 ,, 肖乃衍, 朱专为, 刘翠翠, IntikhabALAM, 陈平华, 卢运海,*福建农林大学农业部福建甘蔗生物学与遗传育种重点实验室 / 作物遗传育种与综合利用教育部重点实验室 / 作物科学学院, 福建福州 350002
,, 肖乃衍, 朱专为, 刘翠翠, IntikhabALAM, 陈平华, 卢运海,*福建农林大学农业部福建甘蔗生物学与遗传育种重点实验室 / 作物遗传育种与综合利用教育部重点实验室 / 作物科学学院, 福建福州 350002Development and Characterization of SSR Markers from the Whole Genome Sequences of Saccharum officinarum (LA-purple)
WANG Heng-Bo,, XIAO Nai-Yan, ZHU Zhuan-Wei, LIU Cui-Cui, Intikhab ALAM, CHEN Ping-Hua, LU Yun-Hai,*Key Laboratory of Ministry of Agriculture for Sugarcane Biology and Genetic Breeding (Fujian), Key Laboratory of Ministry of Education for Genetics, Breeding and Multiple Utilization of Crops, College of Crop Science, Fujian Agriculture and Forestry University, Fuzhou 350002, Fujian, China通讯作者:
第一联系人:
收稿日期:2017-11-20接受日期:2018-03-26网络出版日期:2018-04-09
| 基金资助: |
Received:2017-11-20Accepted:2018-03-26Online:2018-04-09
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (1234KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王恒波, 肖乃衍, 朱专为, 刘翠翠, IntikhabALAM, 陈平华, 卢运海. 基于甘蔗热带种(LA-purple)全基因组序列的SSR开发及特征分析[J]. 作物学报, 2018, 44(9): 1400-1410. doi:10.3724/SP.J.1006.2018.01400
WANG Heng-Bo, XIAO Nai-Yan, ZHU Zhuan-Wei, LIU Cui-Cui, Intikhab ALAM, CHEN Ping-Hua, LU Yun-Hai.
甘蔗为禾本科(Poaceae)甘蔗属(Sacchrum L.)植物, 是一种多年生、可宿根栽培的C4作物, 具有生物量高、二氧化碳补偿点低等优点, 是世界上最重要的糖料作物之一, 是人类食糖的重要来源[1], 在我国农业经济中占有重要地位[2]。现代甘蔗栽培品种绝大多数都是由热带种(Saccharum officinarum L., 2n = 80, x = 10)和割手密(Saccharum spontaneum L., 2n = 40~128, x = 8)种间杂交而来, 且与热带种进行了多次回交[3], 其染色体数目在2n = 100~130之间, 其中80%~90%的染色体来自于热带种, 10%~20%来自于割手密, 而染色体的5%~17%为2个种间的染色体重组类型[4,5]。由于甘蔗的高度多倍体及非整倍体的复杂遗传背景, 使得甘蔗的遗传、育种及基因组测序等都面临巨大的困难和挑战[5,6]。
简单重复序列(simple sequence repeats, SSR), 也被称为微卫星(microsatellites)序列, 是指由1~6个核苷酸组成的不同类型基序多次重复而形成的相对较短、广泛分布于真核生物基因组上的DNA序 列[7,8]。它们在基因组上虽然是随机分布[9], 但更偏向于低重复、富含基因的区域[10]。每一个SSR序列在DNA复制时产生错误的比率较高, 因而可以在种内或种间产生大量的SSR序列长度的变异[11]。作为分子标记, SSR具有多态性高、重复性好、操作简便及共显性等优点, 因而被广泛应用于各种动、植物的品种指纹图谱鉴定、遗传图谱构建及目标性状分子标记筛选等领域[12]。
SSR标记在甘蔗属植物上也得到了较为广泛的应用[13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30], 已有数个关于在甘蔗上开发SSR标记的报道, 如Oliveira等[20]从甘蔗SUCEST (sugarcane EST database)数据库中找出2005个含有SSR的EST序列, 对其中的342个EST-SSRs进行了PCR分析, 发现有224个(65.5%)呈多态性; Singh等[27]从2个甘蔗栽培品种的4085个EST序列中鉴定出351个EST-SSRs, 对其中227个进行了PCR分析, 发现有134个呈多态性; Shamshad等[28]从NCBI数据库中获得10 000个EST序列(累计长4201 kb), 从中鉴定出406个SSRs, 对其中的63个进行了PCR分析, 发现有42个具有多态性。但是目前可公开获得的甘蔗属的SSR分子标记数量仍然十分有限, 大部分甘蔗属的SSR标记的引物序列依然没有公开, 而传统的SSR标记开发存在诸多缺点, 耗费人力、物力、且效率低下, 尤其是对于多倍体的甘蔗; 加之, 甘蔗又为同源多倍体, 其基因组大小预测为10 Gb[31], 远远高于其他禾本科作物, 而甘蔗的“高贵化”育种策略造成现代甘蔗品种遗传基础狭窄[3,4,5,6]。因此, 甘蔗SSR标记的开发和应用比较缓慢, 现有的SSR标记数目有限, 不能满足在甘蔗属植物上开展各项研究的需要, 严重制约了甘蔗的分子遗传研究进展[22]。
本研究旨在挖掘热带种(LA-purple)的基因组数据, 利用生物信息学方法, 分析全基因组基因序列上SSR位点的数量和分布规律, 包括基序结构和类型、比例、重复次数, 进而设计和合成SSR引物, 开发多态的SSR分子标记, 为未来甘蔗属不同种的遗传多样性分析、遗传图谱构建、重要农艺性状形成的遗传机制研究及开展分子育种研究提供SSR标记数据库支撑。
1 材料与方法
1.1 材料
所有材料均为甘蔗属材料, 包括2个大茎野生种(S. robustum)、4个热带种(S. officinarum)、4个割手密(S. spontaneum)和2个栽培品种(Saccharum hybrid), 除科5来自广州甘蔗糖业研究所海南甘蔗育种场外, 其他材料都来自国家甘蔗种质资源圃(National Germplasm Repository of Sugarcane)(表1)。1.2 基因组序列的来源
福建农林大学基因组与生物技术研究中心Ming Ray教授团队采用第三代高通量测序技术, 从头组装, 对热带种(LA-purple)进行了全基因组序列测定工作(数据暂未公布), 并对获得序列上的基因进行了预测。本研究采用的序列(genomic gene sequence)为其中的 255 398个基因, 累计总长1 029 222 285 bp, 平均每个基因的长度为4030 bp。Table 1
表1
表1供试甘蔗种质资源名称及来源
Table 1
| 序号 Code | 名称 Name | 类型 Type | 倍性 Ploidy |
|---|---|---|---|
| 1 | 57NG208 | 大茎野生种 S. robustum | |
| 2 | NG77-004 | 大茎野生种 S. robustum | |
| 3 | 云南75-2-11 YN75-2-11 | 割手密 S. spontaneum | 八倍体 Octoploid |
| 4 | 福建89-1-1 FJ89-1-1 | 割手密 S. spontaneum | 九倍体 Nonaploid |
| 5 | 广东21 GD21 | 割手密 S. spontaneum | 十倍体 Decaploid |
| 6 | 贵州78-2-28 GZ78-2-28 | 割手密 S. spontaneum | 十二倍体 Dodecaploid |
| 7 | 黑车里本 Black cheribon | 热带种 S. officinarum | 八倍体 Octoploid |
| 8 | 路达士 Loethers | 热带种 S. officinarum | 八倍体 Octoploid |
| 9 | 克里斯塔林娜 Crystalina | 热带种 S. officinarum | 八倍体 Octoploid |
| 10 | 拔地拉 Badila | 热带种 S. officinarum | 八倍体 Octoploid |
| 11 | 桂糖35 GT35 | 栽培种 Saccharun hybrid | |
| 12 | 科5 K5 | 栽培种 Saccharun hybrid |
新窗口打开|下载CSV
1.3 SSR位点的查找与SSR引物的开发
本研究采用Perl语言编写的MISA (microsatellite identification tool)软件扫描甘蔗基因组序列, 查找序列中的SSR位点, 该软件下载自http://pgrc.ipk- gatersleben.de/misa/, 以MISA工具高通量识别和查找简单重复序列。该软件还提供一个与批量设计引物Primer 3的接口工具, 把MISA识别出来的SSR序列转为Primer 3需要的格式, 从而方便批量设计引物。SSR位点查找标准条件为, 将核苷酸重复基序(motif)分为二(dinucleotide repeats, DNRs)、三(trinucleotide repeats, TNRs)、四(tetranucleotide repeats, TtNRs)、五(pentanucleotide repeats, PNRs)、六(hexanucleotide repeats, HNRs), 其最低重复数分别定为6、5、4、3、3; 剔除掉重复的SSR引物设计位点, 对SSR位点两侧保守序列设计引物, 用Primer 3 (http://frodo.wi.mit.edu/primer3/)在线设计引物, 引物参数设计为primer length 18~28 bp; annealing temperature 55~65℃; amplicon size 100~500 bp; GC content 45%~65% [27]。1.4 DNA的提取、PCR扩增及电泳分析
在幼苗期采集幼嫩叶片, 用液氮研磨后, 按照CTAB方法[32]提取基因组DNA。提取所有参试材料的基因组DNA后, 用NanoDrop 2000超微量紫外分光光度计(Thermo Fisher Scientific)检测基因组DNA的纯度和浓度, 以1×TE (上海生工生物)将其稀释至25 ng μL-1。于-20℃冰箱保存, 用于后续实验。PCR反应体系含25 ng μL-1 DNA样品2.0 μL、10×PCR buffer (Mg2+ plus) 2.5 μL、25 mmol L-1 dNTPs 1.2 μL、10 μmol L-1引物各0.5 μL、0.5 U μL-1Taq酶 0.1 μL, 最后用ddH2O补足25 μL。PCR扩增程序为94℃预变性5 min; 94℃变性30 s, 65℃退火30 s, 72℃延伸30 s, 共10个循环, 每个循环退火温度降低0.7℃; 94℃变性30 s, 55℃退火30 s, 72℃延伸30 s, 共25个循环; 最后72℃延伸7 min, 4℃保存。Taq酶、dNTP等试剂购自北京康为世纪生物科技有限公司。参照Liu等[23]方法, 所有PCR产物在1.5%的高强度琼脂糖凝胶中分离, 120 V恒压下, 电泳1.5 h, 染色、照相及保存。1.5 统计分析
参照梅嘉洺等[33] SSR电泳图谱统计分析方法, 针对每一对SSR引物, 首先在12个甘蔗属材料上PCR产物的电泳图谱整体分析比较, 鉴定出所有变异类型, 依次被命名为a、b、c、d、e、f等。然后针对每个变异类型(可被看作1个分子标记)统计其在12个材料中的分布情况, 属于该变异类型的记为1, 不属于该变异类型的记为0, 根据统计的结果建立原始数据(0, 1)矩阵。利用NTSYS-pc 2.1软件中的子程序SIMQUAL对矩阵进行样本间的相似性系数(SM)计算, 然后用子程序SAHN中的非加权类平均法(UPGMA)进行聚类分析, 最后用Tree plot绘制树状聚类图。2 结果与分析
2.1 SSR位点的数量、类型及频率分布
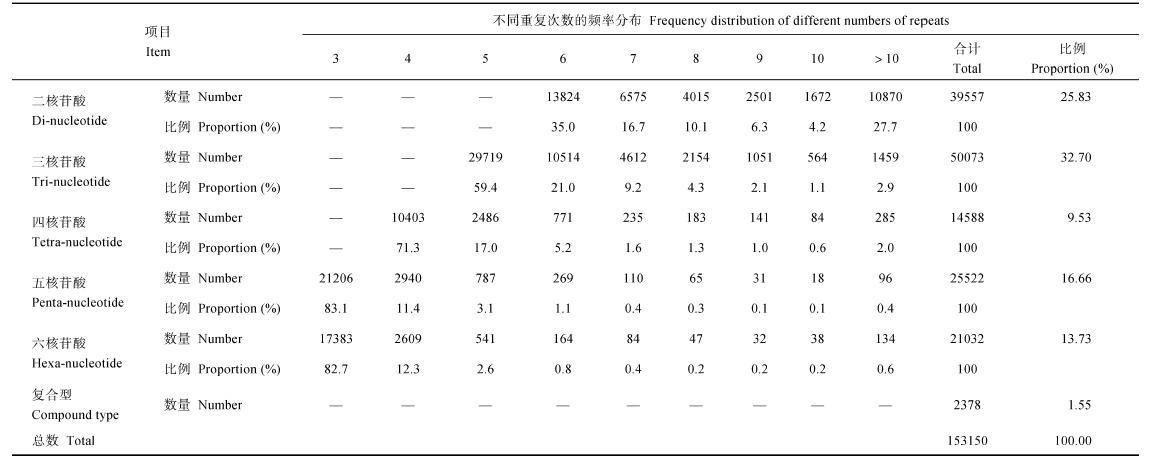
通过MISA软件扫描热带种LA-purple的255 398个基因序列(累计总长为1 029 222 285 bp)来查找二、三、四、五、六碱基5种基序类型的SSR, 共找到153 150个SSR位点, 其中三核苷酸重复基序类型出现的频率最高, 共找出50 072个位点, 占总数的32.70%; 二核苷酸重复基序类型次之, 共找出39 556个位点, 占总数的25.83%, 两者合计占总数的58.53%。而四、五、六核苷酸类型及复合型所占的比例相对较低, 分别为9.53%、16.66%、13.73%和1.55%, 合计占总数的41.47%。平均每1.67个基因或6720 bp含有1个SSR位点(表2)。Table 2
Table 2Frequency distribtiom of various types of simple sequence repent(SSR)motifs with different numbers of repeats among the genes of S.officinarum LA-purple genome
 |
新窗口打开|下载CSV
针对每个SSR核苷酸重复基序类型, 重复次数越少, 出现的频率就越高。在二核苷酸重复基序类型中, 35.0%的SSR位点含有6个重复, 72.3%的SSR位点的重复次数集中在6~10之间; 在三核苷酸重复基序类型中, 59.4%的SSR位点含有5个重复, 97.1%的SSR位点的重复次数集中在5~10之间; 在四核苷酸重复基序类型中, 71.3%的SSR位点含有4个重复, 99.8%的SSR位点的重复次数集中在4~10之间; 在五核苷酸重复基序类型中, 83.1%的SSR位点含有3个重复, 99.6%的SSR位点的重复次数集中在3~10之间; 在六核苷酸重复基序类型中, 82.7%的SSR位点含有3个重复, 99.4%的SSR位点的重复次数集中在3~10之间(表2)。
在二核苷酸重复基序类型中, TA/AT所占的比例最高, 为41.3%; 而CT/GA、TC/AG、TG/AG、GT/CA和CG/GC所占比例相对较低, 分别为17.9%、14.5%、11.5%、10.2%和4.6% (图1)。在三核苷酸重复基序类型中, TGT/ACA所占比例最高, 为15.6%; CGC/GCG、GGC/CCG和GCC/CGG所占比例次之, 分别为10.8%、9.3%和9.2%; 而其余类型单个所占比例分布在0.5%~4.0%之间(图2)。由于四、五、六核苷酸重复基序类型呈现指数上升(分别为252、1020和4092), 因而使其不同类型基序所出现的频率远远低于二、三核苷酸重复基序。
图1
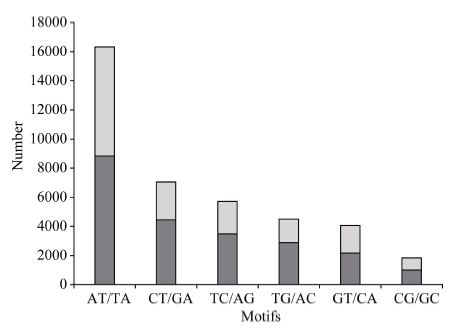 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1二核苷酸重复基序的次数分布
Fig. 1Frequency distribution of dinucleotide repeat motifs
图2
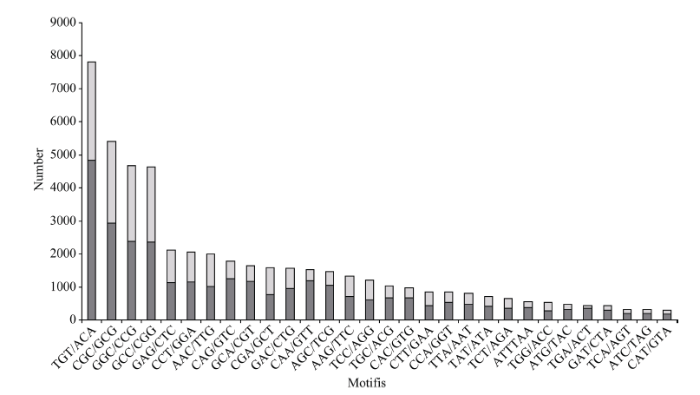 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2三核苷酸重复基序的次数分布
Fig. 2Frequency distribution of trinucleotide repeat motifs
2.2 开发的SSR标记在甘蔗属不同种间的扩增效率和多态性验证
为了验证上述从热带种(LA-purple)全基因组基因序列上查找出的SSR位点的扩增效率和多态性, 从二核苷酸SSR位点中选择绝对数量最多的AT/TA重复基序类型开发引物, 根据Schl?tterer[34]的研究结果, SSR位点基序重复次数越多, 多态性表现越好, 鉴于此, 本研究选取基序重复次数在60~90范围内的100个SSR位点开发引物, 对其进行了引物设计和合成, 然后利用合成的100对引物对12个甘蔗属遗传材料的DNA进行PCR扩增和多态性分析, 结果显示, 共有84对引物能够给出清晰的PCR扩增条带, 而其余16对引物没有给出条带或者给出的条带不够清晰, 有52对引物在12个实验材料上呈现多态性, 每对引物给出2~7种扩增类型, 共给出159个扩增类型(平均每一对引物给出3.1种扩增类型), 其中有27对引物在2个甘蔗栽培品种(科5和桂糖35)之间给出多态性扩增(表3)。图3仅展示了其中4对不同SSR引物在12个供试甘蔗属材料上的代表性PCR分析图谱, 其中除引物SoLaSSR096外, 其他3对引物均给出清晰的PCR扩增条带, 并且SoLaSSR007和SoLaSSR070在12个试验材料上呈现多态性。图3
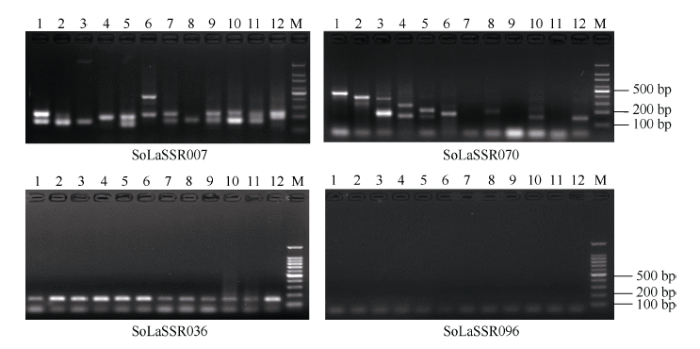 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图34对不同SSR引物在12个供试甘蔗属材料上的代表性PCR扩增图谱
1: 57NG208; 2: NG77-004; 3: 云南75-2-11; 4: 福建89-1-1; 5: 广东21; 6: 贵州78-2-28; 7: 黑车里本; 8: 路达士; 9: 克里斯塔林娜; 10: 拔地拉; 11: 桂糖35; 12: 科5; M: 100 bp DNA ladder。
Fig. 3Representative PCR amplification patterns of four pairs of SSR primers of AT/TA motif in 12 tested Saccharum clones
1: 57NG208; 2: NG77-004; 3: YN75-2-11; 4: FJ89-1-1; 5: GD21; 6: GZ78-2-28; 7: Black cheribon; 8: Loethers; 9: Crystalina; 10: Badila; 11: GT35; 12: K5; M: 100 bp DNA ladder.
Table 3
Table 3SSR primers information of sugarcane with amplified polymorphism
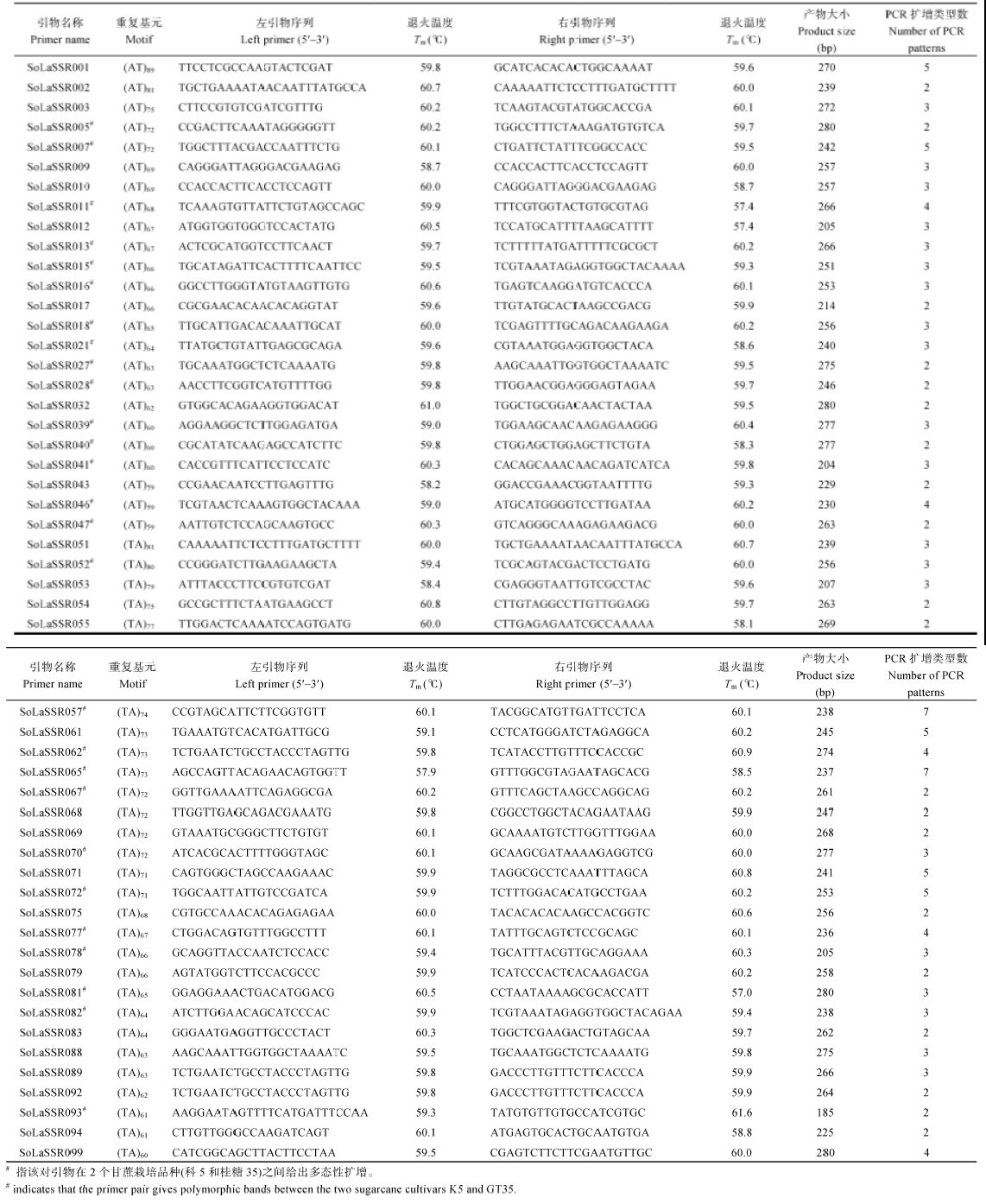 |
新窗口打开|下载CSV
基于上述52对SSR引物所给出的159种PCR扩增类型, 对12个甘蔗属材料进行了UPGMA聚类分析, 从图4可以看出, 供试材料之间的遗传相似系数分布在0.64~0.86之间, 其中热带种材料之间的相似性系数为0.64~0.77, 2个大茎野生种材料之间的相似性系数为0.66, 割手密材料之间的相似性系数为0.77~0.87, 2个甘蔗栽培品种(科5和桂糖35)之间的遗传相似系数为0.715。整体来看, 12个试验材料没有明显按照种类分组, 但4个割手密材料(云南75-2-11、福建89-1-1、广东21和贵78-2-28)被聚合到同一个小组, 而2个栽培品种与3个热带种(路达士、克里斯塔林娜和拔地拉)聚在一个大组, 表明栽培品种与热带种血缘关系较近。
图4
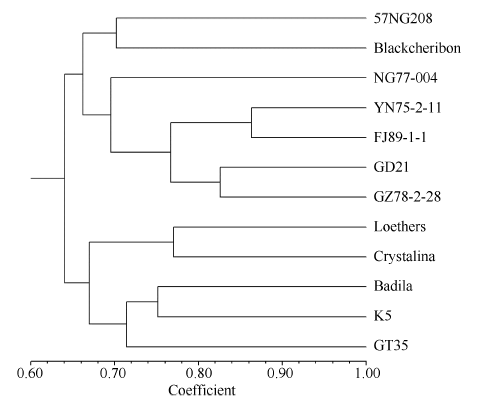 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4基于SSR分子标记的12个甘蔗属材料的UPGMA聚类分析
Fig. 4UPGMA dendrogram of 12 Saccharum clones based on SSR markers
3 讨论
随着测序技术的快速发展, 越来越多植物基因组序列被测序并公布于众, 这给全基因组水平上查找SSR位点、大规模开发SSR标记创造了极好的条件[35]。到目前为止, 基于基因组测序数据来查找、开发SSR标记的报道越来越多[36,37,38,39,40,41,42,43,44,45,46,47,48,49,50], 比如, 拟南芥基因组上平均每1.14 kb就有1个SSR位点[36], 水稻基因组上平均每3.6 kb就有1个SSR位点[37], 甘蓝基因组上平均每4 kb就有1个SSR位点[39], 大豆基因组上平均每4.5 kb就有1个SSR位点[41]等。本研究从甘蔗热带种(LA-purple)全基因组的255 398个预测基因序列(累计总长为1 029 222 285 bp)中共找到153 150个SSR位点, 平均每1.67个基因或6720 bp含有1个SSR位点。由于甘蔗热带种(LA-purple)的全基因组测序工作还没有结束, 本研究只用到其部分基因序列, 因此该结果还不能与上述其他植物上的SSR数据直接比较, 但足以证明SSR在甘蔗热带种(LA-purple)基因组上的广泛分布。在SSR基序的类型方面, 本研究结果显示甘蔗热带种全基因组基因上三核苷酸重复最多(32.70%), 二核苷酸重复次之(25.83%)。这可能是因为本研究找出的SSR位点位于基因内部, 富集了基因上可翻译成蛋白质序列的三核苷酸重复序列。这个结果和基于表达序列标签(EST)中开发SSR (EST-SSR)的结果相一致[35,37,42], 虽然大多数基因组序列上查找SSR位点的结果都显示基因组上均以二核苷酸重复的SSR位点最多, 而三核苷酸重复的SSR位点次之[36,46-47]。
在SSR基序的结构方面, 本研究结果显示甘蔗热带种基因组基因序列中的二核苷酸重复基序SSR类型以AT/TA基序结构出现次数最高, 该结果与烟草[47]、玉米[44]、大豆[38]、可可[45]、高粱[42]的研究结果一致, 但是与水稻、二穗短柄草等AG/TC结构出现次数最高的结果不一致[42]; 三核苷酸重复基序类型以TGT/ACA出现次数最多, 其次是CCG/CGG类型, 这与拟南芥[36]、玉米[44]、水稻[36,42]、高粱[42]、二穗短柄草[42]相类似。产生这种结果很可能是和碱基对所含氢键有关, 即改变GC键需要的能量要高于AT键所需, 因此, DNA复制时滑移产生的SSR重复序列, 需要较少能量的AT结构类型就比较容易产生[47]。
在查出的SSR位点的遗传变异水平方面, 本研究在数量最多的AT/TA重复基序类型中, 选取了100个基序重复次数最高(60~90)的SSR位点, 分析它们在12个甘蔗属材料上的PCR扩增及多态性情况。结果显示, 共有84对引物给出期望的PCR扩增条带, 其中有52对引物在12个实验材料上呈现多态性, 每对给出2~7种扩增类型(平均每一对引物给出3.1种扩增类型); 有27对引物在2个甘蔗栽培品种(科5和桂糖35)之间给出多态性扩增, 这27对引物可以直接用来分析已经制成的“科5×桂糖35”杂交后代分离群体, 参与甘蔗遗传连锁图谱的构建。这个扩增结果及多态性引物的比例可望在今后使用优化的PCR扩增条件及改良的电泳条件(如聚丙烯酰胺电泳技术)而得到进一步的改进和提高, 特别在对本研究找出的SSR位点进行大规模的PCR扩增测试和SSR标记开发时, 需要结合优化的实验条件以便获得最好的效果。
本研究鉴定出的SSR位点全部来自甘蔗热带种基因组基因序列, 这将有利于在甘蔗上寻找与性状显著关联的功能SSR标记。但是由于基因在进化过程中的保守性, 这些基于基因序列的SSR位点(特别是那些位于外显子区域)的多态性会低于那些来自基因组上非编码区的SSR。此外, 热带种基因组为同源多倍体, 现代甘蔗品种80%~90%的染色体都来自热带种, 这些特征都会降低SSR位点的多态性以及PCR扩增质量。因此, 未来在热带种全基因组序列被公布之后, 需要进一步开发非编码区的SSR位点, 同时也要开发来自于甘蔗属内其他种(特别是割手密)的SSR标记, 以便在甘蔗上开发大量的高质量多态性SSR标记。这些新开发的以及前人[13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]已经开发的SSR标记, 需要在改良的实验条件下统一分析、鉴定和去重复, 从而筛选出一批扩增效果好、扩增谱简单、数量充足的多态性SSR标记, 可以被广泛应用在甘蔗及其近缘种的种质资源鉴定、遗传多样性分析、遗传连锁图谱构建及QTL定位等方面的研究, 为甘蔗的品种鉴定、种质资源的管理和利用、重要农艺性状的遗传研究及分子辅助育种等提供良好的条件。
4 结论
甘蔗热带种(LA-purple)全基因组预测基因序列对开发高质量、多态性SSR标记具有巨大的潜力, 为甘蔗及其近缘种的品系鉴定、遗传多样性分析、遗传图谱构建及重要性状的遗传机制解析等提供了重要的分子标记库支撑。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 3]
[本文引用: 2]
DOI:10.1038/nrg1348URLPMID:15153996 [本文引用: 1]

Few genetic markers, if any, have found such widespread use as microsatellites, or simple/short tandem repeats. Features such as hypervariability and ubiquitous occurrence explain their usefulness, but these features also pose several questions. For example, why are microsatellites so abundant, why are they so polymorphic and by what mechanism do they mutate? Most importantly, what governs the intricate balance between the frequent genesis and expansion of simple repetitive arrays, and the fact that microsatellite repeats rarely reach appreciable lengths? In other words, how do microsatellites evolve?
DOI:10.1016/j.tibtech.2007.07.013URLPMID:17945369 [本文引用: 1]
During recent decades, microsatellites have become the most popular source of genetic markers. More recently, the availability of enormous sequence data for a large number of eukaryotic genomes has accelerated research aimed at understanding the origin and functions of microsatellites and searching for new applications. This review presents recent developments of in silico mining of microsatellites to reveal various facets of the distribution and dynamics of microsatellites in eukaryotic genomes. Two aspects of microsatellite search strategies – using a suitable search tool and accessing a relevant microsatellite database – have been explored. Judicious microsatellite mining not only helps in addressing biological questions but also facilitates better exploitation of microsatellites for diverse applications.
[本文引用: 1]
DOI:10.1063/1.363554URLPMID:11799393 [本文引用: 1]
Microsatellites are a ubiquitous class of simple repetitive DNA sequence. An excess of such repetitive tracts has been described in all eukaryotes analyzed and is thought to result from the mutational effects of replication slippage. Large-scale genomic and EST sequencing provides the opportunity to evaluate the abundance and relative distribution of microsatellites between transcribed and nontranscribed regions and the relationship of these features to haploid genome size. Although this has been studied in microbial and animal genomes, information in plants is limited. We assessed microsatellite frequency in plant species with a 50-fold range in genome size that is mostly attributable to the recent amplification of repetitive DNA. Among species, the overall frequency of microsatellites was inversely related to genome size and to the proportion of repetitive DNA but remained constant in the transcribed portion of the genome. This indicates that most microsatellites reside in regions pre-dating the recent genome expansion in many plants. The microsatellite frequency was higher in transcribed regions, especially in the untranslated portions, than in genomic DNA. Contrary to previous reports suggesting a preferential mechanism for the origin of microsatellites from repetitive DNA in both animals and plants, our findings show a significant association with the low-copy fraction of plant genomes.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
DOI:10.1139/g08-105URLPMID:19234567 [本文引用: 3]
Expressed sequence tags (ESTs) offer the opportunity to exploit single, low-copy, conserved sequence motifs for the development of simple sequence repeats (SSRs). The authors have examined the Sugarcane Expressed Sequence Tag database for the presence of SSRs. To test the utility of EST-derived SSR markers, a total of 342 EST-SSRs, which represent a subset of over 2005 SSR-containing sequences that were located in the sugarcane EST database, could be designed from the nonredundant SSR-positive ESTs for possible use as potential genic markers. These EST-SSR markers were used to screen 18 sugarcane (Saccharum spp.) varieties. A high proportion (65.5%) of the above EST-SSRs, which gave amplified fragments of foreseen size, detected polymorphism. The number of alleles ranged from 2 to 24 with an average of 7.55 alleles per locus, while polymorphism information content values ranged from 0.16 to 0.94, with an average of 0.73. The ability of each set of EST-SSR markers to discriminate between varieties was generally higher than the polymorphism information content analysis. When tested for functionality, 82.1% of these 224 EST-SSRs were found to be functional, showing homology to known genes. As the EST-SSRs are within the expressed portion of the genome, they are likely to be associated to a particular gene of interest, improving their utility for genetic mapping; identification of quantitative trait loci, and comparative genomics studies of sugarcane. The development of new EST-SSR markers will have important implications for the genetic analysis and exploitation of the genetic resources of sugarcane and related species and will provide a more direct estimate of functional diversity.
DOI:10.1007/s00122-008-0902-4URLPMID:18946655 [本文引用: 2]
Genomic microsatellite markers are capable of revealing high degree of polymorphism. Sugarcane ( Saccharum sp.), having a complex polyploid genome requires more number of such informative markers for various applications in genetics and breeding. With the objective of generating a large set of microsatellite markers designated as Sugarcane Enriched Genomic MicroSatellite (SEGMS), 6,318 clones from genomic libraries of two hybrid sugarcane cultivars enriched with 18 different microsatellite repeat-motifs were sequenced to generate 4.16 Mb high-quality sequences. Microsatellites were identified in 1,261 of the 5,742 non-redundant clones that accounted for 22% enrichment of the libraries. Retro-transposon association was observed for 23.1% of the identified microsatellites. The utility of the microsatellite containing genomic sequences were demonstrated by higher primer designing potential (90%) and PCR amplification efficiency (87.4%). A total of 1,315 markers including 567 class I microsatellite markers were designed and placed in the public domain for unrestricted use. The level of polymorphism detected by these markers among sugarcane species, genera, and varieties was 88.6%, while cross-transferability rate was 93.2% within Saccharum complex and 25% to cereals. Cloning and sequencing of size variant amplicons revealed that the variation in the number of repeat-units was the main source of SEGMS fragment length polymorphism. High level of polymorphism and wide range of genetic diversity (0.16-0.82 with an average of 0.44) assayed with the SEGMS markers suggested their usefulness in various genotyping applications in sugarcane.
[本文引用: 3]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
DOI:10.1007/s12355-012-0168-7URL [本文引用: 2]
AbstractMolecular diversity within and between Saccharum species clones and elite commercial hybrid varieties was studied using intersimple sequence repeat (ISSR) and simple sequence repeat (SSR) markers. The present study was performed to characterize 81 sugarcane genotypes. A total of 13 ISSR primers used and produced 65 amplified fragments, of which 63 (96.502%) were polymorphic. The Polymorphic Information Content (PIC) value ranged from 0.11 (UBC824) to 0.45 (UBC825) primers with an average value of 0.28. The primer UBC 817 and UBC 825 exhibited highest resolving power (Rp) value 3.8 among thirteen primers. Genetic similarity (GS) by Jaccard’s similarity coefficient ranged from 0.23 to 0.95 with a mean of 0.59. Of the 79 alleles amplified by 28 SSRs primers showed 76 alleles, which were found to be polymorphic (96.202%). The PIC value ranged from 0.06 (VSICRAD4) to 0.55 (VSICRAD26) primers with an average value of 0.17. The primer VSICRAD23 exhibited highest resolving power (Rp) value 4.3 among 28 primers. The GS by Jaccard’s similarity coefficient ranged from 0.11 to 0.91 with a mean of 0.51. Dendrograms constructed using the UPGMA cluster analysis revealed low level of correlation between genetic similarities based on pedigree and DNA profiles. The ISSR and SSR amplification proved to be valuable method for assessing genetic diversity among sugarcane complex and their related wild varieties and for identification of the cultivars.
[本文引用: 2]
[本文引用: 2]
[本文引用: 4]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
DOI:10.1186/1471-2164-11-261URLPMID:2882929 [本文引用: 1]
pAbstract/p pBackground/p pSugarcane (itSaccharum /itspp.) has become an increasingly important crop for its leading role in biofuel production. The high sugar content species itS. officinarum /itis an octoploid without known diploid or tetraploid progenitors. Commercial sugarcane cultivars are hybrids between itS. officinarum /itand wild species itS. spontaneum /itwith ploidy at ~12. The complex autopolyploid sugarcane genome has not been characterized at the DNA sequence level./p pResults/p pThe microsynteny between sugarcane and sorghum was assessed by comparing 454 pyrosequences of 20 sugarcane bacterial artificial chromosomes (BACs) with sorghum sequences. These 20 BACs were selected by hybridization of 1961 single copy sorghum overgo probes to the sugarcane BAC library with one sugarcane BAC corresponding to each of the 20 sorghum chromosome arms. The genic regions of the sugarcane BACs shared an average of 95.2% sequence identity with sorghum, and the sorghum genome was used as a template to order sequence contigs covering 78.2% of the 20 BAC sequences. About 53.1% of the sugarcane BAC sequences are aligned with sorghum sequence. The unaligned regions contain non-coding and repetitive sequences. Within the aligned sequences, 209 genes were annotated in sugarcane and 202 in sorghum. Seventeen genes appeared to be sugarcane-specific and all validated by sugarcane ESTs, while 12 appeared sorghum-specific but only one validated by sorghum ESTs. Twelve of the 17 sugarcane-specific genes have no match in the non-redundant protein database in GenBank, perhaps encoding proteins for sugarcane-specific processes. The sorghum orthologous regions appeared to have expanded relative to sugarcane, mostly by the increase of retrotransposons./p pConclusions/p pThe sugarcane and sorghum genomes are mostly collinear in the genic regions, and the sorghum genome can be used as a template for assembling much of the genic DNA of the autopolyploid sugarcane genome. The comparable gene density between sugarcane BACs and corresponding sorghum sequences defied the notion that polyploidy species might have faster pace of gene loss due to the redundancy of multiple alleles at each locus./p
DOI:10.1093/nar/8.19.4321URLPMID:324241 [本文引用: 1]
Abstract A method is presented for the rapid isolation of high molecular weight plant DNA (50,000 base pairs or more in length) which is free of contaminants which interfere with complete digestion by restriction endonucleases. The procedure yields total cellular DNA (i.e. nuclear, chloroplast, and mitochondrial DNA). The technique is ideal for the rapid isolation of small amounts of DNA from many different species and is also useful for large scale isolations.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s004120000120URLPMID:11072791 [本文引用: 1]
Within the past decade microsatellites have developed into one of the most popular genetic markers. Despite the widespread use of microsatellite analysis, an integral picture of the mutational dynamics of microsatellite DNA is just beginning to emerge. Here, I review both generally agreed and controversial results about the mutational dynamics of microsatellite DNA.
DOI:10.1186/1471-2105-9-374URLPMID:2562394 [本文引用: 2]
pAbstract/p pBackground/p pSimple Sequence Repeat (SSR) or microsatellite markers are valuable for genetic research. Experimental methods to develop SSR markers are laborious, time consuming and expensive. itIn silico /itapproaches have become a practicable and relatively inexpensive alternative during the last decade, although testing putative SSR markers still is time consuming and expensive. In many species only a relatively small percentage of SSR markers turn out to be polymorphic. This is particularly true for markers derived from expressed sequence tags (ESTs). In EST databases a large redundancy of sequences is present, which may contain information on length-polymorphisms in the SSR they contain, and whether they have been derived from heterozygotes or from different genotypes. Up to now, although a number of programs have been developed to identify SSRs in EST sequences, no software can detect putatively polymorphic SSRs./p pResults/p pWe have developed PolySSR, a new pipeline to identify polymorphic SSRs rather than just SSRs. Sequence information is obtained from public EST databases derived from heterozygous individuals and/or at least two different genotypes. The pipeline includes PCR-primer design for the putatively polymorphic SSR markers, taking into account Single Nucleotide Polymorphisms (SNPs) in the flanking regions, thereby improving the success rate of the potential markers. A large number of polymorphic SSRs were identified using publicly available EST sequences of potato, tomato, rice, itArabidopsis/it, itBrassica /itand chicken./p pThe SSRs obtained were divided into long and short based on the number of times the motif was repeated. Surprisingly, the frequency of polymorphic SSRs was much higher in the short SSRs./p pConclusion/p pPolySSR is a very effective tool to identify polymorphic SSRs. Using PolySSR, several hundred putative markers were developed and stored in a searchable database. Validation experiments showed that almost all markers that were indicated as putatively polymorphic by polySSR were indeed polymorphic. This greatly improves the efficiency of marker development, especially in species where there are low levels of polymorphism, like tomato. When combined with the new sequencing technologies PolySSR will have a big impact on the development of polymorphic SSRs in any species./p pPolySSR and the polymorphic SSR marker database are available from urlhttp://www.bioinformatics.nl/tools/polyssr//url./p
[本文引用: 5]
DOI:10.1093/dnares/dsm005URLPMID:2779893 [本文引用: 3]
Microsatellite (MS) polymorphism is an important source of genetic diversity, providing support for map-based cloning and molecular breeding. We have developed a new database that contains 52 845 polymorphic MS loci between indica and japonica, composed of ample Class II MS markers, and integrated 18 828 MS loci from IRGSP and genetic markers from RGP. Based on genetic marker positions on the rice genome (http://rise.genomics.org.cn/rice2/index.jsp ), we determined the approximate genetic distances of these MS loci and validated 100 randomly selected markers experimentally with 90% success rate. In addition, we recorded polymorphic MS positions in indica cv. 9311 that is the most important paternal parent of the two-line hybrid rice in China. Our database will undoubtedly facilitate the application of MS markers in genetic researches and marker-assisted breeding. The data set is freely available from www.wigs.zju.edu.cn/achievment/polySSR.<br>
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 2]
[本文引用: 7]
[本文引用: 7]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
[本文引用: 4]
[本文引用: 4]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}