 ,*河南农业大学农学院 / 河南粮食作物协同创新中心 / 小麦玉米作物学国家重点实验室, 河南郑州 450046
,*河南农业大学农学院 / 河南粮食作物协同创新中心 / 小麦玉米作物学国家重点实验室, 河南郑州 450046Transcriptome Analysis of Premature Senescence Induced by Pollination-prevention in Maize
WU Lian-Cheng**, LI Pei**, TIAN Lei**, WANG Shun-Xi**, LI Ming-Na**, WANG Yu-Yu**, WANG Sai**, CHEN Yan-Hui,*College of Agronomy, Henan Agricultural University / Collaborative Innovation Center of Henan Grain Crops / National Key Laboratory of Wheat and Maize Crop Science, Zhengzhou 450046, Henan, China通讯作者:
第一联系人:
收稿日期:2018-01-2接受日期:2018-07-20网络出版日期:2018-07-30
| 基金资助: |
Received:2018-01-2Accepted:2018-07-20Online:2018-07-30
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (2319KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
吴连成, 李沛, 田磊, 王顺喜, 李明娜, 王宇宇, 王赛, 陈彦惠. 阻断授粉诱导玉米叶片提前衰老的转录组分析[J]. 作物学报, 2018, 44(11): 1661-1672. doi:10.3724/SP.J.1006.2018.01661
WU Lian-Cheng, LI Pei, TIAN Lei, WANG Shun-Xi, LI Ming-Na, WANG Yu-Yu, WANG Sai, CHEN Yan-Hui.
玉米(Zea mays L.)是我国重要的粮食、饲料和经济作物, 在耕地资源日益减少和人口数量不断增加的双重压力下, 继续提高玉米产量依然是当前和今后一段时间迫切需要解决的实际问题。衰老发生在玉米生长发育的最后阶段, 叶片衰老会导致植物光合活性逐步丧失, 影响玉米产量。因此, 适当延迟玉米叶片衰老可提高玉米植株的碳同化作用, 进而提高玉米产量[1]。在玉米籽粒成熟后期, 叶片及时衰老又可以促进营养物质向籽粒库器官运输, 提高营养物质利用率, 同时还将有利于玉米籽粒快速脱水, 提升玉米品质。近年来, 已报道拟南芥等物种中关于衰老的相关研究, 但在玉米中报道相对较少[2,3,4,5,6,7,8,9]。Ceppi等[10]研究发现, 玉米自交系B73在阻断授粉诱导条件下会产生早衰(premature senescence)现象。Zhang等[11]则对玉米自交系齐319叶片自然衰老分子机制探讨认为, 玉米与拟南芥的叶片衰老基本相似, 但玉米叶片自然衰老与诱导衰老在基因调控网络上存在差异。虽然拟南芥等物种中关于衰老相关基因(senescence-associated genes, SAGs)的报道较多[4,6,12-20], 但玉米中的SAGs[21]仍有待进一步挖掘, 其机理尚不完善。因此, 利用一些在阻断授粉诱导条件下表现出衰老明显提前的遗传材料, 从转录组水平研究提前衰老的分子机制, 可以为玉米分子育种提供重要的理论依据。豫816是本课题组选育出的优良玉米自交系。阻断授粉诱导条件下, 该自交系表现出明显提前衰老的现象, 是研究阻断授粉诱导玉米早衰的良好材料。设置授粉(pollination, POL)和非授粉(non-pollination, Non-POL) 2个处理, 对豫816表型性状、叶绿素含量及基因差异表达情况进行研究, 鉴定不同时期、处理条件下的差异表达基因(differentially expressed genes, DEG), 探究阻断授粉诱导玉米提前衰老的分子调控机制, 为玉米新品种选育提供重要理论依据。
1 材料与方法
1.1 植物材料与处理方法
2015年6月, 将豫816种植在河南农业大学科教园区试验田(郑州), 4 m行长, 行距0.6 m, 株距0.3 m。水肥管理及病虫草害防治按照大田进行。在玉米雌穗吐丝前, 选取生长一致的植株挂牌编号, 并将所有试验材料的雌穗套袋, 观察记录各试验单株的吐丝期。玉米吐丝后6 d, 将需要授粉的单株进行统一饱和授粉; 非授粉单株所有雌穗均套袋, 确保不接受外来花粉。在吐丝后10 d (days after silking, DAS)和24 d, 利用SPAD-502 Chlorophyll Meter型叶绿素含量测定仪测定授粉和非授粉植株穗位叶中部的叶绿素相对含量(SPAD), 利用DPS7.0软件检验测定结果差异显著性。在选取RNA-seq分析样本的前一天, 将所用工具进行高温高压灭菌处理, 烘箱中烘干备用, 在测定当天上午9:00取样, 部位与测定叶绿素的部位一致。每5个单株组成一个生物学重复, 每个材料3个生物学重复。以锡箔纸包裹样品, 标记后放入液氮速冻, 保存在-80℃超低温冰箱内, 用于后续试验。
1.2 RNA-seq文库的构建与测序
取每个时期3次生物学重复, 每个生物学重复由5株混合构成。采用TRIzol Reagent (Invitrogen, 美国)试剂, 参照说明书操作规程提取叶片总RNA。质检合格后送华大基因公司构建RNA-seq文库, 并在Illumina HiSeq2000平台上测序。1.3 转录组数据分析
为保证数据质量, 在分析前过滤原始数据以减少数据噪音。过滤后保留下来的clean reads与玉米B73参考基因组v3比对, 获得的Mapped Data用于下一步分析。为鉴定不同生物学重复数据间的关系, 对不同生物学重复数据进行主成分分析(principal component analysis, PCA)。根据测序得到的基因表达水平的RPKM (reads per kilobase per million mapped reads)值[22], 通过Cufflinks[23]软件进行基因差异表达分析。差异显著基因筛选标准为|log2 (Fold Change)|≥1且P-value<0.05。然后利用MapMan[24]功能分类系统对DEGs进行Pathway富集分析。1.4 RT-qPCR分析
利用HiScript Q RT SuperMix for qPCR (Vazyme, 南京)试剂, 参照说明书操作流程反转录合成cDNA。荧光定量PCR反应中采用SYBR Green I Master Mix (BioTeke, 北京)试剂, 仪器为LightCycler 480 Instrument II (Roche, 瑞士)。以18SrRNA gene (F: 5°-CCTGCGGCTTAATTGACTC-3°和R: 5°-GTTAGC AGGCTGAGGTCTCG-3°)为内参基因, 采用2-ΔΔCt方法[25]计算基因的相对表达量, 每个样品重复3次。RT-qPCR用到的引物均通过Primer-BLAST (https:// www.ncbi.nlm.nih.gov/tools/primer-blast/)在线工具设计, 引物序列见表1。Table 1
表1
表1RT-qPCR分析中用到的引物
Table 1
| 基因编号 Gene ID | 正向引物 Forward primer (5°-3°) | 反向引物 Reverse primer (5°-3°) |
|---|---|---|
| GRMZM2G033493 | AATGCAACGGAGCCAACAAT | TTTGTGACAGCTTCGTTCGG |
| GRMZM2G035243 | CCAGCCATCCGTCTATCCAT | TCTAATCTTGCAGCGCGAAC |
| GRMZM2G109070 | GTGTACTACGAGAGGTCCGG | AAAGCCCCAAAACGCATCTT |
| GRMZM2G117198 | GGACACATGTTCGGGTATGC | ATTGGTCACTGTCTCGTCGT |
| GRMZM2G339563 | ATCGTTCTTCAAGGCCAGGA | CATCTCGCGCTTTGAAAGGA |
| GRMZM5G801627 | TGTGCAGGCGACCATGTATA | CATCAACTCAAGACGCCGTT |
| GRMZM2G088053 | AGAGTGAGGCCCAAGATGAC | CTCAGCCTCTCCATCCTCAG |
| GRMZM2G109627 | CGAGGATAACTGCAACGGTG | GTCGTGCAGCTGATGAGAAG |
| GRMZM2G062129 | AGCAAGTCTGATGGCTCACT | AGCCAACCCTTGACTAGCAT |
| GRMZM2G064962 | CGAAACACCACGATCCAAGG | ATGTAGACTGCCTCCCACAC |
新窗口打开|下载CSV
2 结果与分析
2.1 叶片衰老表型及叶绿素含量变化
为了调查豫816在正常授粉和阻断授粉条件下植株间的表型差异, 分别在吐丝后6 d、10 d、24 d和27 d观察叶片衰老情况。对比发现, 吐丝后6~24 d, 授粉植株叶片呈现绿色且表型基本一致, 但在吐丝后24 d非授粉植株叶片大面积变黄。至吐丝后27 d, 非授粉植株叶片全部变黄并逐渐枯萎, 而授粉植株叶片仍保持绿色, 且生长状况良好(图1)。此外, 就非授粉植株的单个叶片衰老情况而言, 起初叶片主叶脉基部轻微发红, 随后叶脉红色程度加深并向叶尖方向扩展; 与此同时, 叶片也开始失绿变黄, 最终干枯合拢。就非授粉植株的穗位叶及其以上叶片而言, 顶部叶衰老速度较快, 穗位叶完全变黄历时最长; 从穗位叶主叶脉开始泛红至整株叶片变黄历时8~10 d。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1授粉和非授粉条件下豫816表型变化
图中DAS表示吐丝后天数; POL表示授粉; Non-POL表示非授粉。
Fig. 1Phenotypic changes of Yu 816 under POL and Non-POL conditions
DAS: days after silking; POL: pollination; Non-POL: non-pollination.
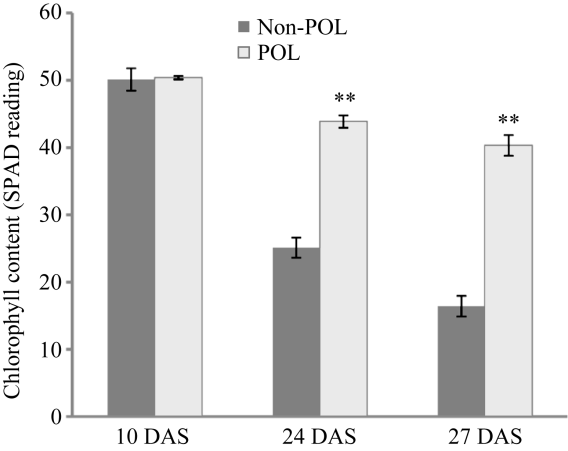
叶绿素降解是叶片衰老进程中最早出现的明显标志之一, 因此, 可通过测定叶片叶绿素相对含量(即SPAD值)来间接衡量叶片衰老程度。随着植株衰老进程, 授粉和非授粉植株的穗位叶均表现出预期的叶绿素含量下降的现象(图2)。授粉植株与非授粉植株间穗位叶的SPAD值差异在吐丝后10 d不显著, 而在吐丝后24 d和27 d均达到极显著水平。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2非授粉和授粉植株穗位叶叶绿素含量变化
**表示非授粉(Non-POL)和授粉(POL)植株穗位叶叶绿素含量之间差异极显著(P < 0.01)。
Fig. 2Chlorophyll content changes of ear leaves in non-pollination(Non-POL) and pollination (POL) plants
** indicates extremely significant difference at P<0.01. DAS: days after silking.
2.2 玉米叶片转录组测序质量及reads分析
为了分析阻断授粉诱导衰老所导致的转录水平变化, 分别对授粉处理组和非授粉处理组吐丝后10 d和24 d的样品构建4个文库, 并在Illumina HiSeq2000平台上测序, 每个样品3次生物学重复。去除low quality reads后, 各样品均有超过91%的clean reads。将这些clean reads与玉米B73基因组参考序列v3比对, 各样品均有超过80%的unique reads进入后续分析(表2)。Table 2
表2
表2样品reads数分布情况
Table 2
| 样本 Sample | 原始数据 Raw data count | 过滤后的 reads数量 Count after filter | 过滤后 reads占比 Reads keep rate (%) | 比对上的reads数量Mapped reads | 比对上的 reads占比 Mapped reads rate (%) | 在参考序列上有 唯一比对位置的 reads数量 Unique mapped reads | 有唯一比对位置的reads占比 Unique mapped reads (%) |
|---|---|---|---|---|---|---|---|
| 10DASN_r1 | 7442024 | 7291875 | 97.98 | 6731287 | 92.31 | 5480898 | 81.42 |
| 10DASN_r2 | 9376032 | 9181843 | 97.93 | 8479906 | 92.36 | 7106564 | 83.80 |
| 10DASY_r1 | 7941949 | 7777455 | 97.93 | 7134198 | 91.73 | 5833995 | 81.78 |
| 10DASY_r2 | 8834814 | 8644115 | 97.84 | 7960889 | 92.10 | 6607957 | 83.01 |
| 24DASN_r1 | 9382351 | 9163732 | 97.67 | 8367413 | 91.31 | 7079381 | 84.61 |
| 24DASN_r2 | 8201533 | 8054678 | 98.21 | 7433771 | 92.29 | 6190232 | 83.27 |
| 24DASY_r1 | 8599542 | 8432296 | 98.06 | 7728019 | 91.65 | 6398312 | 82.79 |
| 24DASY_r2 | 9545411 | 9339804 | 97.85 | 8557539 | 91.62 | 7181523 | 83.92 |
| 24DASY_r3 | 8206919 | 8027609 | 97.82 | 7381500 | 91.95 | 6095486 | 82.58 |
新窗口打开|下载CSV
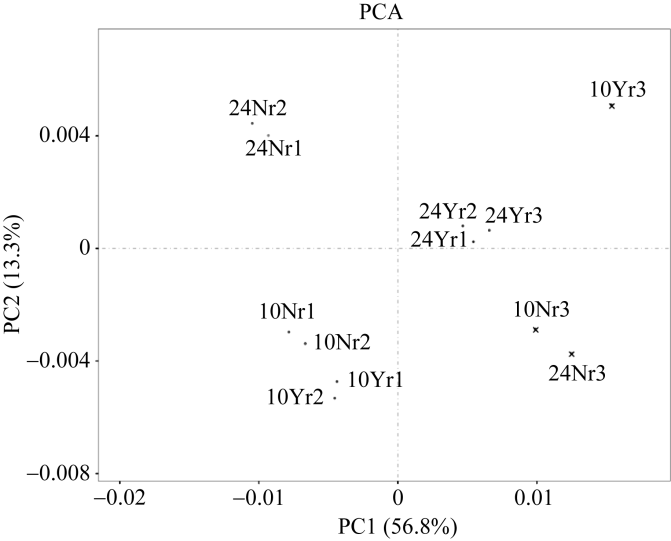
为鉴定不同生物学重复间数据的关系, 对不同生物学重复数据进行主成分分析(图3)。其中, 授粉后10 d的第3个生物学重复, 非授粉10 d和24 d的第3个重复(即图3中的10Yr3、10Nr3和24Nr3)偏离其正常范围, 故将其剔除; 其余生物学重复分布比较集中, 表明重复间及测序结果间有较好的重现性。同时, 授粉和非授粉植株在吐丝后10 d分布比较集中, 说明其转录模式相似, 而吐丝后24 d转录模式产生差异。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3生物学重复间主成分分析
表中N表示非授粉处理; Y表示授粉处理; r表示生物学重复。
Fig. 3Principal component analysis (PCA) for all samples including three replicates
N: non-pollination treatment; Y: pollination treatment; r: biological replicates.
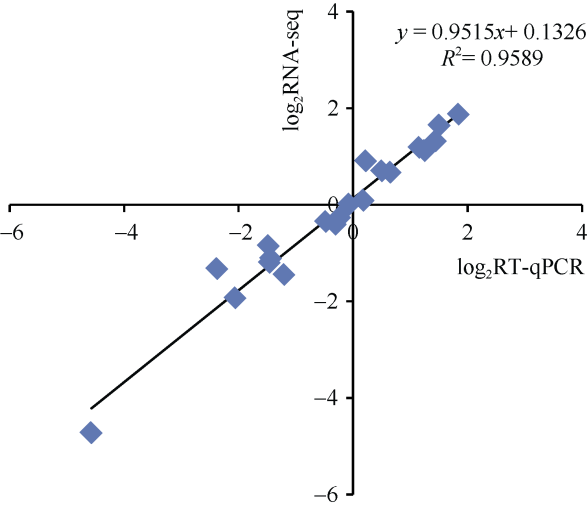
2.3 实时荧光定量PCR
随机选取10个差异表达基因, 利用实时荧光定量PCR检测10 DAS和24 DAS两时期的基因表达情况。如图4所示, 两平台数据间的相关系数达0.96, 说明RNA-seq数据可靠, 可进行相关的转录组分析。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4差异表达基因RT-qPCR验证
Fig. 4Validation of DEGs by RT-qPCR
2.4 差异表达基因的分析
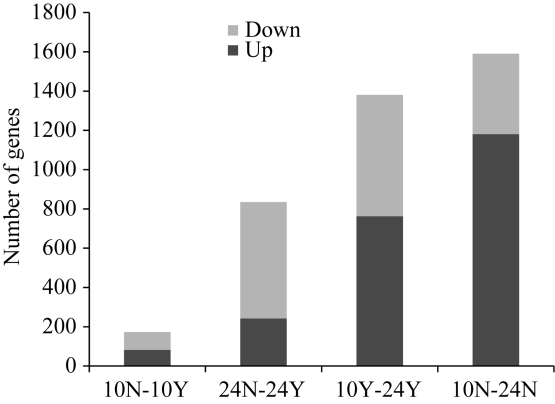
与非授粉植株相比, 吐丝后10 d, 173个基因在授粉植株中差异表达, 其中47.40% (82/173)上调表达, 52.60% (91/173)下调表达; 吐丝后24 d, 835个基因在授粉植株中差异表达, 其中28.98% (242/835)上调表达, 71.02% (593/835)下调表达, 下调表达基因数量是上调表达基因数量的2.45倍。在正常授粉条件下, 吐丝后24 d与吐丝后10 d相比, 有1381个基因差异表达, 其中55.25% (763/1381)上调表达, 44.75% (618/1381)下调表达; 在非授粉条件下, 吐丝后24 d与吐丝后10 d相比, 有1591个基因差异表达, 其中74.23% (1181/1591)上调表达, 25.77% (410/1591)下调表达, 上调表达基因数量是下调基因数量的2.88倍(图5)。图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5差异表达基因的上调下调数目
“10N-10Y”代表吐丝后10 d授粉处理组与非授粉处理组, “24N-24Y”代表吐丝后24 d授粉处理组与非授粉处理组, “10Y-24Y”代表授粉处理组吐丝后24 d与授粉植株吐丝后10 d, “10N-24N”代表非授粉处理组吐丝后24 d与非授粉处理吐丝后10 d。
Fig. 5Number of up- and down-regulated differentially expressed genes (DEGs)
“10N-10Y” represents pollination treatment group at 10 DAS vs non-pollination treatment group at 10 DAS; “24N-24Y” represents pollination treatment group at 24 DAS vs non-pollination treatment group at 24 DAS; “10Y-24Y” means pollination treatment group at 24 DAS vs pollination treatment group at 10 DAS; “10N-24N” means non-pollination treatment group at 24 DAS vs non-pollination treatment group at 10 DAS.
借助Venn图对不同处理、时期的差异表达基因进行分析(图6), 其中吐丝后10 d, 授粉处理组与同期非授粉处理组有22个(12.7%)特有的差异表达基因; 而吐丝后24 d, 授粉处理组与同期非授粉处理间有153个(18.3%)特有的差异表达基因, 结合豫816植株表型变化情况, 表明在吐丝后10 d, 豫816阻断授粉的处理效应开始显现, 但此时仅从叶片表型上尚不能辨别出差异, 至吐丝后24 d, 特有的差异表达基因数量明显增加, 叶片颜色明显变黄, 阻断授粉处理效应增强。授粉植株在吐丝后10 d与24 d间有871个(63.0%)特有的差异表达基因; 非授粉植株在吐丝后10 d与24 d间有579个(36.4%)特有的差异表达基因。
图6
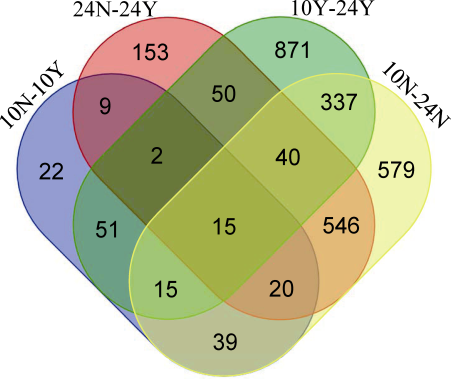 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6差异表达基因Venn图
维恩图中非重叠区的数字表示在一个成对比较中特有的差异表达基因数目, 重叠区的数字表示在不同成对比较中共有的差异表达基因数目。
Fig. 6Venn diagram of DEGs identified in POL and Non-POL plants
The number in non-overlapping section indicates that of special DEGs in one pairwise comparison; the number in overlapping section of DEGs means commonly expressed in different pairwise comparisons.
2.5 差异表达基因GO功能富集分析
GO功能富集分析(图7)表明, 吐丝后10 d, 授粉处理与非授粉处理间的差异表达基因主要富集在刺激响应(response to stimulus)和代谢进程(metabolic process); 吐丝后24 d, 授粉处理与非授粉处理间的差异表达基因显著富集在光合作用(photosynthesis)过程。而授粉植株在吐丝后10 d和24 d间的差异表达基因显著富集在刺激响应(response to stimulus)、胁迫响应(response to stress)、激素刺激响应(response to hormone stimulus)和次级代谢(secondary metabolic process)等生物过程; 非授粉植株在吐丝后10 d与24 d间的差异表达基因显著富集在生物过程调控(regulation of biological process)和细胞过程调控(regulation of cellular process)。图7
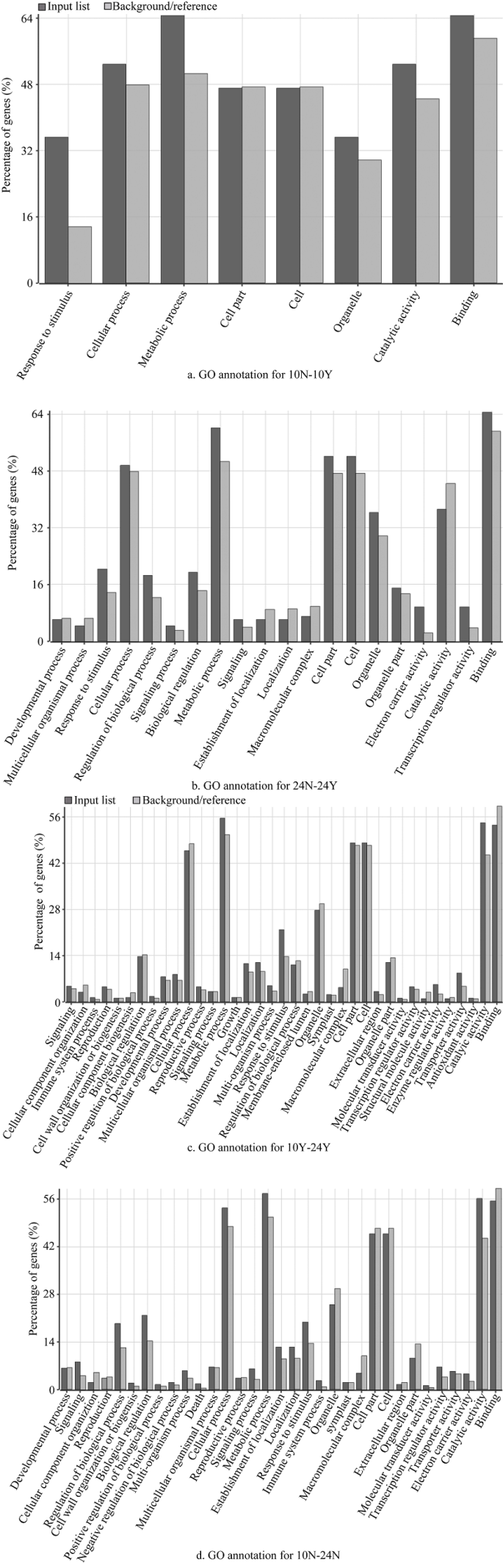 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7差异表达基因的GO功能分析
Fig. 7GO analysis of differentially expressed genes
2.6 差异表达基因Pathway富集分析
利用MapMan工具分别对授粉植株吐丝后10 d与24 d间的差异表达基因(即图8中的“d10Y_d24Y”)和吐丝后24 d授粉植株与非授粉植株间的差异表达基因(即图8中的“d24N_d24Y”)进行Pathway富集分析(图8)。授粉植株吐丝后10 d与24 d间的差异表达上调基因主要富集在光合作用(photosynthesis, PS)、蛋白质降解(protein degradation)以及RNA转录调控(RNA regulation of transcription)路径; 下调基因主要富集在转运(transport)、激素代谢(hormonemetabolism)等路径。吐丝后24 d, 授粉与非授粉植株间的差异表达上调基因主要富集在转运(transport)、胁迫(stress)、信号传导(signalling)、脂质代谢(lipid metabolism)、激素代谢(hormone metabolism)和细胞壁相关进程(cell wall); 其下调基因主要富集在四吡咯合成(tetrapyrrole synthesis)、光合作用(PS)、蛋白质合成(protein synthesis )等路径。图8
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8差异表达基因的Pathway富集分析
Fig. 8Pathway enriched analysis of DEGs
通过对吐丝后10 d豫816授粉处理和同期非授粉处理间的差异表达基因进行Pathway富集分析, 发现了一些可能参与阻断授粉诱导玉米提前衰老的基因(表3)。这些基因主要编码核酸外切酶(exonuclease family protein); 光合作用过程中的葡萄糖-6-磷酸转运蛋白(GPT2); 糖酵解途径中的甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehy-drogenase C2); 脂质转运蛋白(lipid-transfer protein, LTP); 质子泵(H+-ATPase); RNA转录调控过程中具有CCT结构域的B-box型锌指蛋白(B-box type zinc finger protein with CCT domain); 生长素相关蛋白(auxin associated family protein), STP4糖转运蛋白(sugar transporter 4), S-腺苷蛋氨酸甲基转移酶(S- adenosyl-L-methionine-dependent methyltransferases superfamily protein), CBS膜蛋白(CBS domain containing membrane protein); 具有转录后修饰作用的蛋白磷酸酶(protein phosphatase 2C family protein)、SIP3互作蛋白(SOS3-interacting protein 3)和SIP4互作蛋白, 钙结合蛋白(calcium-binding EF-hand family protein), 四吡咯合成过程中的原叶绿素酸脂氧化还原酶(protochlorophyllide oxidoreductase A, PORA); 铝诱导蛋白(aluminium induced protein); 半乳糖醛酸转移酶(galacturonosyltransferase-like 2); 葡糖基转移酶(glycosyltransferase family 61 protein)等。其中, 基因GRMZM2G127846、GRMZM2G 123896、GRMZM2G172214和GRMZM2G412601是吐丝后10 d这一时期所特有的差异表达基因。
Table 3
表3
表3吐丝后10 d参与阻断授粉诱导玉米提前衰老相关基因
Table 3
| 玉米基因编号 Maize gene ID | 差异倍数 FC (10N/10Y) | 基因功能 Gene function |
|---|---|---|
| GRMZM2G127846 | 9.51 | Exonuclease family protein |
| GRMZM2G009223 | 6.56 | Glucose-6-phosphate/phosphate translocator 2 (GPT2) |
| GRMZM2G339562 | 6.17 | Response to low sulfur 4 (LSU4) |
| GRMZM2G087254 | 5.78 | APS reductase 3 (APR3) |
| GRMZM2G071630 | 3.90 | Glyceraldehyde-3-phosphate dehydrogenase C2 |
| GRMZM2G345700 | 3.83 | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
| GRMZM2G139874 | 3.66 | Cinnamate-4-hydroxylase (C4H) |
| GRMZM2G000264 | 3.33 | H(+)-ATPase 11 |
| GRMZM2G079613 | 3.23 | Tetratricopeptide repeat-like superfamily protein |
| GRMZM2G144346 | 0.35 | B-box type zinc finger protein with CCT domain |
| GRMZM2G123896 | 0.33 | Dormancy/auxin associated family protein |
| GRMZM2G051151 | 0.31 | Oxidative stress 3 (OXS3) |
| GRMZM2G106792 | 0.30 | NDR1/HIN1-like 2 |
| GRMZM5G801949 | 0.28 | Sugar transporter 4 (STP4) |
| 玉米基因编号 Maize gene ID | 差异倍数 FC (10N/10Y) | 基因功能 Gene function |
| GRMZM2G068510 | 0.24 | S-adenosyl-L-methionine-dependent methyltransferases superfamily protein |
| GRMZM2G172214 | 0.22 | CBS domain containing membrane protein |
| AC208201.3_FG002 | 0.20 | Protein phosphatase 2C family protein |
| GRMZM2G412601 | 0.19 | SOS3-interacting protein 3 (SIP3) |
| GRMZM2G088819 | 0.16 | Calcium-binding EF-hand family protein |
| GRMZM2G084958 | 0.15 | Protochlorophyllide oxidoreductase A (PORA) |
| GRMZM2G478553 | 0.15 | RING/U-box superfamily protein (ATL3) |
| GRMZM2G142802 | 0.14 | Aluminium induced protein |
| GRMZM2G149024 | 0.12 | Galacturonosyltransferase-like 2 |
| GRMZM2G177050 | 0.11 | SOS3-interacting protein 4 (SIP4) |
| GRMZM2G131055 | 0.08 | Glycosyltransferase family 61 protein |
新窗口打开|下载CSV
同样地, 通过对吐丝后24 d豫816授粉处理与同期非授粉处理间的差异表达基因分析, 发现了一些参与碳水化合物代谢的基因(表4)。它们主要有: ①糖代谢过程中参与海藻糖(trehalose)生物合成的TPS基因(GRMZM2G001304, GRMZM2G068943)和TPP基因(GRMZM2G112830, GRMZM2G099860); ②淀粉生物合成过程中编码腺苷二磷酸葡萄糖焦磷酸化酶(ADP-glucose pyrophosphorylase, AGPase)的GRMZM2G106213基因, 编码淀粉合酶(starch synthase 2, SS2)的GRMZM2G348551基因; ③蔗糖生物合成过程中编码磷酸甘油酸激酶(phosphoglycerate kinase)的GRMZM2G089136基因, 编码甘油醛-3-磷酸脱氢酶(glyceraldehyde 3-phosphate dehydrogenase, GAP-DH)的GRMZM2G104632基因, 编码果糖二磷酸醛缩酶(fructose-bisphosphate aldolase)的GRMZM2G345493、GRMZM2G155253、GRMZM2G 089365和GRMZM2G046284基因, 编码果糖-1,6-二磷酸酶(fructose-1,6-bisphosphatase)的GRMZM5G 836250和GRMZM2G306732基因, 编码蔗糖磷酸合酶(sucrose phosphate synthase)的GRMZM5G875238基因; ④蔗糖降解过程中编码类果糖激酶(fructokinase-like, FLN)的GRMZM2G466780和GRMZM2G103843基因; ⑤既参与淀粉生物合成又参与蔗糖生物合成/降解过程中编码葡萄糖-6-磷酸异构酶(glucose-6-phosphate isomerase)的GRMZM2 G140614和GRMZM2G076075基因。
Table 4
表4
表4吐丝后24 d豫816穗位叶中碳水化合物代谢基因
Table 4
| 玉米基因编号 Maize gene ID | 差异倍数 FC (24N/24Y) | 基因功能 Gene function |
|---|---|---|
| GRMZM2G001304 | 0.19 | Trehalose-6-phosphate synthase (TPS) |
| GRMZM2G068943 | 0.22 | Trehalose-6-phosphate synthase (TPS) |
| GRMZM2G112830 | 4.55 | Trehalose-phosphatase (TPP) |
| GRMZM2G099860 | 4.04 | Trehalose-phosphatase (TPP) |
| GRMZM2G140614 | 2.75 | Glucose-6-phosphate isomerase |
| GRMZM2G076075 | 6.43 | Glucose-6-phosphate isomerase |
| GRMZM2G106213 | 40.55 | ADP glucose pyrophosphorylase (AGPase) |
| GRMZM2G348551 | 6.92 | Starch synthase 2 (SS2) |
| GRMZM2G089136 | 0.07 | Phosphoglycerate kinase |
| GRMZM2G104632 | 2.86 | Glyceraldehyde 3-phosphate dehydrogenase (GAP-DH) |
| GRMZM2G345493 | 0.09 | Fructose-bisphosphate aldolase |
| GRMZM2G155253 | 0.05 | Fructose-bisphosphate aldolase |
| 玉米基因编号 Maize gene ID | 差异倍数 FC (24N/24Y) | 基因功能 Gene function |
| GRMZM2G089365 | 8.92 | Fructose-bisphosphate aldolase |
| GRMZM2G046284 | 0.16 | Fructose-bisphosphate aldolase |
| GRMZM5G836250 | 0.11 | Fructose-1,6-bisphosphatase |
| GRMZM2G306732 | 0.10 | Fructose-1,6-bisphosphatase |
| GRMZM5G875238 | 0.16 | Sucrose phosphate synthase |
| GRMZM2G466780 | 0.20 | Fructokinase-like 1 (FLN1) |
| GRMZM2G103843 | 0.28 | Fructokinase-like 2 (FLN2) |
新窗口打开|下载CSV
3 讨论
衰老是玉米生长发育的最后阶段, 玉米叶片衰老的程度与产量形成[26,27]密切相关。在玉米衰老过程中, 植株自身衰老反应被激活, 叶片内部的细胞和生物大分子结构发生变化[28], 引起叶片细胞的完整性逐渐消失, 叶片逐渐变黄, 进而促进叶片及茎秆中积累的营养物质转移至发育中的玉米籽粒。本研究基于RNA-seq技术, 通过对授粉和阻断授粉条件下的豫816叶片进行转录组分析发现, 授粉植株吐丝后10 d与24 d间差异表达上调基因主要富集在光合作用、蛋白质降解以及RNA转录调控路径; 下调基因主要富集在转运、激素代谢等路径。吐丝后24 d, 授粉与非授粉植株间的差异表达上调基因主要富集在转运、胁迫、信号传导、脂质代谢、激素代谢和细胞壁相关进程; 下调基因主要富集在四吡咯合成、光合作用、蛋白质合成等路径。表明阻断授粉诱导的玉米提前衰老是由多条代谢途径共同参与作用的结果。Zhang等[11]对玉米自交系齐319穗位叶片的转录组分析发现, 在玉米叶片自然衰老早期, 上调基因主要参与了芳香族氨基酸的生物合成、转运、细胞多糖的生物合成和细胞壁大分子物质的分解代谢; 而在叶片自然衰老后期, 上调基因主要参与氨基酸代谢、转运, 细胞凋亡和刺激响应。我们推测玉米自然衰老与阻断授粉诱导的玉米提前衰老之间可能存在某些共有途径。本研究中所使用的豫816材料在阻断授粉处理条件下, 其衰老反应启动和衰老速率相对于正常授粉处理明显提前和加快。本研究在10 DAS时期的材料中鉴定到了一些参与阻断授粉诱导玉米提前衰老的基因。这些基因参与的生物过程主要有RNA降解、光合作用、木质素生物合成、转录调控、糖转运、翻译后修饰、蛋白质去磷酸化和钙信号传导等生物过程, 这些基因在阻断授粉诱导豫816提前衰老中可能起到重要作用。豫816衰老的早期阶段涉及到多个生物过程, 表明叶片衰老是一个涉及不同水平调控的高度复杂生物进程, 这为我们系统全面理解叶片衰老机理提供了有益参考。
植物体内的糖类物质如海藻糖(trehalose)、蔗糖(sucrose)、葡萄糖(glucose)、果糖及其衍生物不仅具有作为代谢物质和细胞结构物质的特性, 而且具有类似激素那样的调控活性, 调节植物包括衰老在内的诸多基础生物进程[29]。其中, 海藻糖-6-磷酸(trehalose-6-phosphate, Tre6P)是植物海藻糖生物合成的中间物, “Suc-Tre6P关系模型”[30,31]认为Tre6P是一种重要的信号代谢物, 且负反馈调节蔗糖水平, 维持植物体内蔗糖水平在一个最佳范围。Wingler等[32]研究发现, 衰老叶片中Tre6P含量积累至正常成熟叶片的15倍时, 参与响应高碳水平供给情况下衰老的适时启动。Tre6P含量随碳可利用性(carbon availability)的升高而增加, 并且在幼小发育组织中抑制蔗糖非发酵相关激酶1 (sucrose non-fermenting related kinase-1, SnRK1)的催化活性, 调节可利用碳用于快速生长。本研究在24 DAS材料中就发现了一些参与海藻糖生物合成、淀粉生物合成/降解等碳水化合物代谢的基因, 表明这些代谢进程可能在玉米衰老启动和发展过程中起重要作用。
本文仅就授粉和阻断授粉处理条件下10 DAS和24 DAS差异表达基因初步探讨, 植物激素(如脱落酸、乙烯、茉莉酸、生长素、细胞分裂素等)[33,34,35,36,37,38]和转录因子家族(WRKY、NAC、MYB)[15,39-40]等因素在玉米衰老早期阶段发挥的重要作用仍有待进一步研究。
4 结论
不同时期不同处理转录水平上存在较大差异。吐丝后10 d, 授粉植株和阻断授粉植株都具有较好的持绿性, 差异表达基因较少; 而吐丝后24 d, 授粉植株持绿性仍旧良好, 但阻断授粉植株的叶片开始大面积变黄, 并且授粉植株与阻断授粉植株间存在较多的差异表达基因。这些差异表达基因主要富集在刺激响应、激素代谢、信号传导等方面, 其调控路径主要涉及转运、光合作用和碳水化合物代谢等过程。相对于正常授粉植株, 阻断授粉诱导条件下的豫816植株体内碳水化合物代谢、光合作用等进程可能受到影响, 造成玉米衰老反应的启动, 导致衰老过程的提前和加快。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1016/S1360-1385(00)01655-1URLPMID:10871899 [本文引用: 1]

Senescence is the last stage of leaf development and one type of programmed cell death that occurs in plants. The relationships among senescence programs that are induced by a variety of factors have been addressed at a molecular level in recent studies. Furthermore, an overlap between the pathogen-response and senescence programs is beginning to be characterized. The complexity of the senescence program is also evident in studies of senescence-specific gene regulation and the role of photosynthesis and plant hormones in senescence regulation. New molecular-genetic approaches are expected to be useful in unraveling the molecular mechanisms of the leaf senescence program.
DOI:10.1146/annurev.arplant.57.032905.105316URL [本文引用: 1]
DOI:10.1093/jxb/ers238URL [本文引用: 1]
[本文引用: 2]
[本文引用: 1]
DOI:10.1007/s11103-016-0532-1URLPMID:27586543 [本文引用: 2]
This piece of the submission is being sent via mail.Leaf senescence is essential for the nutrient economy of crops and is executed by so-called senescence-associated genes (SAGs). Here we explored th
DOI:10.3724/SP.J.1006.2013.00093URL [本文引用: 1]
In order to explore the changes of photochemistry activity and the contribution of photochemistry activity to the maintenance of photosynthetic capacity in maize leaves during senescence, the chlorophyll content, photosynthetic rate, PSI and PSII activities during senescence of leaves in the stay green maize (
DOI:10.3724/SP.J.1006.2013.00093URL [本文引用: 1]
In order to explore the changes of photochemistry activity and the contribution of photochemistry activity to the maintenance of photosynthetic capacity in maize leaves during senescence, the chlorophyll content, photosynthetic rate, PSI and PSII activities during senescence of leaves in the stay green maize (
DOI:10.3724/SP.J.1006.2017.00051URL [本文引用: 1]
水稻叶片早衰直接影响作物的光合效率,减少产量,降低品质。深入研究早衰的分子机制对控制和延缓衰老具有重要意义。本文报道了一个早衰突变体psls1(premature senescence leaf with spots)的基因定位及克隆结果。突变体在发育到七叶期以后,叶片自下而上叶绿素含量下降,过氧化氢过量积累,突变体叶片逐渐黄化至枯萎;其他农艺性状如株高、分蘖数、主穗长、结实率和穗粒数也相应变差。电镜观察进一步发现,psls1衰老叶片中叶绿体降解、类囊体基粒片层模糊,嗜锇颗粒明显增多。遗传分析表明,psls1受1对隐性基因控制,利用psls1×IRAT129杂交组合F2分离群体中的1690个早衰个体,将基因PSLS1定位在第7染色体分子标记ZS-3和ZS-8之间89 kb的范围内。测序研究发现,区间内一个编码铁氧还依赖的谷氨酸合酶基因LOC_Os07g46460的第2外显子末位的G被替换为A,导致转录本的错误剪切,突变体的c DNA缺失了57 bp碱基片段。在突变体中该基因表达量下降,谷氨酸合酶活性降低,其产物谷氨酸含量显著下降、其他氨基酸代谢紊乱。水培低氮处理下可诱发突变体psls1早衰。研究结果表明,由于PSLS1突变使得谷氨酸合酶失活,氮代谢异常而导致突变体psls1早衰。
DOI:10.3724/SP.J.1006.2017.00051URL [本文引用: 1]
水稻叶片早衰直接影响作物的光合效率,减少产量,降低品质。深入研究早衰的分子机制对控制和延缓衰老具有重要意义。本文报道了一个早衰突变体psls1(premature senescence leaf with spots)的基因定位及克隆结果。突变体在发育到七叶期以后,叶片自下而上叶绿素含量下降,过氧化氢过量积累,突变体叶片逐渐黄化至枯萎;其他农艺性状如株高、分蘖数、主穗长、结实率和穗粒数也相应变差。电镜观察进一步发现,psls1衰老叶片中叶绿体降解、类囊体基粒片层模糊,嗜锇颗粒明显增多。遗传分析表明,psls1受1对隐性基因控制,利用psls1×IRAT129杂交组合F2分离群体中的1690个早衰个体,将基因PSLS1定位在第7染色体分子标记ZS-3和ZS-8之间89 kb的范围内。测序研究发现,区间内一个编码铁氧还依赖的谷氨酸合酶基因LOC_Os07g46460的第2外显子末位的G被替换为A,导致转录本的错误剪切,突变体的c DNA缺失了57 bp碱基片段。在突变体中该基因表达量下降,谷氨酸合酶活性降低,其产物谷氨酸含量显著下降、其他氨基酸代谢紊乱。水培低氮处理下可诱发突变体psls1早衰。研究结果表明,由于PSLS1突变使得谷氨酸合酶失活,氮代谢异常而导致突变体psls1早衰。
[本文引用: 1]
[本文引用: 1]
DOI:10.1104/pp.85.3.720URLPMID:16665767 [本文引用: 1]
Objective of research was (a) to evaluate the influence of pollination-prevention on various metabolic parameters of the two maize inbreds B73 and B14A and their F1, and (b) to gain information on the inheritance of leaf senescence, in response to pollination-prevention. The results show that the visual pattern of leaf senescence, in response to prevention of ear pollination, contrasts markedly between the two inbred lines. Relative to control plants, prevention of ear pollination, causes a premature senescence in B73 and B73 B14A plants, while leaves of unpollinated B14A remain green and similar in appearance to pollinated controls. Furthermore, prevention of ear pollination induces a sizable reduction of dry matter accumulation of all above-ground material and changes in various metabolic parameters. An accumulation of sucrose in the leaves of unpollinated B73 and B73 B14A plants is correlated with the development of premature senescence. Finally, the genetic analysis supports suggestions that a single dominant gene is responsible for the differences observed, in the visual pattern of leaf senescence, in response to prevention of ear pollination.
DOI:10.1371/journal.pone.0115617URLPMID:25532107 [本文引用: 2]
Leaf senescence is an important biological process that contributes to grain yield in crops. To study the molecular mechanisms underlying natural leaf senescence, we harvested three different developmental ear leaves of maize, mature leaves (ML), early senescent leaves (ESL), and later senescent leaves (LSL), and analyzed transcriptional changes using RNA-sequencing. Three sets of data, ESL vs. ML, LSL vs. ML, and LSL vs. ESL, were compared, respectively. In total, 4,552 genes were identified as differentially expressed. Functional classification placed these genes into 18 categories including protein metabolism, transporters, and signal transduction. At the early stage of leaf senescence, genes involved in aromatic amino acids (AAAs) biosynthetic process and transport, cellular polysaccharide biosynthetic process, and the cell wall macromolecule catabolic process, were up-regulated. Whereas, genes involved in amino acid metabolism, transport, apoptosis, and response to stimulus were up-regulated at the late stage of leaf senescence. Further analyses reveals that the transport-related genes at the early stage of leaf senescence potentially take part in enzyme and amino acid transport and the genes upregulated at the late stage are involved in sugar transport, indicating nutrient recycling mainly takes place at the late stage of leaf senescence. Comparison between the data of natural leaf senescence in this study and previously reported data for Arabidopsis implies that the mechanisms of leaf senescence in maize are basically similar to those inArabidopsis. A comparison of natural and induced leaf senescence in maize was performed. Athough many basic biological processes involved in senescence occur in both types of leaf senescence, 78.07% of differentially expressed genes in natural leaf senescence were not identifiable in induced leaf senescence, suggesting that differences in gene regulatory network may exist between these two leaf senescence programs. Thus, this study provides important information for understanding the mechanism of leaf senescence in maize.
.
[本文引用: 1]
DOI:10.1073/pnas.1321568111URL
DOI:10.1093/jxb/eru054URLPMID:24600015 [本文引用: 1]
The process of is induced by an extensive range of developmental and environmental signals and controlled by multiple, cross-linking pathways, many of which overlap with plant stress-response signals. Elucidation of this complex regulation requires a step beyond a traditional one-gene-at-a-time analysis. Application of a more global analysis using statistical and mathematical tools of systems biology is an approach that is being applied to address this problem. A variety of modelling methods applicable to the analysis of current and future data are reviewed and discussed using some -specific examples. Network modelling with a transcriptome time course followed by testing predictions with gene-expression data illustrates the application of systems biology tools.
DOI:10.1371/journal.pone.0094210URLPMID:24709840
Abstract As a novel sugar transporter family, SWEETs play important roles in plant growth and development. Here, we characterized a SWEET gene named OsSWEET5 through its overexpression in rice. Heterologous expression assay indicated that OsSWEET5 encoded a galactose transporter in yeast. OsSWEET5-overexpressing plants displayed the phenotypes of growth retardation and precocious senescence at seedling stage. GC-MS analysis showed that the sugar levels were largely altered in the leaves of the OsSWEET5-overexpressing plants. Molecular analysis revealed that these phenotypes might be due to the transcriptional changes of the genes involved in sugar metabolism and transport. In addition, the transgenic plants showed a lower level of auxin with altered transcription of genes involved in auxin signaling and translocation pathways. However, no obvious phenotype was observed between the amiRNA-OsSWEET5 transgenic lines and WT plants, which could be a result of the functional redundancy of the galactose transporters in rice. Taken together, our findings suggest that OsSWEET5 plays a crucial role in regulating the crosstalk between sugar and auxin in rice.
DOI:10.1073/pnas.1522840113URLPMID:26831097
Drought stress is an important environmental factor limiting plant productivity. In this study, we screened drought-resistant transgenic plants from 65 promoter-pyrabactin resistance 1-like (PYL) abscisic acid (ABA) receptor gene combinations and discovered that pRD29A::PYL9 transgenic lines showed dramatically increased drought resistance and drought-induced leaf senescence in both Arabidopsis and rice. Previous studies suggested that ABA promotes senescence by causing ethylene production. However, we found that ABA promotes leaf senescence in an ethylene-independent manner by activating sucrose nonfermenting 1-related protein kinase 2s (SnRK2s), which subsequently phosphorylate ABA-responsive element-binding factors (ABFs) and Related to ABA-Insensitive 3/VP1 (RAV1) transcription factors. The phosphorylated ABFs and RAV1 up-regulate the expression of senescence-associated genes, partly by up-regulating the expression of Oresara 1. The pyl9 and ABA-insensitive 1-1 single mutants, pyl8-1pyl9 double mutant, and snrk2.2/3/6 triple mutant showed reduced ABA-induced leaf senescence relative to the WT, whereas pRD29A::PYL9 transgenic plants showed enhanced ABA-induced leaf senescence. We found that leaf senescence may benefit drought resistance by helping to generate an osmotic potential gradient, which is increased in pRD29A::PYL9 transgenic plants and causes water to preferentially flow to developing tissues. Our results uncover the molecular mechanism of ABA-induced leaf senescence and suggest an important role of PYL9 and leaf senescence in promoting resistance to extreme drought stress.
DOI:10.1104/pp.17.00542URLPMID:28500268
Abstract It is well known that abscisic acid (ABA)-induced leaf senescence and premature leaf senescence negatively affect the yield of rice ( Oryza sativa ). However, the molecular mechanism underlying this relationship, especially the upstream transcriptional network that modulates ABA level during leaf senescence, remains largely unknown. Here, we demonstrate a rice NAC transcription factor, OsNAC2, that participates in ABA-induced leaf senescence. Overexpression of OsNAC2 dramatically accelerated leaf senescence, whereas its knockdown lines showed a delay in leaf senescence. Chromatin immunoprecipitation-quantitative PCR, dual-luciferase, and yeast one-hybrid assays demonstrated that OsNAC2 directly activates expression of chlorophyll degradation genes, OsSGR and OsNYC3 Moreover, ectopic expression of OsNAC2 leads to an increase in ABA levels via directly up-regulating expression of ABA biosynthetic genes ( OsNCED3 and OsZEP1 ) as well as down-regulating the ABA catabolic gene ( OsABA8ox1 ). Interestingly, OsNAC2 is upregulated by a lower level of ABA but downregulated by a higher level of ABA, indicating a feedback repression of OsNAC2 by ABA. Additionally, reduced OsNAC2 expression leads to about 10% increase in the grain yield of RNAi lines. The novel ABA-NAC-SAGs regulatory module might provide a new insight into the molecular action of ABA to enhance leaf senescence and elucidates the transcriptional network of ABA production during leaf senescence in rice. 2017 American Society of Plant Biologists. All Rights Reserved.
.
[本文引用: 1]
DOI:10.1093/jxb/eri103URL [本文引用: 1]
DOI:10.1038/nmeth.1226URLPMID:18516045 [本文引用: 1]
High read number is relevant for RNA-Seq because our ability to and phage lambda templates (Fig. 2c). These standards comprised reliably detect and measure rare, yet physiologically relevant, RNA long (~10,000 nt), intermediate (~1,500 nt) and short (~300 nt) species (those with abundances of 1-10 RNAs per cell) depends on transcripts, and they were designed to span the range of abundance the number of independent pieces of evidence (sequence reads) (~0.5-50,000 transcripts per cell) typically observed in natural obtained for transcripts from each gene. This constraint influenced transcriptomes. RNA-Seq data for the standards were linear across our sequencing strategy, choice of instrument and choice of the a dynamic range of five orders of magnitude in RNA concentra- 25-bp read length.tion. Sequence coverage over test transcripts was highly reproducibleThe sensitivity of RNA-Seq will be a function of both molar con- and quite uniform (Supplementary Fig. 1c). At current practical centration and transcript length. We therefore quantified transcript sequencing capacity and cost (~40 M mapped reads), transcript levels in reads per kilobase of exon model per million mapped reads detection was robust at 1.0 RPKM and above for a typical 2-kilo- (RPKM) (Fig. 1a,c). The RPKM measure of read density reflects base (kb) mRNA (~80 individual sequence reads resulting in a P the molar concentration of a transcript in the starting sample by value <10-16). Beyond simple detection confidence, we analyzed the normalizing for RNA length and for the total read number in the impact of different amounts of sequencing on our ability to measure measurement. This facilitates transparent comparison of transcript the concentration of a given transcript class (defined on the basis of levels both within and between samples.RPKM) within 5% (Fig. 2d). When these RNA standards are usedin conjunction with information on cellular RNA content, abso- Examination of a well-characterized locuslute transcript levels per cell can also be calculated. For example, on Data from a 21-million-read transcriptome measurement of adult the basis of literature values for the mRNA content of a liver cell19 mouse skeletal muscle (Fig. 1b,c) illustrate some key characteris- and the RNA standards, we estimated that 3 RPKM corresponds to tics of our results. Myf6 (also known as Mrf4) is a much-studied about one transcript per liver cell. For C2C12 tissue culture cells, for myogenic transcription factor gene that is expressed specifically and modestly in mus- cle, as expected, but silent in liver and brain.
DOI:10.1038/nprot.2012.016URL [本文引用: 1]
DOI:10.1104/pp.105.060459URL [本文引用: 1]
DOI:10.1006/meth.2001.1262URL [本文引用: 1]
DOI:10.1111/pce.2006.29.issue-3URL [本文引用: 1]
DOI:10.1007/s11103-013-0013-8URL [本文引用: 1]
DOI:10.1016/j.tcb.2009.07.007URLPMID:2755611 [本文引用: 1]
The accumulation of cellular damage is a feature common to all aging cells and leads to decreased ability of the organism to survive. The overall rate at which damage accumulates is influenced by conserved metabolic factors (longevity pathways and regulatory proteins) that control lifespan through adjusting mechanisms for maintenance and repair. Autophagy, the major catabolic process of eukaryotic cells that degrades and recycles damaged macromolecules and organelles, is implicated in aging and in the incidence of diverse age-related pathologies. Recent evidence has revealed that autophagic activity is required for lifespan extension in various long-lived mutant organisms, and that numerous autophagy-related genes or proteins are directly regulated by longevity pathways. These findings support the emerging view that autophagy is a central regulatory mechanism for aging in diverse eukaryotic species.
DOI:10.1016/j.tplants.2015.10.009URLPMID:26603980 [本文引用: 1]
With the rapidly expanding world population, food production must be substantially increased in the coming several decades to meet increasing global demands for food. Source ink interactions modulate carbon (C) assimilation, translocation, partitioning, and storage throughout the plant and have a pivotal role in determining the productivity of harvestable organs. Crop yield is dependent on the source ink relation, which is influenced by environmental changes. Understanding mechanisms by which plants control source activity and sink strength in the context of global climate changes, such as water limitation and high temperature, is of agronomic significance for the development of efficient breeding programs for improving crops. Many examples have demonstrated that manipulation of source activity and sink strength could significantly enhance crop productivity. However, crop productivity is determined by the combinatorial functions of multiple genes involved in signaling and metabolic pathways, which are still not well understood. Additionally, yield increase has not been evaluated in field trials in most studies. Therefore, more work is needed for the translation of basic findings to open field trials.
DOI:10.1093/jxb/ert457URL [本文引用: 1]
DOI:10.1104/pp.16.00417URLPMID:27482078 [本文引用: 1]
Trehalose 6-phosphate (Tre6P), the intermediate of trehalose biosynthesis, is an essential signal metabolite in plants, linking growth and development to carbon status. The Suc-Tre6P nexus model postulates that Tre6P is both a signal and negative feedback regulator of Suc levels, forming part of a mechanism to maintain Suc levels within an optimal range and functionally comparable to the insulin-glucagon system for regulating blood Glc levels in animals. The target range and sensitivity of the Tre6P-Suc feedback control circuit can be adjusted according to the cell type, developmental stage, and environmental conditions. In source leaves, Tre6P modulates Suc levels by affecting Suc synthesis, whereas in sink organs it regulates Suc consumption. In illuminated leaves, Tre6P influences the partitioning of photoassimilates between Suc, organic acids, and amino acids via posttranslational regulation of phosphoenolpyruvate carboxylase and nitrate reductase. At night, Tre6P regulates the remobilization of leaf starch reserves to Suc, potentially linking starch turnover in source leaves to carbon demand from developing sink organs. Use of Suc for growth in developing tissues is strongly influenced by the antagonistic activities of two protein kinases: SUC-NON-FERMENTING-1-RELATED KINASE1 (SnRK1) and TARGET OF RAPAMYCIN (TOR). The relationship between Tre6P and SnRK1 in developing tissues is complex and not yet fully resolved, involving both direct and indirect mechanisms, and positive and negative effects. No direct connection between Tre6P and TOR has yet been described. The roles of Tre6P in abiotic stress tolerance and stomatal regulation are also discussed.
DOI:10.1104/pp.111.191908URL [本文引用: 1]
.
[本文引用: 1]
DOI:10.1104/pp.113.218115URL [本文引用: 1]
DOI:10.3389/fpls.2014.00650URLPMID:4243489 [本文引用: 1]
Abstract Leaf senescence is the last stage of leaf development and is accompanied by cell death. In contrast to senescence in individual organisms that leads to death, leaf senescence is associated with dynamic processes that include the translocation of nutrients from old leaves to newly developing or storage tissues within the same plant. The onset of leaf senescence is largely regulated by age and internal and external stimuli, which include the plant hormone ethylene. Earlier studies have documented the important role of ethylene in the regulation of leaf senescence. The production of ethylene coincides with the onset of leaf senescence, whereas the application of ethylene to plants induces precocious leaf senescence. Recently, many studies have described the components of ethylene signaling and biosynthetic pathways that are involved in modulating the onset of leaf senescence. Particularly, transcription factors (TFs) integrate ethylene signals with those from environmental and developmental factors to accelerate or delay leaf senescence. This review aims to discuss the regulatory cascade involving ethylene and TFs in the regulation of onset of leaf senescence.
DOI:10.1093/jxb/ern035URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.pbi.2016.06.002URLPMID:27314623 [本文引用: 1]
Leaf senescence is finely tuned by many regulatory factors such as NAC (NAM/ATAF/CUC) transcription factors (TFs). NACs comprise one of the largest families of TFs in plants, many of which are differentially regulated during leaf senescence and play a major role in leaf senescence. Recent studies advanced our understanding on the structural and functional features of NAC TFs including target binding specificities of the N-terminal DNA binding domain and dynamic interaction of the C-terminal intrinsically disordered domain with other proteins. NAC TFs control otherNACs and also interact with NACs or other TFs to fine-tune the expression of target genes. These studies clearly demonstrated the highly complex characteristics of NAC regulatory networks, which are dynamically regulated temporally and spatially and effectively integrate multiple developmental and environmental signals.
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}