-suppressed expression of the genes is known to be involved in flowering regulation, especially in the Ehd1/Hd3a pathway and a series of RFT homologs. RID1 seems to be independent of the circadian clock. A model was proposed to place RID1 in the molecular pathways of flowering regulation in rice, for which there are two indispensable elements. In the first, RID1 is controlling the phase transition and initiation of floral induction. In the other, the Hd3a/RFL1/FTL complex acts as the immediate inducer of flowering. Loss of function in either element would cause never-flowering. Once the phase transition is induced with the activation of RID1, flowering signal is transduced and regulated through the various pathways and eventually integrated with FT-like proteins to induce flowering.
DOI:10.1093/jxb/erl206URLPMID:17307745 [本文引用: 3]
Abstract The INDETERMINATE1 gene, ID1, encodes a putative transcription factor that plays an important role in regulating the transition to flowering in maize. Mutant id1 plants have a prolonged vegetative growth phase and fail to make normal flowers. The ID1 gene, which encodes a nuclear-localized zinc finger protein, is expressed exclusively in immature leaves, suggesting that ID1 regulates a leaf-derived floral inductive signal. It is shown by western analysis with anti-ID1-specific antibody that ID1 co-localizes with ID1 mRNA in developing, immature leaves and, similarly, is absent in mature, photosynthetically active leaf blades, as well as the shoot apical meristem. Immunolocalization with anti-ID1 antibody shows that ID1 protein is detected in the nuclei of all cell types in immature leaves. Examination of plants grown in different day/night cycles revealed that ID1 gene expression and protein levels are largely unaffected by variations in light and dark, and that mRNA and protein levels do not follow a circadian pattern. The absence of ID1 expression in greening leaf tips coincides with the sink-to-source transition of developing leaves. It was found that ID1 levels are down-regulated in mature albino leaves similarly as in normal green leaves, suggesting that ID1 activity is controlled developmentally and is not affected by the sink/source status of the leaf or the inability of a mature leaf to engage in photosynthesis. The finding that ID1 expression is developmentally regulated and is unperturbed by external stimuli such as photoperiod supports the supposition that ID1 acts through the autonomous floral inductive pathway in maize.
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/ng.2484URLPMID:23242369 [本文引用: 1]
Maize kernel oil is a valuable source of nutrition. Here we extensively examine the genetic architecture of maize oil biosynthesis in a genome-wide association study using 1.03 million SNPs characterized in 368 maize inbred lines, including 'high-oil' lines. We identified 74 loci significantly associated with kernel oil concentration and fatty acid composition (P < 1.8 x 10(-6)), which we subsequently examined using expression quantitative trait loci (QTL) mapping, linkage mapping and coexpression analysis. More than half of the identified loci localized in mapped QTL intervals, and one-third of the candidate genes were annotated as enzymes in the oil metabolic pathway. The 26 loci associated with oil concentration could explain up to 83% of the phenotypic variation using a simple additive model. Our results provide insights into the genetic basis of oil biosynthesis in maize kernels and may facilitate marker-based breeding for oil quantity and quality.
[本文引用: 1]
URLPMID:2548561 [本文引用: 2]
[本文引用: 1]
DOI:10.1007/s000180050186URLPMID:9676577 [本文引用: 1]
Abstract Several classes of zinc-finger motifs are present in transcription factors and function as parts of DNA-binding and protein-protein interaction domains. Most of the known classes of zinc-finger motifs earlier identified in other eucaryotes have also been found in a number of (putative) transcription factors in plants. In addition, some novel classes of zinc fingers have been identified in plants. Many of these proteins have been implicated in the regulation of important biological processes that are unique to plants, such as flower development, light-regulated morphogenesis and pathogen responses. Thus, plants seem to have adopted pre-existing prototype zinc-finger motifs as well as generated new zinc-finger domains to adapt them to various regulatory processes. Detailed analyses of TFIIIA-type plant zinc-finger proteins revealed unique manners of interactions with target DNA sequences, i.e. recognition of spacing, suggesting that plants have developed unique mechanisms even when proto-type functional motifs were adopted. In this review, attempts were made to summarize the current knowledge of (putative) zinc-finger transcription factors according to a structure-based classification, in view of their involvement in specific regulatory processes and interaction with target DNA sequences.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1104/pp.108.125542URLPMID:18790997 [本文引用: 1]
Recent research into the flowering of rice (Oryza sativa) has revealed both unique and conserved genetic pathways in the photoperiodic control of flowering compared with those in Arabidopsis (Arabidopsis thaliana). We discovered an early heading date2 (ehd2) mutant that shows extremely late flowering under both short- and long-day conditions in line with a background deficient in Heading date1 (Hd1), a rice CONSTANS ortholog that belongs to the conserved pathway. This phenotype in the ehd2 mutants suggests that Ehd2 is pivotal for the floral transition in rice. Map-based cloning revealed that Ehd1 encodes a putative transcription factor with zinc finger motifs orthologous to the INDETERMINATE1 (ID1) gene, which promotes flowering in maize (Zea mays). Ehd1 mRNA in rice tissues accumulated most abundantly in developing leaves, but was present at very low levels around the shoot apex and in roots, patterns that are similar to those of ID1. To assign the position of Ehd2 within the flowering pathway of rice, we compared transcript levels of previously isolated flowering-time genes, such as Ehd1, a member of the unique pathway, Hd3a, and Rice FT-like1 (RFT1; rice florigens), between the wild-type plants and the ehd2 mutants. Severely reduced expression of these genes in ehdl under both short- and long-day conditions suggests that Ehd2 acts as a flowering promoter mainly by up-regulating Ehd1 and by up-regulating the downstream Hd3a and RFT1 genes in the unique genetic network of photoperiodic flowering in rice.
DOI:10.1111/nph.13765URLPMID:26593156 [本文引用: 1]
The number of leaves and their distributions on plants are critical factors determining plant architecture in maize (Zea mays), and leaf number is frequently used as a measure of flowering time, a trait that is key to local environmental adaptation.Here, using a large set of 866 maize‐teosinteBC2S3recombinant inbred lines genotyped by using 1902838 single nucleotide polymorphism markers, we conducted a comprehensive genetic dissection to assess the genetic architecture of leaf number and its genetic relationship to flowering time.We demonstrated that the two components of total leaf number, the number of leaves above (LA) and below (LB) the primary ear, were under relatively independent genetic control and might be subject to differential directional selection during maize domestication and improvement. Furthermore, we revealed that flowering time and leaf number are commonly regulated at a moderate level. The pleiotropy of the genesZCN8,dlf1andZmCCTon leaf number and flowering time were validated by near‐isogenic line analysis. Through fine mapping,qLA1‐1, a major‐effect locus that specifically affectsLA, was delimited to a region with severe recombination suppression derived from teosinte.This study provides important insights into the genetic basis of traits affecting plant architecture and adaptation. The genetic independence of LA from LB enables the optimization of leaf number for ideal plant architecture breeding in maize. The number of leaves and their distributions on plants are critical factors determining plant architecture in maize (Zea mays), and leaf number is frequently used as a measure of flowering time, a trait that is key to local environmental adaptation. Here, using a large set of 866 maize‐teosinteBC2S3recombinant inbred lines genotyped by using 1902838 single nucleotide polymorphism markers, we conducted a comprehensive genetic dissection to assess the genetic architecture of leaf number and its genetic relationship to flowering time. We demonstrated that the two components of total leaf number, the number of leaves above (LA) and below (LB) the primary ear, were under relatively independent genetic control and might be subject to differential directional selection during maize domestication and improvement. Furthermore, we revealed that flowering time and leaf number are commonly regulated at a moderate level. The pleiotropy of the genesZCN8,dlf1andZmCCTon leaf number and flowering time were validated by near‐isogenic line analysis. Through fine mapping,qLA1‐1, a major‐effect locus that specifically affectsLA, was delimited to a region with severe recombination suppression derived from teosinte. This study provides important insights into the genetic basis of traits affecting plant architecture and adaptation. The genetic independence of LA from LB enables the optimization of leaf number for ideal plant architecture breeding in maize.
[本文引用: 1]
DOI:10.1111/nph.13602URLPMID:26256266 [本文引用: 1]
Summary In higher plants, gravitropism proceeds through three sequential steps in the responding organs: perception of gravity signals, signal transduction and asymmetric cell elongation. Light and temperature also influence the gravitropic orientation of plant organs. A series of Arabidopsis shoot gravitropism ( sgr ) mutants has been shown to exhibit disturbed shoot gravitropism. SGR5 is functionally distinct from other SGR members in that it mediates the early events of gravitropic responses in inflorescence stems. Here, we demonstrated that SGR5 alternative splicing produces two protein variants (SGR5α and SGR5β) in modulating the gravitropic response of inflorescence stems at high temperatures. SGR5β inhibits SGR5α function by forming non-DNA-binding heterodimers. Transgenic plants overexpressing SGR5 β (35S: SGR5 β) exhibit reduced gravitropic growth of inflorescence stems, as observed in the SGR5 -deficient sgr5-5 mutant. Interestingly, SGR5 alternative splicing is accelerated at high temperatures, resulting in the high-level accumulation of SGR5 β transcripts. When plants were exposed to high temperatures, whereas gravitropic curvature was reduced in Col-0 inflorescence stems, it was uninfluenced in the inflorescence stems of 35S: SGR5 β transgenic plants and sgr5-5 mutant. We propose that the thermoresponsive alternative splicing of SGR5 provides an adaptation strategy by which plants protect the shoots from hot air under high temperature stress in natural habitats.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}